

ABSTRACT
Oncolytic viruses (OV) preferentially kill cancer cells due in part to defects in their antiviral responses upon exposure to type I interferons (IFNs). However, IFN responsiveness of some tumor cells confers resistance to OV treatment. The human type I IFNs include one IFN-β and multiple IFN-α subtypes that share the same receptor but are capable of differentially inducing biological responses. The role of individual IFN subtypes in promoting tumor cell resistance to OV is addressed here. Two human IFNs which have been produced for clinical use, IFN-α2a and IFN-β, were compared for activity in protecting human head and neck squamous cell carcinoma (HNSCC) lines from oncolysis by vesicular stomatitis virus (VSV). Susceptibility of HNSCC lines to killing by VSV varied. VSV infection induced increased production of IFN-β in resistant HNSCC cells. When added exogenously, IFN-β was significantly more effective at protecting HNSCC cells from VSV oncolysis than was IFN-α2a. In contrast, normal keratinocytes and endothelial cells were protected equivalently by both IFN subtypes. Differential responsiveness of tumor cells to IFN-α and -β was further supported by the finding that autocrine IFN-β but not IFN-α promoted survival of HNSCC cells during persistent VSV infection. Therefore, IFN-α and -β differentially affect VSV oncolysis, justifying the evaluation and comparison of IFN subtypes for use in combination with VSV therapy. Pairing VSV with IFN-α2a may enhance selectivity of oncolytic VSV therapy for HNSCC by inhibiting VSV replication in normal cells without a corresponding inhibition in cancer cells.
IMPORTANCE There has been a great deal of progress in the development of oncolytic viruses. However, a major problem is that individual cancers vary in their sensitivity to oncolytic viruses. In many cases this is due to differences in their production and response to interferons (IFNs). The experiments described here compared the responses of head and neck squamous cell carcinoma cell lines to two IFN subtypes, IFN-α2a and IFN-β, in protection from oncolytic vesicular stomatitis virus. We found that IFN-α2a was significantly less protective for cancer cells than was IFN-β, whereas normal cells were equivalently protected by both IFNs. These results suggest that from a therapeutic standpoint, selectivity for cancer versus normal cells may be enhanced by pairing VSV with IFN-α2a.
INTRODUCTION
The use of viruses to selectively kill cancer cells (oncolytic virotherapy) is a promising alternative therapy for cancer (1). The basis for this treatment approach is that cancer cells frequently have defective antiviral responses that develop as a consequence of cellular transformation (2–5). As a result, they are more susceptible than their normal cellular counterparts to infection and apoptotic death induced by cytopathic viruses (6, 7). Vesicular stomatitis virus (VSV), a negative-strand RNA virus of the family Rhabdoviridae, is being investigated as an oncolytic agent for the treatment of prostate (6, 8), skin (9, 10), colorectal (2, 11, 12), pancreatic (13), brain (14, 15), and other cancers. A variety of attenuated VSV strains have been engineered to express heterologous genes to increase selectivity for tumor cells, to augment tumor cell killing, or to enhance antitumor immunity (reviewed in reference 16). Recombinant VSV strains have produced encouraging preclinical results for a broad range of tumor types (17–22), and VSV expressing the human beta interferon (IFN-β) gene currently is undergoing a phase I clinical trial for the treatment of hepatocellular carcinoma (http://www.clinicaltrials.gov/ct2/show/study/NCT01628640).
Because normal cells respond to viral infection by mounting a protective type I IFN response while many cancer cells do not share this capability (7), the selectivity of VSV for cancer cells has been improved by combination treatment with type I IFN, for example, by incorporating the gene for IFN-β into the VSV genome or by the use of mutant or engineered VSV strains that induce robust IFN production (2, 6, 18). Wild-type VSV inhibits host antiviral responses by globally suppressing host-directed gene expression due to the activity of the viral matrix (M) protein (2, 23–25). M protein mutant strains of VSV, such as M51R VSV, are severely defective for inhibition of the host antiviral response but are not defective for the production of progeny virions or the ability to induce apoptosis (2, 23, 26). Therefore, in contrast to wild-type (wt) VSV, M protein mutant VSV strains effectively induce type I IFN production by normal cells. This promotes selective killing of IFN-sensitive cancer cells (2, 6, 9, 18), as does engineering wt VSV to produce murine or human IFN-β (17, 18, 27, 28).
One of the challenges to oncolytic virus therapy is that individual cancers vary in their sensitivity to oncolytic viruses even when these cancers arise from the same tissue type. Cancer cell resistance to VSV and other oncolytic viruses often is due to partial or full retention of type I IFN responsiveness and/or to constitutive expression of IFN-stimulated antiviral genes (6, 13, 29–34). The human type I IFN family includes 12 subtypes of IFN-α and one subtype of IFN-β, all of which share significant amino acid homology (35). IFN-β and IFN-α bind to the same receptor, which is composed of two subunits, a low-affinity IFNARI and a high-affinity IFNAR2. Formation of a ternary complex activates JAK/STAT signaling and the transcription of hundreds of IFN-stimulated genes (ISGs), which establish an antiviral state (36). Type I IFN also has antiproliferative, proapoptotic, antiangiogenic, and immunoregulatory activities in addition to antiviral activity (37, 38). Although no unique functions have been defined for IFN-α relative to those of IFN-β (39), distinct IFN subtypes are capable of differentially inducing biological responses despite signaling through a shared receptor (40, 41). Although all type I IFN subtypes are similarly effective at inducing antiviral gene expression, IFN-β regulates cellular functions such as growth and apoptosis at concentrations that are orders of magnitude lower than those for any of the IFN-α subtypes (38). This is due in part to the capacity of IFN-β to bind with higher affinity and form more stable ternary complexes with IFNAR1 and IFNAR2 (42–44). Differential immunoregulatory capacities also have been noted for IFN-β and -α and among IFN-α subtypes (41, 45, 46). In addition to ligand binding affinity, differential induction of biological responses by IFN-β and -α has been attributed to IFNAR-1 and -2 expression levels, receptor trafficking (degradation versus recycling), and the presence of cell- and tissue-specific regulatory factors (38, 45, 47–49).
Because IFN-α and -β are not necessarily equivalent in their ability to induce biological responses in a given pathological setting, here we have addressed the role of IFN subtypes in regulating cancer cell resistance to virus-induced oncolysis. The goal of the current study was to compare the relative abilities of IFN-α2a and IFN-β, both of which are currently in clinical use, to protect head and neck squamous cell carcinoma (HNSCC) cells from VSV-induced killing. Human squamous cell carcinoma offers the prospect of therapeutic intervention by a direct intratumoral route and has been used extensively for evaluation of various OV treatments (50). We found that HNSCC lines are more effectively protected from VSV-induced cytolysis by IFN-β than by IFN-α2a when treated with equivalent units of IFN bioactivity. Importantly, this was not the case for nonmalignant cells, which responded similarly to both IFN subtypes. In addition, we found that IFN-β but not IFN-α was key to maintaining a state of persistent infection (PI) of HNSCC cells with M51R VSV. These results demonstrate that with regard to enhancing the selectivity of VSV for cancer cells, combining OV treatment with IFN-α2a rather than IFN-β may be more effective, and they highlight a need for further investigation of IFN subtypes as agents to improve selectivity of OV for cancer cells.
MATERIALS AND METHODS
Reagents.
