

ABSTRACT
Human cytomegalovirus is a widespread pathogen of major medical importance. It causes significant morbidity and mortality in immunocompromised individuals, and congenital infections can result in severe disabilities or stillbirth. Development of a vaccine is prioritized, but no candidate is close to release. Although correlations of viral genetic variability with pathogenicity are suspected, knowledge about the strain diversity of the 235-kb genome is still limited. In this study, 96 full-length human cytomegalovirus genomes from clinical isolates were characterized, quadrupling the amount of information available for full-genome analysis. These data provide the first high-resolution map of human cytomegalovirus interhost diversity and evolution. We show that cytomegalovirus is significantly more divergent than all other human herpesviruses and highlight hot spots of diversity in the genome. Importantly, 75% of strains are not genetically intact but contain disruptive mutations in a diverse set of 26 genes, including the immunomodulatory genes UL40 and UL111A. These mutants are independent of culture passage artifacts and circulate in natural populations. Pervasive recombination, which is linked to the widespread occurrence of multiple infections, was found throughout the genome. The recombination density was significantly higher than those of other human herpesviruses and correlated with strain diversity. While the overall effects of strong purifying selection on virus evolution are apparent, evidence of diversifying selection was found in several genes encoding proteins that interact with the host immune system, including UL18, UL40, UL142, and UL147. These residues may present phylogenetic signatures of past and ongoing virus-host interactions.
IMPORTANCE Human cytomegalovirus has the largest genome of all viruses that infect humans. Currently, there is a great interest in establishing associations between genetic variants and strain pathogenicity of this herpesvirus. Since the number of publicly available full-genome sequences is limited, knowledge about strain diversity is highly fragmented and biased toward a small set of loci. Combined with our previous work, we have now contributed 101 complete genome sequences. We have used these data to conduct the first high-resolution analysis of interhost genome diversity, providing an unbiased and comprehensive overview of cytomegalovirus variability. These data are of major value to the development of novel antivirals and a vaccine and to identify potential targets for genotype-phenotype experiments. Furthermore, these data have enabled a thorough study of the evolutionary processes that have shaped cytomegalovirus diversity.
INTRODUCTION
Human cytomegalovirus (HCMV), the prototype member of the herpesvirus subfamily Betaherpesvirinae, is a widespread and important pathogen. Seroprevalence in the adult population ranges from 45% to 100% (1). After primary infection, HCMV establishes a lifelong, latent infection in myeloid progenitor cells (2). This virus causes mild to no symptoms in immunocompetent individuals but is responsible for considerable morbidity and mortality in immunocompromised individuals such as AIDS patients and transplant recipients (3). Furthermore, infection of the developing fetus can lead to sensorineural hearing loss, neurodevelopmental delay, or stillbirth, making HCMV a notorious congenital pathogen in both developed and developing countries (4). In the United States alone, total health care costs related to HCMV exceed $4.4 billion annually. Consequently, HCMV has been included among high-priority targets in vaccine prioritization reports by the U.S. Institute of Medicine (5). Several vaccine candidates are currently in early development, but licensure is not forthcoming (6).
With a length of 235 kb, HCMV has the longest genome of any known virus infecting humans (7). It is composed of a linear double-stranded DNA (dsDNA) helix and is structured in the characteristic herpesvirus class E architecture, combining two unique regions (unique long [UL] and unique short [US]) that are both flanked by a pair of inverted repeats (terminal repeat long/internal repeat long [TRL/IRL] and internal repeat short/terminal repeat short [IRS/TRS]). UL and US regions can be inserted in both directions between repeats, giving rise to four genome isomers (8). Genetic and antigenic heterogeneity of HCMV isolates was reported early on in cytomegalovirus (CMV) research by neutralization, hybridization, and restriction fragment length polymorphism assays (9–11). This was further confirmed by full-genome analyses of a few clinical isolates (7, 12). PCR sequencing of several hypervariable loci indicated that these loci exist as separate clusters of polymorphisms or genotypes (reviewed in references 13–15). These findings have spiked the interest of clinical virologists in identifying potential correlations between genetic variants and the pathogenic potential of different isolates. Several studies have found some evidence to correlate specific genotypes with disease outcome, investigating polymorphisms in the UL55 (glycoprotein B) (16–19), UL73 (glycoprotein N) (20–22), UL75 (glycoprotein H) (23), UL144 (tumor necrosis factor alpha [TNF-α]-like receptor) (24–26), and UL146 and UL147 (viral CXCL chemokines) (27, 28) genes. Others, however, found no evidence of these relationships (29–32). Overall, these studies have focused on one or, at best, a few genes at a time, ignoring the influence of other variable regions in the genome. Furthermore, variations in more conserved genes could also have a major impact on strain phenotype, as shown for the UL18 gene, encoding a major histocompatibility complex class I (MHC-I) homolog (33, 34). In the near future, more comprehensive approaches that characterize complete viral genomes will become feasible and, if applied to sufficiently large and well-defined patient cohorts, should provide clearer insights into viral determinants of infection outcome.
The introduction of next-generation sequencing (NGS) nearly a decade ago has drastically altered the genomics field and has already shown its promise in the characterization of both inter- and intrahost HCMV diversity (reviewed in reference 35). Despite these recent developments, the number of publicly available complete genomic sequences from clinically representative, low-passage-number HCMV strains is still limited. Considering the established diversity in several genes, there is a clear need to characterize more complete genomic sequences from clinical isolates. In this study, we provide the first high-resolution map of HCMV diversity and evolution through the characterization of 96 additional isolates, quadrupling the amount of publicly available full-genome sequence information. From these data, a wide extent of gene-disrupting mutations in clinical isolates becomes apparent, independent of passage artifacts. Furthermore, we corroborate the important role of recombination in HCMV evolution and identify signatures of selective pressure acting on individual protein residues. This study provides an important compendium of data concerning strain diversity that will be of outstanding value for future research efforts into understanding viral pathogenesis and developing antivirals and vaccines against this important pathogen.
