

ABSTRACT
Many viruses utilize viral or cellular chromatin machinery for efficient infection. Baculoviruses encode a conserved protamine-like protein, P6.9. This protein plays essential roles in various viral physiological processes during infection. However, the mechanism by which P6.9 regulates transcription remains unknown. In this study, 7 phosphorylated species of P6.9 were resolved in Sf9 cells infected with the baculovirus type species Autographa californica multiple nucleopolyhedrovirus (AcMNPV). Mass spectrometry identified 22 phosphorylation and 10 methylation sites but no acetylation sites in P6.9. Immunofluorescence demonstrated that the P6.9 and virus-encoded serine/threonine kinase PK1 exhibited similar distribution patterns in infected cells, and coimmunoprecipitation confirmed the interaction between them. Upon pk1 deletion, nucleocapsid assembly and polyhedron formation were interrupted and the transcription of viral very late genes was downregulated. Interestingly, we found that the 3 most phosphorylated P6.9 species vanished from Sf9 cells transfected with the pk1 deletion mutant, suggesting that PK1 is involved in the hyperphosphorylation of P6.9. Mass spectrometry suggested that the phosphorylation of the 7 Ser/Thr and 5 Arg residues in P6.9 was PK1 dependent. Replacement of the 7 Ser/Thr residues with Ala resulted in a P6.9 phosphorylation pattern similar to that of the pk1 deletion mutant. Importantly, the decreases in the transcription level of viral very late genes and viral infectivity were consistent. Our findings reveal that P6.9 hyperphosphorylation is a precondition for the maximal hyperexpression of baculovirus very late genes and provide the first experimental insights into the function of the baculovirus protamine-like protein and the related protein kinase in epigenetics.
IMPORTANCE Diverse posttranslational modifications (PTMs) of histones constitute a code that creates binding platforms that recruit transcription factors to regulate gene expression. Many viruses also utilize host- or virus-induced chromatin machinery to promote efficient infections. Baculoviruses encode a protamine-like protein, P6.9, which is required for a variety of processes in the infection cycle. Currently, P6.9's PTM sites and its regulating factors remain unknown. Here, we found that P6.9 could be categorized as unphosphorylated, hypophosphorylated, and hyperphosphorylated species and that a virus-encoded serine/threonine kinase, PK1, was essential for P6.9 hyperphosphorylation. Abundant PTM sites on P6.9 were identified, among which 7 Ser/Thr phosphorylated sites were PK1 dependent. Mutation of these Ser/Thr sites reduced very late viral gene transcription and viral infectivity, indicating that the PK1-mediated P6.9 hyperphosphorylation contributes to viral proliferation. These data suggest that a code exists in the sophisticated PTM of viral protamine-like proteins and participates in viral gene transcription.
INTRODUCTION
In eukaryotic cells, genomic DNA is packaged into nucleosomes, the structural unit of chromatin, by wrapping the DNA around histone octamers (1). A diverse array of posttranslational modifications (PTMs) takes place on the tail domains of histones, including acetylation, methylation, phosphorylation, ubiquitination, and ADP-ribosylation. Because the tail domains protrude from the surface of the chromatin and provide binding sites for other protein factors, the presence of single or combinations of PTMs constitutes a code that enables histones to serve as highly selective binding platforms for the association of specific transcription regulatory proteins that determine the transcriptional state of genes (2). Protamines, which may have evolved from H1-like histones (3), are a diverse family of small arginine-rich proteins that are synthesized in late-stage spermatids of many animals and plants. Because of their high Arg content, positively charged protamines can replace histones and form stable complexes with negatively charged DNA to condense the spermatid genome into a genetically inactive state during spermatogenesis (3). Protamines also carry various PTMs that may be involved in extensive cellular, epigenetic, and chromatin changes (4).
Many viral pathogens, such as simian virus 40, adenovirus, and white spot syndrome virus, utilize these basic DNA-binding proteins (histones or protamines) to maintain their genomes in viral chromatin and/or package their genomes into nucleocapsids (5–7). The PTM changes in the histone may be involved in the process of viral infection (8). Baculoviruses are a group of large DNA viruses that specifically infect insects and play a major role in the control of natural populations of holometabolic insects, including the orders Lepidoptera, Hymenoptera, and Diptera (9). Autographa californica multiple nucleopolyhedrovirus (AcMNPV), the type species of baculovirus, encodes a protamine-like protein named P6.9 (10). The P6.9 orthologs appear to be conserved in all baculovirus genomes, suggesting a pivotal role for P6.9 in the virus life cycle. Like other protamines, P6.9 has two structural features. The first is a series of small DNA-anchoring domains containing three or more Arg or Lys residues per domain; these positively charged residues can neutralize and bind the phosphodiester backbone of DNA. The second is the presence of multiple Ser and Thr residues (10 Ser and 7 Thr residues in the 55-amino-acid AcMNPV P6.9) that are thought to function as phosphorylation sites (11); phosphorylation can weaken the P6.9-DNA interaction by neutralizing part of the positive charges of P6.9 (12). Consistent with its structural features, P6.9 can be phosphorylated, and its phosphorylation cycle is important for the condensation of the viral genome into preassembled capsids and the release of viral DNA from the nucleocapsids (13).
In addition to viral DNA packaging and release, P6.9 is associated with chromatin dynamics and gene transcription (14, 15). Similar to the function of protamine during sperm development in humans, viral DNA first utilizes host histones or nucleosomes to form a chromatin-like structure during the early infection stages, and then, in the late infection stages, P6.9 replaces the histone and becomes a component of an electron-dense, chromatin-like structure named the virogenic stroma (VS) (14). The VS is the site for AcMNPV gene transcription, DNA replication, DNA packaging, and nucleocapsid assembly (16). Interestingly, in contrast to the function of protamines as a general transcriptional suppressor through the condensation of DNA, P6.9 promotes viral gene transcription during the late stage of AcMNPV infection (15).
The gene expression of baculoviruses is regulated mainly at the transcriptional level, resulting in the sequential expression of the immediate-early, early, late, and very late genes (17). The immediate-early and early viral genes are transcribed by the host RNA polymerase II, whereas the late and very late genes are transcribed by a virus-encoded RNA polymerase (18). During the very late stage of AcMNPV infection, the very late genes (18 to 76 h postinfection [p.i.]), polyhedrin (polh) and p10, are hyperexpressed; at 48 h p.i., the transcripts compose approximately 24% and 7.5% of the total cellular RNA, respectively (19). The very late proteins are associated with the occlusion of the virions. During the very late stage, newly assembled nucleocapsids are retained in the nuclei and are enveloped to form occlusion-derived virions (ODVs). Subsequently, ODVs are occluded by the major matrix protein, polyhedrin (POLH), to form occlusion bodies (OBs). P10 forms fibrillar structures and appears to be required for the proper assembly of the polyhedron envelope (20). The embedded ODVs are highly stable under most normal environmental conditions and can remain infectious for several years (21). This allows the use of baculoviruses as viral biopesticides for the control of agricultural and forest pests (22). Additionally, baculoviruses have been exploited as expression vectors since 1983 due to their characteristic of very late gene hyperexpression (23), and many thousands of recombinant proteins have been produced successfully using this mainstream technology (24). However, the molecular mechanism of very late gene hyperexpression in baculoviruses has not been completely elucidated.
Protein kinase 1 (PK1) is a virus-encoded serine/threonine kinase. P6.9 is predicted to be a substrate of PK1, because PK1 can phosphorylate histones and protamine in vitro (25–27). PK1 is involved in the transcription of the baculovirus very late genes (28). Deleting pk1 results in the failure of nucleocapsid assembly and occlusion body formation (27). However, the interrelationship between PK1 and the phosphorylation of P6.9 has not been elucidated.
In this paper, we identified 7 phosphorylated species and 1 unphosphorylated species of P6.9 in AcMNPV-infected cells using acetic acid-urea polyacrylamide gel electrophoresis (AU-PAGE). Using mass spectrometry, we found that P6.9 has at least 13 Ser/Thr phosphorylation sites, 9 Arg phosphorylation sites, and 10 methylation sites and that each P6.9 species consists of several isoforms. By analyzing a constructed pk1 deletion mutant, we found that PK1 is involved in the hyperphosphorylation of P6.9 that is responsible for the 3 most phosphorylated P6.9 species in AcMNPV-infected cells. The phosphorylation of 7 Ser/Thr residues in P6.9 is dependent on PK1. Moreover, replacement of the 7 Ser/Thr residues with Ala residues dramatically reduces the transcription of the very late viral genes and viral infectivity. This study is the first to show that the protamine-like protein encoded by the viruses is subjected to PTMs and that sophisticated PTMs can be regulated by a viral kinase.
MATERIALS AND METHODS
Cells and viruses.
The Sf9 insect cell line, clonal isolate 9 from IPLB-Sf21-AE cells that were derived from the fall armyworm Spodoptera frugiperda (29), was cultured at 27°C in TNM-FH medium supplemented with 10% fetal bovine serum, 100 μg/ml penicillin, and 30 μg/ml streptomycin. The pseudo-wild-type AcMNPV vAcWT was described in our previous study (30). The BV titers were determined using a 50% tissue culture infective dose (TCID50) endpoint dilution assay in Sf9 cells (31).
Construction of viruses and plasmids.
The bacmid bMON14272 (Invitrogen), which contains the genome of AcMNPV (strain E2) (32), was used to generate a pk1-null virus by homologous recombination in Escherichia coli as previously described (30). In AcMNPV, the genes pp78/83 and pk1 overlap and are reverse transcribed, with the pp78/83 transcription start site located at nucleotide (nt) 431 of the pk1 open reading frame (ORF) (19, 33). Thus, to generate a complete pk1 knockout virus and to avoid any impact on pp78/83 expression, a 1,575-bp region containing part of the pp78/83 and pk1 coding regions of bMON14272 (nt 5,936 to 7,510) was replaced with a 1,038-bp chloramphenicol resistance (CmR) gene cassette, and then a copy of pp78/83 was inserted into the mini-attTn7 site via site-specific Tn7 transposition (34). A brief description follows.
