

ABSTRACT
Defective interfering RNAs (DI-RNAs) of the viral genome can form during infections of negative-strand RNA viruses and outgrow full-length viral genomes, thereby modulating the severity and duration of infection. Here we document the frequent de novo generation of copy-back DI-RNAs from independent rescue events both for a vaccine measles virus (vac2) and for a wild-type measles virus (IC323) as early as passage 1 after virus rescue. Moreover, vaccine and wild-type C-protein-deficient (C-protein-knockout [CKO]) measles viruses generated about 10 times more DI-RNAs than parental virus, suggesting that C enhances the processivity of the viral polymerase. We obtained the nucleotide sequences of 65 individual DI-RNAs, identified breakpoints and reinitiation sites, and predicted their structural features. Several DI-RNAs possessed clusters of A-to-G or U-to-C transitions. Sequences flanking these mutation sites were characteristic of those favored by adenosine deaminase acting on RNA-1 (ADAR1), which catalyzes in double-stranded RNA the C-6 deamination of adenosine to produce inosine, which is recognized as guanosine, a process known as A-to-I RNA editing. In individual DI-RNAs the transitions were of the same type and occurred on both sides of the breakpoint. These patterns of mutations suggest that ADAR1 edits unencapsidated DI-RNAs that form double-strand RNA structures. Encapsidated DI-RNAs were incorporated into virus particles, which reduced the infectivity of virus stocks. The CKO phenotype was dominant: DI-RNAs derived from vac2 with a CKO suppressed the replication of vac2, as shown by coinfections of interferon-incompetent lymphatic cells with viruses expressing different fluorescent reporter proteins. In contrast, coinfection with a C-protein-expressing virus did not counteract the suppressive phenotype of DI-RNAs.
IMPORTANCE Recombinant measles viruses (MVs) are in clinical trials as cancer therapeutics and as vectored vaccines for HIV-AIDS and other infectious diseases. The efficacy of MV-based vectors depends on their replication proficiency and immune activation capacity. Here we document that copy-back defective interfering RNAs (DI-RNAs) are generated by recombinant vaccine and wild-type MVs immediately after rescue. The MV C protein interferes with DI-RNA generation and may enhance the processivity of the viral polymerase. We frequently detected clusters of A-to-G or U-to-C transitions and noted that sequences flanking individual mutations contain motifs favoring recognition by the adenosine deaminase acting on RNA-1 (ADAR1). The consistent type of transitions on the DI-RNAs indicates that these are direct substrates for editing by ADAR1. The ADAR1-mediated biased hypermutation events are consistent with the protein kinase R (PKR)-ADAR1 balancing model of innate immunity activation. We show by coinfection that the C-defective phenotype is dominant.
INTRODUCTION
Measles virus (MV) is a negative-strand RNA virus of the Paramyxoviridae family with a genome comprising 15,894 nucleotides (nt). The MV genome encodes six genes from which eight proteins are expressed: nucleoprotein (N), phosphoprotein (P), matrix protein (M), fusion protein (F), hemagglutinin (H), and large protein (L; the catalytic subunit of the viral RNA-dependent RNA polymerase), as well as the host response-modulating V and C proteins (1–3).
MV replication occurs in the cytoplasm of infected cells and involves a helical ribonucleocapsid (RNP) formed by viral genomic RNA and N protein. The polymerase complex, consisting of the L and P proteins, recognizes the 3′ end of the RNA and generates a complementary nascent RNA strand that is cotranscriptionally encapsidated (4). Both genomic RNA and antigenomic RNA can serve as a template for replication, whereas only the genomic RNA serves as a template for the transcription of capped and polyadenylated mRNAs (1).
The polymerase of negative-strand RNA viruses is error prone and introduces nucleotide substitutions at fairly high frequencies (about 1 substitution in 10,000 nt) (5–8). Defective genomes are also generated, including abortive genomes and 5′ copy-back defective interfering (DI) RNAs (DI-RNAs) (9). DI-RNAs are generated when the polymerase prematurely terminates genome synthesis and then—when it is still bound to the truncated genome—binds back and reinitiates replication on this RNA, thereby generating a genome-antigenome hybrid with perfectly complementary ends. These complementary ends can form stable panhandle duplex RNA structures, as shown for MV and Sendai virus (SeV), a related paramyxovirus (9, 10).
These double-stranded RNA (dsRNA) structures are potent inducers of the cellular stress and innate immune response (11). They trigger the activation of innate receptors, including retinoic acid-inducible gene I (RIG-I [12]), melanoma differentiation-associated gene-5 (mda-5 [13]), and protein kinase R (PKR [14]). DI-RNAs contain the 3′-terminal promoter sequence and therefore can be replicated into their complementary counterpart, which also contains the promoter sequence at its 3′ end. DI-RNAs, therefore, like the genome-antigenome pair, form a replication system, which, due to its reduced length and therefore increased replication frequency, can interfere with full-length RNA replication and temporarily outgrow full-length RNA (15).
We previously reported the generation of viral DI-RNAs in C-protein-deficient (C-protein-knockout [CKO]) Moraten vaccine MV (MV-CKO) and showed that DI-RNAs induce the PKR-mediated stress and interferon (IFN) responses (14). Here we provide mechanistic insights into the generation of DI-RNAs by a vaccine MV (vac2)-derived infectious cDNA (16) and a wild-type MV (IC323) infectious cDNA (17) immediately after virus rescue. We describe the molecular characteristics of DI-RNAs from C-protein-expressing and C-defective viruses and find structural similarities between DI-RNAs derived from all viruses. Our analysis of multiple independent virus rescues indicated that deficiency in C-protein expression leads to a 10-fold increase in the abundance of DI-RNAs. Although initially demonstrated in serially undiluted virus infections performed at a high multiplicity (18–20), we detected DI-RNAs in stocks as early as passage 1 (P1) after virus rescue. We found clusters of A-to-G or U-to-C transitions in several DI-RNAs in a sequence context characteristic of the mutations introduced by cellular adenosine deaminase acting on RNA-1 (ADAR1) (21–23).
MATERIALS AND METHODS
Cells and viruses.
Vero, Vero-hSLAM (a kind gift from Y. Yanagi [24]), and HeLa cells were cultivated in Dulbecco's high-glucose modified Eagle's medium (D-MEM; HyClone; GE Healthcare Life Sciences, Logan, UT) supplemented with 5% (vol/vol) fetal bovine serum (FBS; Gibco, Life Technologies, Carlsbad, CA). Vero-hSLAM cells were grown in the presence of Geneticin (G418; Corning, Tewksbury, MA) at a final concentration of 0.4 mg/ml. 293-3-46 cells (25), used for virus rescue, were cultivated in D-MEM supplemented with 10% (vol/vol) FBS and 1 mg/ml G418. The mantle cell lymphoma cell line Granta-519 (26) was cultivated in RPMI 1640 (HyClone) supplemented with 10% (vol/vol) FBS.
Recombinant MVs were IC323 (isogenic to the wild-type IC-B strain [17]), IC323-CKO, vac2 (isogenic to the Moraten vaccine strain [16]), vac2-CKO, vac2(GFP) (expressing enhanced green fluorescent protein [eGFP] from an additional transcription unit downstream of H [27]), vac2-CKO(GFP), vac2(mCherryNLS), and vac2-CKO(mCherryNLS). Viruses with the CKO have specific mutations introducing early stop codons in the C open reading frame, as described previously (16).
Generation of mCherryNLS-expressing MVvac2 cDNA.
A construct encoding mCherry with a nuclear localization signal (NLS), mCherryNLS, kindly provided by L. Enquist (28), was cloned into p(+)MV323(GFP)N to replace GFP, yielding p(+)MV323(mCherryNLS)N (Crystal Mendoza, unpublished data). mCherryNLS was amplified from this donor vector by PCR using the following primers (Integrated DNA Technologies [IDT], Coralville, IA), where the restriction sites are underlined: mCherryNLS EcoRI/MluI fwd (TATAGAATTCCCATCAACGCGTTACGCCACCATG) and mCherryNLS NotI/AatII rev (ATATGCGGCCGCGTTCGAGACGTCAGTTTATCTGGATCC). The PCR product was cloned into pCR3 (Life Technologies, Invitrogen, Carlsbad, CA) using the EcoRI/NotI restriction sites and then excised with MluI/AatII (New England BioLabs, Ipswich, MA). The GFP-encoding sequence in pB(+)MVvac2(GFP)H and pB(+)MVvac2-CKO(GFP)H was then replaced with the mCherryNLS fragment to yield pB(+)MVvac2(mCherryNLS)H and pB(+)MVvac2-CKO(mCherryNLS)H, respectively.
Virus rescue and stock production.
Recombinant viruses were generated as described previously (25). Briefly, 293-3-46 cells were transfected with the relevant plasmid carrying full-length viral cDNA and pEMC-La, expressing the viral polymerase. After a 42°C heat shock and 2 days of incubation, cells were transferred to 100-mm dishes with Vero or Vero-hSLAM cells and incubated until syncytium formation was observed. Single syncytia were picked and used to inoculate Vero or Vero-hSLAM cells in 6-well plates. Cells and supernatants were harvested when the maximum cytopathic effect (CPE) was achieved (2 to 4 days), yielding passage 0 stocks. These stocks were used to generate passage 1 stocks by infecting multiple 150-mm dishes of Vero or Vero-hSLAM cells. Virus stocks were grown at 32°C (vac2) or 37°C (IC323). Cells were harvested at the time of the maximum CPE by scraping them into 1 ml Opti-MEM medium (Gibco, Life Technologies, Carlsbad, CA) per 150-mm dish, followed by three freeze-thaw cycles (liquid nitrogen and 37°C). Cleared lysates were aliquoted and stored at −80°C. Viral titers on Vero-hSLAM cells were determined using the 50% tissue culture infectious dose (TCID50) method. For further passages, Vero cells in 150-mm dishes were infected at a multiplicity of infection (MOI) of 0.03 and harvested, and titers were determined.
Virion purification.