MATERIALS AND METHODS
Patient samples, virus culture, and DNA purification.
Both the KU Leuven and University Hospitals Leuven Ethical Committees approved the study protocol (protocol number S55970). A total of 100 samples were collected from different HCMV patients. An overview of all samples included in this study is given in Table S1 in the supplemental material. Samples were collected at the University Hospitals Leuven (n = 81) and Saint-Pierre University Hospital (n = 13) in Belgium and at the Institute of Hematology and Blood Transfusion in the Czech Republic (n = 6). Virus culture, DNA purification, and amplification were executed as described previously (36). Briefly, samples were inoculated onto E1SM fibroblasts and cultured for the number of passages listed in Table S1 in the supplemental material. When isolated foci of cytopathic effects became apparent, viral DNA was isolated by Triton X-100-mediated lysis and micrococcal nuclease digestion of cellular DNA. After DNA extraction, viral DNA was amplified by multiple-displacement amplification.
Sequencing and assembly of genome sequences.
Library preparation for 454 and Illumina sequencing was performed as described previously (36). Libraries were sequenced on the 454 GS FLX (Roche) and GAIIx and HiSeq2000 (Illumina) platforms (see Table S1 in the supplemental material). Full-genome consensus sequences were derived by using an approach that has been discussed extensively, with some modifications (36). This approach consisted of de novo assembly, scaffolding of contigs on HCMV reference sequences, and construction of a hybrid reference combining contig and background reference sequences. Finally, the genome consensus sequence of the strain under study was derived by mapping of sequence reads onto this hybrid reference. The whole assembly procedure was performed by using CLC Genomics Workbench v6.0.2 (Qiagen). Sequence reads were quality trimmed by using a base-calling error probability cutoff of 0.05 and a maximum of 2 ambiguities in each read. After de novo assembly with standard settings, a reference sequence was selected based on BLAST analyses of all contigs of >1 kb. Subsequently, all de novo contigs (or the 2,000 longest contigs when there were >2,000 contigs) were assembled with the selected reference sequence (“assemble sequences to reference” with standard settings), and the hybrid reference was derived by using the “extract consensus sequence” option, whereby areas without coverage in the assembly were filled from the reference sequence. Sequence reads were then mapped to the hybrid reference with standard settings and the “create stand-alone read mappings” option. The process of consensus extraction and sequence read mapping was repeated until the number of reads mapping to the consensus stopped increasing. The final assembly was visualized with Tablet v1.12.12.05, manually inspected, and corrected if necessary (37). At this point, most genomes still had a problematic assembly quality in the internal repeat area. Assemblies were then cut at these regions, and the separate contigs were extended and eventually joined by iterative mapping of sequence reads. Remaining uncertainties were resolved via PCR amplification and Sanger sequencing, as described previously (36). Data concerning the number of sequence reads mapping to the final genome consensus and average read depth are summarized in Table S1 in the supplemental material.
Sequence alignment and genome annotation.
A DNA sequence alignment of all 101 in-house-derived and 27 additional full-genome sequences was constructed with MAFFT v7.158b, option FFT-NS-i (maximum of 1,000 cycles) (38). Previously reported strains that were used in this study are listed in Table S1 in the supplemental material. Full-genome sequences of strains AD169, Towne, and Davis were omitted from all analyses since they are derived from highly passaged laboratory strains with obscure passage histories. It has been well established that these strains are genetically severely altered by these procedures (39–41). Alignment inspection and editing were done with MEGA6 (42). NCBI GenBank annotations of reference strain Merlin were identified in the alignment, and individual open reading frame (ORF) alignments were excised and realigned at the codon level by using the RevTrans v2.0 server with MAFFT v6.240 (43). As a service to the HCMV research community, we have shared fasta files containing all 170 gene alignments used for all gene-specific analyses. Strains containing gene-disrupting mutations were omitted on a gene-by-gene basis. The alignments can be downloaded at http://www.regatools.be/hcmv_gene_alignments.tar.gz. Genome annotations for NCBI GenBank entries were transferred from a genetically intact reference strain (BE/9/2010) by using RATT, with a word size of 30, a cluster size of 400, a maximum extend cluster of 500, and an identity cutoff of 40 (44). ORFs refractory to transfer because of sequence variability or disruptive mutations were manually annotated by referral to the ORF-specific alignments.
Recent studies of HCMV transcription and translation at the full-genome level have hinted at the expression of a much more complicated pattern of RNAs and proteins than the 170 gene products that are currently annotated in the NCBI reference sequence for Merlin (45–47). These findings await further experimentation to firmly establish the expression of these additional products and their conservation in different strains. In our diversity analyses, we have therefore not yet included these putative genes, which often (partially) overlap previously annotated genes.
Analysis of ORF-disrupting mutations.
Disruption of ORFs in specific strains was evaluated in the ORF-specific alignments. All mutations that disrupted ORF integrity compared to the majority of strains were noted (see Table S2 in the supplemental material). These mutations include indels that cause a frameshift leading to a completely altered protein sequence and/or premature termination; deletions including the original start codon or splice sites; and substitutions eliminating start codons, introducing stop codons, or affecting splice sites. Mutations that were shown previously to be artifacts of culture passage were omitted. Furthermore, if original clinical specimens were available, genes containing disruptive mutations were characterized by PCR and Sanger sequencing as described previously (36) (primer sequences and annealing temperatures are listed in Table S3 in the supplemental material).
Detection and analysis of tandem repeats.