Human IFN-α2a (11101-1) and IFN-β (11410) were purchased from PBL Assay Science (Piscataway, NJ). Interferons were aliquoted and stored at −80°C. Antibodies against IFN-α (MAB413) and IFN-β (AB1431) were purchased from Merck Millipore (Billerica, MA).
Cell propagation and virus infections.
Recombinant wild-type (rwt) VSV expressing wt M protein and enhanced green fluorescent protein (rwt VSV-eGFP) or recombinant VSV expressing the M51R mutant M protein and eGFP (M51R VSV-eGFP) were isolated from infectious cDNA clones as described previously (51). Viruses were grown in BHK cells, supernatants containing progeny virions were harvested, and the titers were determined using a plaque assay (26). BHK cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS) and 2 mM l-glutamine. SQ20B, JSQ-3, and SCC61 head and neck tumor cells were a gift from Ezra Cohen (University of San Diego). SQ20B cells were grown in Dulbecco's modified Eagle's medium nutrient mixture F12 (DMEM F12) with 10% FBS, 2 mM l-glutamine, and penicillin-streptomycin (P/S). JSQ-3 and SCC61 cells were grown in DMEM F12 with 10% FBS, 2 mM l-glutamine, P/S, and 0.4 μg/ml of hydrocortisone. RKO cells (kindly provided by John Stewart, Wake Forest University) were cultured in McCoy's 5A medium with 10% FBS, 2 mM l-glutamine, and P/S. Normal human epidermal keratinocyte (NHEK) cells were purchased from Lonza and cultured in KGM-Gold bullet kit medium (192060). Human microvascular endothelial cells (HMVEC) were purchased from Lonza and cultured in EGM Bullet kit medium (cc-3124).
Establishment of persistent VSV infection.
SQ20B, JSQ-3, or SCC61 cells were grown to 70% confluence in 10-cm dishes and infected with M51R VSV at a multiplicity of infection (MOI) of 0.1, 1.0, and 10. After 3 to 4 days, the majority of the cells were dead. The cells that survived the initial infection (approximately 1 to 20% depending on the cell line and dose of virus) were maintained in culture by supplementing with fresh medium and growing to confluence. Cells were split and reseeded repeatedly to maintain the cultures. Cells and cell culture media were collected every 5 passages and frozen at −80°C.
Cell viability assays.
Uninfected or persistently infected tumor cells were seeded at a density of 4 × 104 to 5 × 104 cells/well in a 24-well plate. After a 24-h recovery period, cells were infected with rwt-eGFP or M51R VSV-eGFP at an MOI of 0.1 or 10 PFU per cell in 100 μl of serum-free media for 45 min. Cells were washed once with phosphate-buffered saline (PBS) and covered with 500 μl of medium. Cell viability was assessed at 48 h postinfection by MTT [3-(4,5-dimethylthiazol-2-yl)2 2,5-diphenyl tetrazolium bromide] assay according to the manufacturer's instructions (Roche). Samples were tested in triplicate, and each experiment was performed independently at least 3 times.
IFN protection against viral killing.
Uninfected or persistently infected tumor cells were seeded in 24-well plates at the following densities: JSQ-3 and SCC61 cells at 1.5 ×104 cells/well, SQ20B at 3.3 × 104 cells/well, and RKO cells at 4 × 104 cells/well. Twenty-four hours later, cells were pretreated with 100 IU/ml or 1,000 IU/ml of IFN-α2a or IFN-β. Cells were infected after an additional 24 h with viruses at an MOI of 0.1 or 10 PFU per cell. After 48 h, cell viability was measured by MTT assay as described above.
IFN antibody pretreatment.
Two experimental protocols were used as follows. (i) Uninfected or persistently infected tumor cells were seeded as described above for IFN protection assays. Twenty-four hours later, cells were pretreated with antibody against IFN-α, IFN-β, or both, at a concentration of 500 U/ml, to neutralize low-level, constitutive IFN that was detected in mock-infected cells. Eighteen hours posttreatment, cells were infected with M51R VSV-eGFP at an MOI of 0.1 in the continued presence of the same dose of anti-IFN antibody. At 24 h postinfection, medium samples were collected for plaque assay to determine viral titers, and cells were examined by fluorescence microscopy. The percentage of eGFP-expressing cells was determined using a Nikon Eclipse TE300 epifluorescence microscope. Fluorescent and bright-field images were captured digitally, and the percentage of eGFP-expressing cells per image was determined. At least 100 cells were counted per sample condition. MTT assays were performed on some cultures at 72 h postinfection to evaluate cell viability. (ii) SQ20B cells or JSQ-3 cells persistently infected with M51R VSV were seeded in quintuplicate in 12-well dishes. Twenty-four hours later, antibody against IFN-α or IFN-β (500 U/ml) was added to the culture media. Viable cells from each experimental condition were counted using trypan blue dye exclusion on days 0, 1, 2, 3, and 4 after antibody addition.
Treatment of persistently infected tumor cells with exogenous IFN.
Persistently infected JSQ-3 cells were seeded at a density of 4 × 104 to 5 × 104 cells/well in a 24-well dish. Twenty-four hours later, IFN-β or IFN-α2a (1,000 U/ml) was added to the culture media. Cells were maintained in culture, adding the same concentration of IFN with each passage. After 10 passages, culture medium was collected and tested for the presence of VSV by plaque assay and by adding the culture medium to fresh BHK culture medium at a 1:1 dilution. When VSV was no longer detectable by either method, the addition of IFN to the cultures was stopped. After several passages in the absence of IFN, the cells were tested for susceptibility to VSV infection as described above for cell viability assays.
ELISA.
IFN-β and IFN-α levels present in cell supernatants at 24 h postinfection were determined using VeriKine human IFN-β (41410-1) and IFN-α (41100-1) enzyme-linked immunosorbent assay (ELISA) kits from PBL Assay Science.
Statistics.
All experiments were repeated 3 times, except where indicated, and the means ± standard deviations (SD) are reported. For comparison of 2 treatment groups, the data were analyzed by unpaired Student's t test, and statistical comparisons were considered significant for P < 0.05. For ELISA data shown in Fig. 2a, which had large variability, log transformations were performed prior to making comparisons between groups. For analysis of variance (ANOVA) among multiple treatment groups, the data were analyzed by one-way ANOVA with Tukey's method for adjusting for multiple comparisons. For analyses comparing groups over time (see Fig. 6), two-way ANOVA models were fit with group and time and the group-by-time interaction. The group-by-time interaction term was examined in these models to determine whether there were differences in the change in the outcome (slope) over time. Finally, in the models examining differences over time, we performed pairwise comparisons of groups at 4 days using unpaired t tests to determine whether the groups differed on the last observed time point. All analyses were performed using SAS, version 9.3 (Cary, NC).
FIG 2.