Vero cells (in 10 150-mm dishes) were infected with passage 1 vac2 or vac2-CKO at an MOI of 0.05. Cells were harvested at 48 h postinfection and scraped into 1 ml Opti-MEM medium per dish, followed by three freeze-thaw cycles. Cleared lysates (total volume, about 15 ml) were mixed with Coomassie blue (Brilliant Blue G; final concentration, ∼150 μg/ml; Bio-Rad, Hercules, CA), and the mixture was laid onto a 20%/60% (wt/vol) sucrose cushion (in TNE buffer [50 mM Tris-HCl {pH 7.4}, 100 mM NaCl, 0.1 mM EDTA]) and centrifuged in an SW32Ti rotor (Beckman Coulter, Indianapolis, IN) at 75,000 × g at 4°C for 4 h. The visible blue band at the 20%-60% interface was collected with a 16.5-gauge needle and syringe in a total volume of about 6 ml, diluted with sterile phosphate-buffered saline (PBS; HyClone) supplemented with 0.5 mM MgCl2 (PBS-MgCl2) to a final volume of 15 ml, and then laid onto a 20 to 60% (wt/vol) discontinuous sucrose gradient (in TNE buffer, 5% intervals). Centrifugation was to equilibrium (75,000 × g at 4°C for 16 h) using the SW32Ti rotor. Virions that accumulated in the 30 to 40% region were collected in a total volume of 6 ml, diluted with sterile PBS-MgCl2, and concentrated using Amicon Ultra filter tubes (EMD Millipore, Billerica, MA) with a molecular mass exclusion of 100 kDa (3,500 × g, 4°C) in a tabletop centrifuge. The concentrate was washed once with 15 ml PBS-MgCl2, concentrated to a final volume of 1 ml, and stored at −80°C. Titers were determined by the TCID50 method. Prior to further analysis, purified stocks were subjected to treatment with Benzonase (EMD Millipore, Billerica, MA) at a final concentration of 12.5 U/μl for 2 h at 37°C in 10 mM Tris, pH 8.0, buffer containing 1 mM MgCl2 and10 mM NaCl.
RNA protection assay.
One million TCID50s of purified vac2 or vac2-CKO virions were treated with combinations of nonionic detergent (Triton X-100), protease (trypsin), and nuclease (Benzonase). First, 1% (vol/vol) Triton X-100 (Sigma-Aldrich, St. Louis, MO) was added to virions in reaction buffer (10 mM Tris, pH 8.0, 10 mM NaCl, 1 mM MgCl2). Next, samples were heated to 80°C for 15 min and cooled on ice for 10 min. Then, 0.1 mg/ml trypsin (Worthington, Lakewood, NJ) was added and the samples were incubated at 37°C for 15 min. To inactivate the trypsin, 3 mg/ml soybean trypsin inhibitor (Worthington, Lakewood, NJ) was added and the samples were incubated at 25°C for 30 min. Next, 2.5 U/μl Benzonase was added and the samples were incubated at 37°C for 2.5 h. Nuclease activity was inhibited by addition of 10 mM EDTA, before sample aliquots were subjected to protein analysis or RNA extraction.
RNA extraction, RT-PCR, and PCR analysis.
Vero or Vero-hSLAM cells were infected with viruses at an MOI of 0.1 or left uninfected and harvested at 30 to 48 h postinfection. Total RNA was extracted using either an RNeasy minikit (Qiagen, Valencia, CA) or the TRIzol reagent (Ambion, Life Technologies, Carlsbad, CA) according to the suppliers' instructions and stored at −80°C. One microgram of total RNA was subjected to reverse transcription-PCR (RT-PCR) using SuperScript III reverse transcriptase (Life Technologies, Carlsbad, CA) with a mixture of MV-specific primers (IDT), where restriction sites are underlined: negative-polarity primers A1SnaBI (GCGCGCTACGTAAAAGCTGGGAATAGAAACTTCG), A2vac2-NotI (TAGAGAGAGCGGCCGCTCTGGTGTAAGTCTAGTATCAGA), A2323-NotI (TAGAGAGAGCGGCCGCTCTGGTGTAAGTCTAGTACTAGA), A3NotI (TAGAGAGAGCGGCCGCGATCTAGAATTGGCGGAAACC), and A4NotI (TAGAGAGAGCGGCCGCTCCCTGATGGTCTATGATAATG) and positive-polarity primer B1 (ATGACAGATCTCAAGGCTAAC). Briefly, RNA was mixed with primers and deoxynucleoside triphosphates and subjected to heat denaturation at 98°C for 10 min, followed by incubation on ice for 10 min. After addition of the enzyme mix, samples were incubated at 55°C for 60 min and then at 70°C for 15 min. cDNAs were stored at −20°C. An aliquot of the cDNA (1 μl) was further amplified using GoTaq polymerase (Promega, Madison, WI) and DI-RNA-specific primer combinations (primers A1/A2, A1/A3, and A1/A4) or a control primer pair for standard genomes (primers A1/B1). Samples were analyzed by electrophoresis using 1% (wt/vol) agarose gels and a 100-bp or 1-kb DNA ladder as a size standard (Promega, Madison, WI).
Cloning and analysis of DI-RNAs.
PCR products amplified from the cDNA of MV-infected cells were purified (PCR purification kit; Qiagen, Valencia, CA), digested with SnaBI and NotI-HF (New England BioLabs, Ipswich, MA), purified again, and then ligated with EcoRV/NotI-linearized pCR3 (Invitrogen, Life Technologies, Carlsbad, CA). Ligation products were transformed into OneShot TOP10 cells (Life Technologies, Carlsbad, CA), and the bacteria were plated on LB-ampicillin-agar plates and incubated at 37°C for 12 to 20 h. Single colonies were picked and resuspended in 10 μl H2O, and 5 μl of the suspension was subjected to colony PCR using the primers (IDT) T7fwd (TAATACGACTCACTATAGGG) and BGHrev (TAGAAGGCACAGTCGAGG) and a 2× PCR mix (Promega, Madison, WI). Clones with individual inserts, as analyzed by 1% agarose gel electrophoresis, were selected and grown in 2 ml LB-ampicillin, and plasmids were extracted using a plasmid minikit (Qiagen, Valencia, CA). Sequences were determined by Sanger sequencing using the T7fwd and BGHrev primers and analyzed by alignment to MV reference sequences with GenBank accession numbers AF266287 for vac2 and AB016162 for IC323.
Protein gel electrophoresis and immunoblot analysis.
Protein extracts of infected cells (MOI = 0.1, 30 to 48 h postinfection) were generated, and protein concentrations were determined as described previously (14). Protein (25 μg) was fractionated using 12% polyacrylamide gels and transferred to Immobilon-P membranes (EMD Millipore, Billerica, MA) using a semidry protocol. Membranes were blocked with 5% (wt/vol) bovine serum albumin (Sigma-Aldrich, St. Louis, MO) in Tris-buffered saline (Bio-Rad, Hercules, CA) for 1 h and incubated with primary antibodies at 4°C for 12 to 16 h. Membranes were washed three times with PBS with 0.5% (vol/vol) Tween 20 (PBST) for 5 to 10 min, incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for 1 h, washed again three times with PBST, and incubated with SuperSignal West Pico chemiluminescent substrate (Pierce, Thermo Fisher Scientific, Rockford, IL). Membranes were exposed to HyBlot films (Denville, South Plainfield, NJ) for various amounts of time.
Antibodies.
Rabbit peptide antisera recognizing MV N (N505 antiserum), MV C (C2 antiserum), and MV H (Hcyt antiserum) were described previously (16, 29, 30) and were used at dilutions of 1:5,000 for primary antibody incubation. Mouse monoclonal anti-GFP antibody (eBioscience, San Diego, CA) was used at a 1:1,000 dilution. HRP-conjugated antiactin (Sigma-Aldrich, St. Louis, MO) was used at a 1:25,000 dilution. Rabbit and mouse secondary antibodies conjugated with HRP (Jackson ImmunoResearch, West Grove, PA, and EMD Millipore, Billerica, MA, respectively) were used at 1:10,000 dilutions. For immunofluorescence, mouse anti-dsRNA J2 (English and Scientific Consulting, Szirák, Hungary [31]), anti-MV N505 antiserum, and secondary antibodies antimouse Alexa 488, antimouse Alexa 594, and antirabbit Alexa 405 (Molecular Probes, Life Technologies, Carlsbad, CA) were used as described previously (14).
Northern blot analysis.
A 250-bp fragment of the L trailer region of MV (nt 15641 to 15890) was cloned into the pCR3 vector between the HindIII and XhoI restriction sites (amplification primers [IDT], MV15641-HindIII fwd [AGACCCAAGCTTTATCTTCGTTAAGAATCT] and MV15890 XhoI rev [TGCATGCTCGAGGACAAAGCTGGGAATAGA], where the restriction sites are underlined). Positive and negative single-stranded RNA (ssRNA) probes were generated from HindIII- or XhoI-linearized plasmids using SP6 or T7 RNA polymerase of a digoxigenin (DIG) RNA labeling kit (Roche Diagnostics, Indianapolis, IN). Total RNA (3 μg) or an amount of RNA corresponding to 1 × 106 TCID50s of purified virions was subjected to 1% agarose-2% formaldehyde gel electrophoresis, with DIG-labeled RNA molecular weight marker I (Roche Diagnostics, Indianapolis, IN) being included as described in the DIG Northern starter kit manual (Roche Diagnostics, Indianapolis, IN). RNA was transferred to nylon membranes (Roche Diagnostics, Indianapolis, IN) by bottom-up capillary transfer using 20× SSC (3.0 M NaCl, 0.3 M sodium citrate, pH 7.0) as the buffer. RNAs were cross-linked with 125 mJ of 254-nm UV light before incubation with RNA probes (100 mg/ml) at 68°C for 16 to 20 h under constant agitation. Probe detection was performed according to the instructions in the supplier's manual (Roche Diagnostics, Indianapolis, IN).
Confocal immunofluorescence analysis.
Infection of HeLa cells and immunostaining for dsRNA and MV N protein were performed as described previously (14). Slides were analyzed using an LSM 780 inverted laser scanning confocal microscope and ZEN 2012 (Black Edition, v8.0) software (Carl Zeiss Microscopy, Jena, Germany).
Flow cytometry analysis of coinfections.
