

ABSTRACT
Off-therapy control of viremia by HIV-infected individuals has been associated with two likely players: a restricted viral reservoir and an efficient cell-mediated immune response. We previously showed that a combination of highly suppressive antiretroviral therapy and two experimental drugs, i.e., auranofin and buthionine sulfoximine, was able to reduce the viral reservoir, elicit efficient cell-mediated antiviral responses, and induce intermittent posttherapy viral load control in chronically SIVmac251-infected macaques. We here show that the macaques that had received this drug combination and then stopped antiretroviral therapy were also able to maintain low numbers of activated CD4+ T cells at viral rebound. Moreover, these macaques consistently displayed low-level simian immunodeficiency virus (SIV) diversity, which was in line with the strong and broadly reactive cell-mediated immune responses against conserved Gag antigens. Extended follow-up showed that the two macaques that had received the complete drug combination remained healthy and did not develop AIDS in 2 years of follow-up after therapy suspension. This disease-free survival is longer than twice the average time of progression to AIDS in SIVmac251-infected rhesus macaques. These results suggest that limited numbers of activated T cells at viral rebound and subsequent development of broadly reactive cell-mediated responses may be interrelated in reducing the viral reservoir.
IMPORTANCE The HIV reservoir in CD4+ T cells represents one main obstacle to HIV eradication. Recent studies, however, show that a drastic reduction of this reservoir is insufficient for inducing a functional cure of AIDS. In the present work, we thoroughly studied and subjected to long-term follow-up two macaques showing intermittent control of the virus following suspension of antiretroviral therapy plus an experimental antireservoir treatment, i.e., the gold salt auranofin and the investigational chemotherapeutic agent buthionione sulfoximine (BSO). We found that these drugs were able to decrease the number of activated CD4+ T cells, which are preferential targets for HIV infection. Then, efficient immune responses against the virus were developed in the macaques, which remained healthy during 2 years of follow-up. This result may furnish another building block for future attempts to cure HIV/AIDS.
INTRODUCTION
A functional cure is a state “in which the virus is not eliminated but is controlled effectively by antiviral immune responses so that drug treatment can be withdrawn for prolonged periods of time” (1, 2). Controlled studies in monkeys infected with simian immunodeficiency virus (SIV) or simian-human immunodeficiency virus (SHIV) and anecdotal reports on human immunodeficiency virus type 1 (HIV-1)-infected humans, such as the “Boston patients” and the “Mississippi baby,” have shown that reduction of the viral reservoir, or inhibition of its formation, is a crucial factor for controlling viral load in the absence of antiretroviral therapy (ART) but is not its only determinant (3–6). These reports suggest that without complete eradication of the viral reservoir, viral load control in the absence of therapy is transient or incomplete. Thus, efficient immune responses are likely pivotal to obtain a long-lasting effect on viral load in the chronic phase of the disease, although they may not be essential in posttherapy controllers treated during acute infection (7).
One missing link between restriction of the viral reservoir and development of efficient immune responses could be modulation of immune activation. In this context, some of us focused attention on auranofin, a gold-based compound used to decrease immune activation in individuals with rheumatoid arthritis (4, 8, 9). Auranofin decreases immune activation, likely by causing downmodulation of the costimulatory molecule CD28 in T cells (8). Downmodulation of CD28 is accompanied by a decreased life span of central and transitional memory T cells (TCM and TTM cells) encompassing the viral reservoir (4, 8). In a first in vivo study, a combination of ART and auranofin induced, in the posttherapy follow-up, a peak in viral load, reminiscent of a novel acute infection, followed by a significant yet moderate decrease in the posttherapy viral load set point (4). A subsequent study reproduced these effects, and in some animals, the initial viral load peak and the related immune activation were blunted by a short cycle of ART containing maraviroc, a drug that also impacts immune activation (10). Following the second treatment interruption, these macaques showed intermittent control of viremia to undetectable levels, which was, however, lost in the long term. By adding to the auranofin-containing ART regimen buthionine sulfoximine (BSO) (originally intended to kill the infected cells), an intermittent posttherapy control of viral load to undetectable levels was obtained in the macaques that had received this treatment, and this control was not lost during the entire follow-up period (11). Surprisingly, this functional cure-like condition showed dependence on an unexpected development of CD8+ cell-mediated immune responses (11), but the mechanism behind the immune responses evoked by auranofin and BSO has remained elusive.
The working hypothesis behind the present study is that without these experimental treatments, immune hyperactivation at viral rebound following suspension of ART might spark nonefficient immunodominant responses, thus creating a sort of “immunological junk” rendering the immune system inefficient. There is growing consensus that not all of the antiviral immune responses contribute to immune control of the virus and that some clones directed against conserved epitopes may efficiently mediate viral clearance (12, 13). Limitation of immune activation might therefore contribute to viral load control.
Here, we show that in the aforementioned macaques treated with ART, auranofin, and BSO (11), immune activation is limited during the posttherapy viral rebound. This phenomenon was associated with broadly reactive immune responses against conserved Gag epitopes and limited SIV evolution and replication throughout the follow-up. These results might provide some clues for curing HIV in humans.
MATERIALS AND METHODS
Animal treatment.
Animals were housed at Bioqual, Inc., according to standards and guidelines as set forth in the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC), following approval by the Institutional Animal Care and Use Committee (IACUC). No treatments were administered during the follow-up displayed in the present article, apart from short-term antibiotic prophylaxes in case animals in proximal cages developed bacterial infections. Details on previous treatments, dosages, and rationale have been extensively provided previously (10, 11). and the relevant information is summarized in Table 1 and Fig. 1. Control macaques were selected on the basis of availability of frozen peripheral blood mononuclear cell (PBMC) samples.
TABLE 1.
Baseline characteristics of the macaques in the present study
| Animal | Treatment group | MHC I |
|---|---|---|
| 4890 | ART-auranofin-BSO | Mamu A*02*, Mamu B*17* |
| P157 | ART-auranofin-BSO | Mamu A*11* |
| 4416 | ART-auranofin | Mamu A*02* |
| P185 | ART-auranofin | Mamu B*04* |
| P252 | ART-BSO | Mamu A*02*, Mamu B*03* |
| P177 | ART-auranofin + ART at rebound | Mamu A*01*, Mamu B*01* |
FIG 1.

Schematic representation of the treatments to which the macaques in the present study were subjected following viral load suppression through ART. Chronically SIVmac251-infected macaques were previously treated with a highly intensified ART regimen (i.e., tenofovir, emtricitabine, raltegravir, ritonavir-boosted darunavir, and maraviroc) to suppress viral loads and mimic the condition of HIV-positive individuals under ART (10). Following viral load suppression, auranofin was added to ART, with or without BSO and/or short ART cycles at posttherapy viral rebound (11). These intermittent ART cycles were originally aimed at inducing an immunity against the virus (2, 4) Gray and black arrows indicate, respectively, the timing of BSO administration and of viral rebound following the last treatment suspension.
Separation of PBMCs, isolation of primary T cells, and in vitro cell cultivation.
To test the effects of the combination of auranofin and BSO on survival of activated CD4+ and CD8+ T cells, we devised a set of in vitro experiments. PBMCs from healthy donors were isolated from whole blood by density gradient centrifugation using Ficoll-Paque. CD4+ and CD8+ T cells were isolated by positive selection with specific isolation kits (Miltenyi Biotec). Both isolated CD4+ and CD8+ T cells were cultured in 12-well plates at a concentration of 1 × 106 cells/ml per well. The culturing medium employed was RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and, in the appropriate wells, further complemented with auranofin (250 nM), BSO (250 μM), or a combination of the two. After 24 h (day 1), cells were activated with phytohemagglutinin (PHA) (2 μg/ml) or anti-CD3 (α-CD3)/α-CD28 antibodies (5 μg/ml; BioLegend, San Diego, CA, USA). For α-CD3/α-CD28-mediated activation, cells were transferred to a plate precoated with the antibodies for 12 h before use. In all experiments, one cell culture was left nonactivated to serve as a control. Cells were maintained in an incubator at 37°C in a 5% CO2 atmosphere.
Flow cytometric analysis.