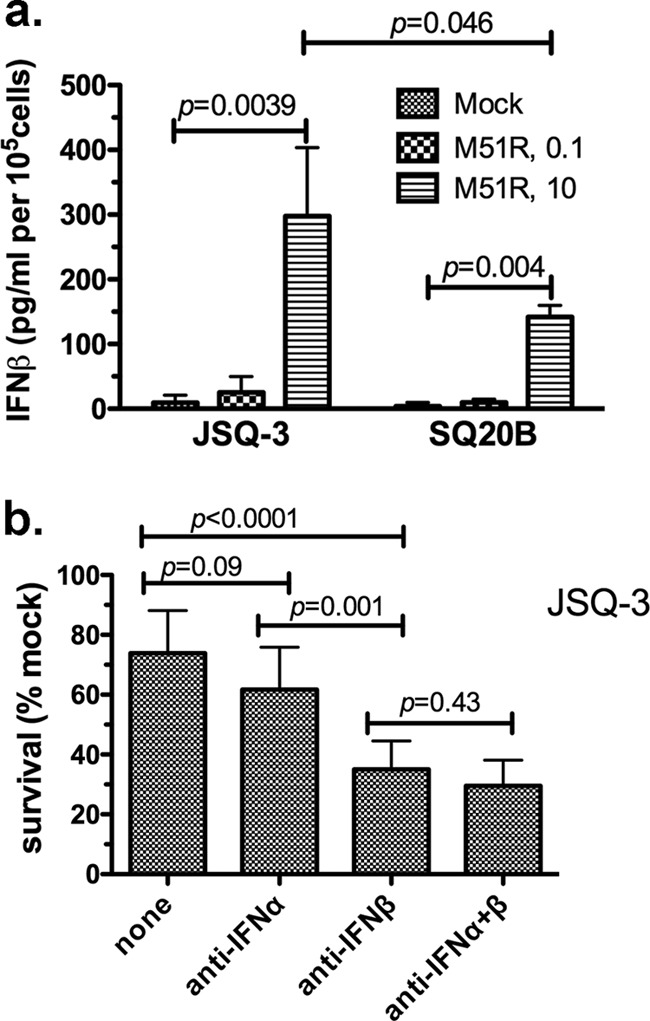
Production and response to type I IFN inhibition by tumor cells infected with M51R VSV. (a) IFN-β levels were measured by ELISA using supernatants taken from JSQ-3 or SQ20B cells 24 h after infection with M51R VSV at the indicated MOIs. Results are expressed as picograms/milliliter of IFN-β per 1 × 105 cells. The means ± SD from 3 individual experiments is shown. (b) Neutralizing antibodies to IFN-β, IFN-α, or a combination of the two antibodies were added to JSQ-3 cells 18 h prior to the addition of M51R VSV (MOI, 0.1) to the cultures. P values were determined by unpaired Student's t test.
FIG 6.

IFN-β maintains the state of persistent infection in tumor cells. (a) SQ20B cells that had established persistent M51R VSV infections (PI-SQ20B) were reinfected at the indicated passage (p) number with M51R VSV at an MOI of 0.1. p0 indicates cells that were infected for the first time (not persistently infected). Neutralizing antibodies to IFN-α, IFN-β, or a combination of the two antibodies were added to some cultures 18 h prior to the addition of virus. At 48 h after reinfection with M51R VSV at the indicated MOI, viability was measured by MTT assay. Results are expressed as the percentage of cells, relative to the number of mock-infected cells, that survived reinfection. Three passages are shown. The results from all three passages were pooled for analysis of variance. The following survival differences (P < 0.05) were found for the treatment groups: (i) anti-IFN-β and anti-IFN-α plus anti-IFN-β groups had decreased survival relative to the no-antibody groups, and (ii) anti-IFN-β groups had decreased survival relative to the anti-IFN-α groups. (b) Persistently infected JSQ-3 cells were maintained continuously in the presence of exogenous IFN-β. When virus was no longer detectable in the cultures, the cells were reinfected with M51R VSV and viability was measured 48 h later. Persistently infected SQ20B (PI-SQ20B) (c) or JSQ-3 (PI-JSQ-3) (d) cells were treated with neutralizing anti-IFN-β or anti-IFN-α antibodies, and viability was measured over 4 days. The number of viable cells per well was determined by light microscopy using the criteria of trypan blue exclusion. The means ± SD from 3 determinations per time point are shown. The following survival differences (P < 0.05) at day 4 were found: (i) the no-antibody group survived in greater numbers than the anti-IFN-β group, and the anti-IFN-α group survived in greater numbers than the anti-IFN-β group for both cell lines. (c and d) Differences in group-by-time interaction (slope) (P < 0.05) were the following: the no-antibody group survived in greater numbers than the anti-IFN-α group, which survived in greater numbers than the anti-IFN-β group (c); the no-antibody group survived in greater numbers than the anti-IFN-β group, and the anti-IFN-α group survived in greater numbers than the anti-IFN-β group (d).
RESULTS
Differential sensitivity of tumor and normal cells to killing by VSV.
The purpose of the experiments shown in Fig. 1 was to evaluate the sensitivity of three HNSCC lines (JSQ-3, SCC61, and SQ20B) to VSV infection and cytolysis. For comparison, RKO, a colorectal tumor cell line that is highly sensitive to the oncolytic effects of VSV (12), normal human keratinocytes (NHEK), and microvascular endothelial cells (HMVEC) were tested. Cells were mock infected or infected with either recombinant M51R VSV (Fig. 1a and c) or recombinant wild-type VSV (rwt VSV) strains that express eGFP (Fig. 1b and d). Susceptibility was measured by quantifying the percentage of cells that were GFP+ (Fig. 1a and b) and the percentage of cells that survived the infection (Fig. 1c and d). As shown in Fig. 1a and c, differences in susceptibility to infection and killing by M51R VSV were apparent at an MOI of 0.1. The order of resistance was the following: JSQ-3 was more resistant than SCC61, and SCC61 was more resistant than SQ20B (P < 0.05). When exposed to rwt VSV, the HNSCC cell lines showed the same pattern of resistance under multiple-cycle infection (MOI of 0.1) (P < 0.05) (Fig. 1b). For both viruses, the susceptibility differences among tumor cells lost significance under single-cycle infection (MOI of 10). Importantly, the normal cells, NHEK cells and HMVEC, were more resistant to infection and oncolysis by M51R VSV at both MOI than were any of the tumor cells (P < 0.05 for normal cells versus tumor cells in all comparisons) (Fig. 1a and c). In contrast, for rwt VSV infection, selectivity for tumor cells over normal cells essentially was lost, as illustrated by a similar sensitivity of all cells to killing by the wt virus (Fig. 1d). These results show that M51R VSV preferentially infects and kills HNSCC over normal cells, illustrating the tumor-selective basis for oncolytic treatment with this virus. As shown previously (12), the positive-control RKO colon carcinoma cells were highly sensitive to infection and killing by both M51R VSV and rwt VSV regardless of MOI.
FIG 1.

Sensitivity of HNSCC lines to M51R VSV and rwt VSV. HNSCC cell lines (JSQ-3, SCC61, and SQ20B) or normal endothelial cells or keratinocytes (HMVEC and NHEK cells, respectively) were infected with eGFP-expressing M51R VSV (a and c) or rwt VSV (b and d) at the indicated MOIs. RKO, a colorectal line that is highly sensitive to VSV, served as a control. The percentage of eGFP-expressing cells, quantified by fluorescence microscopy at 24 h postinfection, is shown in panels a and b. Cell viability as measured by MTT assay at 48 h postinfection is shown in panels c and d. Results are expressed as percentages of cells relative to the number of mock-infected cells that survived infection. The means ± SD from at least 3 individual experiments is shown. *, P < 0.05 for three HNSCC lines, all comparisons; **, P < 0.05 for two normal cell lines versus three HNSCC lines, all comparisons at each MOI. P values were determined by one-way ANOVA with Tukey's posttest.
VSV-infected tumor cell production and response to IFN inhibition.