Granta-519 cells (5 × 105) were infected with various combinations of GFP- and mCherryNLS-expressing vac2 and vac2-CKO at an MOI of 1 for each virus and fixed with 4% paraformaldehyde (Santa Cruz Biotechnology, Santa Cruz, CA) at 48 h postinfection, and fluorescent reporter expression was analyzed using a FACSCalibur cytometer (BD Biosciences, San Jose, CA) and FlowJo software (Tree Star Inc., Ashland, OR).
RESULTS
DI-RNAs are generated at a high frequency immediately after virus rescue.
Copy-back DI-RNAs have two characteristic features: their breakpoint, that is, the last nucleotide incorporated by the polymerase into the progeny genomic RNA before leaving the template antigenome strand (Fig. 1A, left end of black bar), and the reinitiation site, that is, the position of polymerase entry on the progeny RNA. Polymerase reentry results in the synthesis of a cRNA strand of opposite polarity (Fig. 1A, left end of bottom gray bar). Even if such RNA molecules have the potential of forming a dsRNA structure with a hairpin loop (Fig. 1B), they usually do not because the replicating RNAs are cotranscriptionally encapsidated. Copy-back DI-RNA can be specifically amplified by PCR from the cDNA of infected cells by the use of primer pairs in which one primer binds to the copy-back sequence (Fig. 1C, primer A1) and the second primer (primer A2, A3, or A4) binds to the DI sequence downstream of the breakpoint. Although only parts of the DI-RNA sequences are amplified with these primers (the 5′ and 3′ termini outside the primer binding sites are not amplified), the amplified sequence permits calculation of the length of the DI-RNA. Importantly, these primer combinations cannot yield a specific PCR product with standard genomes, since there they have the same polarity.
FIG 1.
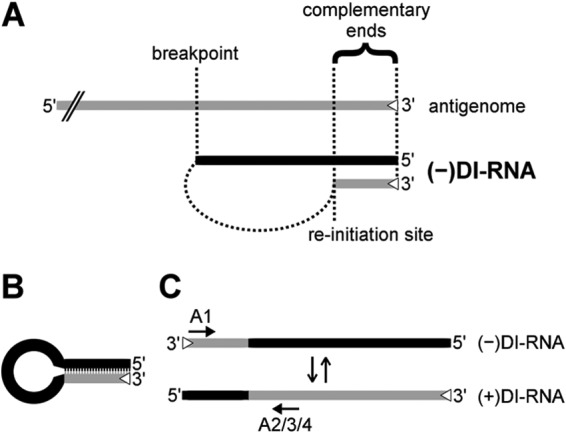
Schematic drawings of 5′ copy-back DI-RNA generation, predicted secondary structure, and replication. (A) 5′ copy-back DI-RNAs are hybrids of truncated genome (black) and antigenome (gray) sequences. During replication, viral polymerase initiates RNA synthesis at the 3′ antigenome promoter (trailer; open triangle) and prematurely terminates synthesis at the breakpoint. The polymerase then reinitiates synthesis on the newly generated strand and finishes replication by recopying the antigenomic 3′ end. The two ends of the DI-RNA are therefore perfectly complementary to each other. (B) Panhandle structure that may be formed through hybridization of the complementary ends of a copy-back DI-RNA in the absence of sufficient N protein. (C) 5′ copy-back DI-RNA replication. The genome DI-RNA [(−)DI-RNA] (top) contains the antigenome promoter in the copy-back sequence (open triangle). Therefore, it can be replicated by the viral polymerase into a complementary antigenome DI-RNA [(+)DI-RNA], which now contains the antigenome promoter at the opposite end. The polarity switch within one DI-RNA molecule allows specific amplification using combinations of primers (primer A1 with either primer A2, A3, or A4; arrows) that would bind in the same direction on a regular antigenome molecule.
To gain insights into the frequency with which DI-RNAs are generated, we analyzed individual viral plaques obtained during the rescue of vaccine (vac2; Fig. 2A, top left) or wild-type (IC323; Fig. 2A, top right) recombinant MV. In addition, we also isolated plaques of mutant viruses unable to express the C protein (vac2-CKO and IC323-CKO; Fig. 2A, bottom left and right, respectively). We generated passage 1 (P1) stocks from these plaques and used these to do infections at a low MOI of 0.1.
FIG 2.
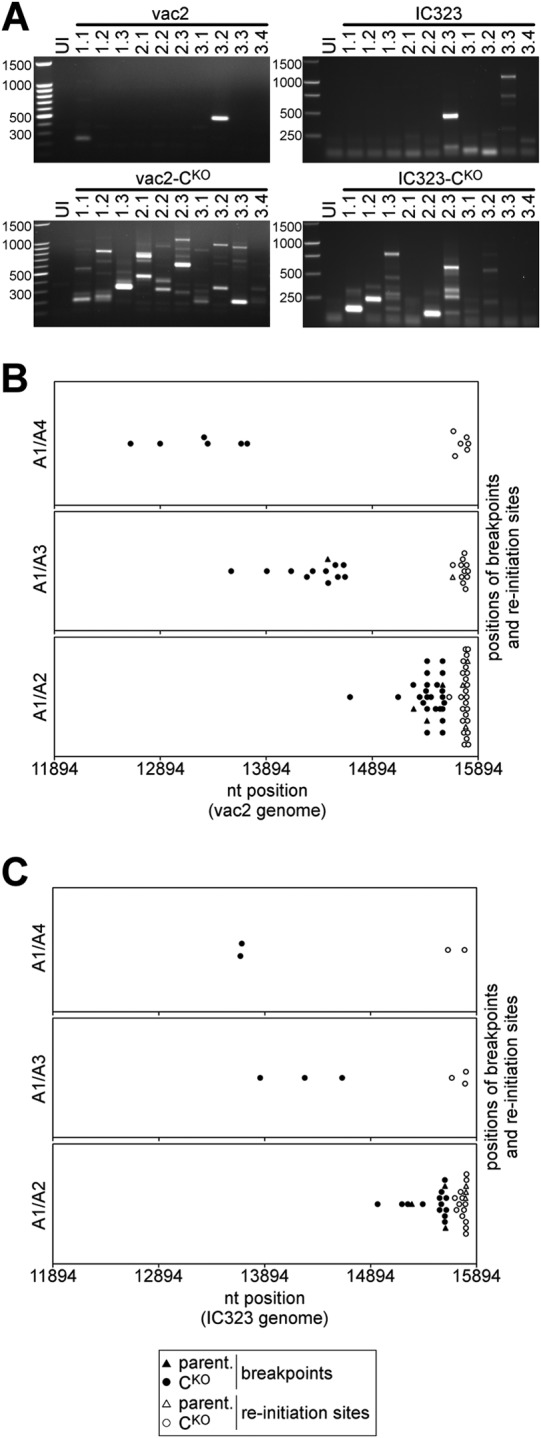
Molecular characterization of DI-RNAs generated by vaccine and wild-type MVs. (A) Analysis of DI-RNAs expressed by multiple clones of vaccine MV (vac2) and wild-type MV (IC323) able to express C protein or not (CKO). Plaques were generated in different rescue experiments, amplified, and analyzed by DI-RNA-specific RT-PCR. Uninfected cells (UI) were included as a negative control. The first number above each lane is the experiment number, and the second number is the plaque number. cDNAs were amplified with the A1/A2 primer combination and analyzed by agarose gel electrophoresis. The numbers on the left of each panel indicate the sizes of the marker bands (in base pairs). (B) Nucleotide positions of the breakpoints and reinitiation sites mapped in DI-RNAs derived from vac2 and vac2-CKO (Table 1). The combinations of primers used for amplification are indicated on the left of the three panels. (C) Breakpoint and reinitiation site analysis of IC323 and IC323-CKO viruses (Table 2).
We designed primers binding to different positions in the L gene, allowing amplification of DI-RNA sequences from less than 0.5 kb to up to 5 kb. Using the A1/A2 primer combination, DI-RNAs were readily detected in at least 2 of 10 individual plaques of C-expressing vac2 (Fig. 2A, top left) and IC323 viruses (Fig. 2A, top right). DI-RNAs were prominent in nearly all plaques of both mutant CKO viruses: multiple DI-RNAs were identified in each vac2-CKO plaque (Fig. 2A, bottom left), and 7 of 10 IC323-CKO plaques (Fig. 2A, bottom right) showed at least one DI-RNA. A unique DI-RNA pattern was detected in each plaque.
Molecular characterization of DI-RNAs from vaccine and wild-type MV strains.
To further characterize the DI-RNAs, we cloned their cDNAs and obtained the sequences of 65 independent clones. Table 1 summarizes the data for vaccine-derived DI-RNAs: 44 sequences were obtained, and 4 of these were derived from parental vac2 (top four lines) and 40 were derived from mutant vac2-CKO. Table 2 summarizes the data for IC323 virus-derived DI-RNAs: 21 sequences were obtained, and 3 of these were derived from parental IC323 (top three lines) and 18 were derived from mutant IC323-CKO. The breakpoints and reinitiation sites for all DI sequences are summarized in Fig. 2B for vac2 and in Fig. 2C for IC323 viruses. Breakpoints were distributed broadly (Fig. 2B and C). Their frequency of detection depended on the location of the primer used for amplification (primer A2, A3, or A4) and was inversely proportional to the distance from the 5′ end of the genome (Fig. 2B and C; compare the top, middle, and bottom panels). The average breakpoint position for vac2-derived DI-RNAs was nucleotide 14,843, with a standard deviation of 831 nucleotides. For IC323-derived DI-RNAs, the average breakpoint was nt 15,122 ± 681. In contrast, deduced reinitiation sites for most DI sequences clustered in a narrow region within the last 200 nucleotides of the MV genome (for vac2, nt 15,758 ± 43; for IC323, nt 15,758 ± 50) close to the region containing the sequence of primer A1 used for amplification. The total length of the DI-RNAs deduced from the positions of breakpoints and reinitiation sites (Tables 1 and 2, DI-RNA length columns) ranged from about 400 nt to about 3.5 kb. Interestingly, a few identical DI sequences appeared in virus plaques generated from independent rescue experiments. Identical DI-RNAs were assigned to the same class, labeled with the letters A to E (Tables 1 and 2, Clone class columns).
TABLE 1.