After 72 h of culture (day 3), CD4+ and CD8+ T cells cultured in vitro were collected and stained for viability and expression levels of the alpha chain of the interleukin-2 (IL-2) receptor (i.e., CD25), a marker chosen to confirm the results of previous experiments (14). Briefly, the cells (3 × 105 per sample) were initially surface stained with saturating concentrations of an α-CD25 antibody (phycoerythrin [PE] conjugated; BD Biosciences, Franklin Lakes, NJ, USA) for 20 min at 4°C in magnetically activated cell sorting (MACS) buffer. Afterwards, cells were washed in phosphate-buffered saline (PBS), stained with annexin V (allophycocyanin [APC] conjugated; Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature in an appropriate buffer provided by the manufacturer, and eventually resuspended in 1% paraformaldehyde in PBS. Fluorescence was acquired flow cytometrically (BD, Franklin Lakes, NJ, USA) and analyzed using the FACSDiva software (BD, Franklin Lakes, NJ, USA).
For measurement of the number of activated T cells in vivo, 3 × 105 macaque PBMCs were stained for 20 min at 4°C in MACS buffer using anti-CD3 (fluorescein isothiocyanate [FITC] conjugated), anti-CD4 (PE conjugated), anti-CD8 (peridinin chlorophyll protein [PerCP]-Cy5 conjugated), and anti-HLA-DR (APC conjugated), as previously described (10). All antibodies were purchased from BD (Franklin Lakes, NJ, USA). The flow cytometric analysis was conducted on a FACSCanto II instrument (BD, Franklin Lakes, NJ, USA).
For measurement of T cell-dependent NK cell effector responses, 1 1 × 105 to 1 × 105 macaque PBMCs were stimulated with SIVmac239 Gag peptides (complete set of 15-mer peptides overlapping by 11 amino acids and spanning the entire protein; NIH AIDS Research and Reference Reagent Program, Germantown, MD) for 24 h as previously described (15, 16). Rhesus cytokine-producing NK cells were identified as live CD3− CD8+ CD159a+ CD56+ cells. Nonstimulated and staphylococcal enterotoxin B (SEB) (5 μl/ml; Sigma)-treated tubes were used as negative and positive controls, respectively.
MTS assay.
In our in vitro experiments, we eventually measured the overall cell viability after 96 h of culture (day 4) with a colorimetric assay employing the tetrazolium compound 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (inner salt) (MTS). The MTS assay was performed with the CellTiter 96 AQueous One Solution cell proliferation assay system (Promega) according to the manufacturer's instructions. Briefly, 100 μl of medium/well was transferred to a 96-well plate and incubated with 20 μl of a substrate solution (One Solution reagent) for 3 h in a CO2 (5%) incubator at 37°C. A blank well, containing culture medium and One Solution reagent at the same ratio, was included for each experiment. Absorbance levels were recorded at 490 nm, and, following subtraction of the blank, data were normalized over the levels of absorbance of the control well.
Measurement of intracellular GSH levels.
To assess persistence of γ-glutamylcysteine synthetase inhibition by BSO in vivo, intracellular glutathione/glutathione disulfide (GSH/GSSG) was measured with a glutathione assay kit (Sigma-Aldrich Italia, Milan, Italy) as previously described (8). Briefly, PBMC samples (106 cells per sample) were deproteinized with a 5% 5-sulfosalicylic acid solution, and their GSH content was then measured using a kinetic assay. In this assay, catalytic amounts of GSH cause a continuous reduction of 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) to TNB, shifting the color of the substrate. The fluorescence of the reaction product (TNB) was measured at 412 nm. Values were expressed as nanomoles of GSH in the original sample.
Measurement of gold levels in plasma.
To assess persistence of auranofin in plasma of treated macaques, we measured the concentration of Au ions, given that Au is the basic mediator of the therapeutic effect of auranofin and there is one Au atom per drug molecule. Biological samples were prepared for analysis of Au content by digesting samples with 1 ml of concentrated HNO3 and heated until clear. Two milliliters of water was added, and the solutions were filtered through a 0.45-μm GHP filter and run on the Varian ICP-MS 820 instrument to determine the concentration of gold in each sample. Standards were prepared using HAuCl4 in 30% HNO3.
IFN-γ ELISpot assay.
Cell-mediated immune responses in macaques were detected by measuring gamma interferon (IFN-γ) secretion in an enzyme-linked immunosorbent spot (ELISpot) assay. Macaque PBMCs were stimulated with Gag peptides of SIVmac239, SIVsm, SIVver, and HIV-1 (these peptides were obtained through the AIDS Research and Reference Reagent Program, National Institutes of Health, Bethesda, MD) and SIVsab (a kind gift of Cristian Apetrei, University of Pittsburgh, Pittsburgh, PA). Peptides were resuspended in dimethyl sulfoxide (DMSO) and pooled to span the whole Gag protein of each viral strain employed in the analysis. Specifically, the following pools were used: two pools for SIVmac239 Gag (pool 1, amino acids 1 to 263; pool 2, amino acids 253 to 510), two pools for SIVsab (pool 1, amino acids 1 to 283; pool 2, amino acids 273 to 556), two pools for SIVver (pool 1, amino acids 1 to 267; pool 2, amino acids 257 to 520), one pool for SIVsm (amino acids 1 to 500), and one pool for HIV-1 group M subtype C (amino acids 1 to 490). Additionally, four smaller pools were employed for the analysis of the conserved regions of the SIVmac239 Gag: conserved pool 1 (amino acids 149 to 171), conserved pool 2 (amino acids 225 to 251), conserved pool 3 (amino acids 257 to 371), and conserved pool 4 (amino acids 392 to 436). All assays were performed with the ELISpotPRO kit for detection of monkey IFN-γ (Mabtech AB, Nacka Strand, Sweden) according to the manufacturer's instructions.
Briefly, 1.5 × 105 Ficoll-isolated macaque PBMCs were added to 96-well plates precoated with an anti-human/monkey IFN-γ antibody (monoclonal antibody [MAb] GZ-4). Cells were resuspended in RPMI 1640 plus 10% FBS with or without 2 μg/ml of each peptide pool or, alternatively, with concanavalin (2 μg/ml) or α-CD3 (0.1 μg/ml) as positive controls. Triplicate wells were employed for each experimental condition. After 48 h of incubation at 37°C with 5% CO2, the cells were rinsed from the plates, and a biotinylated anti-human/monkey IFN-γ antibody (MAb 7-B6-1; Mabtech) was added to the wells. The plates were then washed with PBS and incubated with the substrate solution (5-bromo-4-chloro-3-indolylphosphate [BCIP]/nitroblue tetrazolium [NBT]-plus). Spot-forming cells (SFC) were counted using an automated reader (Immunospot reader; CTL Analyzers, LLC, Cleveland, OH). Data were expressed as the average number of SFC/106 cells ± standard error of the mean (SEM) after subtracting from individual values the average number of background spots detected in the negative controls.
Detection of neutralizing antibodies.
To assess the humoral response against the virus, the levels of neutralizing antibodies in sera from different time points during the study were measured as described previously (11). The TZM-bl assay system was employed for the analyses (17), using a neutralization-sensitive (SIVmac251.6) and a neutralization-resistant (SIVmac251.30) pseudotyped virus. Virus pseudotyped with murine leukemia virus Env (SVA-MLV) was included to assess non-SIV-specific neutralizing activity in the sera. Briefly, serial dilutions of sera from the indicated time points were preincubated with the viruses (ca. 150,000 relative light unit equivalents) for 1 h at 37°C. Following addition of TZM-bl target cells, the cultures were incubated for 48 h and then lysed and assayed for luciferase activity. Neutralization titers are the sample dilution at which relative luminescence units (RLU) were reduced by 50% (ID50) compared to RLU in virus control wells after subtraction of background RLU in cell control wells. In several instances, ID50 endpoint titers against SIVmac251.6 were not achieved in the dilution series employed in the assays. Thus, an 80% neutralization titer (ID80) was calculated to expose the relative potency of the neutralizing antibody response against this virus.
Quantitative assay for SIVmac251 viral RNA levels.