Tandem repeats (TRs) were identified in the reference strain Merlin genome sequence, using a method similar to the one described previously for herpes simplex virus 1 (HSV-1) (48). To avoid duplicate detection of identical TRs in TRL/TRS and IRL/IRS regions, the Merlin sequence was trimmed of its terminal repeat sequences. Perfect repeats with a period size (length of one repeat unit) of 1 to 6 were identified with MIcroSAtellite identification tool v1.0 (MISA) (http://pgrc.ipk-gatersleben.de/misa/). Homopolymers (period size of 1) were reported when they were longer than 5 copies (>5 nucleotides [nt]), TRs with a period size of 2 to 6 were reported when the total repeat length was >9 nt (5 copies with a period size of 2, 4 copies with a period size of 3, 3 copies with a period size of 4, and 2 copies with period sizes of 5 and 6). Compound repeats identified by MISA were divided into their individual constituent repeats. When these repeats were overlapping, the longest repeat was retained. Longer and nonperfect repeats were identified by using Tandem Repeat Finder v4.07b (TRF) with alignment weights 2, 5, and 5 for matches, mismatches, and indels, respectively, a minimum score of 40, and a maximum period size of 500 (49). If TRF repeats contained overlap, only the highest-scoring repeat was retained. MISA and TRF TR sets were then combined, retaining the longest TR in case of overlap. Subsequently, conservation of TRs identified in Merlin was assessed by referral to the multiple alignment of 124 complete HCMV genome sequences (see Table S1 in the supplemental material). TRs that showed overlap with the Merlin TRs and fulfilled the MISA or TRF criteria described above were identified as orthologous repeats. TRs that did not have orthologous repeats in >50% of strains were omitted from further analyses. For all TRs, period size, copy number, position in the genome (coding RNA/noncoding RNA [ncRNA]/intron/intergenic and UL/US/IRL/IRS), and repeat type (homopolymer, period of 1 nt; microsatellite, period of 2 to 9 nt; minisatellite, period of >9 nt) were recorded. TRs were reported to be conserved if >50% of strains had identical sequences and copy numbers.
Phylogenetic analyses.
To maximize the amount of genetic information included in our analyses, strains Toledo, TB40/E, 6397, and HAN2 were omitted from the full-genome alignment, along with the previously excluded strains AD169, Towne, and Davis (see Table S1 in the supplemental material). These strains all contain large genome deletions and/or rearrangements that interfere with a proper alignment. Since sites containing gaps in one or more strains are omitted from several diversity calculations, this would lead to the loss of important sequence information. Genome-wide diversity estimates are thus based on a set of 124 full-genome sequences, 101 of which were sequenced in our laboratory (see Table S1 in the supplemental material). For analyses at the gene level, the complete set of 128 low-passage-number strains was used, although strains mutated in a specific gene were omitted on a gene-by-gene basis (see Table S2 in the supplemental material).
Genome-wide diversity statistics were calculated by using DnaSP v5.10 and MEGA6 (42, 50). Nucleotide diversity (π), the number of polymorphic sites, and the average number of nucleotide differences were calculated by using the DNA polymorphism option of DnaSP, excluding gapped sites. A sliding window of π along the genome alignment was constructed with a window size of 500 nt and a step size of 100 nt. To compare HCMV diversity to those of the other human herpesviruses, the overall mean distance (Jukes-Cantor model) and transition/transversion ratio were calculated for genome alignments of available strain sequences of all human herpesviruses by using the overall mean distance option of MEGA6, with pairwise deletion of gapped sites.
Phylogenetic network analyses were performed with SplitsTree v4.13.1 (51). Neighbor-net split networks were constructed by using uncorrected p-distances and excluding gap sites. Network construction using the Jukes-Cantor model instead of p-distances yielded similar network topologies. Recombination was further studied with the BootScan function of SimPlot v3.5.1, using a window size of 2,000 nt, a step size of 500 nt, gap stripping, empirical transition/transversion ratio, neighbor-joining tree construction with the Kimura two-parameter model, and 100 bootstrap replicates (52). Recombination estimates for HCMV, HSV-1, varicella-zoster virus (VZV), and Epstein-Barr virus (EBV) were compared by analyzing an equal set of 9 full-genome sequences (the total number of strains available for EBV). Strains of HCMV, HSV-1, and VZV were chosen to best cover total diversity based on a split network of all strains (see Fig. 6A and 7). Recombination breakpoints were analyzed by using Recombination Detection Program (RDP) v3.44 (53). This program combines several recombination detection algorithms. Detection of breakpoints with RDP, GENECONV, Chimera, MaxChi, and 3Seq were combined with secondary detection with BootScan and SiScan (only for testing of breakpoints identified by previously used methods). Analyses were run with the linear-sequences option, checking of alignment consistency, and automatic masking of identical sequences. Breakpoints were reported if they were detected by at least two independent methods and the Bonferroni-corrected P value was <0.05. Duplicate breakpoints were counted only once; uncertain breakpoints were omitted. Gene-level recombination was analyzed by using three separate approaches. First, evidence for recombination inside a gene was assessed with the Phi-test included in the SplitsTree package (54). This is a simple and robust test that determines whether recombination signals are detected in the alignment. Next, the genetic algorithm for recombination detection (GARD) reported whether recombination was present and identified presumable recombination breakpoints (55). GARD was run via the Datamonkey Web server of the HyPhy package (56, 57). Finally, recombination breakpoints were further identified by using RDP3 as described above. RDP3-detected breakpoints were used for calculations of breakpoint density, reporting the number of breakpoints per kilobase for each gene.
FIG 6.

Widespread recombination between HCMV strains. Recombination between separate HCMV strains was analyzed. (A) Neighbor-net split network of 124 full-genome sequences showing numerous reticulate connections that are indicative of recombination. The Phi-test for recombination gave strong statistical evidence for recombination. Countries of isolation of different strains are represented with the international two-letter country codes at the beginning of strain names (CN, China; KR, South Korea; IT, Italy; US, United States; other codes are defined in the legend to Fig. 5). Asian strains JHC and HAN are highlighted with a gray background. (B) BootScan analysis of 9 strains highlighted in the split network. Strain BE/25/2010 (highlighted in black) was used as a reference strain.
FIG 7.