The CmR gene cassette was PCR amplified from pUC18-Cm (30) with primer pair CmRU/CmRD (all PCR primers are listed in Table 1) and cloned into pUC18 (TaKaRa) to generate pUC18-CmR. Two homologous fragments flanking the deletion region were PCR amplified from bMON14272 using primer pairs pp78/83-U/pp78/83-D and pk1-U/pk1-D. The products were cloned successively into pUC18-CmR to generate pUC18-US-CmR-DS, and this plasmid was digested to obtain a linear fragment that contained the CmR gene flanked by the two homologous regions. The linear fragment was electrotransformed into DH10Bac (Invitrogen), resulting in the replacement of the target deletion region of bMON14272 with the CmR gene cassette via Red/ET homologous recombination as previously described (30). The resulting bacmid was named bpp78/83-pk1KO.
TABLE 1.
Primer sequences used in this study
| Primer | Sequence |
|---|---|
| CmRU | 5′-CCCGGGCCCTTTCGTCTTCGAATAAATACC-3′ |
| CmRD | 5′-TCTAGATAAACCAGCAATAGACATAAGCG-3′ |
| pp78/83-U | 5′-GAGCTCTTAAGCGCTAGATTCTGTGCGTT-3′ |
| pp78/83-D | 5′-CCCGGGGAATTAAAATCGGGCACAGTTAGA-3′ |
| pk1-U | 5′-TCTAGAAACATACAAGTTGCTAACCGGC-3′ |
| pk1-D | 5′-CTGCAGGGTTATTAAAGATTTTGGTAGACAATGT-3′ |
| R-pp78/83-U | 5′-GAGCTCCGTTCGAACAGTTCATCGATC-3′ |
| R-pp78/83-D | 5′-CTGCAGCTAAAATACTCCAACGTGCCG-3′ |
| pk1(M)Ft | 5′-TTCGTCATTGCCACCACAAATGCTACGCTGCAAACGCTGG-3′ |
| pk1(M)Fs | 5′-AATGCTACGCTGCAAACGCTGG-3′ |
| pk1(M)Rt | 5′-TCGTAGATATGAATCTGTACAATGTGGTGGCAATGACGAA-3′ |
| pk1(M)Rs | 5′-TCGTAGATATGAATCTGTACAA-3′ |
| pk1-FLAG-D | 5′-CATTTTTACTTATCGTCGTCATCCTTGTAATCCGACAAAAACTCATGTTTTATAATTTGTTTGT-3′ |
| FLAG-pk1-U | 5′-GATTACAAGGATGACGACGATAAGTAAAAATGCCACTTGTTTTACGAGTAGAATTC-3′ |
| R-pp78/83-pk1-D | 5′-CTGCAGCTGCAGCATGCACATAGTATGTCGGATAAA-3′ |
| ppk1-FLAG-U | 5′-GGATCCGCCACCATGGCCACCACAAATGCTACGCTGC-3′ |
| ppk1-FLAG-D | 5′-CTCGAGCTTATCGTCGTCATCCTTGTAATCCTTATCGTCGTCATCCTTGTAATC-3′ |
| p6.9-U | 5′-GGATCCATGGTTTATCGTCGCCGTCGC-3′ |
| p6.9-HA-D | 5′-GGCGTAATCTGGGACGTCGTATGGGTAATAGTAGCGTGTTCTGTAA-3′ |
| HA-p6.9-U | 5′-GAACACGCTACTATTACCCATACGACGTCCCAGATTACGCCTACCCATACGACGTCCCAGATTACGCC-3′ |
| p6.9-D | 5′-CTCGAGTTAGGCGTAATCTGGGACGTCGTATGGGTA-3′ |
| Pro-P6.9-U | 5′-CGGAATTCACCCGCTCAGTCGGATGT-3′ |
| Pro-P6.9-D | 5′-CGAGCTCGTTTAAATTGTGTAATTTATGTAG-3′ |
| q-polh-U | 5′-TTAGGTGCCGTTATCAAGA-3′ |
| q-polh-D | 5′-GCCACTAGGTAGTTGTCT-3′ |
| q-ie1-U | 5′-TGTGATAAACAACCCAACGA-3′ |
| q-ie1-D | 5′-GTTAACGAGTTGACGCTTGC-3′ |
| q-vp39-U | 5′-TTGCGCAACGACTTTATACC-3′ |
| q-vp39-D | 5′-TAGACGGCTATTCCTCCACC-3′ |
| q-lef6-U | 5′-TAGATTCTCCTCGCTTGG-3′ |
| q-lef6-D | 5′- ATTTCGCCATCTTCTAAATT-3′ |
| q-18s-U | 5′-TACCGATTGAATGATTTAGTGAGG-3′ |
| q-18s-D | 5′-TACGGAAACCTTGTTACGACTTT-3′ |
| q-gp41-U | 5′-CGTAGTGGTAGTAATCGCCGC-3′ |
| q-gp41-D | 5′-AGTCGAGTCGCGTCGCTTT-3′ |
A 2,359-bp fragment containing the pp78/83 cassette was PCR amplified from bMON14272 using primers R-pp78/83-U and R-pp78/83-D and then was cloned into pEASY-T3 (TransGen Biotech). Because the fragment contains the partial coding region of pk1, the start codon of PK1 was mutated to ATT with primers pk1(M)Ft, pk1(M)Fs, pk1(M)Rt, and pk1(M)Rs as previously described (35) to eliminate the expression of a potentially functional PK1. The mutated pp78/83 cassette was cloned into pFB1-PH-GFP to generate pFB1-pp78/83(M)-PH-GFP. pFB1-PH-GFP was constructed by inserting a polh gene under the control of its native promoter and an enhanced green fluorescent protein (egfp) gene under the control of an AcMNPV immediate-early gene promoter into the pFastBac1 plasmid (Invitrogen) (36). Finally, the fragment pp78/83(M)-PH-GFP was inserted into the mini-attTn7 site of bpp78/83-pk1KO via site-specific transposition as previously described (30). The resulting pk1 deletion AcMNPV mutant was designated vPK1KO.
Similarly, a repair virus, vPK1:FLAG, was generated by inserting the gene cassettes of both pp78/83 and pk1 into bpp78/83-pk1KO. The fragment containing pp78/83 and pk1 was amplified from bMON14272 by overlap extension PCR with primers R-pp78/83-U, pk1-FLAG-D, FLAG-pk1-U, and R-pp78/83-pk1-D and then cloned into pFB1-PH-GFP. To facilitate PK1 detection, the pk1 ORF was tagged with a FLAG-coding sequence at its 3′ end in vPK1:FLAG.
Recombinant viruses containing mutations in the p6.9 gene was generated by introducing modified forms of p6.9 into bP6.9KO (nt 86924 to 86971 deleted) (11). The p6.9 mutants with 7 residues (S10, S17, T18, S41, S43, T45, and S49) mutated to Ala were generated by total gene synthesis technology and then cloned into plasmid pJWF (Beijing Genomics Institute). The P6.9 native promoter was PCR amplified from bMON14272 by primers Pro-P6.9-U/Pro-P6.9-D and inserted into pUC18-SV40 (37) to generate pUC-Pro-SV40. The p6.9 mutant was cloned into pUC-Pro-SV40 to generate pUC-Pro-P6.9:7M. Pro-P6.9:7M was digested from the plasmid described above and inserted into pFB1-PH-GFP to generate the final plasmid, pFB1-P6.9:7M-PG. pFB1-P6.9:7M-PG then was transformed into bP6.9KO to generate the P6.9 mutation recombinant virus vP6.9:7M. A series of P6.9 mutation viruses (vPK1:1M) were constructed as described for vP6.9:7M, in which each of the 7 PK1-dependent Ser/Thr phosphorylation sites was separately mutated to Ala.
To determine whether PK1 interacts with P6.9 in the absence of viral infection, the transient expression plasmids pIB-PK1:FLAG and pIB-P6.9:3HA were constructed as follows. The pk1 ORF with a FLAG coding sequence at its 3′ end was PCR amplified from bMON14272 with primers ppk1-FLAG-U and ppk1-FLAG-D. The p6.9 ORF with a 3× hemagglutinin (HA) coding sequence at its 3′ end was amplified from vP6.9:HA (11) by overlap extension PCR with primers p6.9-U, p6.9-HA-D, HA-p6.9-U, and p6.9-D. The two products then were cloned into the transient expression vector pIB/V5-His (Invitrogen), generating pIB-PK1:FLAG and pIB-P6.9:3HA, respectively.
All of the constructs were verified by PCR analysis and Sanger sequencing. Bacmid DNAs and plasmids were isolated with the Large-Construct kit (Qiagen) and the E.Z.N.A. plasmid minikit I (Omega), respectively, and quantified by determining the optical density.
AU-PAGE and immunoblotting.
To separate the different phosphorylated species of P6.9, AU–15% PAGE (width, 16 cm; height, 20 cm) was performed as previously described (11). After electrophoresis, the proteins were transferred onto a 0.22-μm nitrocellulose membrane (Whatman) in 0.7% glacial acetic acid transfer buffer (38). The membrane was immunoblotted with an anti-P6.9 antibody (1:1,000) (11). A donkey anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (1:5,000; Amersham Biosciences) was used as the secondary antibody.
Mass spectrometry.
P6.9 species were separated by AU–15% PAGE as described above. After Coomassie brilliant blue staining and excision from the gel, each species was digested with chymotrypsin (final concentration, 0.05 μg/μl; Nα-p-tosyl-l-lysine chloromethyl ketone treated, mass spectrometry [MS] grade; Thermo Scientific) in 100 mM Tris-HCl (pH 8.0) and 10 mM CaCl2 overnight at 37°C. The in-gel digestion procedure was performed mainly as previously described (39), although the reduction and alkylation steps were skipped. A total of 10 μl of the digested aliquots was loaded onto a precolumn and then analyzed via an analytical column (EASY-Spray column; 15 cm by 75 μm; C18; 3 μm) using nano-liquid chromatography (LC) (Thermo Scientific EASY-nLC 1000 system). The nano-LC flow rate was 300 nl/min. The gradient used was the following (solvent A, 0.1% formic acid; solvent B, 0.1% formic acid, acetonitrile): 0 min, 98% A and 2% B; 2 min, 95% A and 5% B; 32 min, 65% A and 35% B; 35 min, 2% A and 98% B; 40 min, 2% A and 98% B.