We next inquired if relative susceptibility or resistance of tumor cells to VSV-induced oncolysis involved type I IFN. The production of IFN-β and -α was measured after M51R VSV infection of JSQ-3 and SQ20B cells, the most resistant and susceptible HNSCC lines, respectively. IFN-β was detected in the supernatants of both JSQ-3 and SQ20B cells, and levels were increased in response to infection with M51R VSV. At an MOI of 10, both SQ20B and JSQ-3 produced significantly more IFN-β than mock-infected cells (P = 0.004 and P = 0.0039, respectively) (Fig. 2a). The more resistant JSQ-3 cells produced slightly more IFN-β than the SQ20B cells infected with M51R VSV at an MOI of 10 (P = 0.046). IFN-β was not detected significantly above mock infection levels by either cell line infected at an MOI of 0.1. Neither cell line infected with rwt VSV secreted IFN-β, reflecting the ability of this strain to suppress the host response. IFN-α was not detected in the supernatants of any of the infected cell lines (data not shown).
To determine if production of type I IFN by JSQ-3 cells contributed to their resistance to M51R VSV infection at an MOI of 0.1, the cells were pretreated with neutralizing antibodies against IFN-β, IFN-α, or a combination of anti-IFN-α and anti-IFN-β (Fig. 2b). Antibodies were added 18 h prior to infection with M51R VSV. Fewer cells survived the infection in the presence of anti-IFN-β neutralizing antibody (approximately 35% survival) compared to the control with no antibody present (approximately 75% survival) (P < 0.0001). In contrast, the addition of anti-IFN-α antibody did not significantly increase susceptibility to virus-induced killing compared to that of no-antibody treatment (P = 0.09). A combination of the two antibodies did not change susceptibility relative to that of anti-IFN-β alone (P = 0.43). Anti-IFN-β antibodies protected JSQ3 cells significantly more than the anti-IFN-α antibodies (P = 0.001). Collectively the data shown in Fig. 2 indicate that HNSCC cells retain some capacity to produce and respond to IFN-β but not IFN-α, and this contributes to their resistance to M51R VSV-induced killing.
Differential responsiveness of tumor cells infected with VSV to IFN pretreatment.
The results shown in Fig. 2 suggest that autocrine IFN-β plays a greater role in protecting JSQ-3 cells from VSV infection than does IFN-α. To further address the possibility that JSQ-3 cells respond differentially to IFN-β and -α, we determined the extent to which pretreatment with exogenous IFNs would confer protection from VSV-induced cytolysis. Because JSQ-3 cells are more sensitive to rwt VSV than to M51R VSV (Fig. 1), and because rwt VSV blocks endogenous IFN production, this virus was chosen to evaluate protection by exogenous IFN treatment of JSQ-3 cells, NHEK cells, and HMVEC. Cells were primed for 18 h with either IFN-α2a or IFN-β at 100 or 1,000 U/ml prior to infection. At 48 h, the percentage of cells that survived was measured by MTT assay (Fig. 3a to c). Similar to the data shown in Fig. 1, approximately 30% of JSQ-3 cells survived the rwt virus infection in the absence of added IFN (Fig. 3a). Cell survival following pretreatment with IFN-α2a at both 100 U/ml and 1,000 U/ml was not significantly different compared to that of the control (P = 0.9 and P = 0.16, respectively). In contrast, cell survival following pretreatment with IFN-β was significantly greater than that of the control at both 100 U/ml (P = 0.0165) and 1,000 U/ml (P = 0.0004). In addition, survival at 100 and 1,000 U/ml IFN-β was significantly greater than survival at the same doses of IFN-α (P = 0.0132 and P = 0.004, respectively). Notably, this differential protective effect was not observed in nonmalignant cells. In the absence of added IFN, both normal cell types, NHEK and HMVEC, were susceptible to rwt virus-induced cytolysis, likely due to inhibition of the host antiviral response by wt M protein (Fig. 3b and c, first bar). In contrast to JSQ-3 cells, both doses of IFN-β and IFN-α2a protected normal cells to a significant extent (P < 0.05 for all comparisons versus the control, no IFN added) with no significant difference between IFN-α and -β (Fig. 3b and c).
FIG 3.

Protective effect of exogenous IFNs on tumor and normal cells infected with rwt VSV. (a) JSQ-3 cells, (b) NHEK cells, and (c) HMVEC were preincubated for 18 h with recombinant human IFN-α2a or IFN-β at either 100 or 1,000 U/ml and then infected with rwt VSV at an MOI of 10. Viability was measured at 48 h by MTT assay. Results shown are the means ± SD from at least 3 experiments per treatment. *, P < 0.05 for 100 U/ml IFN-β versus 100 U/ml IFN-α; *, P < 0.05 1,000 U/ml IFN-β versus 1,000 U/ml IFN-α, determined by one-way ANOVA with Tukey's posttest. (d) Viral titers in supernatants from JSQ-3 cells and HMVEC cells were measured at 24 h by plaque assay. Two independent experiments for each cell type and virus are indicated by open and closed symbols.
As an additional test for the protective effects of IFN, supernatants were collected at 24 h postinfection and virus titers were measured by plaque assay (Fig. 3d). For JSQ-3 cells (circles), both doses of IFN-β more effectively reduced virus titers than did the same doses of IFN-α2a (Fig. 3d), similar to the results of cell viability assays (Fig. 3a). For HMVEC (squares), although virus titers were 2 log lower than those of JSQ-3, IFN-α2a and -β were similarly effective in reducing virus production. NHEK cells were protected by pretreatment with IFNs in a manner similar to that of HMVEC, but results were variable in order of magnitude of the response over different lots of cells (data not shown). These data show that JSQ-3 cells are capable of responding to IFN and that IFN-β is more protective against VSV infection and cytolysis than IFN-α2a. In contrast, IFN-β and -α2a protected normal cells to a similar extent. The results with normal cells provide evidence that differential protection of tumor cells by IFN subtypes was not due to differences in specific activities or functional integrity of the IFN-β and IFN-α2a preparations.
The ability of IFN-α2a and IFN-β to protect the more susceptible cancer cells, SCC61, SQ20B, and RKO, from infection and killing was measured for both rwt and M51R viruses (Fig. 4). Both viruses were evaluated due to increased sensitivity of these cell lines to infection and killing relative to that of JSQ-3 (Fig. 1). IFN-β killed mock-infected SQ20B cells at 1,000 U/ml (data not shown), precluding measurement of protection at that dose. With the exception of SQ20B cells treated with 1,000 U/ml IFN-β, IFN treatment alone did not exert toxic effects on the tumor cells. For both SCC61 and SQ20B, interferon pretreatment protected the cells from killing by both M51R and rwt VSV relative to that of the control (no interferon pretreatment). However, when equivalent doses of IFN-α and -β were compared, IFN-β was significantly more protective against virus-induced killing than was IFN-α2a (P < 0.001 for all comparisons). Similarly, in RKO colon cancer cells, which were highly susceptible to VSV, as shown previously (12), IFN-β at 1,000 U/ml exerted a significantly greater level of protection from killing by M51R VSV than did IFN-α at 1,000 U/ml (P < 0.05) (Fig. 4a). IFN-β induced only a modest level of protection against rwt VSV, while IFN-α had no protective effect (Fig. 4b). Consistent with the survival data, IFN-β also reduced virus titers to a greater extent than IFN-α2a at equivalent doses, and this pattern was consistent for both viruses in all three cell lines. Similar results were obtained with IFN-α2b, an allelic variant of IFN-α2 that is also in clinical use (data not shown). We conclude that tumor cells responded differently to IFN-β and -α2a; exogenous IFN-β was more protective than was IFN-α2a when tumor cells were challenged with oncolytic VSV. Importantly, this was not the case for normal keratinocytes and endothelial cells, which were protected equivalently by IFN-α2a and IFN-β.