Diversity and recombination in HSV-1, VZV, and EBV. Using the full-genome sequences listed in Table S1 in the supplemental material, neighbor-net split networks were constructed for HSV-1, VZV, and EBV strains. In all three cases, statistically significant evidence for recombination was detected. Countries of isolation of different strains are represented with the international two-letter country codes at the beginning of strain names (KE, Kenya; JP, Japan; HK, Hong Kong; NG, Nigeria; GH, Ghana; CA, Canada; RU, Russia; NL, the Netherlands; MA, Morocco; MX, Mexico; other codes are defined in the legends to Fig. 5 and Fig. 6). In all three networks, distinct clusters are recognizable. Strains chosen for recombination analysis with RDP3 are underlined.
To assess the overall selection type acting on a gene, estimates of the ratio of nonsynonymous substitutions per nonsynonymous site (dN) to synonymous substitutions per synonymous site (dS) were made with MEGA6, using the Nei-Gojobori method (Jukes-Cantor) with 1,000 bootstrap replicates, treating gaps by pairwise deletion. Individual sites under positive or negative selection were further assessed by using the Datamonkey Web server of the HyPhy package. After inference of a nucleotide substitution model, recombination was detected by using GARD. Further analyses were based on either neighbor-joining trees of the complete gene when no recombination was detected or GARD-inferred trees of the separate recombination fragments. Subsequently, evidence of positive and negative selection at the codon level was assessed by using the SLAC, FEL, FUBAR, and MEME algorithms (58–60). The RCSB Protein Data Bank (PDB) was queried for structural data for HCMV proteins with residues under positive selection. Such a structure was available only for pUL18 (PDB accession number 3D2U). Positively selected residues were visualized on the structure with UCSF Chimera v1.9 (61).
Statistical analyses.
All statistical analyses were performed by using RStudio v0.98.1073. Comparisons of gene diversity (dN), recombination density (breakpoints per kilobase), and selection density (percentage of codons under positive or negative selection) over different gene families, conservation groups, and functions were performed with Kruskal-Wallis one-way analysis of variance (KWt), and pairwise comparisons were performed with pairwise Wilcoxon rank sum tests (WRSts) with Holm correction for multiple testing. Nonparametric tests were chosen since there was a large difference in the sizes of the groups.
Nucleotide sequence accession numbers.
All full-genome consensus sequences derived from this study were submitted to the NCBI GenBank database under accession numbers KP745633 to KP745728.
RESULTS
High-throughput sequencing of complete genomes from clinical HCMV isolates.
To efficiently characterize the genetic diversity of a large set of complete genomic sequences derived from clinical HCMV isolates, we recently described a method that combines limited virus culturing and virion DNA purification with multiple-displacement amplification and NGS (36). We showed that this procedure was able to generate highly pure viral DNA suitable for NGS analysis and validated that strain consensus sequences were representative of the original virus populations in the clinical isolates. Here, we implemented this method to characterize complete genomes in a group of 100 clinical HCMV isolates (see Table S1 in the supplemental material). These isolates were collected from Belgian (n = 94) and Czech (n = 6) individuals infected with HCMV, including healthy adults, immunosuppressed patients, and congenitally infected infants.
DNA sequence reads were generated by using a combination of the 454 GS FLX and Illumina NGS platforms. In Table S1 in the supplemental material, some basic genome assembly statistics are listed. We successfully derived a full-genome consensus sequence for 96/100 strains, with the average read depth ranging from 35 to 3,315 (median, 2,031) and the proportion of reads mapping to the consensus ranging from 1% to 98% (median, 89%). While eight isolates were successfully sequenced at read percentages of <10%, sample purity was >50% in the majority of isolates (77/96). For three strains, we could not determine the full-genome consensus because coverage was too low or unevenly distributed, leaving too many sequence gaps for finishing through Sanger sequencing. For one strain, coverage was adequate, but the isolate clearly consisted of multiple genome variants, and a single, meaningful consensus sequence could not be obtained, nor was it possible to segregate the constituting variants, since NGS data provide no connection between variants at different variable loci. The predominance of a single genome variant along the entire genome for all other strains suggests that these sequences constitute a contiguous genome.
As reported previously, genome assembly consisted of de novo assembly, scaffolding on a reference sequence, and subsequent iterative mapping of NGS reads on the genome scaffold (36, 62). Terminal repeats were omitted from the scaffold since these repeats are identical to the internal repeats. This approach was mostly successful, but assembly of the internal repeat regions (IRL/IRS) generally required additional consideration, since de novo contigs tended to break at these regions. Because of the high sequence variability of the internal repeat regions (Fig. 1), reference-assisted iterative mapping usually did not solve this issue. Therefore, contigs were split, sequence reads were assembled on both contigs separately, and contigs were enlarged by the 50% read overhangs at contig ends. This process was reiterated until contigs could be joined. The transition between unique and repeat regions needed to be determined manually to correctly add the terminal repeat sequences at both genome ends. Transitions can be recognized from NGS read assemblies through the concomitant mapping of transition-crossing reads from different genome isomers with inverted UL and US directions. While the position of the US-IRS junction is stable, presumably because it is located inside the TRS1/IRS1 reading frame, the UL-IRL junction is located in a noncoding region and is positioned differently in separate strains. Strain BE/5/2010 had an unusual layout for the IRL/IRS region, with the IRL repeat starting after the IRS repeat and being completely encompassed by it.
FIG 1.

Diversity and evolution of the HCMV genome. Shown is an overview of genetic diversity and evolutionary pressure along the HCMV genome. The genome is divided into four panels. Each panel consists of four separate tracks. In the top track, nucleotide diversity is calculated in a sliding window of 500 nt with a step size of 100 nt. As only ungapped residues are included in each window, the distance between two data points may vary in areas with many indels. In the second track, the extents of recombination and positive selection are assessed for each gene. Displayed above the center of the appropriate gene (bottom track), dark and light blue bars represent recombination breakpoint density and the percentage of codons under positive selection, respectively. For optimal resolution, values were cut off at 10 breakpoints (brp)/kb and 4% codons under positive selection. Green and red bars in the third track indicate the genome positions of conserved and variable tandem repeats. The bottom track annotates genes and other genome elements in four layers. The first two layers show genes carried on the forward strand, and the last two layers show genes carried on the reverse strand. Spliced exons are connected with thin black lines. Genomic inverted repeats (TRL, IRL/IRS, and TRS) and long noncoding RNAs are represented in black; genes are colored on a scale from green to red, indicating the frequency of ORF-disrupting mutations in separate clinical isolates.