A hybrid linear ion trap-Orbitrap mass spectrometer (LTQ Orbitrap Elite; Thermo Scientific) with a Nanospray Flex ion source was coupled to the outlet of the nano-LC. The fractions eluted from the nano-LC were infused into the electrospray source. Electrospray ionization was performed at optimal conditions of 1.8 kV and 250°C source temperature. All spectra were obtained in the positive-ion mode. Full-scan MS was recorded with a resolution of 60,000 (at m/z 400), and the precursor ion scan was recorded over the m/z range of 350 to 1,800. The top 10 precursor ions from the full-scan MS were fragmented by collision-induced dissociation (CID) and electron-transfer dissociation (ETD) in turn prior to collection of the MS/MS data. A database search was performed with Proteome Discoverer 1.3 (Thermo Scientific) using the SEQUEST program (40) with the amino acid sequence of AcMNPV P6.9. The search parameters included the following: chymotrypsin enzyme specificity, allowing for two missed cleavage sites; dynamic modification of S/T/R phosphorylation (79.966 Da), S/T acetylation (42.047 Da), and R mono-/dimethylation (14.016/28.031 Da); precursor ion mass tolerance, ±10 ppm; fragment ion mass tolerance, ±1.2 Da; ETD, ±0.7 Da; and false discovery rate (FDR) of all peptide and protein identifications, 1% to 5%. To evaluate the accuracy of phosphorylation site localizations, the phosphoRS algorithm was employed (41). For each phosphorylation site, pRS probabilities above 75% indicated that the site actually was phosphorylated. Other PTM sites were confirmed by manual validation.
Time course analysis of PK1 expression.
Sf9 cells (1 × 106) were infected with vPK1:FLAG at a multiplicity of infection (MOI) of 5 TCID50/cell. At the indicated time points p.i., the cells were washed with phosphate-buffered saline (PBS; 135 mM NaCl, 4.7 mM KCl, 10 mM Na2HPO4, and 2 mM NaH2PO4, pH 7.4) and collected by centrifugation at 3,000 × g for 10 min at 4°C. Immunoblotting was performed as described elsewhere (42). A mouse monoclonal anti-FLAG antibody (1:2,000; Abmart) was used as the primary antibody to detect PK1.
Transfection.
Sf9 cells (1 × 106) were seeded in a 35-mm dish and allowed to adhere for 2 h. One microgram of bacmid DNA or 2 μg of plasmid DNA was mixed with 2 μl Cellfectin II reagent (Invitrogen) according to the manufacturer's protocol. The DNA-lipid mixture was incubated at room temperature for 1 h and then added to the cells. After 5 h, the DNA-lipid mixture was replaced with fresh medium. Time zero was defined as the time when the fresh medium was added.
Immunofluorescence.
Sf9 cells (5 × 105) were infected with vPK1:FLAG at an MOI of 5 TCID50/cell or transfected with 2 μg bacmid DNA of vPK1:FLAG or vPK1KO. At different time points p.i. or p.t., the cells were processed for confocal laser scanning microscopy as previously described (43). The mouse monoclonal anti-FLAG antibody (1:200) and the rabbit polyclonal anti-P6.9 antibody (1:200) (11) were used as the primary antibodies in combination. An Alexa Fluor 555-conjugated donkey anti-mouse antibody (1:200; Invitrogen/Molecular Probes) and an Alexa Fluor 647-conjugated goat anti-rabbit antibody (1:200; Invitrogen/Molecular Probes) were used as the secondary antibodies. Prior to observation, the cells were stained with Hoechst 33342 (Invitrogen) and washed with PBS. The labeled cells were visualized with a Zeiss 7 Duo NLO laser scanning confocal microscope (Carl Zeiss; Germany).
Immunoprecipitation.
Sf9 cells (1 × 107) were infected with vPK1:FLAG at an MOI of 5 TCID50/cell or cotransfected twice with 9 μg of pIB-PK1:FLAG and 9 μg of pIB-P6.9:3HA. At 48 h p.i./p.t., the cells were harvested, washed with PBS, and lysed in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS; Thermo Scientific) supplemented with 2 μg/ml cOmplete EDTA-free protease inhibitor cocktail (Roche). Immunoprecipitation was performed as previously described (44), and the immunocomplexes were resolved by SDS–15% PAGE. Immunoblotting was performed with the mouse monoclonal anti-FLAG antibody or the rabbit polyclonal anti-P6.9 antibody. Sf9 cells infected with vAcWT (MOI of 5 TCID50/cell) and cells cotransfected with pIB-V5/His and pIB-P6.9:3HA were used as negative controls.
Flow cytometry.
Sf9 cells were transfected with the bacmid of vPK1KO or vPK1:FLAG. At 48 h p.t., the transfected cells were washed once with PBS, harvested, and resuspended in PBS. The transfected cells were selected using flow cytometry (MoFlo XDP; Beckman Coulter, United States) based on green fluorescent protein (GFP) fluorescence. GFP fluorescence was excited at 488 nm, and emission was measured with a 530-nm/540-nm band pass filter. Mock-transfected cells served as a negative control for autofluorescence.
qPCR.
Sf9 cells (1 × 106) were transfected with 1 μg of bacmid DNA and harvested at 24 h p.t. Total DNA was isolated using a Universal Genomic DNA extraction kit (TaKaRa) and digested with 20 U of DpnI (New England BioLabs) at 37°C. Viral DNA replication was determined by real-time quantitative PCR (qPCR) as previously described (45) with primers for the gp41 gene in the AcMNPV genome. The number of viral DNA genome copies in each sample was calculated by using a standard curve generated from a dilution series of vAcWT DNA.
RNA extraction and qRT-PCR.
Sf9 cells (1 × 106) were transfected with 1 μg bacmid DNA of vP6.9:7M, vP6.9:1M, vP6.9KO, or vP6.9:HA. Total RNA isolation and cDNA synthesis were performed as previously described (37). Quantitative reverse transcription-PCR (qRT-PCR) was performed using TransStart Green qPCR SuperMix (TransGen Biotech) according to the manufacturer's protocol. Five genes (ie1, lef6 vp39, polh, and p10) were chosen to investigate the transcriptional levels. The qPCR data of viral genes were normalized to host 18S rRNA (15), and the relative expression of genes was calculated using the 2−ΔΔCT method (46).
Purification and fractionation of BVs and ODVs.
Sf9 cells were infected with vPK1:FLAG. BVs and ODVs were purified and fractionated into envelope and nucleocapsid fractions as previously described (43). Immunoblotting was performed with the mouse monoclonal anti-FLAG antibody, a monoclonal anti-GP64 AcC6 antibody (1:3,000; eBioscience), a rabbit polyclonal anti-AcMNPV ODV-E25 antibody (1:2,000) (47), or a polyclonal anti-AcMNPV VP39 antibody (1:1,000) (48). A donkey anti-rabbit HRP-conjugated antibody (1:10,000; Amersham Biosciences) or a goat anti-mouse HRP-conjugated antibody (1:5,000; Pierce) was used as the secondary antibody.
IEM.
Sf9 cells (1 × 106) were infected with vPK1:FLAG at an MOI of 5 TCID50/cell and prepared for immunoelectron microscopy (IEM) as previously described (44). Ultrathin sections were immunolabeled and stained as previously described (49). Immunolabeling was performed with the mouse monoclonal anti-FLAG antibody (1:100) followed by incubation with the secondary antibody, a goat anti-mouse IgG that was conjugated to 10-nm gold particles (1:50; Sigma-Aldrich). The samples were visualized with a JEOL JEM-1400 transmission electron microscope (JEOL; Japan) at an accelerating voltage of 120 kV.
RESULTS
Identification of the P6.9 PTM sites.
P6.9 is a small, arginine-rich, highly positively charged protein. Approximately 6 to 8 phosphorylated P6.9 species in AcMNPV-infected cells can be resolved as ladders using AU-PAGE (11, 50). AU-PAGE resolves P6.9 species based on their charge. One phosphate group decreases the positive charge of P6.9 by one; therefore, each slower-migrating rung represents an increased phosphorylation occupancy. To clarify the phosphorylation states of P6.9, we reexamined the phosphorylated species in Sf9 cells infected with an AcMNPV construct (vPK1:FLAG). The gene content of vPK1:FLAG is almost identical to that of wild-type AcMNPV; however, its PK1 was tagged with a FLAG tag at its C terminus (see details below). A ladder composed of 8 species of P6.9 was observed in vPK1:FLAG-infected cells (Fig. 1A). In contrast, only one P6.9 species was detected in budded virions (BVs). BVs are formed via budding of newly assembled nucleocapsids from the plasma membrane and are highly infectious to most insect tissues during the early infection of baculoviruses (9). The P6.9 species in BVs is unphosphorylated (50). The P6.9 species in BVs had the same mobility as the fastest P6.9 species in the vPK1:FLAG-infected cells, indicating that the P6.9 species in the vPK1:FLAG-infected cells are composed of 7 phosphorylated and 1 unphosphorylated species.
FIG 1.

PTM sites of P6.9. (A) Immunoblotting for P6.9 in vPK1:FLAG-infected cells. vPK1:FLAG-infected cells were harvested at 48 h p.i. P6.9 species were separated by AU-PAGE into 8 rungs based on their charge states and detected by immunoblotting with the anti-P6.9 antibody. (B) The P6.9 species were stained with Coomassie brilliant blue and numbered 0 for the unphosphorylated species (including the BV) and 1 to 7 for the phosphorylated species. (C) All of the identified phosphorylation and methylation sites of P6.9 are indicated in a combined schematic. The phosphorylated residues are indicated by lowercase letters; the methylated residues are indicated by capital letters. vPK1:FLAG, vPK1:FLAG-infected cells; BV, BVs purified from the supernatants of vPK1:FLAG-infected cells; S, serine; T, threonine; R, arginine.