To quantify viral replication in vivo, plasma SIVmac251 RNA levels were measured by real-time nucleic acid sequence-based amplification (NASBA) assay, as previously described (18–20). The interassay coefficient of variation of the technique was 19% (11), well within the limits of the typical interassay variability of SIV RNA detection techniques (see, e.g., references 10 and 21). The values obtained were periodically validated using two different real-time-PCR techniques as previously described in (10, 22).
Quantitative assay for SIVmac251 viral DNA.
In order to have a surrogate marker for the viral reservoir, total viral DNA was measured by real-time PCR in two different centers, i.e., Bioqual, Inc., and ABL. For the main analyses, conducted at Bioqual, Inc., DNA was extracted with the phenol-chloroform method as previously described (10, 11). Quantification was performed by amplifying a region of the gag gene by real-time PCR in a 7700 sequence detection system (Applied Biosystems, Foster City, CA, USA). The detection limit of this assay is two copies of viral DNA/5 × 105 cells. Technical details and validations of the assay have been provided previously (10).
The analyses at ABL were conducted as described previously (19, 23). Briefly, genomic DNAs from tissues were isolated with the DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol, except for a modification at the DNA elution step (19). The SIV gag gene and the macaque albumin gene sequence (the latter used as an internal control) were amplified by real-time TaqMan qPCR technology in a 7500 real-time system (Applied Biosystems). The normalized value of the SIV viral DNA load was calculated as SIV DNA copy number/macaque albumin gene copy number × 2 × 106 and expressed as the number of SIV viral DNA copies per 106 PBMCs (23).
Viral sequencing.
Viral sequencing was performed to monitor viral evolution (sequencing of the C2-V3 region of gp130) and to monitor immune escape (sequencing of gag).
For the former, RNA was extracted from plasma samples, and the SIV env gene was then reverse transcribed and PCR amplified from the plasma RNA with the SIV versions of the primers described previously (24). Pyrosequencing of the C2-V3 region was performed on PCR amplicons, i.e., amplified from gp130 env DNA fragments. Briefly, nested PCR products for sequencing were generated using the external products described above as templates with custom-designed fusion oligonucleotides that contained the 454 adaptor sequences (Roche Lib-A primer A and primer B), followed by a 10-base-pair multiplex identifier (MID) sequence to permit sample pooling and finally SIV template-specific oligonucleotides. Samples were quantified by fluorometry with the Quant-iT PicoGreen double-stranded DNA (dsDNA) assay kit (Life Technologies), pooled in equimolar concentrations, and sequenced on a 454 GS Junior System (Roche Diagnostics) using the GS Junior titanium sequencing chemistry. The resulting reads were trimmed to exclude the MIDs and primer sequence, and low-quality reads were filtered using the GS run processor according to length and quality scores.
Sequencing of gag was performed as described previously (25). Briefly viral RNA in plasma was isolated with the QIAamp MinElute virus spin kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Viral RNA was reverse transcribed and amplified using the one-step reverse transcription-PCR kit (Qiagen), generating amplicons spanning SIVmac239. Bulk Sanger sequencing was performed on an ABI 3730xl DNA analyzer using ET terminator technology (GE Healthcare). Sequences were assembled against the SIVmac239 reference sequence (GenBank accession number M33262) and conceptually translated using CodonCode Aligner version 3.7.1.1 (CodonCode Corporation) and MacVector 11.1 trial version (MacVector, Inc.).
Bioinformatic analyses.
Multisequence alignments of reads of the C2-V3 region and neighbor-joining phylogenetic trees were constructed using BioEdit and ClustalX and using MUSCLE (PMID 15034147), and MEGA5 (PMID 21546353) for maximum-likelihood analyses.
For multisequence alignments of the Gag proteins from different primate lentiviruses, sequences were retrieved from the NCBI website and aligned with Clustal Omega (EMBL-EBI), and sequence distances were visualized using phylogeny.fr (26).
Statistical analyses.
Differences between variables were calculated using parametric tests (Student's t test for comparisons between two groups and analysis of variance [ANOVA] followed by an appropriate posttest for multiple comparisons). An appropriate transformation was applied to restore normality, where necessary. Repeated-measures tests were used when analyzing matched observations. Trends were analyzed by regression analysis, followed by the extra sum-of-squares F posttest. Calculations were conducted using the software GraphPad Prism 5.00.288 (GraphPad Software, Inc., San Diego, CA, USA).
Multivariate analysis was used to study the contributions of independent predictors to the posttherapy viral load set point and the difference between the pre- and posttherapy viral load set points (Δ viral set point). The analysis was conducted with the IBM SPSS software (v. 21; IBM, Armonk, NY, USA), using a type I regression parameter and, as dependent variables, the posttherapy viral set point (calculated as the median of the available measurements) and the Δ viral set point (calculated as a log difference between the pre- and posttherapy viral set points). The choice of the potentially predicting parameters was based on extensive literature analysis. Possibly independent predictors were considered to be the number of drugs simultaneously employed, the administration of peculiar drugs such as mefloquine or the α-CD8 monoclonal antibody, the total duration of therapy (i.e., the number of days during which macaques were exposed to drugs), the number of therapeutic cycles, and the pretherapy CD4 nadir. A “therapy” was considered to be a sequence of therapeutic cycles where the time between neighboring treatments did not exceed 90 days.
RESULTS
Auranofin and BSO kill activated T lymphocytes in vitro.
In previous studies (4, 8), we showed that auranofin induced apoptosis and downmodulation of the markers of long-lived phenotypes, i.e., CD27 and CD28, in resting TCM and TTM cells. Addition of BSO, however, did not enhance annexin V stainability or CD27 downmodulation in nonactivated central and transitional memory T cells (data not shown).
Based on a previous report showing that the combination of auranofin and BSO impacted T-cell activation in vitro more than auranofin alone (14), we decided to explore this path by examining the effects of BSO on the life span of lymphocytes treated with auranofin for 24 h and subsequently activated. When activated with α-CD3 and α-CD28 antibodies for 48 h, human CD4+ and CD8+ T cells that had received auranofin and BSO displayed lower expression of CD25, as previously reported (14), and higher annexin V stainability (Fig. 2A; see Fig. S1 in the supplemental material) than controls left untreated or treated with single agents. This effect was paralleled by failure of the two-drug-treated cells to form rosettes (Fig. 2B; see Fig. S1 in the supplemental material). CD4+ and CD8+ T cells treated with auranofin alone showed partial failure to express CD25 and form rosettes, whereas BSO alone had little or no effect on these phenomena (Fig. 2A and B; see Fig. S1 in the supplemental material). The ability of auranofin to reduce activation induced by α-CD3 and α-CD28 stimulation is consistent with the downmodulating effect of this drug on the expression of CD28 previously reported in T lymphocytes (8).
FIG 2.

Treatment with auranofin (AF) and buthionine sulfoximine (BSO) reduces CD25 levels and activation of T lymphocytes in vitro. CD4+ and CD8+ T cells were isolated from whole blood and left untreated or treated with AF (250 nM), BSO (250 μM), or a combination of the two. At 24 h posttreatment, cells were activated with α-CD3 and α-CD28 antibodies. (A) Levels of CD25 and annexin V measured at 48 h postactivation (data are from CD4+ T cells). (B) Photographs taken at 72 h postactivation (CD4+ T-cell cultures). (C and D) Viability of CD4+ (C) (6 donors) and CD8+ (D) (3 donors) T cells, as measured with the MTS assay at 72 h postactivation. Values are means ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CD4+ and CD8+ T cells treated with auranofin and BSO eventually exhibited very low cell viability at 72 h (Fig. 2C and D). Auranofin alone decreased cell viability at 72 h, but this effect was weaker than that induced by the two-drug combination (Fig. 2C and D). BSO alone did not exert any significant effect on cell viability.
Similar results were obtained when CD4+ and CD8+ T cells were activated with phytohemagglutinin (PHA) (see Fig. S2 in the supplemental material). We conclude that T lymphocytes treated simultaneously with auranofin and BSO fail in the normal activation process and die.
Auranofin and BSO decrease T-lymphocyte activation in vivo.