FIG 4.

Protective effect of exogenous IFNs on SCC-61, SQ-20B, and RKO cells infected with VSV. SCC61 (a), SQ20B (b), or RKO (c) cells were preincubated for 18 h with recombinant human IFN-α2a or IFN-β (100 U/ml or 1,000 U/ml) and then infected with M51R VSV (left) or rwt VSV (middle) at the indicated MOIs. Viability was measured at 48 h. Results shown are the means ± SD from 4 experiments per treatment. *, P < 0.05 for 100 U/ml IFN-β versus 100 U/ml IFN-α and P < 0.05 for 1,000 U/ml IFN-β versus 1,000 U/ml IFN-α, each determined by one-way ANOVA with Tukey's posttest. Viral titers in supernatants (right) were measured at 24 h by plaque assay. Two independent experiments for SCC61 and SQ20B and three independent experiments for RKO infected with M51R VSV (circles) or rwt VSV (squares) are shown.
Role of IFN subtypes during persistent virus infection of HNSCC cells.
Further evidence for the importance of IFN-β versus IFN-α2a was obtained by analysis of HNSCC cells persistently infected with M51R VSV. HNSCC cells that survived infection with M51R VSV were continuously passaged, yielding a population of persistently infected cells that were resistant to superinfection with VSV, as described previously for neural cells (52). The evidence that the cells were persistently infected was the continuous production of low levels of infectious virus (range, 102 to 105 PFU/ml; data not shown) for multiple passages up to 95 and resistance to superinfection with rwt and M51R viruses. The development of resistance to superinfection as a function of continuous passage of the three HNSCC lines is depicted in Fig. 5. With successive passages, virtually all SQ20B and JSQ-3 tumor cells developed the capacity to survive upon reinfection with rwt and M51R VSV. Similar results were obtained with SCC61, although resistance to infection developed more slowly and the cells preserved a degree of sensitivity to killing by rwt VSV, even after 95 passages.
FIG 5.
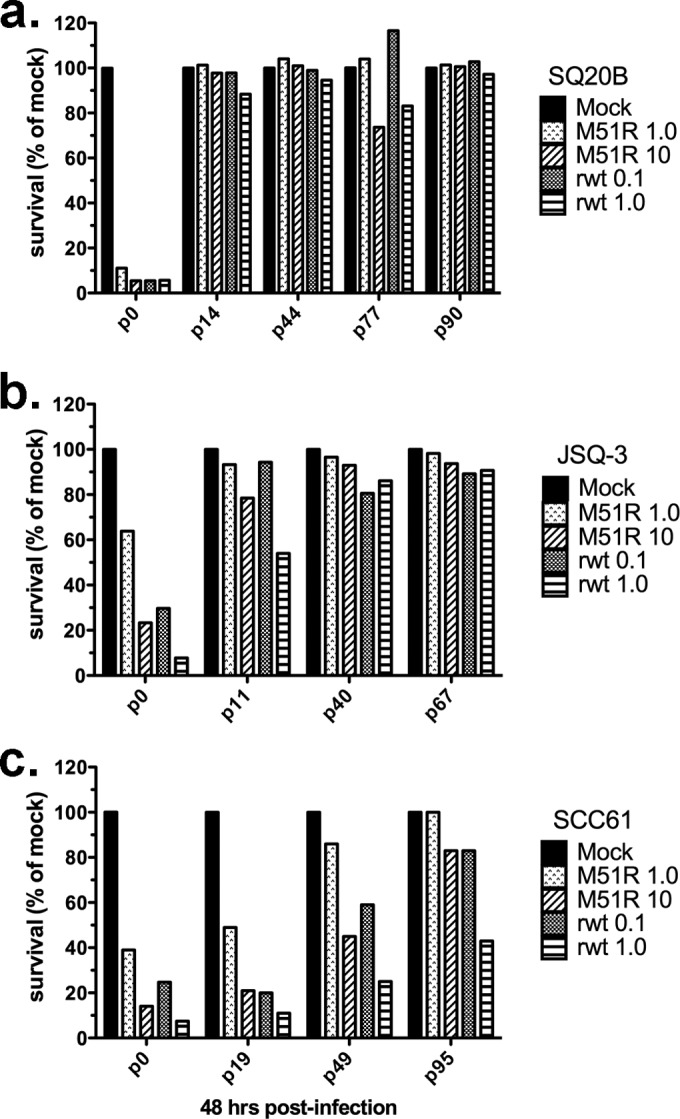
Tumor cells persistently infected with M51R VSV resist killing upon superinfection with M51R or rwt VSV. Tumor cells that had established persistent M51R VSV infections, generated as described in Materials and Methods, were reinfected at the indicated passage (p) number with M51R or rwt VSV at the indicated MOIs. p0 indicates cells that were infected with VSV for the first time (not persistently infected). At 48 h after reinfection, viability was measured by MTT assay. Results are expressed as the percentage of cells, relative to the number of mock-infected cells, that survived reinfection. Data from four (SQ20B) or three (JSQ-3 and SCC61) separate passages are shown.
The role of IFN-α compared to that of IFN-β in the development and maintenance of persistent infection was addressed by treating persistently infected cells with neutralizing antibodies against IFN-α or -β or with exogenous IFN-α or -β. As shown in Fig. 6, pretreatment of persistently infected SQ20B (PI-SQ20B) with anti-IFN-β but not anti-IFN-α decreased their ability to resist superinfection with M51R VSV for each cell passage tested. When the different passages of the M51R VSV-resistant PI-SQ20B cells were analyzed together, pretreatment with anti-IFN-α did not have a significant effect compared to no pretreatment, whereas pretreatment with anti-IFN-β significantly decreased survival from reinfection with M51R VSV compared to no pretreatment or with pretreatment with anti-IFN-α (P < 0.0001 for both comparisons). Adding anti-IFN-α to the pretreatment in combination with anti-IFN-β did not significantly enhance its effect. A similar effect was obtained in the more M51R-VSV-susceptible PI-SCC61 cells (data not shown).
The number of live, persistently infected SQ20B and JSQ-3 cells per well was measured over the course of 4 days after treatment with neutralizing anti-IFN antibodies (Fig. 6c and d). The number of viable control cells incubated in the absence of antibody increased during the time course as the cells continued to divide. Anti-IFN-β treatment (triangles) led to a decline in viability of PI-SQ20B (Fig. 6c) and PI-JSQ-3 (Fig. 6d) cells, whereas treatment with anti-IFN-α had a nominal effect on survival of PI-SQ20B cells and PI-JSQ-3 cells (squares). The slope over time was different for the three conditions in both cell lines (P < 0.0001). The IFN-β treatment slope was significantly different compared not only to the control but also to the IFN-α treatment in both SQ20B and JSQ-3 cells (P < 0.0001 for all 4 comparisons).