Although P6.9 is phosphorylated in AcMNPV-infected cells (11, 50), the exact phosphorylation sites have not been determined. In the present study, the 8 P6.9 species separated by AU-PAGE (no. 0 to 7; Fig. 1B) were analyzed by mass spectrometry utilizing both CID and ETD fragmentation techniques to determine the phosphorylation sites.
Mass spectrometry provided an unambiguous assignment of the phosphorylation sites. Altogether, there were 13 phosphorylated Ser/Thr sites, among which 8 were Ser residues (S9, S10, S17, S23, S24, S41, S43, and S49) and 5 were Thr residues (T11, T13, T14, T45, and T18) (Fig. 1C; also see Table S1 in the supplemental material). As expected, no phosphorylation site was detected in the BV or no. 0 P6.9 species. By comparing the detected phosphorylation sites of the P6.9 species (no. 1 to 7 in Fig. 1C), we found that most of the phosphorylation sites were located at the P6.9 N terminus. S9, T13, T14, S23, and S24 could be phosphorylated in all 7 species. In contrast, S10 and T11 could be phosphorylated in only 6 species (the exception was no. 1), and the phosphorylation of S17 and T18 was detected only in no. 7. Only 4 residues at the C terminus (S41, S43, T45, and S49) were identified as phosphorylated. The S41 phosphorylation was identified only in no. 3 and no. 5, the S43 phosphorylation was identified in no. 1 to 6, the T45 phosphorylation was observed only in no. 2 and no. 3, and the S49 phosphorylation was detected in no. 1 to 4.
In addition to the Ser/Thr phosphorylation sites, 9 Arg sites (R6, R7, R8, R19, R21, R22, R42, R47, and R48) were phosphorylated. The phosphorylation of R7, R8, R22, and R42 was detected in no. 1 to 6, and phosphorylated R21 was detected in no. 2 to 7, excluding no. 4. However, the occurrence of R6, R19, R47, and R48 phosphorylation was relatively low. Phosphorylated R6 was detected only in no. 3 and no. 7, phosphorylated R19 in no. 7, phosphorylated R47 in no. 2 to 4, and phosphorylated R48 in no. 1 to 4 and no. 6.
In addition to phosphorylation modifications, we also searched the peptides for other PTMs that have been found in protamines, such as acetylation and methylation (4). No acetylated sites were detected in any of the P6.9 species. In contrast, 10 methylated sites were identified, including R7, R8, R19, R20, R21, R22, R28, R32, R34, and R38. R28 was monomethylated; R7, R8, R19, R20, R34, and R38 were dimethylated; and R21, R22, and R32 represented both states.
Because the methylation of arginine residues does not alter the charge of a protein (51), the different mobilities of P6.9 species in AU-PAGE should result from phosphorylation. Thus, the phosphorylation levels of P6.9 are incremental because their mobilities decrease from species no. 0 (and BV) to no. 7. Moreover, from the mass spectrometry data, the number of identified phosphorylation sites in each species is larger than expected, which indicates that there are multiple isoforms in each P6.9 species. For example, it is reasonable to infer that only one phosphorylated group is present in P6.9 species no. 1 (Fig. 1C), but 12 phosphorylation sites were identified in this species. This implies that species no. 1 consists of a mixed spectrum representing 12 different monophosphorylated isoforms. Similarly, each of the remaining P6.9 phosphorylated species also are a mixture of isoforms with the same amount of phosphate groups in different combinations. Moreover, if no phosphorylation isoforms exist, only one type of this phosphorylated peptide should have been detected. However, differentially phosphorylated peptides always were detected for the same fragment of P6.9 in each species (see Table S1 in the supplemental material). For example, for the fragment RRRRRSSTGTTY (4 to 15 amino acids [aa]) in species no. 2, 7 differentially phosphorylated peptides were detected, including RRRRrSSTGTTY, RRRrrSSTGTTY, RRRRRsSTGTTY, RRRRRSstGTTY, RRRRRssTGTTY, RRRRRSStGTTY, and RRRRRSSTGtTY (phosphorylation sites are indicated as lowercase letters). These data further support the existence of multiple phosphorylation isoforms in each P6.9 species.
PK1 interacts with P6.9.
As abundant phosphorylated Ser and Thr residues were detected in P6.9, and PK1, which is predicted to be a serine/threonine kinase, has been found to be capable of phosphorylating protamines in vitro (27), we inspected the potential association between P6.9 and PK1. Immunoblotting indicated that PK1 first emerged at 18 h p.i. (Fig. 2A); therefore, immunofluorescence was performed to characterize the distribution patterns of PK1 and P6.9 from 18 to 48 h p.i. Sf9 cells infected with vPK1:FLAG were incubated concurrently with an anti-FLAG antibody and an anti-P6.9 antibody. Immunofluorescence showed that PK1 localized at the nucleus at 18 h p.i. Most of the detected PK1 formed a ring pattern in the ring zone, a peristromal region enclosing the VS (52) (Fig. 2B). From 24 to 48 h p.i., PK1 accumulated at the ring zone, and discrete foci with the presence of PK1 also were observed in the VS. Importantly, P6.9 had a similar distribution pattern during infection. Dye overlay of the green (P6.9) and red (PK1) channels further demonstrated that P6.9 colocalized with PK1, as shown by the yellow channel in the overlay images (Fig. 2B).
FIG 2.

PK1 interacts with P6.9. (A) Time course analysis of PK1 expression. Sf9 cells were mock infected or infected with vPK1:FLAG. At the indicated time points p.i., the cells were collected, resolved by SDS–15% PAGE, and analyzed by immunoblotting with a monoclonal anti-FLAG antibody. Mi, mock-infected cells. (B) Colocalization of PK1 and P6.9. Sf9 cells were infected with vPK1:FLAG. At the indicated time points p.i., the cells were probed with the anti-FLAG and anti-P6.9 antibody and then visualized using an Alexa Fluor 555-conjugated goat anti-mouse antibody (red) and an Alexa Fluor 647-conjugated donkey anti-rabbit antibody (green). The merged lane (yellow) shows that PK1 colocalized with P6.9. Nuclear DNA was stained with Hoechst 33342 (blue). (C) Sf9 cells were infected with vPK1:FLAG or vAcWT. At 48 h p.i., the cells were collected and lysed for immunoprecipitation with protein A/G agarose beads conjugated with an anti-FLAG antibody. The immunoblot was probed with anti-FLAG antibody to detect the expression and immunoprecipitation of PK1, and anti-P6.9 antibody was used to detect the expression and coimmunoprecipitation of P6.9. vAcWT-infected cells were used as a negative control. Input, cell lysates; IP, immunoprecipitation with protein A/G agarose beads conjugated with an anti-FLAG antibody. (D) Sf9 cells were cotransfected with pIB-PK1:FLAG and pIB-P6.9:3HA. Cells cotransfected with pIB/V5-His and pIB-P6.9:3HA were used as negative controls. At 48 h p.t., immunoprecipitation assays were performed as described above.
The interaction between PK1 and P6.9 was further investigated by an immunoprecipitation assay. vPK1:FLAG-infected cells were harvested at 48 h p.i., and the cell lysates were subjected to immunoprecipitation with protein A/G agarose beads conjugated with an anti-FLAG antibody. The presence of PK1 and P6.9 in the immunocomplexes was assessed by immunoblotting with the anti-FLAG or anti-P6.9 antibody. vAcWT-infected cells were used as a negative control. As shown in Fig. 2C, PK1 and P6.9 were detected in the lysates of vPK1:FLAG-infected cells (lane Input). Both P6.9 and PK1 were detected in immunocomplexes precipitated by the anti-FLAG beads (lane IP), indicating that P6.9 could be coimmunoprecipitated with PK1. This result verifies that PK1 can interact with P6.9 in AcMNPV-infected cells.
In a transient-transfection assay, Sf9 cells were cotransfected with pIB-PK1:FLAG and pIB-P6.9:3HA. At 48 h posttransfection (p.t.), the cells were collected and subjected to immunoprecipitation. P6.9 was copurified with PK1 (Fig. 2D), confirming the interaction between PK1 and P6.9 and indicating that this interaction was independent of any other viral factors.
P6.9 undergoes PK1-dependent hyperphosphorylation during viral infection.
To assess the potential involvement of PK1 in P6.9 phosphorylation, a pk1 knockout recombinant virus (vPK1KO) was constructed (Fig. 3A). Simultaneously, a pk1 repair virus named vPK1:FLAG also was generated, in which PK1 was tagged with a FLAG tag at its C terminus (Fig. 3A). The expression of pp78/83 in vPK1KO bacmid-transfected cells was confirmed by immunoblotting with a rabbit polyclonal anti-Pp78/83 antibody (53) (Fig. 3B). To determine the effect of pk1 deletion on viral replication, Sf9 cells were transfected with bacmids of vPK1KO, vPK1:FLAG, or vAcWT. Deletion of pk1 blocked the production of both infectious BVs and polyhedra (Fig. 3C and D), and electron microscopy observed that both nucleocapsid assembly and ODV formation were interrupted (data not shown), consistent with a previous study (27). Moreover, we also found that pk1 deletion specifically downregulated the expression of very late genes and the expression of other viral genes remained unaffected (data not shown), which also have been reported in previous studies (28, 54).
FIG 3.