Vaccine virus-derived DI-RNAs
| Virus | Expt no. and plaque no. | DI clone | Nucleotide positiona |
Clone classb | Length (no. of nucleotides) |
Rule of sixc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Breakpoint | Reinitiation site | Insertions/deletions | dsRNA stem | ssRNA loop | DI-RNA length | |||||
| vac2 | 1.1 | 1/01 | 15560 | 15750 | A | 145 | 190 | 480 | Y | |
| 1.1 | 13/02 | 14475 | 15655 | +2 | 240 | 1,180 | 1,662 | Y | ||
| 3.2 | 2/01 | 15286 | 15796 | 99 | 510 | 708 | Y | |||
| 3.2 | 2/04 | 15412 | 15778 | B | 117 | 366 | 600 | Y | ||
| vac2-CKO | 1.1 | 3/01 | 15575 | 15765 | 130 | 190 | 450 | Y | ||
| 1.1 | 3/05 | 15560 | 15786 | C | 109 | 226 | 444 | Y | ||
| 1.2 | 24/01 | 13306 | 15759 | 136 | 2,453 | 2,725 | N | |||
| 1.2 | 24/03 | 12891 | 15659 | 236 | 2,768 | 3,240 | Y | |||
| 1.2 | 4/01 | 15535 | 15787 | 108 | 252 | 468 | Y | |||
| 1.2 | 4/06 | 15560 | 15792 | D | 103 | 232 | 438 | Y | ||
| 1.2 | 4/07 | 15560 | 15750 | A | 145 | 190 | 480 | Y | ||
| 1.3 | 17/02 | 14570 | 15786 | 109 | 1,216 | 1,434 | Y | |||
| 1.3 | 17/04 | 14129 | 15765 | 130 | 1,636 | 1,896 | Y | |||
| 1.3 | 17/05 | 14624 | 15777 | −3 | 118 | 1,153 | 1,386 | Y | ||
| 1.3 | 17/06 | 14548 | 15754 | 141 | 1,206 | 1,488 | Y | |||
| 1.3 | 25/07 | 13655 | 15741 | 154 | 2,086 | 2,394 | Y | |||
| 1.3 | 5/01 | 15494 | 15735 | +3 | 160 | 241 | 564 | Y | ||
| 1.3 | 5/05 | 15284 | 15750 | 145 | 466 | 756 | Y | |||
| 2.1 | 18/05 | 14639 | 15741 | 154 | 1,102 | 1,410 | Y | |||
| 2.1 | 18/06 | 14277 | 15767 | 128 | 1,490 | 1,746 | Y | |||
| 2.1 | 26/03 | 13713 | 15755 | 140 | 2,042 | 2,322 | Y | |||
| 2.1 | 26/05 | 12612 | 15728 | 167 | 3,116 | 3,450 | Y | |||
| 2.1 | 6/01 | 15344 | 15756 | 139 | 412 | 690 | Y | |||
| 2.1 | 6/05 | 15412 | 15778 | B | 117 | 366 | 600 | Y | ||
| 2.1 | 6/12 | 15402 | 15621 | −107 | 167 | 219 + 107 | 660 | Y | ||
| 2.2 | 19/03 | 13899 | 15791 | 104 | 1,892 | 2,100 | Y | |||
| 2.2 | 7/01 | 15450 | 15794 | 101 | 344 | 546 | Y | |||
| 2.2 | 7/12 | 15377 | 15777 | 118 | 400 | 636 | Y | |||
| 2.3 | 27/10 | 13341 | 15677 | 218 | 2,336 | 2,772 | Y | |||
| 2.3 | 8/01 | 15412 | 15778 | B | 117 | 366 | 600 | Y | ||
| 2.3 | 8/03 | 15139 | 15799 | 96 | 660 | 852 | Y | |||
| 2.3 | 8/07 | 14684 | 15792 | 103 | 1,108 | 1,314 | Y | |||
| 3.1 | 20/05 | 14334 | 15800 | 95 | 1,466 | 1,656 | Y | |||
| 3.1 | 9/02 | 15560 | 15792 | D | 103 | 232 | 438 | Y | ||
| 3.1 | 9/05 | 15557 | 15759 | 136 | 202 | 474 | Y | |||
| 3.1 | 9/11 | 15412 | 15778 | B | 117 | 366 | 600 | Y | ||
| 3.2 | 10/05 | 15412 | 15778 | B | 117 | 366 | 600 | Y | ||
| 3.2 | 10/09 | 15412 | 15802 | 93 | 390 | 576 | Y | |||
| 3.3 | 11/01 | 15560 | 15786 | C | 109 | 226 | 444 | Y | ||
| 3.3 | 21/03 | 14459 | 15735 | 160 | 1,276 | 1,596 | Y | |||
| 3.4 | 12/02 | 15560 | 15750 | A | 145 | 190 | 480 | Y | ||
| 3.4 | 12/05 | 15412 | 15778 | B | 117 | 366 | 600 | Y | ||
| 3.4 | 22/01 | 13563 | 15749 | 146 | 2,186 | 2,478 | Y | |||
| 3.4 | 22/04 | 14478 | 15655 | +5 | 240 | 1,177 | 1,662 | Y | ||
| vac-CKO(GFP) | UCSB (14) | 220 | 15507 | 15797 | 98 | 290 | 486 | Y | ||
Nucleotide positions of breakpoints and reinitiation sites as aligned to the reference sequence for the Moraten vaccine virus (GenBank accession number AF266287).
The letters A to D indicate clone classes when identical DI-RNAs were isolated from independent plaques.
The rule of six indicates whether the calculated total length of the DI-RNA is a multiple of six (yes [Y]) or not (no [N]).
TABLE 2.
Wild-type virus-derived DI-RNAs
| Virus | Expt no. and plaque no. | DI clone | Nucleotide positiona |
Clone classb | Length (no. of nucleotides) |
Rule of sixc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Breakpoint | Reinitiation site | Insertions/deletions | dsRNA stem | ssRNA loop | DI-RNA length | |||||
| IC323 | 2.1 | 29/03 | 15595 | 15793 | E | 102 | 198 | 402 | Y | |
| 2.3 | 55/04 | 15282 | 15788 | 107 | 506 | 720 | Y | |||
| 3.2 | 30/06 | 15600 | 15794 | 101 | 194 | 396 | Y | |||
| IC323-CKO | 1.1 | 57/03 | 15595 | 15793 | E | 102 | 198 | 402 | Y | |
| 1.1 | 64/01 | 14271 | 15659 | 236 | 1,388 | 1,860 | Y | |||
| 1.2 | 33/04 | 15595 | 15793 | E | 102 | 198 | 402 | Y | ||
| 1.2 | 58/01 | 15545 | 15759 | 136 | 214 | 486 | Y | |||
| 1.2 | 58/04d | 15545 | 15759 | 136 | 214 | 486 | Y | |||
| 1.3 | 34/09 | 15562 | 15688 | 207 | 126 | 540 | Y | |||
| 1.3 | 59/05 | 14960 | 15773 | 122 | 813 | 1,057 | N | |||
| 1.3 | 68/01 | 13677 | 15779 | 116 | 2,102 | 2,334 | Y | |||
| 2.2 | 60/01 | 15608 | 15798 | 97 | 190 | 384 | Y | |||
| 2.2 | 60/06e | 15608 | 15798 | 97 | 190 | 384 | Y | |||
| 2.2 | 70/01 | 13664 | 15624 | 271 | 1,960 | 2,502 | Y | |||
| 2.3 | 61/01 | 15190 | 15742 | 153 | 552 | 858 | Y | |||
| 2.3 | 61/06 | 15385 | 15703 | 192 | 318 | 702 | Y | |||
| 2.3 | 65/03 | 13850 | 15792 | 103 | 1,942 | 2,148 | Y | |||
| 3.1 | 66/10 | 14624 | 15784 | +4 | 111 | 1,160 | 1,386 | Y | ||
| 3.2 | 63/03 | 15244 | 15730 | 165 | 486 | 816 | Y | |||
| 3.3 | 40/01 | 15560 | 15786 | 109 | 226 | 444 | Y | |||
| 3.4 | 41/02 | 15595 | 15793 | E | 102 | 198 | 402 | Y | ||
Nucleotide positions of breakpoints and reinitiation sites as aligned to the reference sequence for the IC-B wild-type virus from which IC323 is derived (GenBank accession number AB016162).
The letter E indicates the clone class when identical DI-RNAs were isolated from independent plaques.
The rule of six indicates whether the calculated total length of the DI-RNA is a multiple of six (yes [Y]) or not (no [N]).
This clone differed from 58/01 of the same rescue by a biased hypermutation event.
This clone differed from 60/01 of the same rescue by a biased hypermutation event.
We also assessed whether the DI-RNAs conformed to the rule of six, which stipulates that only paramyxovirus genomes with hexameric lengths are efficiently replicated (10, 32). Indeed, the calculated total DI-RNA length was a multiple of six for 63 out of 65 clones. However, DI-RNA clones 24/01 (vac2-CKO) and 59/05 (IC323-CKO) did not comply with this rule. Interestingly, 6 out of 65 DI-RNAs possessed internal deletions or insertions (Tables 1 and 2, Insertions/deletions columns) that made them compliant with the rule of six (Tables 1 and 2, Rule of six columns). Assuming that DI-RNA generation is a purely random process, about 83% (5 out of 6) of newly generated DI-RNAs are predicted to be noncompliant with the rule of six, but because we found that only about 3% of the DI-RNAs were noncompliant, we conclude that DI-RNAs of hexameric length had a replicative advantage.
Clusters of A-to-G and U-to-C transitions in DI-RNA sequences.
DI-RNAs of negative-strand RNA viruses (19, 33) and even full-length MV genomes from diseased human brains (34) can accumulate clusters of A-to-G or U-to-C transitions, characteristic of the action of the cellular ADAR1 deaminase (35, 36). We detected clusters of A-to-G or U-to-C transitions (biased hypermutation events) in 9 DI-RNA sequences (Fig. 3A and Table 3). The percentage of nucleotides affected by these transitions varied from about 4 to 42% (average 21%). Notably, all identified clones exhibited only one type of transition, and we did not observe a switch in the type of transition at the breakpoint. Figure 3A illustrates one biased hypermutation event: 27 transitions to C among 70 U residues (39%) within the sequence prior to the breakpoint and then an additional 9 transitions to C among 41 U residues (22%) after the reinitiation site. Interestingly, the DI-RNA carried an internal deletion in the copy-back sequence that spanned 107 nt between positions 15,653 and 15,759 (Fig. 3A, dashes). The internal deletion generated a DI-RNA compliant with the rule of six.