We recently conducted a small but controlled study in chronically SIVmac251-infected rhesus macaques (11) aimed at showing the impact of auranofin and BSO (administered before stopping ART) on posttherapy viral load control. Macaques that were progressing to AIDS or that were derived from other studies in which potentially therapeutic strategies had failed were treated with a highly intensified ART, either alone or in combination with auranofin (11). Of the latter animals, (i) two did not receive any further treatment and were kept untreated following therapy suspension, (ii) two received further short ART cycles at viral rebound after therapy suspension to limit viral reseeding, and (iii) two received an eventual cycle of BSO before ART and auranofin were suspended (treatments are summarized in Fig. 1 and Table 1). One of the animals of the second group received BSO during the last of the further short ART cycles, but, in contrast to the schedule applied to the third group, BSO was administered in the absence of auranofin. This schedule allowed us to preliminarily distinguish between the additive and synergistic effects of the two drugs in vivo.
Using frozen PBMC samples from these macaques, we investigated whether the in vitro killing effects of the auranofin-BSO combination on activated T lymphocytes were associated with decreased numbers of activated cells in vivo during the posttherapy viral rebound. As a tool to measure the frequency of activated T lymphocytes, we chose the widely validated activation marker HLA-DR (27). We found that the macaques that had received ART, auranofin, and BSO displayed a lower frequency of HLA-DR+ T lymphocytes at viral rebound (Fig. 3). Subsequently, CD8+ T cells became activated, and their activation levels were maintained throughout the follow-up. In contrast, CD4+ T-cell activation was maintained at lower levels than those for CD8+ T cells, with periodic increases that were transient. In contrast, following suspension of ART plus auranofin or ART plus BSO, there was a higher level of lymphocyte activation at viral rebound that was more marked in the CD4+ T-cell compartment. The activation level in these control macaques was maintained stably over time or, in one macaque, dropped during progression to AIDS (Fig. 3), in line with the immune deficiency and lymphocyte anergy characterizing the later stages of the disease (28, 29). We conclude that monkeys that had received the combination of auranofin and BSO were characterized by low-level T-cell activation at viral rebound.
FIG 3.

Treatment with auranofin and BSO reduces levels of HLA-DR in T lymphocytes at viral rebound. Levels of HLA-DR were measured by flow cytometry and are displayed as percentages of HLA-DR+ CD4+ (squares) and HLA-DR+CD8+ (circles) T cells. (A to C) The posttreatment data refer to the time frame following suspension of ART-auranofin-BSO (A), ART-auranofin (B), and ART-BSO (C). The gray arrows indicate the approximate time of viral rebound. (D) Proportions of HLA-DR+ CD4+ (squares) and HLA-DR+ CD8+ (circles) T cells closest to the viral rebound. Data include an additional macaque subjected to ART plus auranofin without BSO (P177; not shown in the figure). Probit transformation was applied to normalize the percentage values to a scale of −∞ to +∞, suitable for parametric test analyses. *, P < 0.05; **, P < 0.01.
We then aimed at investigating how the drug effects would be exerted following therapy suspension. We therefore measured the persistence of the drug effects during the posttherapy period (Fig. 4). Concentration of glutathione in PBMCs is the most reliable clinical indicator for the activity of BSO, which is an irreversible inhibitor of γ-glutamyl cysteine synthetase (a limiting-rate enzyme in glutathione synthesis) (30). Glutathione was maintained at low levels (similar to those observed in our in vitro treatments [data not shown]) for 1 month after BSO administration (Fig. 4A). At 2 months posttherapy, one of the macaques (4890) experienced a major increase in glutathione, which may be due to a compensatory increase, whereas the other two macaques receiving BSO maintained decreased GSH levels compared to baseline (Fig. 4A).
FIG 4.

In vivo levels of glutathione and gold before, during, and after treatment with auranofin and BSO. (A) Concentration of total glutathione (i.e., GSH/GSSG, the molecule[s] depleted by BSO) in PBMCs of the two macaques (4890 and P157) that received auranofin and BSO (squares) and in the macaque (P252) that received BSO separately from auranofin (circles). The arrow shows the approximate timing of viral rebound following therapy suspension. Data are expressed as means ± SEM (calculated from the values from the macaques that received auranofin and BSO [squares]). (B) Concentration of gold (i.e., the active principle of auranofin) in the plasma of four treated macaques. The dotted line shows the concentration of auranofin employed in the in vitro experiments described in this article. Data are expressed as means ± SEM. For details about dosages and schedule of administration of auranofin and BSO see Fig. 1, Table 1 and reference 11.
Measurement of the plasma levels of the gold ion [Au(I)], i.e., the active principle of auranofin, showed that the metal persisted at therapeutic concentrations for at least 2 months following therapy suspension (Fig. 4B). Persistence of the drugs, or their effects, thus overlapped with the initial posttherapy viral rebound, which is likely to be a key event for the subsequent virus/host dynamics. On the whole, these data show that the combination of auranofin and BSO, but not the two drugs administered separately, decreases the numbers of activated T lymphocytes during viral rebound and that the “functional cure”-like condition is preceded by T-cell activation dynamics different from those in macaques progressing to AIDS.
Decreased numbers of activated T lymphocytes are accompanied by the emergence of broadly reactive anti-Gag cell-mediated responses.
Cell-mediated immune responses against Gag have consistently been shown to negatively correlate with viral load (31–35). As our macaques displayed enhanced levels of immunity against the Gag antigen following treatment with ART, auranofin, and BSO (11) (see Fig. S3 in the supplemental material), we decided to better characterize this immune response by analyzing whether the epitopes targeted were evolutionarily conserved. We reasoned that epitopes which underwent little or no change through the evolution of different lentiviral strains were likely to be fundamental for viral fitness and thus be effective targets, which could account for the long-term intermittent viral load control displayed by the macaques following suspension of ART, auranofin, and BSO. We analyzed the responses of the PBMCs of the macaques that had received ART-auranofin-BSO to stimulation with Gag peptides of distinct primate lentiviruses, such as SIVsm from sooty mangabeys (Cercocebus atys), SIVsab from African green monkeys (Chlorocebus sabaeus), SIVver from vervet monkeys (Chlorocebus pygerythrus), and HIV-1 from humans (subtype C). We found that the responses were cross-reactive to various degrees, with the strongest cross-reactivity being observed with SIVsm (Fig. 5A), in line with the close phylogenetic relationship of this virus with SIVmac (Fig. 5B). In the same phylogenetic tree (Fig. 5B) the SIVsab strain clustered separately from the other strains infecting African green monkeys (i.e., SIVver, SIVgri, and SIVtan), in accordance with the previously described mosaic structure of the genetic material of SIVsab (36). Notably, both macaques also showed cross-reactivity against SIVver and more tenuous responses toward SIVsab and HIV-1, with the latter being more evident in macaque 4890 (Fig. 5A; see Fig. S4 in the supplemental material). Given the ability of PBMCs of rhesus macaques to respond upon stimulation with antigens of viruses related to, yet significantly different from, SIVmac251, we conclude that the immune responses evoked by auranofin and BSO in vivo were broadly cross-reactive.
FIG 5.