The addition of high-dose IFN-β (1,000 U/ml) to the culture media of PI-JSQ-3 cells led to the elimination of VSV after 11 to 22 passages as determined by serial plaque assay measurements on culture media samples. The “cured” cells reverted to the original phenotype with a susceptibility to infection and oncolysis by VSV similar to that of the parental JSQ-3 cells (Fig. 6b). In contrast, repeated addition of high-dose IFN-α did not cure the cells of VSV infection. Therefore, in the persistent infection model, tumor cells demonstrated differential responses to IFN-α and -β, similar to results with cells undergoing primary infection with VSV; in the case of persistent infection, IFN-β but not IFN-α was an essential regulator of PI status.
DISCUSSION
This study demonstrates that head and neck cancer cell carcinoma (HNSCC) cells vary in their sensitivity to killing by VSV, and that responsiveness to type I IFNs in part determines the level of sensitivity. This is similar to results reported previously for HNSCC and for other tumor cell types (13, 16, 30, 31, 53). The striking result in the current study is that IFN-β and -α did not equivalently protect HNSCC cells from VSV-induced killing. In two separate experimental contexts, primary and persistent infection, HNSCC cell lines were less well protected from VSV oncolysis by IFN-α than by IFN-β. First, protection against M51R VSV-induced killing was greater when tumor cells were pretreated with IFN-β than when they were pretreated with equivalent doses of IFN-α2a (Fig. 3 and 4). Second, neutralization of IFN-β but not IFN-α led to death of cells during primary infection (Fig. 2b). Third, we found that survival of tumor cells that were persistently infected with M51R VSV depended on endogenously produced IFN-β, but that this activity was not shared by IFN-α2a (Fig. 6). Specifically, during persistent infection, neutralization of endogenously produced IFN-β led to uncontrolled virus replication and cell death, while addition of exogenous, high-dose IFN-β cured the infection. The finding that IFN-α and -β did not similarly modulate the outcome of persistent infection of HNSCC cells with VSV provides further support for the conclusion drawn from primary cell infections that IFN-induced protection of tumor cells from oncolytic VSV is IFN subtype dependent. In vivo evidence has been presented for VSV persistence as defined by detection of infectious virus, viral genomic RNA, or viral antigen weeks or months after resolution of acute infection (54–56). In theory, persistent infection of tumor cells could potentiate oncolytic therapy by providing a low-level, continuous source of infectious virus, viral RNA, or viral protein for sustained oncolysis and/or activation of antitumor immune responses. However, the relevance of persistent infection for the oncolytic activity of VSV in cancer models remains to be determined.
The idea of combining oncolytic VSV with IFN treatment to improve selectivity is enhanced by the finding that IFN-α is less effective at protecting head and neck cancer cells from VSV oncolysis than is IFN-β, while both IFNs protect normal cells equivalently. It has been demonstrated in multiple systems that IFN-α and -β are not necessarily equivalent in their capacity to induce a particular biological response despite the fact that they signal through the same receptor (reviewed in reference 38). This is illustrated by the substantially more effective induction of antiproliferative and immunoregulatory activities by IFN-β than by IFN-α. For example, IFN-β inhibited the proliferation of fibroblasts and induced chemokine expression in epithelial cells with 50% effective concentrations (EC50s) that were one to three orders of magnitude lower than that of several IFN-α subtypes (38, 43). In contrast, all type I IFNs share the ability to induce protective antiviral responses at concentrations in the low-picomolar range (38, 43). Nevertheless, individual IFN subtypes are not all equally effective at inducing protection against a particular virus in a given biological system. For example, vaccinia virus (VV) vaccine vectors engineered to express IFN-β were significantly attenuated for replication in vitro and in vivo, while VV engineered to express IFN-α4 replicated normally (57). Similarly, IFN-β protected human fibroblasts from herpes simplex virus 1 (HSV-1) and HSV-2 infection more effectively than did 5 different IFN-α subtypes (58), and IFN-α6 was superior to IFN-β and 5 other IFN-α subtypes for protection against influenza virus infection in vivo (59). With regard to VSV, in in vitro studies in which IFNs were normalized against a reference standard for equivalent units of bioactivity, IFN-β, -α8, and -α14 were more effective than other subtypes at inducing an antiviral response in human fibroblasts and epithelial cells (43, 60).
Numerous studies addressing the differential induction of biological activities by type I IFNs have led to a threshold model that explains subtype-specific activities transduced through a shared receptor (reviewed in reference 38). In this model, some biological responses initiated by type I IFNs are high-threshold responses, requiring high-affinity ligand/receptor binding, the formation of stable IFN/IFNAR1/IFNAR2 ternary complexes, and sustained signaling, while others are low-threshold responses that require only low-affinity interactions, transient formation of ternary complexes, and short-term signaling. In this model, the induction of antiviral genes by type I IFN binding to IFNAR1/2 is a low-threshold response that is shared by all type I IFN subtypes and is manifested in a variety of experimental systems by the demonstration of roughly equivalent abilities of IFN-α and -β to protect against virus infection at EC50s in the picomolar range. This is in contrast to the induction of anti-proliferative, apoptotic, and immunoregulatory genes, which are high-threshold responses. The latter responses require higher concentrations of ligand and are driven most effectively by IFN-β, which binds with higher affinity to IFNAR than do any of the IFN-α subtypes, and induces more sustained responses. When interpreted in light of the threshold model, our results suggest that the threshold for transmission of an antiviral signal by type I IFN is raised in some cancer cells compared to the level in normal cells. Specifically, induction of an antiviral response in cancer cells may require higher-affinity interactions with IFNAR1/2 and the formation of more stable ternary complexes than does the same response in normal cells. This could explain the increased capacity of IFN-β to protect HNSCC cells from VSV-induced oncolysis compared to that of IFN-α2a.
One possible mechanism by which the threshold for antiviral activity can be raised in malignant cells is limiting receptor levels. In human fibrosarcoma cells, IFN-α2a was more dependent on IFNAR receptor levels for the induction of several biological responses than was IFN-β (49). In addition, cancer cell sensitivity to the antiproliferative effects of type I IFNs was shown to be regulated by receptor levels (61). Alternatively, an increased level of activity of a negative regulatory factor, such as USP18, a receptor-proximal inhibitor of type I IFN signaling (48), could dampen IFN-α2-mediated responses in cancer cells. For example, USP18 was shown to exert greater inhibitory effects on IFN-α2 signaling than on IFN-β signaling in human amnion-derived WISH cells due to the reduced affinity of IFN-α relative to that of IFN-β for the type I IFN receptor (48).
Similar to our previous findings and those of others (2, 6, 9, 18), M protein mutant VSV, which induces a robust IFN response by normal cells (2, 62), showed selectivity for tumor cells. This property was essentially lost with rwt VSV, presumably due to its capacity to disable the host antiviral response (23). Our finding that exogenous IFN-α preferentially protected normal cells over HNSCC cells from the oncolytic effects of M51R VSV while IFN-β effectively protected both normal and cancer cells implies that at least in this system, IFN-α is a better choice than IFN-β as an agent to further enhance selectivity of the virus for tumor cell killing.