FIG 3.

ADAR1-like hypermutations in DI-RNA clones. (A) Alignment of the sequence of DI-RNA clone 06/12 to the vac2 genome (nt 15639 to 15402) and antigenome (nt 15621 to 15887) sequences (GenBank accession number AF266287). The binding sites of the A1 and A2 primers are underlined (top line and bottom two lines, respectively). Identical nucleotides are shown as dots. The breakpoint and reinitiation site nucleotides are indicated. Deleted nucleotides are shown as dashes. (B) Frequency analysis of nucleotides flanking hypermutated sites in DI-RNA clones (all 9 events detected here; Table 3). (C) Frequency of neighboring nucleotides flanking ADAR1 hypermutation sites defined in vitro (21).
TABLE 3.
ADAR1-mediated hypermutations in DI-RNA clones
| Virus | Expt no. and plaque no. | DI clone | Polaritya | No. of hypermutations observed/no. of total nucleotidesb | % editingc |
|---|---|---|---|---|---|
| vac2-CKO | 2.1 | 06/12 | U → C | 36/111 | 31.5 |
| 2.3 | 08/03 | U → C | 6/141 | 4.3 | |
| 3.1 | 20/05 | U → C | 16/126 | 12.7 | |
| IC323-CKO | 1.2 | 58/04 | U → C | 5/52 | 9.6 |
| 1.3 | 59/03d | A → G | 17/67 | 25.4 | |
| 2.2 | 60/06 | U → C | 8/21 | 38.1 | |
| 3.1 | 62/04d | U → C | 19/54 | 35.2 | |
| 3.1 | 62/06d | A → G | 28/67 | 41.8 | |
| 3.1 | 62/11d | U → C | 11/54 | 20.4 |
Polarity indicates whether the hypermutations were observed as A → G or U → C in the genomic orientation.
The number of mutations observed/number of total nucleotides of the respective type (A or U) in a cluster.
The percentage is the calculated frequency of transitions observed over possible events.
These sequences were derived through semispecific hybridization of primer A2 at position −15670 of the MV genome.
ADAR1 is a cellular enzyme that catalyzes the deamination of adenosines in dsRNA to produce inosine, which is recognized as guanosine (35, 36). The A-to-I editing activity of ADAR1 is affected by the neighboring nucleotides of the A that is deaminated (Fig. 3C) (21). We assessed whether the nucleotides flanking the modification sites in the MV DI-RNAs conformed to the established signature for ADAR1. The neighboring nucleotide frequencies (positions −4 to +4) of all observed hypermutations (Fig. 3B) were in good agreement with the nearest neighbor signature reported by Eggington et al. (21) (Fig. 3C is reconstructed from data available in the supplemental material of the original publication [21]). Most significantly, our data set and that of Eggington et al. (21) show an underrepresentation of C and G residues at the −1 position and the overrepresentation of U at the −2 position. This suggests that ADAR1 may have acted on the DI-RNAs generated during MV infection.
DI-RNAs are encapsidated and protected by lipid envelopes.
To assess whether DI-RNAs are incorporated into viral particles, we generated two virus stocks: one without detectable DI-RNA (vac2; we started with plaque 1.2; Fig. 2A) and the other with high DI-RNA levels (vac2-CKO; we started with plaque 2.1; Fig. 2A). Since >90% of infectious MV remains cell associated, intracellular infectivity was purified using a two-step density gradient protocol (Fig. 4A). The visible bands for vac2 and vac2-CKO particles had similar intensities in both steps, suggesting that the amount of physical particles was similar for the two preparations. However, the infectious titers of the purified virus preparations differed between vac2 and vac2-CKO by about 10-fold (Fig. 4B, right columns), similar to the ratio of the original stocks (Fig. 4B, left columns).
FIG 4.

Analysis of nucleic acids in vac2- and vac2-CKO-infected cells and in purified vac2 and vac2-CKO particles. (A) Two-step gradient purification of MV particles. Virus bands are indicated by brackets. (B) Titers (numbers of TCID50s per milliliter) of virus stocks prior to and after purification. (C) RNA protection assay with purified virions. Samples were treated with the indicated combinations of trypsin, Triton X-100, and Benzonase; immunoblots were against MV H and N proteins; RT-PCR/PCR analysis was performed for DI-RNAs (A1/A2 primers) and full-length RNA (A1/B1 primers). (D) Northern blot analysis of total RNA from about 3 × 105 uninfected (UI; lane 1) or infected (lanes 2 and 3) Vero cells harvested at 32 h postinfection, as well as RNA extracted from 1 × 106 TCID50s of purified (pur.) and Benzonase-treated vac2 (lane 4) and vac2-CKO (lane 5) particles. The positions of the antigenome and L, H-L, and F-H-L mRNAs (+probe) are indicated on the right side of the left panel. The identities of the genomic RNA and the DI-RNAs (−probe) mapped by PCR amplification are indicated on the right side of the right panel. Lane M, molecular size marker. The numbers on the left side of the left panel indicate the sizes of the marker bands (in bases).
To assess whether the viral RNAs were encapsidated and protected by a lipid envelope, purified virions were treated with different combinations of nuclease (Benzonase), protease (trypsin), and nonionic detergent (Triton X-100). To control for enzymatic activities, the viral envelope glycoprotein H and nucleocapsid protein N were analyzed after treatment (Fig. 4C, top two panels). For both parental and CKO virion preparations, trypsin treatment in the absence of detergent led to the proteolytic cleavage of most H protein (Fig. 4C, lanes 4 and 9), whereas N protein was affected only to a small extent, if at all. However, when virions were treated with Triton X-100 prior to trypsinization, both H and N were completely degraded by trypsin (Fig. 4C, lanes 5 and 10).
We also extracted RNA from the samples, reverse transcribed it, and subjected the cDNA to PCR analysis using DI-RNA-specific primers (Fig. 4C, panel A1/A2) and full-length RNA-specific primers (Fig. 4C, panel A1/B1). Benzonase treatment in the absence of Triton X-100 and trypsin affected both DI-RNA- and full-length RNA levels only to a small extent (Fig. 4C, lanes 2 and 7). Treatment of virions with trypsin only prior to nuclease treatment did not affect the strength of the PCR signals (Fig. 4C, lanes 4 and 9). However, when virions were treated with Triton X-100, the viral RNA became accessible to Benzonase, leading to the complete loss of the PCR signals (Fig. 4C, lanes 3, 5, 8, and 10). We conclude that both DI-RNA and full-length RNA are encapsidated by N and that the majority of these ribonucleocapsids (RNPs) are protected by a lipid envelope. Notably, treatment of the RNPs with trypsin was not required when virions were treated with detergent (Fig. 4C, lanes 3 and 8), indicating that RNPs became nuclease sensitive after Triton X-100 treatment.
High levels of DI-RNA genomes are found in particles.
To further characterize the viral RNP, after Benzonase treatment we extracted RNA from 1 × 106 TCID50s of purified virions. As a control, we also extracted total RNA from Vero cells infected with parental and CKO viruses and from uninfected cells. These samples were analyzed by Northern blotting using single-strand RNA probes complementary to the L/trailer region of the antigenome (Fig. 4D, +probe) or genome (Fig. 4D, −probe).
Figure 4D shows an analysis of the antigenomic (left) and genomic (right) RNA from uninfected cells, infected cells, and purified virus particles. As expected, the 16-kb full-length antigenomic and genomic RNAs were detected in all four virus samples. However, whereas the antigenome bands for vac2 and vac2-CKO in infected cells had similar intensities (Fig. 4D, left; compare lanes 2 and 3), vac2-CKO showed less genomic RNA than vac2 (Fig. 4D, right; compare lanes 2 and 3), correlating with the reduced infectivity recovered from vac2-CKO-infected cells compared to the infectivity recovered from vac2-infected cells. The difference in intensity of the genome band was not detected in purified virion samples (Fig. 4D, right; compare lanes 4 and 5), since we loaded RNA from equal numbers of infectious units of both viruses. However, antigenome ratios were altered in purified virions (Fig. 4D, left; compare lanes 4 and 5), indicating that vac2-CKO had incorporated higher levels of the antigenome into virions than vac2. For total RNA samples from infected cells, the antigenome probe also detected bands for L mRNA (about 7 kb) and two bands that corresponded to polycistronic H-L (9 kb) and F-H-L mRNAs (11 kb) (37). As expected, the L, H-L, and F-H-L mRNAs were not detected in the purified viruses.
Using either the antigenome probe or the genome probe, we observed multiple RNA species with a smaller molecular size that were present only in vac2-CKO samples. The sizes of these bands ranged from 0.1 kb to about 7 kb, with most being in the 0.3- to 2-kb range. The sizes of some RNAs were similar to those of specific DI-RNAs of vac2-CKO plaque 1 from experiment 2 (referred to as 2.1), as identified by the PCR cloning approach (Table 1; compare the DI-RNA clone identifiers with those marked in Fig. 4D). Similarly sized RNAs were also detected, but in greater abundance, in purified vac2-CKO virions (Fig. 4D, right, lane 5). We detected more RNA species with the genome probe than with the antigenome probe, consistent with the notion that these RNAs may be truncated genomes. We conclude that DI-RNAs are incorporated into intracellular particles since they are protected by a lipid envelope. The 10-fold reduced infectivity of vac2-CKO correlates with the enhanced incorporation of DI-RNA genomes into intracellular particles.
CKO viruses generate dsRNA very early.
Since DI-RNAs are most efficiently amplified at a high MOI, we sought to limit cell-cell fusion, a process resulting in the bulk transfer of genomes. We therefore carried out infections on Vero cells, rather than on the more fusogenic Vero-hSLAM cells. Because we needed viruses expressing an alternative fluorescent reporter protein for subsequent competition experiments, we generated vac2-based variants expressing mCherry (mCherryNLS [28]). Figure 5A shows protein expression and DI-RNA levels after two passages of the vac2-based viruses expressing either GFP or mCherryNLS. As expected, passage 1 DI-RNA amplification in Vero cells was minimally detectable, if it was detectable at all (lanes P1, A1/A2 panel). However, significant amounts of DI-RNA-specific PCR products were detected for both mutant CKO viruses at passage 2 but not for parental vac2 (lanes P2, A1/A2 panel).