Posttherapy development of broadly reactive anti-Gag immune responses in macaques treated with ART-auranofin-BSO. (A) Cell-mediated immune responses against the Gag antigens of various simian and human lentiviruses. The viruses employed are SIVmac239 (N portion and C portion, light and dark blue, respectively), SIVsm (red), SIVsab (N portion and C portion, ochre and orange, respectively), SIVver (N portion and C portion, light and dark green, respectively), and HIV-1 (purple). (B) Phylogenetic tree constructed on the basis of the Gag amino acid sequences of primate lentiviruses. Highlighted are the viruses used for the ELISpot analyses. Gag sequences from the following viruses were used to generate the tree: SIVsab, host Chlorocebus sabaeus (accession number U04005.1); SIVsm, host Cercocebus atys (accession number AF334679.1); HIV-2, host Homo sapiens (accession number A14062.1); SIVmac239, host Macaca mulatta (accession number AY611495.1); SIVver, host Chlorocebus aethiops (accession number M30931.1); SIVtan, host Chlorocebus tantalus (accession number U58991.1); SIVgri, host Chlorocebus pygerythrus (accession number M66437.1); SIVgor, host Gorilla gorilla (accession number FJ424871.1); HIV-1 (subtype C), host Homo sapiens (accession number AX457080.1); SIVcpz, host Pan troglodytes (accession number JN091691.1); SIVsun, host Cercopithecus solatus (accession number AF131870.1); SIVlhoest, host Cercopithecus lhoesti (accession number AF075269.1); SIVolc, host Procolobus verus (accession number AM713177.1); and SIVcol, host Colobus guereza (accession number AF301156.1). The recently sequenced Gag from ELVgv, an endogenous lentivirus from Galeopterus variegatus (order Dermoptera) (50) was used as an outgroup. The tree was constructed with phylogeny.fr (as described in reference 26) using a Clustal Ω alignment. (C) Cell-mediated immune responses against peptide pools encompassing conserved regions of the C-terminal half of the SIVmac239 Gag protein (i.e., CR3 and CR4; see Text S1 in the supplemental material). The sequences of the conserved regions employed for the analysis are shown in Text S1 in the supplemental material. Immune responses were measured by IFN-γ ELISpot. The ELISpot data are expressed as number of spot-forming cells (SFC) × 106 PBMCs. The ratio of conserved responses over the total is calculated, for each time point, as a sum of the mean number of SFC detected with CR3 and CR4 peptides divided by the mean number of SFC detected with peptides spanning the C-terminal half of the Gag.
To further analyze these broadly reactive responses, our next step was to test whether the highly conserved regions of Gag might be the main target of the immune responses following treatment with auranofin and BSO. Guided by sequence alignments (see Text S1 in the supplemental material), we selected four highly conserved regions of Gag and analyzed the IFN-γ response to peptide pools corresponding to these regions. Two peptide pools (i.e., corresponding to conserved regions CR1 and CR2, spanning amino acids 149 to 171 and 225 to 251, respectively) lay in the N-terminal half and two (i.e., corresponding to conserved regions CR3 and CR4, spanning amino acids 257 to 371 and 389 to 439, respectively) in the C-terminal half of the Gag antigen (note that the C-terminal half of Gag displays a higher degree of evolutionary conservation [see Text S1 in the supplemental material]). Our analyses showed that the strongest responses were evoked by CR3 (Fig. 5C; see Fig. S5 in the supplemental material). Of note, titers of neutralizing antibodies did not show any association with the therapeutic effects observed (see Fig. S6 in the supplemental material).
The emergence of broadly reactive anti-Gag cell-mediated responses is accompanied by IFN-γ responses in natural killer cells.
While this article was being prepared, Scott-Algara et al. (37) reported significant IFN-γ responses in natural killer (NK) cells of posttreatment controllers from the VISCONTI cohort (i.e., individuals who had started ART early during acute infection and had been subjected to treatment interruption). Therefore, we aimed at investigating whether these phenomena might also occur in the macaques that had received auranofin and BSO. Our results showed that macaques that had received auranofin and BSO simultaneously had detectable IFN-γ responses in CD56+ cells upon stimulation with Gag peptides at at least one time point of the posttreatment follow-up (Fig. 6). No responses were observed posttreatment in the macaques that had received auranofin without BSO (Fig. 6). One macaque that had received auranofin and BSO separately (P252) showed, in the posttreatment period, an intermediate frequency of Gag-specific CD56+ IFN-γ+ cells (Fig. 6).
FIG 6.
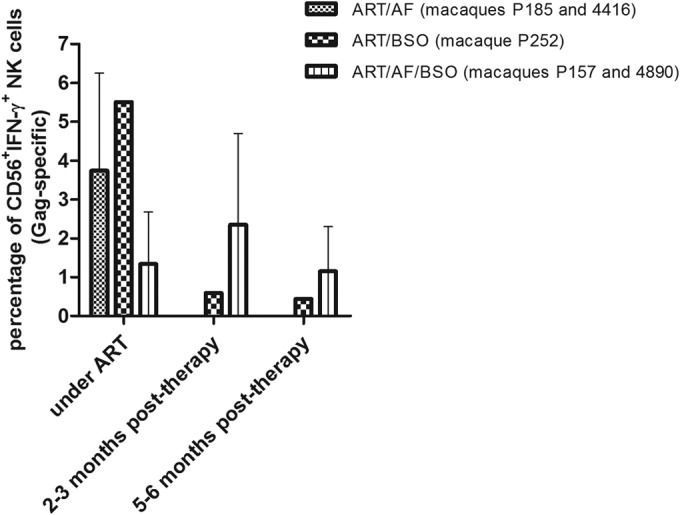
Macaques that had received ART, auranofin, and BSO displayed higher posttherapy levels of Gag-specific, IFN-γ-producing natural killer (NK) cells than control animals. PBMCs were stimulated with SIV-specific Gag peptides for 24 h to evaluate T-cell-dependent production of IFN-γ by CD56+ NK cells. Data are expressed as a mean ± range. Time points were selected on the basis of the documented presence of ELISpot responses and on sample availability.
Extended follow-up of macaques that had received auranofin-BSO.
As mentioned in the previous sections, our earlier study showed an intermittent control of viral load only in those macaques that had been treated with ART, auranofin, and BSO simultaneously, although all macaques that had received auranofin showed some varying degree of control of viral load following treatment suspension (11). To explore the long-term effects of the virus/immune system dynamics induced by treatment with auranofin and BSO, we subjected to extended follow-up the only two macaques from our cohort (11) that had been simultaneously treated with ART, auranofin, and BSO. Thereafter, both macaques had shown intermittent viral load control (11), and no further treatment cycles were administered during the entire follow-up period.
One macaque (P157), which had shown in the posttherapy period a viral set point consistent with elite control (i.e., <5,000 copies of viral RNA/ml [25]), was subjected to CD8+ cell depletion at 42 weeks posttherapy, when viral load in plasma and vaginal liquids was undetectable, as previously described (11). Sequencing of the gag gene following CD8+ T-cell depletion revealed a homogeneous wild-type Gag, identical to the ancestral SIVmac239 Gag sequence (data not shown), thus supporting the aforementioned hypothesis that targeting of the highly conserved regions that are fundamental for correct protein folding limits the virus evolutionary options. In this macaque, however, the original CD8+ T-cell numbers never recovered (before depletion, 1,022 ± 274 [mean ± standard deviation {SD}]; after depletion, 396 ± 204 cells/μl) (see Fig. S7 in the supplemental material), leading to partial loss of immune control despite maintenance of detectable anti-Gag immunity (mean number of spot-forming cells [SFC]/106 PBMCs = 1,072 ± 677 [mean ± SD]). Instead, the number of CD4+ T cells was kept relatively stable after a transient decrease observed during the early stages of the CD8+ T-cell depletion (before depletion, 896 ± 228; after depletion, 886 ± 411, cells/μl [mean ± SD]) (see Fig. S7 in the supplemental material). When we compared the viral loads in the same macaque after CD8+ cell depletion with the pretherapy viral load data for this macaque as measured by the same technique (NASBA), we found that despite the low levels of CD8+ T cells, the viral load was maintained at a level approximately one order of magnitude lower than the pretherapy viral set point (respectively, 4.14 ± 0.17 log10 viral RNA copies/ml versus 5.04 ± 0.20 log10 viral RNA copies/ml [mean ± SEM; P = 0.0165 by two-tailed t test]) (Fig. 7A). The posttherapy viral loads were double-checked with another technique, i.e., real-time PCR (see Table S1 in the supplemental material). At the time of euthanasia, the animal did not show any AIDS-related signs or symptoms, and its weight had increased by 15%, compared to the pretherapy values (data not shown). Our anti-CD8 treatment was aimed at showing a transient increase in viral load, similar to that observed in the model described by Pandrea et al. (38). The unforeseen incomplete recovery of CD8+ T cells and partial loss of viral load control induced us, for both ethical and investigational reasons, not to apply the same depletion to the other animal that had received the combined treatment with ART, auranofin, and BSO.
FIG 7.