The importance of type I IFNs as modulators of oncolytic therapy with VSV was illustrated recently. In one study, VSV-resistant prostate tumor cells became sensitive to infection and cytolysis in vitro and in vivo after treatment with triptolide, a drug that was shown to block type I IFN signaling at the transcriptional level (53). In another study, treatment with specific JAK1/2 inhibitors increased the susceptibility of head and neck cancer cells to VSV-induced oncolysis (33). The current study provides a clinically relevant example of differential IFN-induced bioactivity and illustrates the need for further study on the contribution of individual IFN subtypes to tumor cell resistance to VSV and other oncolytic viruses. We have not addressed the role of IFN-α subtypes other than that of IFN-α2a (and the allelic variant IFN-α2b, which yielded similar results; data not shown) in the current study. The results reported here warrant a similar evaluation of other IFN-α subtypes, as well as the consensus IFN-α (IFN-con1 or CIFN), which has significant antiviral activity against VSV (63, 64). This information will facilitate the design of OV therapies with enhanced selectivity for tumor cells.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant R01AI032983 (D.S.L.) and by the Wake Forest Translational Science Institute KL2 Research Scholar program (M.P.). Services of the Biostatistics Core, the Cell and Virus Vector Core, and the Cellular Imaging Core were supported by National Cancer Institute grant CCSG P30CA012197, Comprehensive Cancer Center of Wake Forest University.
We thank Ezra Cohen (UC San Diego, Moores Cancer Center) for providing the HNSCC cell lines and Margie McKenzie for technical assistance.
REFERENCES
- 1.Russell SJ, Peng KW, Bell JC. 2012. Oncolytic virotherapy. Nat Biotechnol 30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stojdl DF, Lichty BD, ten Oever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275. doi: 10.1016/S1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 3.Colamonici OR, Domanski P, Platanias LC, Diaz MO. 1992. Correlation between interferon (IFN) alpha resistance and deletion of the IFN alpha/beta genes in acute leukemia cell lines suggests selection against the IFN system. Blood 80:744–749. [PubMed] [Google Scholar]
- 4.Wong LH, Krauer KG, Hatzinisiriou I, Estcourt MJ, Hersey P, Tam ND, Edmondson S, Devenish RJ, Ralph SJ. 1997. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem 272:28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 5.Sun WH, Pabon C, Alsayed Y, Huang PP, Jandeska S, Uddin S, Platanias LC, Rosen ST. 1998. Interferon-alpha resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood 91:570–576. [PubMed] [Google Scholar]
- 6.Ahmed M, Cramer SD, Lyles DS. 2004. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 7.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 8.Moussavi M, Tearle H, Fazli L, Bell JC, Jia W, Rennie PS. 2013. Targeting and killing of metastatic cells in the transgenic adenocarcinoma of mouse prostate model with vesicular stomatitis virus. Mol Ther 21:842–848. doi: 10.1038/mt.2012.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wollmann G, Davis JN, Bosenberg MW, van den Pol AN. 2013. Vesicular stomatitis virus variants selectively infect and kill human melanomas but not normal melanocytes. J Virol 87:6644–6659. doi: 10.1128/JVI.03311-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. 2007. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res 67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 11.Huang TG, Ebert O, Shinozaki K, Garcia-Sastre A, Woo SL. 2003. Oncolysis of hepatic metastasis of colorectal cancer by recombinant vesicular stomatitis virus in immune-competent mice. Mol Ther 8:434–440. doi: 10.1016/S1525-0016(03)00204-1. [DOI] [PubMed] [Google Scholar]
- 12.Stewart JH, Ahmed M, Northrup SA, Willingham M, Lyles DS. 2011. Vesicular stomatitis virus as a treatment for colorectal cancer. Cancer Gene Ther 18:837–849. doi: 10.1038/cgt.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J Virol 86:3073–3087. doi: 10.1128/JVI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cary ZD, Willingham MC, Lyles DS. 2011. Oncolytic vesicular stomatitis virus induces apoptosis in U87 glioblastoma cells by a type II death receptor mechanism and induces cell death and tumor clearance in vivo. J Virol 85:5708–5717. doi: 10.1128/JVI.02393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozduman K, Wollmann G, Piepmeier JM, van den Pol AN. 2008. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J Neurosci 28:1882–1893. doi: 10.1523/JNEUROSCI.4905-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hastie E, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol 93:2529–2545. doi: 10.1099/vir.0.046672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurisetty VV, Heiber J, Myers R, Pereira GS, Goodwin JW, Federspiel MJ, Russell SJ, Peng KW, Barber G, Merchan JR. 2014. Preclinical safety and activity of recombinant VSV-IFN-beta in an immunocompetent model of squamous cell carcinoma of the head and neck. Head Neck 36:1619–1627. doi: 10.1002/hed.23502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol 77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez M, Porosnicu M, Markovic D, Barber GN. 2002. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol 76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porosnicu M, Mian A, Barber GN. 2003. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res 63:8366–8376. [PubMed] [Google Scholar]
- 21.Heiber JF, Barber GN. 2011. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J Virol 85:10440–10450. doi: 10.1128/JVI.05408-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naik S, Nace R, Barber GN, Russell SJ. 2012. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-beta. Cancer Gene Ther 19:443–450. doi: 10.1038/cgt.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol 77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Black BL, Lyles DS. 1992. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J Virol 66:4058–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferran MC, Lucas-Lenard JM. 1997. The vesicular stomatitis virus matrix protein inhibits transcription from the human beta interferon promoter. J Virol 71:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopecky SA, Willingham MC, Lyles DS. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J Virol 75:12169–12181. doi: 10.1128/JVI.75.24.12169-12181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willmon CL, Saloura V, Fridlender ZG, Wongthida P, Diaz RM, Thompson J, Kottke T, Federspiel M, Barber G, Albelda SM, Vile RG. 2009. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res 69:7713–7720. doi: 10.1158/0008-5472.CAN-09-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenks N, Myers R, Greiner SM, Thompson J, Mader EK, Greenslade A, Griesmann GE, Federspiel MJ, Rakela J, Borad MJ, Vile RG, Barber GN, Meier TR, Blanco MC, Carlson SK, Russell SJ, Peng KW. 2010. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum Gene Ther 21:451–462. doi: 10.1089/hum.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naik S, Russell SJ. 2009. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther 9:1163–1176. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- 30.Saloura V, Wang LC, Fridlender ZG, Sun J, Cheng G, Kapoor V, Sterman DH, Harty RN, Okumura A, Barber GN, Vile RG, Federspiel MJ, Russell SJ, Litzky L, Albelda SM. 2010. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon responsiveness defines potential efficacy. Hum Gene Ther 21:51–64. doi: 10.1089/hum.2009.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blackham AU, Northrup SA, Willingham M, D'Agostino RB Jr, Lyles DS, Stewart JH. 2013. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery 153:333–343. doi: 10.1016/j.surg.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moerdyk-Schauwecker M, Shah NR, Murphy AM, Hastie E, Mukherjee P, Grdzelishvili VZ. 2013. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology 436:221–234. doi: 10.1016/j.virol.