Construction of AcMNPV mutants. (A) Schematic diagram of the AcMNPV mutants used in this study. bpp78/83-pk1KO was constructed by replacing a 1,575-bp fragment of pp78/83 and pk1 in bMON14272 with a 1,038-bp CmR gene via Red/ET homologous recombination in Escherichia coli. The pp78/83-pk1-rescued virus (vPK1:FLAG) was constructed by transposing both gene cassettes of pp78/83 and pk1 tagged with a FLAG epitope as well as the polh and egfp genes into the mini-attTn7 locus of bpp78/83-pk1KO. vPK1KO was generated by inserting only the pp78/83, polh, and egfp genes into bpp78/83-pk1KO. To prevent the expression of a C-terminus-truncated pk1 mutant in insect cells, the initiation codon (ATG) of pk1 was mutated to ATT. vP6.9:HA contained a p6.9 ORF tagged with an HA epitope sequence as previously described (11). The 7 PK1-dependent Ser/Thr residues were replaced with Ala residues to construct vP6.9:7M. A series of P6.9-mutation viruses (vPK1:1M) were constructed as described for vP6.9:7M, in which each of the 7 PK1-dependent Ser/Thr phosphorylation sites were separately mutated to Ala. GenR, gentamicin resistance; CmR, chloramphenicol resistance. (B) Sf9 cells were transfected with vPK1KO bacmid. At 48 h p.t., the cells were immunoblotted with a rabbit polyclonal anti-Pp78/83 antibody. Cells transfected with vGP64KO bacmid were used as a positive control. (C) Sf9 cells were transfected with bacmids of vAcWT, vPK1KO, or vPK1:FLAG. At the indicated time points p.t., the cells were observed with a fluorescence microscope or a light microscope. (D) Sf9 cells were infected with vPK1:FLAG or vAcWT at an MOI of 5 TCID50/cell or supernatants from vPK1KO bacmid-transfected cells at 96 h p.t. The supernatants were harvested at the indicated time points p.i., and BV titers were determined using TCID50 assays. Each data point represents the average titer from three independent infections. The error bars represent the standard deviations lg TCID50/ml, the logarithm of the TCID50/ml value with base 10.
To investigate any effect of pk1 deletion on P6.9 phosphorylation, the phosphorylation pattern of P6.9 was analyzed. At 48 h p.t., equal numbers of vPK1KO and vPK1:FLAG bacmid-transfected cells were obtained by flow cytometry sorting and subjected to AU-PAGE and immunoblot analysis. Only 4 phosphorylated species and the unphosphorylated species of P6.9 were observed in the vPK1KO bacmid-transfected cells. The 3 slowest-migrating rungs (arrows), which represent the 3 most phosphorylated P6.9 species in vPK1:FLAG bacmid-transfected cells, vanished from the vPK1KO bacmid-transfected cells (Fig. 4A). This finding indicates that P6.9 undergoes PK1-dependent hyperphosphorylation. Notably, the amount of the unphosphorylated P6.9 species was significantly decreased in the vPK1KO bacmid-transfected cells in some samples; however, this decrease did not always occur.
FIG 4.

Hyperphosphorylation of P6.9 is PK1 dependent. (A) At 48 h p.t., vPK1KO and vPK1:FLAG bacmid-transfected cells were purified using flow cytometry, subjected to AU-PAGE, and then immunoblotted with the anti-P6.9 antibody. BV, BVs purified from the supernatants of vPK1:FLAG-infected cells. (B) The patterns of PTM sites on P6.9 in vPK1KO bacmid-transfected cells. vPK1KO bacmid-transfected cells were harvested at 48 h p.t. using flow cytometry and then subjected to AU-PAGE and Coomassie brilliant blue staining. The unphosphorylated species (no. 0) and the phosphorylated species (no. 1 to 4) are indicated to the right of the panel. The identified phosphorylation and methylation sites are shown in a combined schematic. The phosphorylated residues are indicated by lowercase letters; the methylated residues are indicated by capital letters. (C) The phosphorylation pattern of vP6.9:7M. At 48 h p.t., vP6.9:7M bacmid-transfected cells were purified using flow cytometry, subjected to AU-PAGE, and then immunoblotted with the anti-P6.9 antibody.
To further identify which residues are phosphorylated in a PK1-dependent manner, the phosphorylation sites of the 5 P6.9 species in the vPK1KO bacmid-transfected cells were mapped by mass spectrometry. Only 3 Ser residues (S9, S23, and S24), 3 Thr residues (T11, T13, and T14), and 4 Arg residues (R7, R8, R21, and R22) were phosphorylated in vPK1KO bacmid-transfected cells (Fig. 4B); thus, there were significantly fewer phosphorylated residues than in the vPK1:FLAG-infected cells. The lack of detectable phosphorylation of the other 7 Ser/Thr sites (S10, S17, T18, S41, S43, T45, and S49) and 5 Arg sites (R6, R19, R42, R47, and R48) suggests that PK1 is required for their phosphorylation.
Five methylation sites (R21, R22, R37, R47, and R48) were identified in P6.9 in vPK1KO bacmid-transfected cells, and all of the modified Arg residues were dimethylated (Fig. 4B). Notably, R37, R47, and R48 of P6.9 were methylated in vPK1KO bacmid-transfected cells but were not methylated upon vPK1:FLAG infection. Similar to the results in vPK1:FLAG-infected cells, no acetylation site was detected in vPK1KO bacmid-transfected cells. Our data indicate that the PTM patterns of P6.9 species were substantially changed following pk1 deletion. Therefore, PK1 participates in the regulation of P6.9 PTMs.
The PK1-dependent hyperphosphorylation of P6.9 is required for the hyperexpression of very late genes.
To investigate the role of the PK1-dependent hyperphosphorylation of P6.9 in the virus life cycle, we constructed a recombinant AcMNPV named vP6.9:7M, in which the 7 Ser/Thr residues of P6.9 mentioned above were mutated to Ala residues (Fig. 3A). By using vP6.9:7M, we simulated the situation in which PK1 remained intact and further investigated the phosphorylation state of P6.9.
The P6.9 phosphorylation pattern in vP6.9:7M first was analyzed with AU-PAGE and immunoblotting. The hyperphosphorylated species of P6.9, which corresponded to species no. 5 to 7 in vPK1:FLAG, were not detected in vP6.9:7M (Fig. 4C). This result corroborated the absence of the hyperphosphorylated P6.9 species in vPK1KO, suggesting that these PK1-dependent Ser/Thr phosphorylation sites play important roles in P6.9 hyperphosphorylation.
The infectivity of vP6.9:7M was severely impaired, as only a small amount of infection spread was observed up to 96 h p.t. In addition, the number of polyhedron-containing cells in vP6.9:7M bacmid-transfected cells were significantly lower than that in vP6.9:HA bacmid-transfected cells (Fig. 5A). The infectious BV production of vP6.9:7M was reduced by 450-fold compared to that of vP6.9:HA (Fig. 5B). The viral DNA replication in vP6.9:7M bacmid-transfected cells was analyzed by qPCR, and vP6.9:HA and vP6.9KO were used as controls. As shown in Fig. 5C, at 24 h p.t., the amounts of the vP6.9:7M, vP6.9KO, and vP6.9HA genomic DNA were comparable, indicating that neither the mutation nor deletion of P6.9 affected viral DNA synthesis. qRT-PCR then was performed to analyze the transcription kinetics of AcMNPV genes in vP6.9:7M, vP6.9KO, and vP6.9:HA. Five representative viral genes, ie1, lef6, vp39, polh, and p10, were selected. ie1 and lef6 are immediate-early and early genes, respectively, that are transcribed by the host RNA polymerase II. vp39 is a late gene, and polh and p10 are very late genes; these three genes are transcribed by a viral RNA polymerase (9). At 24 h p.t. the transcription levels of ie1, lef6, and vp39 in vP6.9:7M were similar to those in vP6.9:HA, whereas the transcription levels of polh and p10 in vP6.9:7M were reduced to 50% and 59% of that in vP6.9:HA (Fig. 5D). The transcription levels of polh and p10 in cells transfected with bacmid vP6.9KO or vP6.9:7M were comparable. Therefore, our results indicate that the PK1-dependent hyperphosphorylation of P6.9 is important for viral infectivity and also is necessary for the maximal hyperexpression of very late genes in AcMNPV.
FIG 5.

Hyperphosphorylation of P6.9 regulates viral proliferation and transcription of very late genes. (A) Sf9 cells were transfected with 2 μg bacmid DNA of vP6.9:7M or vP6.9:HA. At 24 and 96 h p.t., cells were observed with a fluorescence microscope or a light microscope. (B) Infectious BV production of vP6.9:7M and vP6.9:HA. The supernatants at 96 h p.t. were harvested, and the BV titers were determined with TCID50 assays. Each value represents the averages from three independent transfections. lg TCID50/ml, the logarithm of the TCID50/ml value with base 10. (C) qPCR analysis of viral DNA replication. Sf9 cells (1 × 106) were transfected with 1 μg bacmid DNA of vP6.9:7M, vP6.9KO, or vP6.9:HA. At 24 h p.t., total cellular DNA was extracted, digested with DpnI, and analyzed by qPCR. Error bars indicate standard deviations for three independent experiments. (D) qRT-PCR analysis of viral gene transcription. Sf9 cells (1 × 106) were transfected with 1 μg bacmid DNA of vP6.9:7M, vP6.9KO, or vP6.9:HA. At 24 h p.t., the indicated viral genes were measured by qRT-PCR. The transcription levels were normalized to that of the host 18S rRNA transcripts and are shown as the percentages of the corresponding genes in the vP6.9:HA bacmid-transfected cells. Data were analyzed by Student's t test. *, P < 0.05; **, P < 0.01. Error bars indicate standard deviations from three independent experiments.
To investigate whether the decreases in very late gene transcription and the infectivity of vP6.9:7M resulted from a mutation of a hallmark residue, a series of P6.9 mutant viruses was constructed. The 7 PK1-dependent Ser/Thr phosphorylation residues were separately mutated to Ala residues to construct vP6.9:10A, vP6.9:17A, vP6.9:18A, vP6.9:41A, vP6.9:43A, vP6.9:45A, and vP6.9:49A (Fig. 3A). None of the P6.9 mutants exhibited any decrease in infectivity or level of transcripts of the very late genes (data not shown). These results suggest that there is not a key phosphorylation site among the 7 PK1-dependent Ser/Thr residues and that the hyperphosphorylation of P6.9 enhances very late gene transcription and viral infectivity.
PK1 is a component of the nucleocapsid of BVs and ODVs.