HCMV displays the highest level of genetic diversity of all human herpesviruses.
The overall genetic diversity of the HCMV genome was assessed by aligning our 96 genomes with 28 previously reported sequences (see Table S1 in the supplemental material). This alignment contains 255,248 sites: 223,991 sites are without gaps, and 31,528 of these nongapped sites (14%) are polymorphic. The interstrain nucleotide diversity, π, was estimated to be 0.021, and the average number of nucleotide differences between two genomes is 4,734. This number is higher than the recent estimate of π of 0.015 for murine cytomegalovirus (MCMV), based on 11 complete genomes (63). There is a clear discrepancy with the values for intrastrain nucleotide diversity that were reported for congenitally infected infants (π = 0.18 to 0.25) (64). Care should be taken in directly comparing these estimates, as interstrain diversity is estimated from measuring polymorphisms in separate consensus sequences, while intrastrain diversity is derived from characterizing polymorphisms of a single virus population. However, the large discrepancy suggests that many of the variants that were identified in these intrahost populations are deleterious and are not passed on. This notion is supported by the apparent stability of genotype sequences in patients (30, 65–69).
To put these data in perspective toward the other human herpesviruses, we estimated the overall mean distance and transition/transversion ratio for alignments of all available complete genomic sequences of all nine human herpesviruses (Table 1). Apart from HCMV, only for HSV-1, VZV, and EBV were the numbers of complete genomic sequences adequate for a meaningful estimation of overall diversity. With >0.02 substitutions/site, versus <0.01 substitutions/site, HCMV stands out as being significantly more diverse than these alpha- and gammaherpesviruses (P = 0.012 for comparisons with HSV-1, VZV, and EBV only, or P = 2.5e−06 for comparisons with all human herpesviruses [determined by one-sample t test]). This is not an artifact of the higher number of sequences available for HCMV, since analysis of the overall mean distance in five random, separate subsets gave highly similar results (0.025 to 0.028 substitutions/site). In fact, considering the larger geographical diversity of HSV-1 and VZV strains included in this analysis, the current estimate for HCMV could even be too low (see “HCMV evolution has been shaped by pervasive recombination,” below). There is also a great discrepancy in the estimated transition/transversion ratios between different herpesvirus species. HCMV has a relatively high ratio of 2.53. This is probably a consequence of strong purifying selection removing transversions, which result in more nonsynonymous mutations (see “Positively selected residues provide a genetic fingerprint of the evolutionary arms race between virus and host,” below) (70).
TABLE 1.
Genetic diversity of human herpesviruses
| Subfamily | Speciesa | No. of strainsb | Overall mean distance (substitutions/site) | Standard errorc | Transition/transversion ratio |
|---|---|---|---|---|---|
| Alphaherpesvirinae | HHV-1 (HSV-1) | 26 | 0.0076 | 0.000079 | 1.63 |
| HHV-2 (HSV-2) | 2 | 0.0041 | 0.000083 | 1.29 | |
| HHV-3 (VZV) | 46 | 0.0014 | 0.000040 | 2.01 | |
| Betaherpesvirinae | HHV-5 (HCMV) | 124 | 0.0266 | 0.000100 | 2.53 |
| HHV-6A | 2 | 0.0135 | 0.000380 | 1.88 | |
| HHV-6B | 2 | 0.0070 | 0.000138 | 1.81 | |
| HHV-7 | 2 | 0.0013 | 0.000084 | 2.80 | |
| Gammaherpesvirinae | HHV-4 (EBV) | 9 | 0.0087 | 0.000086 | 1.36 |
| HHV-8 (KSHV) | 3 | 0.0021 | 0.000066 | 1.05 |
The official taxonomic names (human herpesvirus 1 [HHV-1] to HHV-8) are given, followed by common names in parentheses, if available. HSV-1, herpes simplex virus 1; VZV, varicella-zoster virus; HCMV, human cytomegalovirus; EBV, Epstein-Barr virus; KSHV, Kaposi sarcoma-associated herpesvirus.
GenBank accession numbers for strains used in genome alignments are listed in Table S1 in the supplemental material; data for the HSV-1 alignment were reported previously (48).
Standard errors were calculated from 500 bootstrap replicates.
In Fig. 1 (top), π is represented in a sliding window along the HCMV genome. This clearly delineates several diversity hot spots, isolated by long stretches of conserved sequence. To further analyze the heterogeneity of divergence, ratios of nonsynonymous substitutions per nonsynonymous site (dN) were calculated for all 170 genes as a measure of the divergence of the encoded proteins (see Table S4 in the supplemental material). A list of the most divergent genes is provided in Table 2. All 30 genes that have a dN value of >0.025 are listed, along with their gene family, conservation over herpesvirus subfamilies, their confirmed/proposed function(s) (71), and studies that have previously characterized and classified the diversity of these genes. Only 1 of these 30 genes is conserved in all mammalian herpesviruses (UL73, encoding glycoprotein N), while 3 others are conserved within the subfamily Betaherpesvirinae. For 4 of these 30 genes, no previous studies have analyzed diversity in clinical isolates. For 9 others (including 5 of the 6 most variable genes), analyses were limited to partial genomic sequences of <10 clinical isolates (7). This demonstrates the added value of a high-throughput, comprehensive analysis of divergent genome regions. Reciprocally, genes such as UL55 and UL75, encoding glycoproteins B and H, respectively, have been sequenced extensively because of their known functional roles, but they are not in this broad group of the most divergent genes. On the other end of the diversity spectrum, the 25 most conserved genes (dN of <0.002) are listed in Table 3. The majority of these genes (19/25) are conserved in all mammalian herpesviruses (14/25), in betaherpesviruses (3/25), or in beta- and gammaherpesviruses (2/25) (7).