FIG 5.
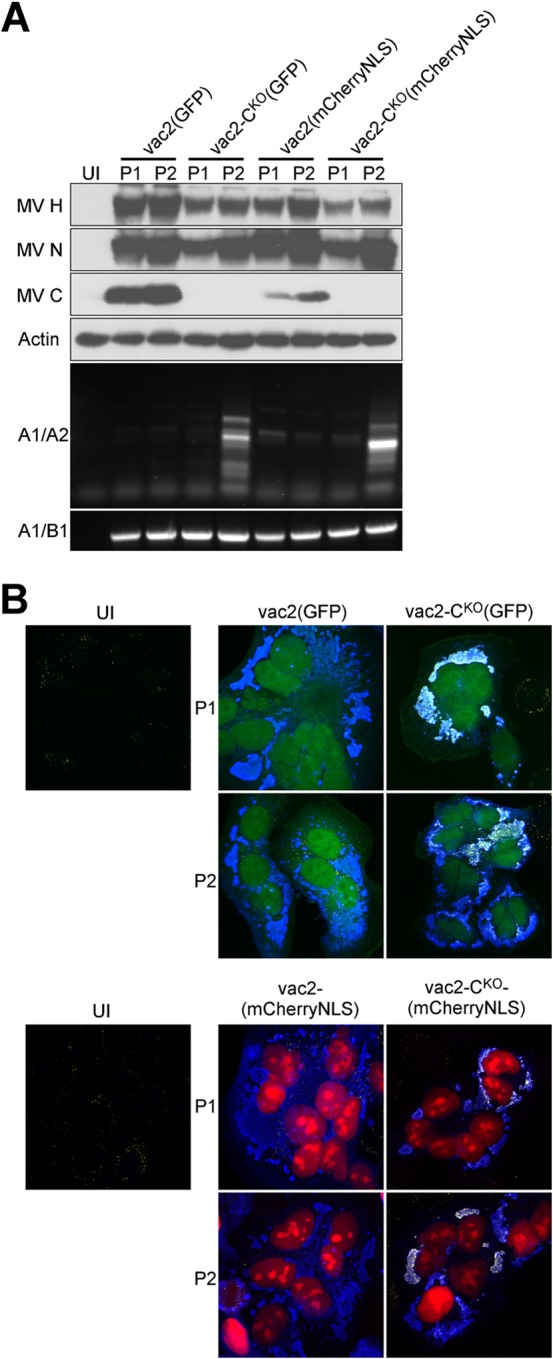
DI-RNA and dsRNA generation by CKO viruses expressing either GFP or mCherryNLS. (A) Western blot and PCR analysis of P1 and P2 of fluorescent reporter-expressing vac2 and vac2-CKO. The antisera used for detection are indicated on the left of the top four panels. Primer pairs used for amplification are indicated on the left of the bottom two panels. UI, uninfected cells. (B) Confocal microscope analysis of HeLa cells infected with fluorescent reporter-expressing vac2 and vac2-CKO. (Top) Autofluorescence of GFP (green); (bottom) autofluorescence of mCherryNLS (red); (all panels) immunofluorescence staining for dsRNA (yellow) and MV N protein (blue). Magnifications, ×40.
Having previously documented the kinetics of dsRNA production of standard and CKO viruses in HeLa cells (14), we infected these cells and performed immunofluorescence staining for viral N protein and dsRNA (Fig. 5B). We found strong dsRNA staining (Fig. 5B, yellow, seen as white when superimposed onto blue) colocalizing with viral replication sites (revealed by N protein; Fig. 5B, blue signal) in passages 1 and 2 of either CKO virus. In contrast, the C-expressing parental vac2 of either passage did not elicit detectable dsRNA staining at viral replication sites. Thus, immunofluorescence staining detects dsRNA before DI-RNA can be detected by PCR amplification.
The CKO phenotype is dominant.
To assess whether C-protein expression in trans can limit DI-RNA amplification, we coinfected cells with virus stocks possessing either high or undetectable DI-RNA levels. These viruses expressed different fluorescent proteins (either GFP or mCherryNLS) for easy replication tracking. We infected interferon response-defective Granta-519 cells with each virus at an equivalent MOI and quantified the GFP and mCherry expression at 48 h postinoculation by fluorescence-activated cell sorter analysis (Fig. 6).
FIG 6.
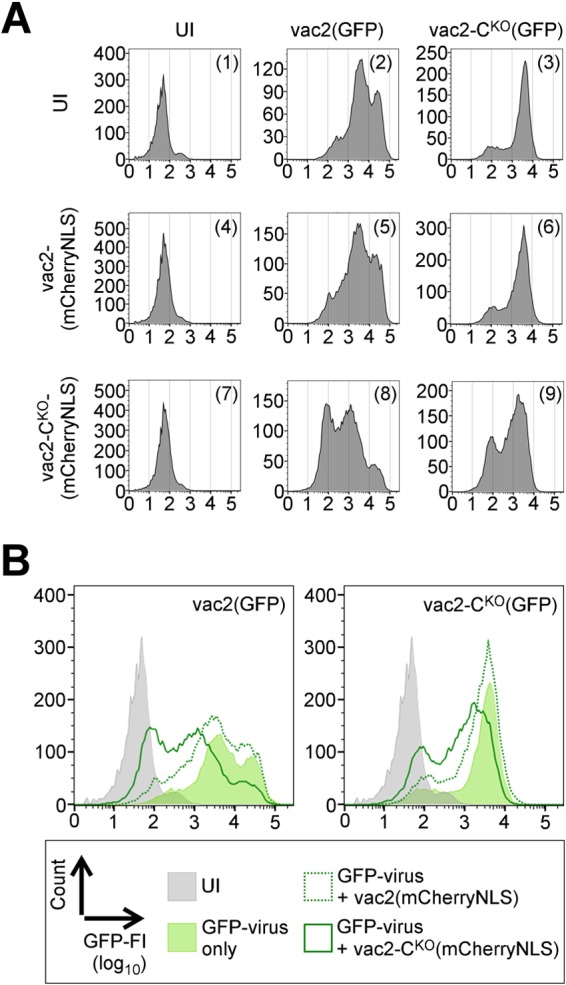
Dominance of the CKO phenotype. (A) GFP fluorescence intensities of cells coinfected with viruses indicated on the top and on the left. UI, uninfected cells. Vertical axis, cell counts (linear scale); horizontal axis, GFP fluorescence intensity (FI; log10 scale). (B) Overlay of graphs from panel A. (Left) Summary of coinfections with vac2(GFP); (right) summary of coinfections with vac2-CKO(GFP). Gray area, uninfected cells (panel 1); light green area, single GFP-virus infection (panel 2 or 3); dotted green line, GFP-virus coinfected with vac2(mCherryNLS) (panel 5 or 6); solid green line, GFP-virus coinfected with vac2-CKO(mCherryNLS) (panel 8 or 9).
Figure 6A shows the levels of GFP expression in cells left uninfected (panel 1) or infected only with either virus expressing GFP [vac2(GFP) in panel 2; vac2-CKO(GFP) in panel 3] or only with either virus expressing mCherryNLS [vac2(mCherryNLS) in panel 4; vac2-CKO(mCherryNLS) in panel 7]. Panels 5, 6, 8, and 9 document the levels of GFP expression in cells coinfected with two viruses, as indicated on the top of each column and on the left of each row. The median fluorescence intensity (MFI) of vac2(GFP) (panel 2) was about 4,500, or 110 times over the background MFI of 41 (panel 1); the MFI of vac2-CKO(GFP) (panel 3) was about 70 times over the background MFI. Infection with mCherryNLS-expressing viruses was not detected in the GFP channel (panels 4 and 7).
We then measured GFP expression levels in cells coinfected with 1:1 mixtures of viruses that were either DI-RNA poor (vac2-derived stocks) or DI-RNA rich (vac2-CKO-derived stocks). We undertook these mixed infections to assess whether DI-RNA-rich stocks interfere with the replication of DI-RNA-poor stocks (dominant negative effects). Alternatively, this experiment might reveal a positive effect of DI-RNA-poor (C-protein-expressing) stocks on the replication of DI-RNA-rich stocks, presumably through trans-complementation by the C protein.
Figure 6B illustrates the results of mixed infection analyses. Figure 6B, left, shows that addition of a DI-RNA-poor stock [vac2(mCherryNLS)line] to another DI-RNA-poor stock [vac2(GFP)] reduced the GFP MFI about 1.5-fold (compare the light green area with the dark green dotted line). Addition of a DI-RNA-rich stock [vac2-CKO(mCherryNLS)] reduced the GFP MFI of the DI-RNA-poor stock more than 8-fold (compare the light green area with the dark green solid line). In contrast, as shown in Fig. 6B, right, addition of a DI-RNA-poor stock [vac2(mCherryNLS)line] to a DI-RNA-rich stock [vac2-CKO(GFP)] reduced the GFP MFP 1.2-fold (compare the light green area with the dark green dotted line), while addition of a DI-RNA-rich stock [vac2-CKO(mCherryNLS)] to another DI-RNA-rich stock had a more pronounced negative effect (a 3.5-fold reduction of the GFP MFI; compare the light green area with the dark green solid line). Because DI-RNA-rich virus stocks in any combination had a negative effect on the growth of the DI-RNA-poor stocks and because DI-RNA-poor stocks which expressed C protein had no positive effect on DI-RNA-rich stocks, we conclude that the CKO effect is dominant.
DISCUSSION
MV C-protein function.
Viruses of the Morbillivirus, Henipavirus, and Respirovirus genera of the Paramyxoviridae family express one or more C proteins (2). However, different studies have revealed alternative functions of C proteins. In the case of MV and Nipah virus, minigenome assays have suggested that C protein downregulates genome replication (38, 39). Inhibition of transcription but not of replication was described for Sendai virus (SeV) in in vitro assays (40), although SeV C protein also is reported to be essential for the maintenance of negative-strand RNA genomes in virus particles (41) and an SeV-CKO mutant did not undergo a shift from early (antigenome-dominated) to late (genome-dominated) replication (42). The generation and accumulation of DI-RNAs in C-defective paramyxoviruses would account for many of these observations.