Serine/threonine kinase activity has been detected in AcMNPV virions (55). Moreover, the results described above showed that the PK1-dependent hyperphosphorylation of P6.9 plays a pivotal role in viral infectivity. Considering that P6.9 phosphorylation is a prerequisite for the release of the viral genomes from the capsids (13), we speculated that PK1 localizes on the nucleocapsids to phosphorylate P6.9. To address this possibility, BVs and ODVs from vPK1:FLAG-infected cells were purified, fractionated into envelope and nucleocapsid fractions, and analyzed by immunoblotting. As expected, PK1 was detected in the nucleocapsid fractions of both BVs and ODVs (Fig. 6A). As controls for different structures of the virions, the major nucleocapsid protein VP39, the BV envelope-specific protein GP64, and the BV/ODV envelope protein ODV-E25 were detected in the expected fractions. This result confirms that PK1 is a nucleocapsid-associated protein and supports the long-speculated idea that nucleocapsid-associated PK1 phosphorylates P6.9 to increase the negative charge of P6.9 and facilitate the release of viral DNA during the nucleocapsid uncoating process after a virion invades a host cell (13).
FIG 6.
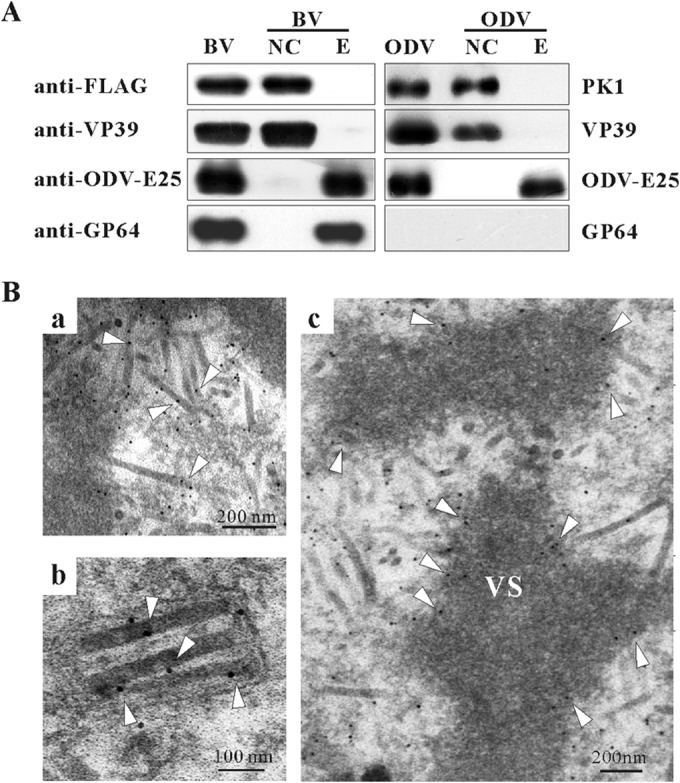
PK1 is a nucleocapsid component of both virions. (A) BVs and ODVs were purified from the supernatants and the pellets of vPK1:FLAG-infected cells, respectively, and fractionated into envelope (E) and nucleocapsid (NC) fractions. Immunoblotting was performed with anti-FLAG antibody to detect PK1, anti-ODV-E25 antibody to detect the BV/ODV envelope protein ODV-E25, anti-VP39 antibody to detect the major capsid protein VP39, and anti-GP64 antibody to detect the BV envelope-specific protein GP64. (B) Immunoelectron microscopy showing the distribution of PK1 in vPK1:FLAG-infected cells. At 60 h p.i., the cells were fixed, dehydrated, embedded in LR white resin, and processed for immunogold labeling with a mouse monoclonal anti-FLAG antibody. Shown are processing nucleocapsids in the VS (a), nucleocapsids of the mature ODVs (b), edge of the electron-dense matte regions of the VS (c). Open triangles point to the specific locations of 10-nm gold particles.
Immunoelectron microscopy (IEM) also was performed to confirm the localization of PK1. Consistent with the virion fractionation assays mentioned above, a considerable number of gold particles (indicative of PK1) were detected in the processing nucleocapsids in the VS (Fig. 6B, image a) and the nucleocapsids of ODVs (Fig. 6B, image b). Gold particles also were observed at the edge of the electron-dense matte regions of the VS (Fig. 6B, image c), implying that PK1 incorporates into the nucleocapsids in this area along with the viral genomes. Few gold particles were detected in vAcWT-infected cells using the anti-FLAG antibody, indicating the specific adsorption of this antibody (data not shown).
DISCUSSION
The baculovirus core protein is a protamine-like protein, and its gene has been found in all baculovirus genomes sequenced. Evidence from prior studies suggest a model in which the core protein is phosphorylated upon the entrance of baculovirus nucleocapsids into the nucleus, resulting in the release of the viral genome and the subsequent initiation of viral transcription and DNA replication (9). The core protein is dephosphorylated during nucleocapsid assembly, resulting in up to 100-fold condensation of the newly synthesized DNA in the preassembled capsid sheath (13, 56). Interestingly, several studies have suggested that this protein has diverse functions and is involved in not only the condensation and release of the viral genomes but also host and viral chromatin dynamics and viral gene transcription (14, 15). Its ortholog in the baculovirus type species AcMNPV, which is named P6.9, has different phosphorylated states (11, 50). However, the sites of phosphorylation and the corresponding kinases have not been determined unambiguously.
Here, we characterized P6.9 PTMs using mass spectrometry. In addition to the identification of abundant phosphorylated Ser/Thr residues, we also found 9 phosphorylated Arg residues on P6.9. The phosphorylation of Arg residues was not described in AcMNPV P6.9 in Oppenheimer and Volkman's study (50). This contradiction may be due to the degradation of phosphoarginine in an extremely low-pH environment (57), because in Oppenheimer and Volkman's study a 6 M HCl solution was used to hydrolyze P6.9 into single amino acids. A similar case also has been reported in another study on VP12, the P6.9 ortholog in Plodia interpunctella granulovirus (PiGV). In VP12, the phosphorylation of Arg residues could be detected by nuclear magnetic resonance but not by phosphoamino acid analysis, in which the protein was degraded in a low-pH solution (58).
In this report, we also found that the virus-encoded protein kinase PK1 regulates the phosphorylation states of P6.9. Using AU-PAGE, we found that the 3 most phosphorylated P6.9 species in vPK1:FLAG bacmid-transfected cells were absent from vPK1KO bacmid-transfected cells. By comparing the phosphorylated residues of P6.9 in vPK1:FLAG and vPK1KO bacmid-transfected cells, we found 7 Ser/Thr residues (S10, S17, T18, S41, S43, T45, and T49) and 5 Arg residues (R6, R19, R42, R47, and R48) that were phosphorylated only in the vPK1:FLAG-infected cells (Fig. 7), suggesting that the phosphorylation of these 12 residues is PK1 dependent. From these results, we propose the following two points. The first is that PK1 enhances the phosphorylation level in the N terminus of P6.9. A total of 3 DNA-anchoring domains, which typically contain 3 to 7 arginine residues, were predicted in the N-terminal region of P6.9 (11). Thus, the hyperphosphorylation of P6.9, especially at its N terminus, may facilitate the disassociation of viral DNA and P6.9, because the more negatively charged phosphate groups that are added on P6.9, the more positive charges of P6.9 that are neutralized. Second, the phosphorylation sites located in the C terminus of P6.9 are PK1 dependent. Seven residues (S41, R42, S43, T45, R47, R48, and S49) in the P6.9 C-terminal region were phosphorylated upon vPK1:FLAG infection but were not phosphorylated in vPK1KO bacmid-transfected cells. Notably, the C terminus of P6.9 is important for AcMNPV proliferation, because deletion of the 11 C-terminal amino acid residues (aa 45 to 55) of P6.9 block BV production (59). PK1 may phosphorylate the P6.9 C terminus either directly or indirectly. There are 3 arginine-serine (RS) repeats in this region, and two Ser and one Arg residue among them were phosphorylated (Fig. 7). The RS repeats in protamines are potential phosphorylation targets of SR protein-specific kinase 1 (60) and topoisomerase I (61). Therefore, it is possible that the C-terminal phosphorylation of P6.9 is carried out by other cellular kinases, which are in turn regulated by PK1.
FIG 7.

Schematic of PTM sites identified in P6.9. Asterisks represent the phosphorylation sites. Circles represent the mono-/dimethylation sites. The rectangle outlines the RS repeats.
Our results indicate that P6.9 hyperphosphorylation is a precondition for the maximal hyperexpression of baculovirus very late genes. This may result from the recruitment of transcription factors and/or the alteration of viral chromatin by hyperphosphorylated P6.9. The recruitment of some transcription factors requires multiple PTMs on histones. For example, the methylation of Lys4 and Lys9 in histone H3 and the methylation of Lys20 in histone H4 help to maintain active marks by allowing the binding of BRAHMA, the enzyme of the remodeling dSWI/SNF complex (62). Due to its role as a basic DNA-binding protein, hyperphosphorylated P6.9 may adopt a similar strategy, in which some combinations of the abundant phosphorylation sites and even the methylation sites may help provide unique binding surfaces for transcription factors. In contrast, the DNA binding ability of P6.9 is reduced to approximately 50% upon phosphorylation (12). Thus, the addition of abundant phosphate groups may weaken the P6.9-DNA interaction by neutralizing part of the positive charges of P6.9. Hyperphosphorylation also may interrupt the potential interaction between P6.9 regions within the monomers, because PTMs regulate protein conformation as well as protein-protein interactions, and the phosphorylation of protamine has been suggested to disturb intraprotamine and interprotamine interactions (63, 64). Hence, the highly compact DNA-P6.9 complex may be further relaxed upon P6.9 hyperphosphorylation, making the corresponding genomic regions more accessible for the very late gene transcription complex. The findings that PK1 can bind to the promoter of polh and that PK1 is a component of the very late gene transcription complex (65) may help further explain the specificity of this PK1-dependent transcription promotion. Notably, another viral protein, protein kinase-interacting protein (PKIP), can stimulate the kinase activity of PK1 (66) and is specifically required for very late gene expression, including POLH and P10 (67). These observations suggest that there is a PKIP-PK1-P6.9-POLH/P10 signaling pathway that specifically enables the hyperexpression of very late genes in baculoviruses.