TABLE 2.
Most divergent HCMV genes
| Gene | dNa | Gene familyb | Gene conservationc | Function(s)d | Referencef |
|---|---|---|---|---|---|
| RL6 | 0.555 | RL11 | No | Latencye | 7 |
| RL5A | 0.516 | RL11 | No | Unknown | 7 |
| RL12 | 0.467 | RL11 | No | Virion protein,e immunomodulatione | 7 |
| UL146 | 0.448 | CXCL | No | Immunomodulation | 67 |
| RL13 | 0.297 | RL11 | No | Cell tropism,e virion protein, immunomodulation,e replication | 7 |
| UL9 | 0.235 | RL11 | No | Viral growth | 7 |
| UL1 | 0.156 | RL11 | No | Virion protein, cell tropism,e assemblye | 98 |
| UL139 | 0.144 | NA | No | Immunomodulatione | 137 |
| UL74 | 0.110 | NA | Beta | Viral spread, assembly, entry, immunomodulation, virion protein | 138 |
| UL11 | 0.106 | RL11 | No | Immunomodulation | 139 |
| UL73 | 0.103 | NA | Core | Entry, virion protein, latencye | 140 |
| UL6 | 0.086 | RL11 | No | Unknown | 98 |
| UL144 | 0.082 | NA | No | Immunomodulation, latency | 141 |
| UL120 | 0.075 | UL120 | No | Unknown | 7 |
| UL20 | 0.068 | NA | No | Immunomodulatione | NA |
| UL8 | 0.064 | RL11 | No | Unknown | 7 |
| UL4 | 0.060 | RL11 | No | Virion protein, latencye | 142 |
| UL7 | 0.056 | RL11 | No | Immunomodulation | 98 |
| UL142 | 0.047 | MHC | No | Immunomodulation | 7 |
| UL147 | 0.046 | CXCL | No | Immunomodulatione | 67 |
| UL37 | 0.041 | NA | Beta | Latency,e replication, apoptosis, gene regulation, immunomodulation, viral growth | 143 |
| UL22A | 0.039 | NA | No | Immunomodulation,e virion protein | NA |
| UL148D | 0.036 | NA | No | Unknown | 144 |
| UL10 | 0.034 | RL11 | No | Viral growth | 98 |
| UL2 | 0.033 | NA | No | Unknown | 7 |
| UL150 | 0.033 | NA | No | Latencye | 145 |
| UL25 | 0.032 | UL25 | No | Virion protein | NA |
| UL133 | 0.028 | NA | No | Assembly,e latencye | 7 |
| US34A | 0.028 | NA | No | Unknown | NA |
| UL33 | 0.027 | GPCR | Beta | Immunomodulation, virion protein, host modulation | 146 |
dN, nonsynonymous substitutions per nonsynonymous site.
NA, not assigned to a gene family.
Gene conservation over different herpesvirus subfamilies. Core, conserved in all mammalian herpesviruses; Beta, conserved in all members of the subfamily Betaherpesvirinae; No, not conserved in all members of the subfamily Betaherpesvirinae.
Functions of the encoded gene products were reported previously (71).
Proposed function, which needs further validation.
Studies characterizing diversity in clinical isolates for each gene. NA indicates that there have been no previous reports analyzing sequence diversity for this gene.
TABLE 3.
Most conserved HCMV genes
| Gene | dNa | Gene familyb | Gene conservationc | Function(s)d |
|---|---|---|---|---|
| UL46 | 0.000 | NA | Core | Virion protein |
| UL85 | 0.000 | NA | Core | Assembly |
| UL103 | 0.000 | NA | Core | Virion protein, assembly |
| US18 | 0.000 | US12 | No | Unknown |
| UL26 | 0.001 | US22 | No | Virion protein, gene regulation, virion stability |
| UL29 | 0.001 | US22 | Beta | Virion protein,e gene regulation, apoptosis, cell tropism |
| UL31 | 0.001 | DURP | Beta | Gene regulation |
| UL35 | 0.001 | UL25 | Beta | Gene regulation,e replication, nucleotide repair, assembly, virion protein |
| UL41A | 0.001 | NA | No | Virion protein |
| UL44 | 0.001 | NA | Core | Host modulation, replication, latencye |
| UL50 | 0.001 | NA | Core | Virion protein,e latency,e assembly |
| UL57 | 0.001 | NA | Core | Replication |
| UL79 | 0.001 | NA | Betagamma | Gene regulation, latencye |
| UL86 | 0.001 | NA | Core | Assembly |
| UL88 | 0.001 | NA | Betagamma | Virion protein, assemblye |
| UL89 | 0.001 | NA | Core | Viral growth, assembly |
| UL96 | 0.001 | NA | Core | Virion protein, assembly |
| UL98 | 0.001 | NA | Core | Nucleotide repair, latencye |
| UL102 | 0.001 | NA | Core | Replication |
| UL104 | 0.001 | NA | Core | Assembly |
| UL105 | 0.001 | NA | Core | Replication, latencye |
| UL114 | 0.001 | NA | Core | Nucleotide repair, replication, latencye |
| US13 | 0.001 | US12 | No | Unknown |
| US24 | 0.001 | US22 | No | Virion protein, gene regulation |
| US31 | 0.001 | US1 | No | Unknown |
dN, nonsynonymous substitutions per nonsynonymous site.
NA, not assigned to a gene family.
Gene conservation over different herpesvirus subfamilies. Core, conserved in all mammalian herpesviruses; Beta, conserved in all members of the subfamily Betaherpesvirinae; Betagamma, conserved in all members of the subfamilies Betaherpesvirinae and Gammaherpesvirinae; No, not conserved in all members of the subfamily Betaherpesvirinae.
Functions of the encoded gene products were reported previously (71).
Proposed function, which needs further validation.