In the absence of C protein, production of full-length genomes would be reduced and defective genomes would be incorporated into virions instead. In addition, the dsRNA structures formed by DI-RNAs are known to activate PKR (14), which induces an arrest in protein synthesis via the cellular stress response, thereby delaying viral replication. While in a preceding study we detected DI-RNA in one MV-CKO stock that had been passaged several times at a low multiplicity of infection (14), here we provide evidence for the generation of DI-RNAs as early as passage 1 following virus rescue. Moreover, we observed about 10 times more DI-RNA in CKO virus infection than in parental virus infection, implying a genetic link between the CKO phenotype and DI-RNA generation. Notably, CKO mutants of SeV and human parainfluenza virus type 1 have also been reported to generate double-stranded RNA and activate PKR, but these studies did not address the origin or the structure of these RNAs (43, 44). Although CKO mutants of several paramyxoviruses can be rescued and are only moderately attenuated in cultured cells (16), they are strongly attenuated in vivo (45, 46). Thus, C proteins of diverse paramyxoviruses may share a similar function, to enhance RNA replication processivity.
The frequency but not the quality of DI-RNA generation is determined by the C protein. Most breakpoints of the DI-RNAs are widely distributed along the MV genome, with the frequency of the appearance of a breakpoint at a specific position being inversely proportional to the distance from the 5′ end of the genome. In contrast, reinitiation sites are clustered in a narrow region close to the 5′ end of the genome. We note, however, that repositioning of the copy-back primer (Fig. 1C, primer A1) to a more internal binding site resulted in the detection of DI-RNAs with more internal reinitiation sites (data not shown).
Our Northern blot analysis showed several RNAs hybridizing with the genome probe (Fig. 4C, right) that did not hybridize with the antigenome probe (Fig. 4C, left) and whose sizes did not correspond to the calculated sizes of any cloned DI-RNA. These species may possibly reflect truncated genomes lacking the copy-back sequence. Indeed, several kinds of defective replication products have been observed for vesicular stomatitis virus (19, 47) and SeV (48, 49).
The CKO phenotype of MV is dominant: a C-protein-defective virus inhibited the replication efficiency of a C-protein-expressing virus in trans. On the other hand, a C-protein-expressing virus did not prevent DI-RNA replication once DI-RNA had been generated. Conceivably, the C protein might function together with the polymerase complex in separating the nascent and template RNA hybrid, or, alternatively, C protein might support the rapid cotranscriptional encapsidation of the nascent RNA with N protein. Future studies will discriminate between these possibilities.
ADAR1-mediated editing and the PKR-ADAR balancing model.
DI-RNAs of negative-strand RNA viruses are potent triggers of innate immune receptors, including PKR, mda-5, and RIG-I (12–14), thereby inducing the expression of type I IFNs in vitro (50) and in vivo (51). The MV DI-RNAs characterized here, if not cotranscriptionally encapsidated, form perfect hairpins throughout the double-stranded structure. These hairpins are ADAR1 substrates in vitro (23). Destabilization of these hairpins through A-to-I editing could account for the proviral function of ADAR1, which was documented in several viral infections and attributed to suppression of activation of innate immune responses (52, 53). For example, in cell lines deficient in ADAR1 expression, MV strongly induces IFN (54), PKR activation (55), and the cellular stress response (56, 57). Furthermore, higher levels of dsRNA accumulate in ADAR1-deficient cells infected with MV than in cells sufficient in ADAR1 expression (14).
Herein we provide evidence for the enzymatic activity of ADAR1 on MV DI-RNAs. We identified several DI-RNA sequences bearing clusters of A-to-G or U-to-C mutations and show through a computational approach that the sequences flanking the edited nucleotides, as well as the average editing frequency, were characteristic of ADAR1, but not ADAR2 (21). Of the three known ADARs, only the IFN-inducible ADAR1 isoform (also called p150) is cytoplasmic (58, 59) and localizes to stress granules (56). These granules are induced during MV-CKO infection and form in close proximity to viral replication sites (14). In contrast, the constitutively expressed isoforms of ADAR1 (p110) and ADAR2 are exclusively found in the cell nuclei (53). Thus, the subcellular cytoplasmic localization of only ADAR1-p150 is correct for editing of MV RNAs, which are exclusively replicated in the cytoplasm (2).
Notably, in individual DI-RNAs the transitions were of the same type (either A to G or U to C) and occurred on both sides of the breakpoint and reinitiation site. This pattern indicates that the generated DI-RNAs were direct substrates for editing by ADAR1 but not the full-length MV genomes. If the latter were the case, the polarity switch between breakpoint and reinitiation site would have also caused the ADAR1 transitions to change within the DI-RNA molecule. Thus, our findings suggest that ADAR1 editing destabilizes unencapsidated DI-RNAs that have dsRNA structures. These unencapsidated DI-RNAs may be the double-stranded RNA that elicits innate immune responses in negative-strand RNA virus infections. ADAR1 A-to-I editing has also been documented in full-length RNA virus genomes (33, 34, 60, 61) and was recently inferred by deep sequencing analyses of several paramyxoviruses (62, 63).
DI-RNAs as a frequent by-product of wild-type and vaccine-lineage infections.
DI-RNAs were originally detected in laboratory strains propagated at a high multiplicity (9) and later also in commercial vaccines (64, 65). Here we establish that DI-RNAs can accumulate in the earliest passage available for analysis following the initial virus rescue. Moreover, we documented DI-RNA generation in a wild-type MV strain, thereby providing evidence that DI-RNAs are not restricted to vaccine-lineage viruses and have the potential to influence virulence.
DI-RNAs may alter the characteristics of MV live-attenuated vaccines and modulate the efficacy of recombinant MV engineered for vaccination against heterologous pathogens or used as oncolytic vectors (27, 66–68). While vaccine producers seek to avoid DI-RNAs in order to standardize efficacy, oncolytic potency may not rely uniquely on efficient viral replication and cell lysis but may also rely on immunostimulatory properties (69, 70). The balance between replication efficiency and immune activation of an oncolytic vector may be an important key to a successful therapy, and immunostimulatory DI-RNAs might be a desirable feature of certain oncolytic vectors. Further studies of the MV C protein and its function may lead to the design of viruses that strike an optimal balance between replication efficiency and immune activation.
ACKNOWLEDGMENTS
This work was supported in part by research grant AI-63476 (to R.C.) and research grants AI-12520 and AI-20611 (to C.E.S.) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, U.S. Public Health Service, and by research fellowship PF791/1-1 from the Deutsche Forschungsgemeinschaft (to C.K.P.). G.M.M., W.E.M., and X.M. were supported in part by the Mayo Graduate School.
We thank Patricia Devaux, Marie Frenzke, Chanakha Navaratnarajah, Crystal Mendoza, and Ryan Donohue for critically reading and discussing the manuscript.
REFERENCES
- 1.Rima BK, Duprex WP. 2009. The measles virus replication cycle. Curr Top Microbiol Immunol 329:77–102. doi: 10.1007/978-3-540-70523-9_5. [DOI] [PubMed] [Google Scholar]
- 2.Lamb RA, Parks GD. 2013. Paramyxoviridae, p 957–995. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Griffin DE. 2013. Measles virus, p 1042–1069. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Longhi S. 2009. Nucleocapsid structure and function. Curr Top Microbiol Immunol 329:103–128. doi: 10.1007/978-3-540-70523-9_6. [DOI] [PubMed] [Google Scholar]
- 5.Schrag SJ, Rota PA, Bellini WJ. 1999. Spontaneous mutation rate of measles virus: direct estimation based on mutations conferring monoclonal antibody resistance. J Virol 73:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol Mol Biol Rev 76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domingo E, Holland JJ. 1997. RNA virus mutations and fitness for survival. Annu Rev Microbiol 51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 8.Moya A, Elena SF, Bracho A, Miralles R, Barrio E. 2000. The evolution of RNA viruses: a population genetics view. Proc Natl Acad Sci U S A 97:6967–6973. doi: 10.1073/pnas.97.13.6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolakofsky D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555. doi: 10.1016/0092-8674(76)90223-3. [DOI] [PubMed] [Google Scholar]
- 10.Calain P, Roux L. 1993. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol 67:4822–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holland JJ, Kennedy SIT, Semler BL, Jones CL, Roux L, Grabau EA. 1980. Defective interfering RNA viruses and the host-cell response, p 137–192. In Fraenkel-Conrat H, Wagner RR (ed), Comprehensive virology, vol 16 Plenum Press, New York, NY. [Google Scholar]
- 12.Baum A, Sachidanandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Runge S, Sparrer KM, Lassig C, Hembach K, Baum A, Garcia-Sastre A, Soding J, Conzelmann KK, Hopfner KP. 2014. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog 10:e1004081. doi: 10.1371/journal.ppat.1004081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfaller CK, Radeke MJ, Cattaneo R, Samuel CE. 2014. Measles virus C protein impairs production of defective copyback double-stranded viral RNA and activation of protein kinase R. J Virol 88:456–468. doi: 10.1128/JVI.02572-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang AS. 1973. Defective interfering viruses. Annu Rev Microbiol 27:101–117. doi: 10.1146/annurev.mi.27.100173.000533. [DOI] [PubMed] [Google Scholar]
- 16.Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. 2007. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 360:72–83. doi: 10.1016/j.virol.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 17.Takeda M, Takeuchi K, Miyajima N, Kobune F, Ami Y, Nagata N, Suzaki Y, Nagai Y, Tashiro M. 2000. Recovery of pathogenic measles virus from cloned cDNA. J Virol 74:6643–6647. doi: 10.1128/JVI.74.14.6643-6647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcus PI, Gaccione C. 1989. Interferon induction by viruses. XIX. Vesicular stomatitis virus—New Jersey: high multiplicity passages generate interferon-inducing, defective-interfering particles. Virology 171:630–633. [DOI] [PubMed] [Google Scholar]
- 19.O'Hara PJ, Nichol ST, Horodyski FM, Holland JJ. 1984. Vesicular stomatitis virus defective interfering particles can contain extensive genomic sequence rearrangements and base substitutions. Cell 36:915–924. doi: 10.1016/0092-8674(84)90041-2. [DOI] [PubMed] [Google Scholar]