Additionally, we did not detect phosphorylation and methylation on the same residue concurrently. If PK1 is involved in the determination of some residues between methylation and phosphorylation, one possible mechanism is that the PTM status of these residues is determined by PK1 phosphorylation on nearby Ser/Thr residues. Previous research showed that modification on one residue of histone can determine the PTMs of a nearby residue. For example, in mammalian histone H3, the phosphorylation of Ser10 results in the inhibition of Lys9 methylation, but it is synergistically coupled with Lys9 and/or Lys14 acetylation during hormonal stimulation and mitogenesis (68). PK1 may adopt a similar regulatory manner to indirectly choose between methylation and phosphorylation of specific residues. We found that the residues which have the potential to be methylated and phosphorylated (R7, R8, R19, R21, and R22) are located within the DNA-anchoring domains. Thus, we speculate that the different modifications of these residues is involved in the subtle regulation of the binding between P6.9 and the DNA molecule.
Based on our mass spectrometry data, we found that each P6.9 species consists of multiple isoforms with different combinations of phosphate groups. Combined, the various charged species and phosphorylation at different sites could produce as many as 13,922 distinct phosphorylated P6.9 isoforms. However, the actual number of the phosphorylated isoforms may be far less than that, because there are composition preferences in the phosphorylation of some residues. For instance, the phosphorylation of S10 was always accompanied by the phosphorylation of S9 or T11 (see Table S1 in the supplemental material). Phosphorylation at different sites might be associated with different roles for a protein. For example, the phosphorylation of residue T11 in the mammalian histone H3 results in the enhanced recruitment of the GCN histone acetyltransferase to the cyclin B1 promoter to acetylate residues K9 and K14; in contrast, the phosphorylation of residues S10 and S28 is associated with chromatin decondensation for transcriptional activation in interphase (69). There are a large number of phosphorylated isoforms of P6.9, implying that P6.9 could be involved in a diversity of processes regulated by phosphorylation. It would be interesting to investigate whether the different permutations of phosphate groups are related to different biological processes.
In this study, we were not able to precisely characterize each single isoform of P6.9. However, on the basis of their different phosphorylation levels, we can briefly classify the P6.9 species into at least 3 groups, including the unphosphorylated species found in BVs and infected cells, the hypophosphorylated species that may be phosphorylated by host kinases, and the hyperphosphorylated species that are PK1 dependent. According to existing data, these 3 groups of P6.9 may perform distinct functions. (i) Newly synthesized viral DNA has to be condensed up to 100-fold during nucleocapsid assembly (56). Thus, the unphosphorylated P6.9 (Fig. 1B, no. 0 and BV in AU-PAGE) should be associated with the encapsidation of viral DNA into procapsids. (ii) According to our observations, as long as the hypophosphorylated P6.9 species exist (Fig. 4B and C, no. 1 to 4 in AU-PAGE), the expression of most of the viral genes is not affected (vPK1KO and vP6.9:7M bacmid-transfected cells). However, when the hypophosphorylated P6.9 species are absent (vP6.9KO bacmid-transfected cells) during the late infection phase, there is a general downregulation of most of the viral genes. These findings suggest that the hypophosphorylated P6.9 species have broad transcription-promotion effects on viral genes, although the functional mechanism of the hypophosphorylated P6.9 species remains to be determined. (iii) As described above, the hyperphosphorylated P6.9 species (Fig. 1B, no. 5 to 7 in AU-PAGE) facilitate very late gene hyperexpression and may help release the viral genome upon virus invasion. Thus, the existence of various P6.9 species and their multiple functions reflect how the virus can sufficiently utilize a protamine or protamine-like protein in each phase of its life cycle. Future studies also should focus on the function of methylation, which may play roles in the replication of baculoviruses.
In summary, our experiments provide novel insights into the regulation of P6.9 PTMs and examine the relationship between the PTM patterns of the viral protamine-like protein and a viral serine/threonine kinase. These results provide valuable information for further explorations into the molecular mechanism by which pathogen protamine-like protein PTMs regulate viral proliferation and gene transcription. Furthermore, because we detected various modification patterns in P6.9, future studies will focus on the potential code and its function on viral protamine-like proteins, which are rarely studied in virology.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Zhihong Hu (Wuhan Institute of Virology) for providing ODV-E25 polyclonal antiserum. We thank Ling Fang for providing mass spectrometry support.
This research was supported by the Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education Program (20120171110011) and the National Nature Science Foundation of China (31370188).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00333-15.
REFERENCES
- 1.Simpson RT, Bustin M. 1976. Histone composition of chromatin subunits studied by immunosedimentation. Biochemistry 15:4305–4312. doi: 10.1021/bi00664a026. [DOI] [PubMed] [Google Scholar]
- 2.Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Balhorn R. 2007. The protamine family of sperm nuclear proteins. Genome Biol 8:227. doi: 10.1186/gb-2007-8-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunner AM, Nanni P, Mansuy IM. 2014. Epigenetic marking of sperm by post-translational modification of histones and protamines. Epigenet Chromatin 7:2. doi: 10.1186/1756-8935-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fareed GC, Davoli D. 1977. Molecular biology of papovaviruses. Annu Rev Biochem 46:471–522. doi: 10.1146/annurev.bi.46.070177.002351. [DOI] [PubMed] [Google Scholar]
- 6.Laver WG. 1970. Isolation of an arginine-rich protein from particles of adenovirus type 2. Virology 41:488–500. doi: 10.1016/0042-6822(70)90170-4. [DOI] [PubMed] [Google Scholar]
- 7.Witteveldt J, Vermeesch AM, Langenhof M, de Lang A, Vlak JM, van Hulten MC. 2005. Nucleocapsid protein VP15 is the basic DNA binding protein of white spot syndrome virus of shrimp. Arch Virol 150:1121–1133. doi: 10.1007/s00705-004-0483-8. [DOI] [PubMed] [Google Scholar]
- 8.Lieberman PM. 2006. Chromatin regulation of virus infection. Trends Microbiol 14:132–140. doi: 10.1016/j.tim.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Rohrmann GF. 2013. Baculovirus molecular biology: 3rd ed. National Library of Medicine, National Center for Biotechnology Information, Bethesda, MD. [PubMed] [Google Scholar]
- 10.Wilson ME, Mainprize TH, Friesen PD, Miller LK. 1987. Location, transcription, and sequence of a baculovirus gene encoding a small arginine-rich polypeptide. J Virol 61:661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, Zhao H, Fang Z, Yuan M, Yang K, Pang Y. 2012. Distribution and phosphorylation of the basic protein P6.9 of Autographa californica nucleopolyhedrovirus. J Virol 86:12217–12227. doi: 10.1128/JVI.00438-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson ME, Consigli RA. 1985. Functions of a protein kinase activity associated with purified capsids of the granulosis virus infecting Plodia interpunctella. Virology 143:526–535. doi: 10.1016/0042-6822(85)90391-5. [DOI] [PubMed] [Google Scholar]
- 13.Funk CJ, Consigli RA. 1993. Phosphate cycling on the basic protein of Plodia interpunctella granulosis virus. Virology 193:396–402. doi: 10.1006/viro.1993.1136. [DOI] [PubMed] [Google Scholar]
- 14.Wilson ME, Miller LK. 1986. Changes in the nucleoprotein complexes of a baculovirus DNA during infection. Virology 151:315–328. doi: 10.1016/0042-6822(86)90052-8. [DOI] [PubMed] [Google Scholar]
- 15.Peng Y, Li K, Pei RJ, Wu CC, Liang CY, Wang Y, Chen XW. 2012. The protamine-like DNA-binding protein P6.9 epigenetically up-regulates Autographa californica multiple nucleopolyhedrovirus gene transcription in the late infection phase. Virol Sin 27:57–68. doi: 10.1007/s12250-012-3229-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser MJ. 1986. Ultrastructural observations of virion maturation in Autographa californica nuclear polyhedrosis virus infected Spodoptera frugiperda cell cultures. J Ultrastruct Mol Struct Res 95:189–195. doi: 10.1016/0889-1605(86)90040-6. [DOI] [Google Scholar]
- 17.Friesen PD. 1997. Regulation of baculovirus early gene expression, p 141–170. In Miller LK. (ed), The baculoviruses. Plenum Press, New York, NY. [Google Scholar]
- 18.Fuchs LY, Woods MS, Weaver RF. 1983. Viral transcription during Autographa californica nuclear polyhedrosis virus infection: a novel RNA polymerase induced in infected Spodoptera frugiperda cells. J Virol 48:641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen YR, Zhong S, Fei Z, Hashimoto Y, Xiang JZ, Zhang S, Blissard GW. 2013. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J Virol 87:6391–6405. doi: 10.1128/JVI.00194-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohrmann GF. 1992. Baculovirus structural proteins. J Gen Virol 73(Part 4):749–761. doi: 10.1099/0022-1317-73-4-749. [DOI] [PubMed] [Google Scholar]
- 21.Bergold GH. 1963. The nature of nuclear polyhedrosis viruses, p 413–455. In Steinhaus EA. (ed), Insect pathology: an advance treatise. Academic Press, New York, NY. [Google Scholar]
- 22.Moscardi F. 1999. Assessment of the application of baculoviruses for control of Lepidoptera. Annu Rev Entomol 44:257–289. doi: 10.1146/annurev.ento.44.1.257. [DOI] [PubMed] [Google Scholar]