Relationships of gene diversity with gene family, gene conservation across herpesviruses, and encoded function(s) were further explored and are visualized for gene families in Fig. 2 (top). Clear and significant differences in gene diversity between separate gene families and gene conservation groups were found (P = 4.8e−06 and P = 2.0e−08 [determined by KWt]) (Fig. 2). Because of the small number of genes in several gene families, pairwise comparisons were significant only for the RL11 family versus the US6 (P = 0.037 by WRSt), US12 (P = 0.0047 by WRSt), and US22 (P = 0.0044 by WRSt) gene families. The RL11 gene family indeed truly stands out for the high number of variable genes. While variability in RL11 genes was described previously, analyses of the most variable members, RL5A, RL6, RL12, and RL13, were limited to a comparison of seven strains (Table 2). Especially for the RL13 gene, it would be of interest to study the functional behavior of different variants, as this gene has been implicated as a growth temperance factor (72) and in immunomodulation (73). Considering the latter function of RL13, we found that the endocytic YxxL motif essential for internalization of IgGs was 100% conserved among clinical isolates. Cytomegalovirus-specific genes were significantly more diverse than genes conserved in all mammalian herpesviruses (P = 8.4e−08 by WRSt), betaherpesvirus genes (P = 0.0024 by WRSt), and genes conserved between beta- and gammaherpesviruses (P = 0.0025 [determined by WRSt]). The UL73 (encoding glycoprotein N) and UL74 (encoding glycoprotein O) genes are outliers within core and betaherpesvirus genes, respectively. Variability in these genes has been widely studied (Table 2). Similarly, we found statistically significant differences in the genetic diversity of genes classified according to the function of the encoded product (P = 9.8e−06 by KWt) (based on the functional classification reported in reference 71). Pairwise comparisons were significant only for immunomodulation genes versus genes encoding assembly (P = 0.0017 by WRSt), gene regulation (P = 0.0031 by WRSt), and replication (P = 0.011 by WRSt) functions. Generally, diverse genes are involved in interactions with the host (immunomodulation, entry, spread, cell tropism, and virion proteins, which include surface glycoproteins), while conserved genes perform core viral functions such as replication, assembly, modulation of the host cell cycle and proteins, gene regulation, cellular trafficking, nucleotide repair, virion stability, latency, and viral growth.
FIG 2.

Variability, recombination, and selection in HCMV gene families. Gene diversity (dN), recombination breakpoint density, and the percentages of codons under positive and negative selection are indicated for HCMV genes within gene families; each dot represents a gene. Only genes belonging to specific gene families are represented. Group averages are designated with horizontal lines.
Tandem repeats in the HCMV genome.
Another important source of sequence variation is the heterogeneity in the copy numbers of adjacently repeated elements or tandem repeats (TRs), caused by recombination or strand slippage replication. Variation in TRs is associated with phenotypic variability, regulation of gene expression, and genetic evolvability in both prokaryotes and eukaryotes (74–76). Furthermore, several studies have found evidence that TR variations may impact strain functionality and pathogenicity in viruses (77–82). The presence of TRs in HCMV was described previously, and TR polymorphisms could be used as epidemiological markers to distinguish clinical isolates (83–85). A comparative analysis of TRs in several members of the family Herpesviridae, based on a single genome sequence for each species, found the highest TR content in the alphaherpesvirus pseudorabies virus (18% of total nucleotides), followed by HSV-1 (9%), EBV (7%), KSHV (4.5%), and VZV and HCMV (3%) (86). To assess the total set of repeats in HCMV genomes, we identified all homopolymers (repeats with a period size of 1 nt), microsatellites (period size of 2 to 9 nt), and minisatellites (period size of >9 nt) in the genome of reference strain Merlin and subsequently searched for orthologous TRs in 123 other HCMV genome sequences. The total set of identified repeats is reported in Table S5 in the supplemental material. In total, 779 TRs were found in the genome of strain Merlin, 23 of which are duplicated or triplicated in TRL and/or TRS inverted repeats. For 683/779 TRs (88%), an orthologous repeat could be found in the majority of the other HCMV strains. These 683 TRs constitute 3.9% of the total nucleotides and are annotated in the HCMV genome in Fig. 1. Only 51 of these orthologous repeat sets (7%) were classified as variable (<50% conservation of repeat sequence and copy number) (Fig. 1). While 81% of the total nucleotides are within protein-coding regions, only 65% of TR nucleotides are found inside genes (Fig. 3A). Reciprocally, there is a clear overrepresentation of TRs in noncoding regions, including the 4 long noncoding RNAs, introns, and intergenic regions. Likewise, the internal repeat regions (IRL-IRS) that make up only 1% of the trimmed Merlin genome contain 8% of TR nucleotides (Fig. 3B). Overall, the level of TR conservation is higher in coding than in noncoding regions (P = 9.9e−09 by Fisher's exact test [FEt]) (Fig. 3C). When different repeat types were analyzed separately, this held true for homopolymers (P = 1.3e−06 by FEt) and minisatellites (P = 0.017 by FEt) but not for microsatellites (P = 0.56 by FEt). Recently, TRs of HSV-1 were analyzed based on a collection of 26 complete genomes by using a similar approach (48). The authors of that study found 584 orthologous TRs in this data set, corresponding to 4.3 TRs/kb (5.4% of nucleotides), which is higher than the 2.9 TRs/kb (3.9% of nucleotides) that we found for HCMV. HSV-1 also has more variable TRs than does HCMV (17% versus 7%). HSV-1 shows a similar overrepresentation of TRs in noncoding regions and in the genomic inverted repeats. Likewise, most variable TRs are located in noncoding regions in both viruses. While HSV-1 has a higher proportion of genes containing TRs (92%, versus 79% for HCMV), this is caused mostly by the large number of homopolymers in HSV-1 genomes (3.4 TRs/kb, versus 1.2 TRs/kb for HCMV). When only the proportion of genes containing micro- and minisatellites is calculated (thus excluding homopolymers), 68% of HCMV genes still contain TRs, while this proportion is decreased to 39% for HSV-1.
FIG 3.