Long-term follow-up of viral load in macaques treated with ART-auranofin-BSO. Posttherapy viral loads are shown as circles. The posttherapy viral set points in the follow-up period after our previous publication (11) are shown by the brackets. Pretherapy viral set points are shown by the dashed horizontal lines and are based on all available measurements starting from 30 weeks before the initiation of ART. Both pre- and posttherapy viral set points are based on NASBA assay measurements (limit of detection, 50 copies of viral RNA/ml of plasma). Viral loads outside the brackets (not included in the analysis) are shown for clarity's sake and are aimed at aiding the reader in the historical understanding of the entire posttherapy follow-up. For macaque P157, the viral loads outside the brackets, and not included in the present analysis, were measured with a different technique (i.e., real-time PCR; limit of detection, 40 copies of viral RNA/ml of plasma [11]). The areas shaded in gray correspond to the periods in which a definition for elite control was applicable (25) and are characterized by an undetectable viral load set point.
This other macaque (4890) displayed, from week 43 posttherapy, a viral set point below the detection limit (Fig. 7B), and this was confirmed by independent testing in three different laboratories (see Table S1 in the supplemental material). Viral DNA in PBMCs, a widely accepted surrogate marker of the viral reservoir (39, 40), also gradually declined over time (see Fig. S8 in the supplemental material). The result of the extended follow-up thus showed a scenario that was, in the long term, very similar to that observed with the other auranofin-BSO-treated macaque (P157), which had shown undetectable or low-level viral DNA before it was subjected to CD8+ cell depletion (11). Moreover, CD4+ T cells of macaque 4890 gradually increased following the initial consumption that had been observed during the acute-infection-like condition at viral rebound following therapy suspension (see Fig. S8 in the supplemental material). Of note, apart from cellular/molecular analyses, the consistent weight increase displayed by macaque 4890 following therapy suspension is in line with the absence of disease progression (see Fig. S8 in the supplemental material). Samples from week 49 to week 92 were subjected to further validations with different assays in distinct locations (see Table S1 in the supplemental material), which confirmed the overall trends.
Treatment with ART-auranofin-BSO is followed by maintenance of limited viral diversity or gradual loss of viral quasispecies.
Because following treatment with ART, auranofin, and BSO, the macaques showed periodic viral blips (Fig. 7), we characterized the virus that emerged during these blips. For this purpose, we performed next-generation 454 pyrosequencing of the viral populations in plasma samples corresponding to selected blips. Based on estimates of viral load or inefficiencies in RNA extractions and/or reverse transcription steps, we estimated that a minimum of 100 SIVmac RNA copies (per plasma sample) were sequenced per bar-coded primer set. Based on >1,000 sequence reads, we obtained 10× oversampling of the SIVmac population at each time point. Finally we sequenced the variable C2/V3 region of the viral envelope gp130 glycoprotein to ensure a high SIVmac diversity. The resulting viral quasispecies can be visualized with neighbor-joining trees (Fig. 8) or similar maximum-likelihood trees (data not shown; sequences are available upon request).
FIG 8.

Viral diversity following suspension of ART-auranofin-BSO. The trees show the viral quasispecies detected in plasma during different viral load peaks following therapy suspension. The time point analyzed and the relative frequency of the given quasispecies during that time point are shown. The analysis was performed by 454 pyrosequencing of the C2-V3 region of the env gene. The figure presents only unique quasispecies over a 1% frequency, i.e., set by the approximate error rate of 454 pyrosequencing and quantity of RNA templates (51).
Macaque P157 displayed only one haplotype per peak after the initial viral rebound. Phylogenetic analysis showed that the dominant SIVmac haplotypes at different peaks were quite genetically distinct (2 to 14 nucleotide substitutions in a 365-nucleotide sequence read), suggesting that the distinct haplotypes were not a consequence of 454 read error and not due to ongoing slow evolution caused by continuous virus replication. Instead, these findings suggest multiple events of reactivation from latency in the interpeak period. Interestingly, during the viral rebound that accompanied CD8+ cell depletion, four main viral quasispecies emerged, suggesting that the diversity of the viral reservoir may have been controlled by the CD8+ cell-mediated immunity.
In macaque 4890, a certain degree of viral diversity appeared during the initial viral rebound (Fig. 8). However, despite an attempt to escape immunity detected during the blip at 40 weeks posttherapy, the number of viral quasispecies decreased, and thereafter, the virus detected during each blip became homogeneous (Fig. 8), in line with the decrease in viral DNA (see Fig. S8 in the supplemental material). Interestingly, at 1 year after therapy suspension, the dominant virus emerging during the blip was very close to the ancestral SIVmac251 sequence, suggesting reactivation from a long-lived reservoir (Fig. 8). Taken together, these data show that inhibition of T-cell activation at viral rebound followed by broadly reactive anti-Gag cell-mediated responses was associated with maintenance of low-level viral diversity.
A retrospective analysis of a cohort of 19 macaques supports the efficacy and safety of auranofin-BSO regimens.
We finally aimed at giving a statistical confirmation of the therapeutic effects observed. We therefore tested, by multivariate analysis, the outcomes of a series of treatments, consisting of antiretroviral drugs, auranofin, BSO, and CD8+ cell depletion, within a cohort of 19 SIVmac251-infected macaques subjected to several related treatments (references 4, 10, and 11 and unpublished data). Treatment outcome was considered in terms of viral set point reduction (Δ viral set point) and posttherapy viral set point. The analysis confirmed that parameters associated with the viral reservoir size (e.g., inhibition of initial reseeding of viral reservoir through short ART cycles at viral rebound; P = 0.033), with immune containment of viral load (absence of CD8+ cell depletion; P = 0.001), and with inhibition of T-cell activation (administration of the complete treatment consisting of 5-drug ART, auranofin, and BSO; P < 0.001) were significantly associated with posttherapy decrease of the viral set point (see Table S2 in the supplemental material). Furthermore, nadir CD4+ T-cell counts (P = 0.002), which identify the viral reservoir (41), but not pretherapy viral loads (P = 0.520), also contributed to the extent of the Δ viral set point (see Table S2 in the supplemental material). This analysis confirms the working hypothesis that a functional cure is contributed by (i) a restricted viral reservoir, (ii) restricted CD4+ T-cell activation, and (iii) efficient CD8+ immune responses. Moreover, ELISpot and sequencing data support the hypothesis that viral load containment is likely to be contributed by the aforementioned parameters and not to be dependent on the genetic background of the animals (see Text S2 in the supplemental material).
DISCUSSION
Based on the results shown in the present article, it is possible to hypothesize that decreased T-cell activation is a key component for a functional cure of HIV/AIDS (42), along with viral reservoir reduction and broadly reactive cell-mediated responses. Based on the evidence presented here, we propose a reconstruction of the events leading to the posttreatment viral load containment described in the present paper, as follows.
(i) Previously published data showed that viral reservoir reduction obtained by inducing selective apoptosis in the memory compartment using a combination of auranofin and ART resulted in a decrease of the viral load set point. However, this did not lead to control of the viral load to undetectable levels (4). That a limited viral reservoir alone is insufficient for obtaining such a condition is also shown by the recent cases of the “Mississippi baby” and the “Boston patients” (5, 6), as well as controlled studies in macaques (3, 4). It appears that the absence of any specific immunity allows a limited number of infected cells to restore disease progression.
(ii) From our retrospective analysis, addition of BSO to auranofin, carrying out further immune-modulating effects by decreasing intracellular glutathione, appeared to be essential for obtaining prolonged, albeit intermittent, posttherapy control of the viral load in our macaques. The in vitro data presented here show a decreased life span of activated T lymphocytes after treatment with auranofin and BSO. In vivo, the auranofin levels and glutathione depletion persisted at least for 1 month after drug withdrawal. Thus, if the viral load peak that we observed during the first viral rebound were a novel acute infection-like condition, as we hypothesized previously (for a review, see reference 43), its timing might fall into the therapeutic window of the drugs, influencing the subsequent course of the infection. In particular, the decrease in activated T lymphocytes that we show in the present study may have limited, at viral rebound, the immune exhaustion linked to immune hyperactivation which occurs during disease progression (44, 45).