- 23.Smith GE, Summers MD, Fraser MJ. 1983. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol Cell Biol 3:2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Oers MM, Pijlman GP, Vlak JM. 2014. Thirty years of baculovirus-insect cell protein expression: from dark horse to mainstream technology. J Gen Virol 96(Part 1):6–23. doi: 10.1099/vir.0.067108-0. [DOI] [PubMed] [Google Scholar]
- 25.Reilly LM, Guarino LA. 1994. The pk-1 gene of Autographa californica multinucleocapsid nuclear polyhedrosis virus encodes a protein kinase. J Gen Virol 75(Part 11):2999–3006. doi: 10.1099/0022-1317-75-11-2999. [DOI] [PubMed] [Google Scholar]
- 26.Bischoff DS, Slavicek JM. 1994. Identification and characterization of a protein kinase gene in the Lymantria dispar multinucleocapsid nuclear polyhedrosis virus. J Virol 68:1728–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang C, Li M, Dai X, Zhao S, Hou Y, Zhang Y, Lan D, Wang Y, Chen X. 2013. Autographa californica multiple nucleopolyhedrovirus PK-1 is essential for nucleocapsid assembly. Virology 443:349–357. doi: 10.1016/j.virol.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Fan X, Thirunavukkarasu K, Weaver RF. 1996. Temperature-sensitive mutations in the protein kinase-1 (pk-1) gene of the Autographa californica nuclear polyhedrosis virus that block very late gene expression. Virology 224:1–9. doi: 10.1006/viro.1996.0500. [DOI] [PubMed] [Google Scholar]
- 29.Vaughn JL, Goodwin RH, Tompkins GJ, McCawley P. 1977. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 13:213–217. doi: 10.1007/BF02615077. [DOI] [PubMed] [Google Scholar]
- 30.Wu W, Lin T, Pan L, Yu M, Li Z, Pang Y, Yang K. 2006. Autographa californica multiple nucleopolyhedrovirus nucleocapsid assembly is interrupted upon deletion of the 38K gene. J Virol 80:11475–11485. doi: 10.1128/JVI.01155-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Reilly DR, Miller LK, Luckow VA. 1992. Baculovirus expression vectors: a laboratory manual. Oxford University Press, New York, NY. [Google Scholar]
- 32.Luckow VA, Lee SC, Barry GF, Olins PO. 1993. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J Virol 67:4566–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Possee RD, Sun TP, Howard SC, Ayres MD, Hill-Perkins M, Gearing KL. 1991. Nucleotide sequence of the Autographa californica nuclear polyhedrosis 9.4 kbp EcoRI-I and -R (polyhedrin gene) region. Virology 185:229–241. doi: 10.1016/0042-6822(91)90770-C. [DOI] [PubMed] [Google Scholar]
- 34.Peters JE, Craig NL. 2001. Tn7: smarter than we thought. Nat Rev Mol Cell Biol 2:806–814. doi: 10.1038/35099006. [DOI] [PubMed] [Google Scholar]
- 35.Chiu J, March PE, Lee R, Tillett D. 2004. Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res 32:e174. doi: 10.1093/nar/gnh172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai X, Stewart TM, Pathakamuri JA, Li Q, Theilmann DA. 2004. Autographa californica multiple nucleopolyhedrovirus exon0 (orf141), which encodes a RING finger protein, is required for efficient production of budded virus. J Virol 78:9633–9644. doi: 10.1128/JVI.78.18.9633-9644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Y, Long Z, Qiu J, Yuan M, Li G, Yang K. 2012. An ac34 deletion mutant of Autographa californica nucleopolyhedrovirus exhibits delayed late gene expression and a lack of virulence in vivo. J Virol 86:10432–10443. doi: 10.1128/JVI.00779-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shechter D, Dormann HL, Allis CD, Hake SB. 2007. Extraction, purification and analysis of histones. Nat Protoc 2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 39.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 40.Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 41.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. 2011. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Russel DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 43.Yuan M, Huang Z, Wei D, Hu Z, Yang K, Pang Y. 2011. Identification of Autographa californica nucleopolyhedrovirus ac93 as a core gene and its requirement for intranuclear microvesicle formation and nuclear egress of nucleocapsids. J Virol 85:11664–11674. doi: 10.1128/JVI.05275-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei D, Wang Y, Zhang X, Hu Z, Yuan M, Yang K. 2014. Autographa californica nucleopolyhedrovirus Ac76: a dimeric type II integral membrane protein that contains an inner nuclear membrane-sorting motif. J Virol 88:1090–1103. doi: 10.1128/JVI.02392-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vanarsdall AL, Okano K, Rohrmann GF. 2005. Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology 331:175–180. doi: 10.1016/j.virol.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 46.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 47.Wang R, Deng F, Hou D, Zhao Y, Guo L, Wang H, Hu Z. 2010. Proteomics of the Autographa californica nucleopolyhedrovirus budded virions. J Virol 84:7233–7242. doi: 10.1128/JVI.00040-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Li Z, Chen W, Pang Y. 2007. Cloning, expression of Autographa californica nucleopolyhedrovirus vp39 gene in Escherichia coli and preparation of its antibody. Biotechnology 17:5–7 http://en.cnki.com.cn/Article_en/CJFDTOTAL-SWJS200703002.htm. [Google Scholar]
- 49.Wu W, Liang H, Kan J, Liu C, Yuan M, Liang C, Yang K, Pang Y. 2008. Autographa californica multiple nucleopolyhedrovirus 38K is a novel nucleocapsid protein that interacts with VP1054, VP39, VP80, and itself. J Virol 82:12356–12364. doi: 10.1128/JVI.00948-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oppenheimer DI, Volkman LE. 1995. Proteolysis of p6.9 induced by cytochalasin D in Autographa californica M nuclear polyhedrosis virus-infected cells. Virology 207:1–11. doi: 10.1006/viro.1995.1046. [DOI] [PubMed] [Google Scholar]
- 51.Tripsianes K, Madl T, Machyna M, Fessas D, Englbrecht C, Fischer U, Neugebauer KM, Sattler M. 2011. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat Struct Mol Biol 18:1414–1420. doi: 10.1038/nsmb.2185. [DOI] [PubMed] [Google Scholar]
- 52.Knudson DL, Harrap KA. 1975. Replication of nuclear polyhedrosis virus in a continuous cell culture of Spodoptera frugiperda: microscopy study of the sequence of events of the virus infection. J Virol 17:254–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Wang Q, Liang C, Song J, Li N, Shi H, Chen X. 2008. Autographa californica multiple nucleopolyhedrovirus nucleocapsid protein BV/ODV-C42 mediates the nuclear entry of P78/83. J Virol 82:4554–4561. doi: 10.1128/JVI.02510-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra G, Chadha P, Chaudhury I, Das RH. 2008. Inhibition of Autographa californica nucleopolyhedrovirus (AcNPV) polyhedrin gene expression by DNAzyme knockout of its serine/threonine kinase (pk1) gene. Virus Res 135:197–201. doi: 10.1016/j.virusres.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 55.Miller LK, Adang MJ, Browne D. 1983. Protein kinase activity associated with the extracellular and occluded forms of the baculovirus Autographa californica nuclear polyhedrosis virus. J Virol 46:275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bud HM, Kelly DC. 1980. An electron microscope study of partially lysed baculovirus nucleocapsids: the intranucleocapsid packaging of viral DNA. J Ultrastruct Res 73:361–368. doi: 10.1016/S0022-5320(80)90095-7. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt A, Trentini DB, Spiess S, Fuhrmann J, Ammerer G, Mechtler K, Clausen T. 2014. Quantitative phosphoproteomics reveals the role of protein arginine phosphorylation in the bacterial stress response. Mol Cell Proteomics 13:537–550. doi: 10.1074/mcp.M113.032292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilson ME, Consigli RA. 1985. Characterization of a protein kinase activity associated with purified capsids of the granulosis virus infecting Plodia interpunctella. Virology 143:516–525. doi: 10.1016/0042-6822(85)90390-3. [DOI] [PubMed] [Google Scholar]
- 59.Wang M, Tuladhar E, Shen S, Wang H, van Oers MM, Vlak JM, Westenberg M. 2010. Specificity of baculovirus P6.9 basic DNA-binding proteins and critical role of the C terminus in virion formation. J Virol 84:8821–8828. doi: 10.1128/JVI.00072-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papoutsopoulou S, Nikolakaki E, Chalepakis G, Kruft V, Chevaillier P, Giannakouros T. 1999. SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res 27:2972–2980. doi: 10.1093/nar/27.14.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lewis JD, Ausio J. 2002. Protamine-like proteins: evidence for a novel chromatin structure. Biochem Cell Biol 80:353–361. doi: 10.1139/o02-083. [DOI] [PubMed] [Google Scholar]
- 62.Beisel C, Imhof A, Greene J, Kremmer E, Sauer F. 2002. Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature 419:857–862. doi: 10.1038/nature01126. [DOI] [PubMed] [Google Scholar]
- 63.Balhorn R. 1982. A model for the structure of chromatin in mammalian sperm. J Cell Biol 93:298–305. doi: 10.1083/jcb.93.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pirhonen A, Linnala-Kankkunen A, Maenpaa PH. 1994. Identification of phosphoseryl residues in protamines from mature mammalian spermatozoa. Biol Reprod 50:981–986. doi: 10.1095/biolreprod50.5.981. [DOI] [PubMed] [Google Scholar]
- 65.Mishra G, Chadha P, Das RH. 2008. Serine/threonine kinase (pk-1) is a component of Autographa californica multiple nucleopolyhedrovirus (AcMNPV) very late gene transcription complex and it phosphorylates a 102kDa polypeptide of the complex. Virus Res 137:147–149. doi: 10.1016/j.virusres.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 66.Fan X, McLachlin JR, Weaver RF. 1998. Identification and characterization of a protein kinase-interacting protein encoded by the Autographa californica nuclear polyhedrosis virus. Virology 240:175–182. doi: 10.1006/viro.1997.8944. [DOI] [PubMed] [Google Scholar]
- 67.McLachlin JR, Yang S, Miller LK. 1998. A baculovirus mutant defective in PKIP, a protein which interacts with a virus-encoded protein kinase. Virology 246:379–391. doi: 10.1006/viro.1998.9210. [DOI] [PubMed] [Google Scholar]
- 68.Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 69.Baek SH. 2011. When signaling kinases meet histones and histone modifiers in the nucleus. Mol Cell 42:274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.