- 20.Huang AS, Baltimore D. 1970. Defective viral particles and viral disease processes. Nature 226:325–327. doi: 10.1038/226325a0. [DOI] [PubMed] [Google Scholar]
- 21.Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wulff BE, Sakurai M, Nishikura K. 2011. Elucidating the inosinome: global approaches to adenosine-to-inosine RNA editing. Nat Rev Genet 12:81–85. doi: 10.1038/nrg2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehmann KA, Bass BL. 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 24.Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol 75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter MA. 1995. Rescue of measles viruses from cloned DNA. EMBO J 14:5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jadayel DM, Lukas J, Nacheva E, Bartkova J, Stranks G, De Schouwer PJ, Lens D, Bartek J, Dyer MJ, Kruger AR, Catovsky D. 1997. Potential role for concurrent abnormalities of the cyclin D1, p16CDKN2 and p15CDKN2B genes in certain B cell non-Hodgkin's lymphomas. Functional studies in a cell line (Granta 519). Leukemia 11:64–72. [DOI] [PubMed] [Google Scholar]
- 27.del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. 2007. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J Virol 81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor MP, Kobiler O, Enquist LW. 2012. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc Natl Acad Sci U S A 109:17046–17051. doi: 10.1073/pnas.1212926109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toth AM, Devaux P, Cattaneo R, Samuel CE. 2009. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J Virol 83:961–968. doi: 10.1128/JVI.01669-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cathomen T, Naim HY, Cattaneo R. 1998. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol 72:1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schonborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res 19:2993–3000. doi: 10.1093/nar/19.11.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rager M, Vongpunsawad S, Duprex WP, Cattaneo R. 2002. Polyploid measles virus with hexameric genome length. EMBO J 21:2364–2372. doi: 10.1093/emboj/21.10.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cattaneo R. 1994. Biased (A → I) hypermutation of animal RNA virus genomes. Curr Opin Genet Dev 4:895–900. doi: 10.1016/0959-437X(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 34.Cattaneo R, Schmid A, Eschle D, Baczko K, ter Meulen V, Billeter MA. 1988. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell 55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. 1994. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci U S A 91:11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bass BL, Weintraub H. 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55:1089–1098. doi: 10.1016/0092-8674(88)90253-X. [DOI] [PubMed] [Google Scholar]
- 37.Cattaneo R, Rebmann G, Schmid A, Baczko K, ter Meulen V, Billeter MA. 1987. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J 6:681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bankamp B, Wilson J, Bellini WJ, Rota PA. 2005. Identification of naturally occurring amino acid variations that affect the ability of the measles virus C protein to regulate genome replication and transcription. Virology 336:120–129. doi: 10.1016/j.virol.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 39.Sleeman K, Bankamp B, Hummel KB, Lo MK, Bellini WJ, Rota PA. 2008. The C, V and W proteins of Nipah virus inhibit minigenome replication. J Gen Virol 89:1300–1308. doi: 10.1099/vir.0.83582-0. [DOI] [PubMed] [Google Scholar]
- 40.Curran J, Marq JB, Kolakofsky D. 1992. The Sendai virus nonstructural C proteins specifically inhibit viral mRNA synthesis. Virology 189:647–656. doi: 10.1016/0042-6822(92)90588-G. [DOI] [PubMed] [Google Scholar]
- 41.Irie T, Nagata N, Yoshida T, Sakaguchi T. 2008. Paramyxovirus Sendai virus C proteins are essential for maintenance of negative-sense RNA genome in virus particles. Virology 374:495–505. doi: 10.1016/j.virol.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 42.Irie T, Okamoto I, Yoshida A, Nagai Y, Sakaguchi T. 2014. Sendai virus C proteins regulate viral genome and antigenome synthesis to dictate the negative genome polarity. J Virol 88:690–698. doi: 10.1128/JVI.02798-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boonyaratanakornkit J, Bartlett E, Schomacker H, Surman S, Akira S, Bae YS, Collins P, Murphy B, Schmidt A. 2011. The C proteins of human parainfluenza virus type 1 limit double-stranded RNA accumulation that would otherwise trigger activation of MDA5 and protein kinase R. J Virol 85:1495–1506. doi: 10.1128/JVI.01297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeuchi K, Komatsu T, Kitagawa Y, Sada K, Gotoh B. 2008. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J Virol 82:10102–10110. doi: 10.1128/JVI.00599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Devaux P, Hodge G, McChesney MB, Cattaneo R. 2008. Attenuation of V- or C-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J Virol 82:5359–5367. doi: 10.1128/JVI.00169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathieu C, Guillaume V, Volchkova VA, Pohl C, Jacquot F, Looi RY, Wong KT, Legras-Lachuer C, Volchkov VE, Lachuer J, Horvat B. 2012. Nonstructural Nipah virus C protein regulates both the early host proinflammatory response and viral virulence. J Virol 86:10766–10775. doi: 10.1128/JVI.01203-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Timm C, Akpinar F, Yin J. 2014. Quantitative characterization of defective virus emergence by deep sequencing. J Virol 88:2623–2632. doi: 10.1128/JVI.02675-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Re GG, Gupta KC, Kingsbury DW. 1983. Sequence of the 5′ end of the Sendai virus genome and its variable representation in complementary form at the 3′ ends of copy-back defective interfering RNA species: identification of the L gene terminus. Virology 130:390–396. doi: 10.1016/0042-6822(83)90093-4. [DOI] [PubMed] [Google Scholar]
- 49.Re GG, Gupta KC, Kingsbury DW. 1983. Genomic and copy-back 3′ termini in Sendai virus defective interfering RNA species. J Virol 45:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shivakoti R, Siwek M, Hauer D, Schultz KL, Griffin DE. 2013. Induction of dendritic cell production of type I and type III interferons by wild-type and vaccine strains of measles virus: role of defective interfering RNAs. J Virol 87:7816–7827. doi: 10.1128/JVI.00261-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mercado-Lopez X, Cotter CR, Kim WK, Sun Y, Munoz L, Tapia K, Lopez CB. 2013. Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 31:5713–5721. doi: 10.1016/j.vaccine.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfaller CK, Li Z, George CX, Samuel CE. 2011. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol 23:573–582. doi: 10.1016/j.coi.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samuel CE. 2011. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Z, Okonski KM, Samuel CE. 2012. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J Virol 86:3787–3794. doi: 10.1128/JVI.06307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toth AM, Li Z, Cattaneo R, Samuel CE. 2009. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem 284:29350–29356. doi: 10.1074/jbc.M109.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okonski KM, Samuel CE. 2013. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J Virol 87:756–766. doi: 10.1128/JVI.02270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.John L, Samuel CE. 2014. Induction of stress granules by interferon and down-regulation by the cellular RNA adenosine deaminase ADAR1. Virology 454-455:299–310. doi: 10.1016/j.virol.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.George CX, John L, Samuel CE. 2014. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J Interferon Cytokine Res 34:437–446. doi: 10.1089/jir.2014.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patterson JB, Samuel CE. 1995. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15:5376–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cattaneo R, Schmid A, Spielhofer P, Kaelin K, Baczko K, ter Meulen V, Pardowitz J, Flanagan S, Rima BK, Udem SA, Billeter MA. 1989. Mutated and hypermutated genes of persistent measles viruses which caused lethal human brain diseases. Virology 173:415–425. doi: 10.1016/0042-6822(89)90554-0. [DOI] [PubMed] [Google Scholar]
- 61.Otani S, Ayata M, Takeuchi K, Takeda M, Shintaku H, Ogura H. 2014. Biased hypermutation occurred frequently in a gene inserted into the IC323 recombinant measles virus during its persistence in the brains of nude mice. Virology 462-463:91–97. doi: 10.1016/j.virol.2014.05.035. [DOI] [PubMed] [Google Scholar]
- 62.van den Hoogen BG, van Boheemen S, de Rijck J, van Nieuwkoop S, Smith DJ, Laksono B, Gultyaev A, Osterhaus AD, Fouchier RA. 2014. Excessive production and extreme editing of human metapneumovirus defective interfering RNA is associated with type I IFN induction. J Gen Virol 95:1625–1633. doi: 10.1099/vir.0.066100-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rima BK, Gatherer D, Young DF, Norsted H, Randall RE, Davison AJ. 2014. Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J Virol 88:3826–3836. doi: 10.1128/JVI.03351-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bellocq C, Mottet G, Roux L. 1990. Wide occurrence of measles virus subgenomic RNAs in attenuated live-virus vaccines. Biologicals 18:337–343. doi: 10.1016/1045-1056(90)90039-3. [DOI] [PubMed] [Google Scholar]
- 65.Calain P, Roux L. 1988. Generation of measles virus defective interfering particles and their presence in a preparation of attenuated live-virus vaccine. J Virol 62:2859–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pfaller CK, Cattaneo R, Schnell MJ. 2015. Reverse genetics of Mononegavirales: how they work, new vaccines, and new cancer therapeutics. Virology 479-480C:331–344. doi: 10.1016/j.virol.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramsauer K, Schwameis M, Firbas C, Mullner M, Putnak RJ, Thomas SJ, Despres P, Tauber E, Jilma B, Tangy F. 2015. Immunogenicity, safety, and tolerability of a recombinant measles-virus-based chikungunya vaccine: a randomised, double-blind, placebo-controlled, active-comparator, first-in-man trial. Lancet Infect Dis 15:519–527. doi: 10.1016/S1473-3099(15)70043-5. [DOI] [PubMed] [Google Scholar]
- 68.Stebbings R, Fevrier M, Li B, Lorin C, Koutsoukos M, Mee E, Rose N, Hall J, Page M, Almond N, Voss G, Tangy F. 2012. Immunogenicity of a recombinant measles-HIV-1 clade B candidate vaccine. PLoS One 7:e50397. doi: 10.1371/journal.pone.0050397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, Vile RG. 2009. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther 20:1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miest TS, Cattaneo R. 2014. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol 12:23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]