(iii) Decreased numbers of activated T cells may at first glance appear to be contradictory to the broadly reactive cell-mediated responses characterizing the intermittent but prolonged control of the infection that ensued. Our result, however, is in line with recent research supporting the concept that activation of immunodominant responses may hamper more efficient responses directed toward conserved Gag epitopes (13). One possibility is that reduction of immune activation at viral rebound may have hampered major histocompatibility complex (MHC)-restricted immunodominant responses toward epitopes capable of escaping the immune response through mutations. An alternative/complementary hypothesis is that during the initial viral rebound, the selective pressure exerted by auranofin and BSO on the SIV-activated T-cell clones would induce the preferential survival of the responses toward the most frequent antigens, i.e., the highly conserved epitopes of Gag, which are less likely to mutate due to structural constraints (46). These immune responses are in line with maintenance of low-level viral diversity over time and failure of the virus to evolve phenotypes distant from those of the aboriginal virus. This may have ensured the prolonged decreased viral set point that we observed.
We are well aware that the present study presents some limitations, such as the fact that it was conducted in macaques, not in humans, and with a relatively low number of animals employed. We address these as follows. (i) The exceptional length of the follow-up, driven by the necessity to assess the long-term efficacy of the therapy (i.e., the only parameter which can yield the promise of a meaningful improvement over current ART for life), significantly increased the costs of the study, hampering the expansion of the numbers at this stage. (ii) The multivariate analysis reported here, which took into account a large cohort of animals that had received a wide array of similar, but yet incomplete, therapeutic interventions, provides strong statistical support for the notion that the posttreatment effects were specifically associated with the auranofin-BSO combination. (iii) This conclusion is also supported by ELISpot and sequencing analyses excluding the contribution of an MHC class I genetic background to the result obtained. (iv) Moreover, when dramatic changes in disease progression are clearly associated with the timing and striking benefit of a treatment, limited numbers have previously been considered to be acceptable (2, 47–49), given their potential role in opening new therapeutic avenues. Finally, our preliminary data indicate that, in contrast to from control animals, macaques that had received ART, auranofin, and BSO displayed detectable posttherapy levels of Gag-specific, IFN-γ-producing natural killer (NK) cells. If this early observation is confirmed with larger animal cohorts, it would represent an interesting resemblance to HIV-positive individuals from the VISCONTI cohort experiencing a functional cure of the disease following therapy suspension. These phenomena will need to be explored by future research.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by ISS intramural grants (recipient, A.S.), ISS travel grants (recipients, I.L.S. and A.S.), NIAID-NIH contract HHSN27201100016C (recipient, D.M.), NSF grant 1058726 (recipient, N.F.), and, in part, the Intramural Research Program of the NIH, National Cancer Institute (recipient, M.R.-G.).
We are grateful to Cristian Apetrei and Egidio Brocca-Cofano for providing SIVver and SIVsab peptides, to Rossella Sgarbanti for technical advice, and to Alessandra Boe, Sarah Spell, and the staff at Bioqual and ABL for technical support. The NIH AIDS Reagent program, DAIDS, NIAID, provided the complete set of SIVmac239 Gag peptides.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00396-15.
REFERENCES
- 1.Trono D, Van Lint C, Rouzioux C, Verdin E, Barré-Sinoussi F, Chun TW, Chomont N. 2010. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 329:174–180. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- 2.Van Rompay KK, Trott KA, Jayashankar K, Geng Y, LaBranche CC, Johnson JA, Landucci G, Lipscomb J, Tarara RP, Canfield DR, Heneine W, Forthal DN, Montefiori D, Abel K. 2012. Prolonged tenofovir treatment of macaques infected with K65R reverse transcriptase mutants of SIV results in the development of antiviral immune responses that control virus replication after drug withdrawal. Retrovirology 9:57. doi: 10.1186/1742-4690-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mavigner M, Watkins B, Lawson B, Lee ST, Chahroudi A, Kean L, Silvestri G. 2014. Persistence of virus reservoirs in ART-treated SHIV-infected rhesus macaques after autologous hematopoietic stem cell transplant. PLoS Pathog 10:e1004406. doi: 10.1371/journal.ppat.1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis MG, DaFonseca S, Chomont N, Palamara AT, Tardugno M, Mai A, Collins M, Wagner WL, Yalley-Ogunro J, Greenhouse J, Chirullo B, Norelli S, Garaci E, Savarino A. 2011. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 25:1347–1356. doi: 10.1097/QAD.0b013e328347bd77. [DOI] [PubMed] [Google Scholar]
- 5.Shah SK, Persaud D, Wendler DS, Taylor HA, Gay H, Kruger M, Grady C. 2014. Research on very early ART in neonates at risk of HIV infection. Lancet Infect Dis 14:797. doi: 10.1016/S1473-3099(14)70893-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henrich TJ, Hanhauser E, Marty FM, Sirignano MN, Keating S, Lee TH, Robles YP, Davis BT, Li JZ, Heisey A, Hill AL, Busch MP, Armand P, Soiffer RJ, Altfeld M, Kuritzkes DR. 2014. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: report of 2 cases. Ann Intern Med 161:319–327. doi: 10.7326/M14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P, Melard A, Prazuck T, Descours B, Guergnon J, Viard JP, Boufassa F, Lambotte O, Goujard C, Meyer L, Costagliola D, Venet A, Pancino G, Autran B, Rouzioux C, ANRS VISCONTI Study Group . 2013. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy. ANRS VISCONTI Study. PLoS Pathog 9:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chirullo B, Sgarbanti R, Limongi D, Shytaj IL, Alvarez D, Das B, Boe A, DaFonseca S, Chomont N, Liotta L, Petricoin EI, Norelli S, Pelosi E, Garaci E, Savarino A, Palamara AT. 2013. A candidate anti-HIV reservoir compound, auranofin, exerts a selective ‘anti-memory’ effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis 4:e944. doi: 10.1038/cddis.2013.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roder C, Thomson MJ. 2015. Auranofin: repurposing an old drug for a golden new age. Drugs R D 15:13–20. doi: 10.1007/s40268-015-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shytaj IL, Norelli S, Chirullo B, Della Corte A, Collins M, Yalley-Ogunro J, Greenhouse J, Iraci N, Acosta EP, Barreca ML, Lewis MG, Savarino A. 2012. A highly intensified ART regimen induces long-term viral suppression and restriction of the viral reservoir in a simian AIDS model. PLoS Pathog 8:e1002774. doi: 10.1371/journal.ppat.1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shytaj IL, Chirullo B, Wagner W, Ferrari MG, Sgarbanti R, Corte AD, LaBranche C, Lopalco L, Palamara AT, Montefiori D, Lewis MG, Garaci E, Savarino A. 2013. Investigational treatment suspension and enhanced cell-mediated immunity at rebound followed by drug-free remission of simian AIDS. Retrovirology 10:71. doi: 10.1186/1742-4690-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murakoshi H, Akahoshi T, Koyanagi M, Chikata T, Naruto T, Maruyama R, Tamura Y, Ishizuka N, Gatanaga H, Oka S, Takiguchi M. 4 March 2015. Clinical control of HIV-1 by cytotoxic T cells specific for multiple conserved epitopes. J Virol doi: 10.1128/JVI.00020-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, Lai J, McHugh HL, Hao H, Zhang H, Margolick JB, Gurer C, Murphy AJ, Valenzuela DM, Yancopoulos GD, Deeks SG, Strowig T, Kumar P, Siliciano JD, Salzberg SL, Flavell RA, Shan L, Siliciano RF. 2015. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 517:381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vint IA, Chain BM, Foreman JC. 1993. The interaction of auranofin and buthionine sulfoximine blocks activation of human peripheral T lymphocytes. Cell Immunol 152:152–161. doi: 10.1006/cimm.1993.1275. [DOI] [PubMed] [Google Scholar]