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Escobar-Zarate D, Liu YP, Suksanpaisan L, Russell SJ, Peng KW. 2013. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther 20:582–589. doi: 10.1038/cgt.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monsurro V, Beghelli S, Wang R, Barbi S, Coin S, Di Pasquale G, Bersani S, Castellucci M, Sorio C, Eleuteri S, Worschech A, Chiorini JA, Pederzoli P, Alter H, Marincola FM, Scarpa A. 2010. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: potential relevance for adenoviral gene therapy. J Transl Med 8:10. doi: 10.1186/1479-5876-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genin P, Vaccaro A, Civas A. 2009. The role of differential expression of human interferon–a genes in antiviral immunity. Cytokine Growth Factor Rev 20:283–295. doi: 10.1016/j.cytogfr.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 36.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 38.Piehler J, Thomas C, Garcia KC, Schreiber G. 2012. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev 250:317–334. doi: 10.1111/imr.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uze G, Schreiber G, Piehler J, Pellegrini S. 2007. The receptor of the type I interferon family. Curr Top Microbiol Immunol 316:71–95. [DOI] [PubMed] [Google Scholar]
- 40.da Silva AJ, Brickelmaier M, Majeau GR, Lukashin AV, Peyman J, Whitty A, Hochman PS. 2002. Comparison of gene expression patterns induced by treatment of human umbilical vein endothelial cells with IFN-alpha 2b vs. IFN-beta 1a: understanding the functional relationship between distinct type I interferons that act through a common receptor. J Interferon Cytokine Res 22:173–188. [DOI] [PubMed] [Google Scholar]
- 41.Gibbert K, Schlaak JF, Yang D, Dittmer U. 2013. IFN-alpha subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol 168:1048–1058. doi: 10.1111/bph.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. 2008. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem 283:32925–32936. doi: 10.1074/jbc.M806019200. [DOI] [PubMed] [Google Scholar]
- 43.Jaks E, Gavutis M, Uze G, Martal J, Piehler J. 2007. Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol 366:525–539. doi: 10.1016/j.jmb.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 44.Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, Uze G, Schreiber G. 2006. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol 26:1888–1897. doi: 10.1128/MCB.26.5.1888-1897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Severa M, Remoli ME, Giacomini E, Ragimbeau J, Lande R, Uze G, Pellegrini S, Coccia EM. 2006. Differential responsiveness to IFN-alpha and IFN-beta of human mature DC through modulation of IFNAR expression. J Leukoc Biol 79:1286–1294. doi: 10.1189/jlb.1205742. [DOI] [PubMed] [Google Scholar]
- 46.Foster GR, Masri SH, David R, Jones M, Datta A, Lombardi G, Runkell L, de Dios C, Sizing I, James MJ, Marelli-Berg FM. 2004. IFN-alpha subtypes differentially affect human T cell motility. J Immunol 173:1663–1670. doi: 10.4049/jimmunol.173.3.1663. [DOI] [PubMed] [Google Scholar]
- 47.Marijanovic Z, Ragimbeau J, van der Heyden J, Uze G, Pellegrini S. 2007. Comparable potency of IFNalpha2 and IFNbeta on immediate JAK/STAT activation but differential down-regulation of IFNAR2. Biochem J 407:141–151. doi: 10.1042/BJ20070605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Francois-Newton V, Livingstone M, Payelle-Brogard B, Uze G, Pellegrini S. 2012. USP18 establishes the transcriptional and anti-proliferative interferon alpha/beta differential. Biochem J 446:509–516. doi: 10.1042/BJ20120541. [DOI] [PubMed] [Google Scholar]
- 49.Moraga I, Harari D, Schreiber G, Uze G, Pellegrini S. 2009. Receptor density is key to the alpha2/beta interferon differential activities. Mol Cell Biol 29:4778–4787. doi: 10.1128/MCB.01808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lott JB. 2012. Oncolytic viruses: a new paradigm for treatment of head and neck cancer. Oral Surg Oral Med Oral Pathol Oral Radiol 113:155–160. doi: 10.1016/j.tripleo.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 51.Whitlow ZW, Connor JH, Lyles DS. 2006. Preferential translation of vesicular stomatitis virus mRNAs is conferred by transcription from the viral genome. J Virol 80:11733–11742. doi: 10.1128/JVI.00971-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Desforges M, Despars G, Berard S, Gosselin M, McKenzie MO, Lyles DS, Talbot PJ, Poliquin L. 2002. Matrix protein mutations contribute to inefficient induction of apoptosis leading to persistent infection of human neural cells by vesicular stomatitis virus. Virology 295:63–73. doi: 10.1006/viro.2001.1329. [DOI] [PubMed] [Google Scholar]
- 53.Ben Yebdri F, Van Grevenynghe J, Tang VA, Goulet ML, Wu JH, Stojdl DF, Hiscott J, Lin R. 2013. Triptolide-mediated inhibition of interferon signaling enhances vesicular stomatitis virus-based oncolysis. Mol Ther 21:2043–2053. doi: 10.1038/mt.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fultz PN, Shadduck JA, Kang CY, Streilein JW. 1982. Vesicular stomatitis virus can establish persistent infections in Syrian hamsters. J Gen Virol 63:493–497. doi: 10.1099/0022-1317-63-2-493. [DOI] [PubMed] [Google Scholar]
- 55.Simon ID, Publicover J, Rose JK. 2007. Replication and propagation of attenuated vesicular stomatitis virus vectors in vivo: vector spread correlates with induction of immune responses and persistence of genomic RNA. J Virol 81:2078–2082. doi: 10.1128/JVI.02525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turner DL, Cauley LS, Khanna KM, Lefrancois L. 2007. Persistent antigen presentation after acute vesicular stomatitis virus infection. J Virol 81:2039–2046. doi: 10.1128/JVI.02167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Day SL, Ramshaw IA, Ramsay AJ, Ranasinghe C. 2008. Differential effects of the type I interferons alpha4, beta, and epsilon on antiviral activity and vaccine efficacy. J Immunol 180:7158–7166. doi: 10.4049/jimmunol.180.11.7158. [DOI] [PubMed] [Google Scholar]
- 58.Harle P, Cull V, Guo L, Papin J, Lawson C, Carr DJ. 2002. Transient transfection of mouse fibroblasts with type I interferon transgenes provides various degrees of protection against herpes simplex virus infection. Antiviral Res 56:39–49. doi: 10.1016/S0166-3542(02)00093-1. [DOI] [PubMed] [Google Scholar]
- 59.James CM, Abdad MY, Mansfield JP, Jacobsen HK, Vind AR, Stumbles PA, Bartlett EJ. 2007. Differential activities of alpha/beta IFN subtypes against influenza virus in vivo and enhancement of specific immune responses in DNA vaccinated mice expressing haemagglutinin and nucleoprotein. Vaccine 25:1856–1867. doi: 10.1016/j.vaccine.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 60.Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, Pestka S, Schreiber G. 2011. Binding and activity of all human alpha interferon subtypes. Cytokine 56:282–289. doi: 10.1016/j.cyto.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 61.Wagner TC, Velichko S, Chesney SK, Biroc S, Harde D, Vogel D, Croze E. 2004. Interferon receptor expression regulates the antiproliferative effects of interferons on cancer cells and solid tumors. Int J Cancer 111:32–42. doi: 10.1002/ijc.20236. [DOI] [PubMed] [Google Scholar]
- 62.Ahmed M, Brzoza KL, Hiltbold EM. 2006. Matrix protein mutant of vesicular stomatitis virus stimulates maturation of myeloid dendritic cells. J Virol 80:2194–2205. doi: 10.1128/JVI.80.5.2194-2205.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blatt LM, Davis JM, Klein SB, Taylor MW. 1996. The biologic activity and molecular characterization of a novel synthetic interferon-alpha species, consensus interferon. J Interferon Cytokine Res 16:489–499. doi: 10.1089/jir.1996.16.489. [DOI] [PubMed] [Google Scholar]
- 64.Ozes ON, Reiter Z, Klein S, Blatt LM, Taylor MW. 1992. A comparison of interferon-Con1 with natural recombinant interferons-alpha: antiviral, antiproliferative, and natural killer-inducing activities. J Interferon Res 12:55–59. doi: 10.1089/jir.1992.12.55. [DOI] [PubMed] [Google Scholar]