- 15.Vargas-Inchaustegui DA, Xiao P, Tuero I, Patterson LJ, Robert-Guroff M. 2012. NK and CD4+ T cell cooperative immune responses correlate with control of disease in a macaque simian immunodeficiency virus infection model. J Immunol 189:1878–1885. doi: 10.4049/jimmunol.1201026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vargas-Inchaustegui DA, Xiao P, Demberg T, Pal R, Robert-Guroff M. 7 May 2015. Therapeutic envelope vaccination in combination with antiretroviral therapy temporarily rescues SIV-specific CD4(+) T cell-dependent NK cell effector responses in chronically infected rhesus macaques. Immunology doi: 10.1111/imm.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol 79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romano JW, Shurtliff RN, Dobratz E, Gibson A, Hickman K, Markham PD, Pal R. 2000. Quantitative evaluation of simian immunodeficiency virus infection using NASBA technology. J Virol Methods 86:61–70. doi: 10.1016/S0166-0934(99)00184-6. [DOI] [PubMed] [Google Scholar]
- 19.Lee EM, Chung HK, Livesay J, Suschak J, Finke L, Hudacik L, Galmin L, Bowen B, Markham P, Cristillo A, Pal R. 2010. Molecular methods for evaluation of virological status of nonhuman primates challenged with simian immunodeficiency or simian-human immunodeficiency viruses. J Virol Methods 163:287–294. doi: 10.1016/j.jviromet.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Dunham RM, Gordon SN, Vaccari M, Piatak M, Huang Y, Deeks SG, Lifson J, Franchini G, McCune JM. 2013. Preclinical evaluation of HIV eradication strategies in the simian immunodeficiency virus-infected rhesus macaque: a pilot study testing inhibition of indoleamine 2,3-dioxygenase. AIDS Res Hum Retroviruses 29:207–214. doi: 10.1089/AID.2012.0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diop OM, Gueye A, Dias-Tavares M, Kornfeld C, Faye A, Ave P, Huerre M, Corbet S, Barre-Sinoussi F, Müller-Trutwin MC. 2000. High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J Virol 74:7538–7547. doi: 10.1128/JVI.74.16.7538-7547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cline AN, Bess JW, Piatak M Jr., Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol 34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 23.Chung HK, Unangst T, Treece J, Weiss D, Markham P. 2008. Development of real-time PCR assays for quantitation of simian betaretrovirus serotype-1, -2, -3, and -5 viral DNA in Asian monkeys. J Virol Methods 152:91–97. doi: 10.1016/j.jviromet.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 24.Ratcliff AN, Shi W, Arts EJ. 2013. HIV-1 resistance to maraviroc conferred by a CD4 binding site mutation in the envelope glycoprotein gp120. J Virol 87:923–934. doi: 10.1128/JVI.01863-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burwitz BJ, Giraldo-Vela JP, Reed J, Newman LP, Bean AT, Nimityongskul FA, Castrovinci PA, Maness NJ, Leon EJ, Rudersdorf R, Sacha JB. 2012. CD8+ and CD4+ cytotoxic T cell escape mutations precede breakthrough SIVmac239 viremia in an elite controller. Retrovirology 9:91. doi: 10.1186/1742-4690-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cockerham LR, Siliciano JD, Sinclair E, O'Doherty U, Palmer S, Yukl SA, Strain MC, Chomont N, Hecht FM, Siliciano RF, Richman DD, Deeks SG. 2014. CD4+ and CD8+ T cell activation are associated with HIV DNA in resting CD4+ T Cells. PLoS One 9:e110731. doi: 10.1371/journal.pone.0110731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boasso A, Shearer GM. 2008. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol 126:235–242. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borthwick NJ, Bofill M, Gombert WM, Akbar AN, Medina E, Sagawa K, Lipman MC, Johnson MA, Janossy G. 1994. Lymphocyte activation in HIV-1 infection. II. Functional defects of CD28− T cells. AIDS 8:431–441. [DOI] [PubMed] [Google Scholar]
- 30.Griffith OW, Meister A. 1979. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J Biol Chem 254:7558–7560. [PubMed] [Google Scholar]
- 31.Stephenson KE, Li H, Walker BD, Michael NL, Barouch DH. 2012. Gag-specific cellular immunity determines in vitro viral inhibition and in vivo virologic control following simian immunodeficiency virus challenges of vaccinated rhesus monkeys. J Virol 86:9583–9589. doi: 10.1128/JVI.00996-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards BH, Bansal A, Sabbaj S, Bakari J, Mulligan MJ, Goepfert PA. 2002. Magnitude of functional CD8+ T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol 76:2298–2305. doi: 10.1128/jvi.76.5.2298-2305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuñiga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J Virol 80:3122–3125. doi: 10.1128/JVI.80.6.3122-3125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 35.Julg B, Williams KL, Reddy S, Bishop K, Qi Y, Carrington M, Goulder PJ, Ndung'u T, Walker BD. 2010. Enhanced anti-HIV functional activity associated with Gag-specific CD8 T-cell responses. J Virol 84:5540–5549. doi: 10.1128/JVI.02031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin MJ, Hui H, Robertson DL, Müller MC, Barré-Sinoussi F, Hirsch VM, Allan JS, Shaw GM, Sharp PM, Hahn BH. 1994. Mosaic genome structure of simian immunodeficiency virus from west African green monkeys. EMBO J 13:2935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott-Algara D, Didier C, Arnold V, Cummings JS, Sáez-Cirión A, Boufassa F, Rouzioux C, Hocqueloux L, Lambotte O. 2015. Post-treatment controllers have particular NK cells with high anti-HIV capacity: VISCONTI Study, abstr 52 Abstr. Conf Retroviruses Opportun Infect. [Google Scholar]
- 38.Pandrea I, Gaufin T, Gautam R, Kristoff J, Mandell D, Montefiori D, Keele BF, Ribeiro RM, Veazey RS, Apetrei C. 2011 Functional cure of SIVagm infection in rhesus macaques results in complete recovery of CD4+ T cells and is reverted by CD8+ cell depletion. PLoS Pathog 7:e1002170. doi: 10.1371/journal.ppat.1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lafeuillade A, Poggi C, Chadapaud S, Hittinger G, Khiri H, Halfon P. 2001. Impact of immune interventions on proviral HIV-1 DNA decay in patients receiving highly active antiretroviral therapy. HIV Med 2:189–194. doi: 10.1046/j.1468-1293.2001.00065.x. [DOI] [PubMed] [Google Scholar]
- 40.Cockerham LR, Deeks SG. 2014. Biomarker reveals HIV's hidden reservoir. eLife 3:e04742. doi: 10.7554/eLife.04742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sékaly RP. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatano H. 2013. Immune activation and HIV persistence: considerations for novel therapeutic interventions. Curr Opin HIV AIDS 8:211–216. doi: 10.1097/COH.0b013e32835f9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shytaj IL, Savarino A. 2013. A cure for AIDS: a matter of timing? Retrovirology 10:145. doi: 10.1186/1742-4690-10-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narváez AB, Hunt P, Martin JN, Kahn JO, Levy J, McGrath MS, Hecht FM. 2004. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 104:942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 45.Xu H, Wang X, Lackner AA, Veazey RS. 2013. CD8 down-regulation and functional impairment of SIV-specific cytotoxic T lymphocytes in lymphoid and mucosal tissues during SIV infection. J Leukoc Biol 93:943–950. doi: 10.1189/jlb.1112580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bacchetti P, Deeks SG, McCune JM. 2011. Breaking free of sample size dogma to perform innovative translational research. Sci Transl Med 3:87ps24. doi: 10.1126/scitranslmed.3001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O, Blau IW, Hofmann WK, Thiel E. 2009. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med 360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 49.Olsen DB, Davies ME, Handt L, Koeplinger K, Zhang NR, Ludmerer SW, Graham D, Liverton N, MacCoss M, Hazuda D, Carroll SS. 2011. Sustained viral response in a hepatitis C virus-infected chimpanzee via a combination of direct-acting antiviral agents. Antimicrob Agents Chemother 55:937–939. doi: 10.1128/AAC.00990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hron T, Fábryová H, Pa Es J, Elleder D. 2014. Endogenous lentivirus in Malayan colugo (Galeopterus variegatus), a close relative of primates. Retrovirology 11:84. doi: 10.1186/s12977-014-0084-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Archer J, Weber J, Henry K, Winner D, Gibson R, Lee L, Paxinos E, Arts EJ, Robertson DL, Mimms L, Quiñones-Mateu ME. 2012. Use of four next-generation sequencing platforms to determine HIV-1 coreceptor tropism. PLoS One 7:e49602. doi: 10.1371/journal.pone.0049602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.