Tandem repeats (TRs) in the HCMV genome. TRs in reference strain Merlin were identified, and orthologous repeats were searched for in a data set of 124 complete HCMV genomes. Only TRs with orthologs in >50% of strains were included in the analysis. (A) TR nucleotide content (9,008 nt) in coding and noncoding (intergenic, intron, and ncRNA) regions compared to the distribution of total nucleotides (231,784 nt) over these regions. (B) Similar to panel A, where TR and total nucleotide distributions over unique long (UL), unique short (US), and internal repeat long and short (IRL-IRS) genome regions are compared. Percentages in panels A and B do not add up to 100% because of rounding errors. (C) Conservation of TRs between coding and noncoding TRs and between different TR types (homopolymers, microsatellites, and minisatellites). TRs were reported to be conserved if >50% of strains had identical repeat sequences and copy numbers.
TR polymorphisms in noncoding regions might have a profound impact on gene regulation and expression by altering binding sites for regulatory proteins, chromatin structure, transcript stability and transcription, splicing, or translation efficiency (87–90). However, we can only speculate about their effects based on sequence data alone. Overall, selection seems to have constrained the presence of unstable TR elements inside coding regions, illustrated by the discrepancy of TR frequencies between coding and noncoding regions (Fig. 3A) and the higher level of conservation of TRs in coding regions (Fig. 3C). Therefore, it is conceivable that some TR variations in coding regions might have specific functions or provide the virus with greater adaptability because of their intrinsic instability (so-called “evolutionary tuning knobs” [91]). Mutation rates in TRs can be up to 100,000 times higher than those in other parts of the genome. Therefore, we assessed the potential impact of TR variation in coding regions on the encoded proteins (see Table S5 in the supplemental material). Nine out of the 13 variable TRs inside protein-coding regions constitute variability in the longer and nonperfect TRs, as determined by TRF analysis. Diversity in these minisatellites comprises mostly variations in repeat sequence and period length, caused by nucleotide divergence in these areas. The UL50 and UL111A genes contain variable trinucleotide microsatellites, causing amino acid stretches of various lengths (Table 4). Homopolymer length variation is present in the RL12 and UL1 genes, but it does not cause frameshifts and results only in amino acid divergence. In fact, none of these 13 variable TRs in protein-coding regions cause frameshifts that disrupt ORF integrity. Because TRs cataloged as conserved (>50% of strains with conserved repeat sequence and copy number) could also contain variation in a minority of strains, we analyzed an additional set of 53 protein-encoding TRs with variations in period length or copy number below the 50% threshold (see Table S5 in the supplemental material). These TRs comprised 26 homopolymers, 5 microsatellites, and 22 minisatellites. While most of these TR variations either were conserved or led to amino acid variations and indels without a clear repetitive character at the protein level, seven TRs caused the occurrence of repetitive single-amino-acid stretches of various lengths (Table 4). These stretches contain mostly small and hydrophilic amino acids, suggesting selective constraints toward these residues in coding TRs (92). It is assumed that TRs inside coding regions form flexible, unstructured, and hydrophilic loops (75). These amino acid loops might be involved in protein-protein interactions that could be altered by changes in loop length. For example, the variable proline stretch in UL50 might have functional consequences for the efficiency of nuclear egress of HCMV capsids. Together with pUL53, pUL50 forms the nuclear egress complex (NEC). A random screen for dominant negative mutants of M50 (the MCMV homolog of UL50) identified this proline-rich motif to be essential for nuclear egress, and this finding was confirmed for HCMV UL50 (93). The authors of that study suggested that this motif likely controlled a binding site for a NEC interaction partner. The latter could be HCMV pUL97, a protein kinase that was recently found to phosphorylate S216, a site neighboring the proline stretch. This phosphorylation modulates NEC localization and nuclear egress (94, 95). It might be of interest to assess the effect of the large length heterogeneity (4 to 12 residues) in the proline motif on nuclear egress efficiency. Finally, homopolymer length variation in the UL111A and UL133 genes caused frameshifts that resulted in premature ORF termination in strains BE/16/2010 and BE/17/2010 (UL111A) and in strain BE/2/2012 (UL133) (see Tables S2 and S5 in the supplemental material).
TABLE 4.
Tandem repeats encoding variable repetitive elements at the protein level
| nt positionsa | Gene | Period size (nt) | Copy no. (range) | Repetitive element in protein (copy no. [range]) |
|---|---|---|---|---|
| 27690–27696 | UL22A | 1 | 6–9 | Glycine stretch (1–3) |
| 73888–73902 | UL50 | 3 | 3–11 | Proline stretch (4–12) |
| 99616–99627 | UL69 | 3 | 3–9 | Proline stretch (4–12) |
| 118052–118063 | UL80/UL80.5 | 3 | 2–6 | Serine stretch (4–7) |
| 150242–150253 | UL102 | 3 | 2–5 | Serine stretch (3–4) |
| 161081–161092 | UL111A | 3 | 1–8 | Threonine stretch (1–8) |
| 162890–162901 | UL112 | 3 | 3–5 | Glycine stretch (3–5) |
| 162925–162951 | UL112 | 12–15 | 1.9–2.3 | Glycine stretch (7–9) |
| 178548–178559 | UL132 | 3 | 2–8 | Glutamate stretch (2–8) |
Positions in the genome of reference strain Merlin.
Wild-type HCMV strains contain ORF-disrupting mutations in a wide range of nonessential genes.
The accumulation of gene-disrupting mutations in cell culture-passaged HCMV strains is a well-described phenomenon (35, 96). As first suggested for isolates of koi herpesvirus, some disrupting mutations might also occur in vivo (97). Recently, a few studies have indicated that some HCMV mutants may indeed be present in clinical isolates prior to culture passage (36, 62, 98). In particular, strains JP (62) and BE/21/2010 (36) were sequenced directly from clinical material and displayed disruptive mutations in the RL5A and UL111A genes and in the RL5A, UL9, and UL150 genes, respectively. Furthermore, we showed that RL5A, UL1, UL9, and UL111A mutants identified in three additional strains after limited passaging were also present in the original clinical isolate (36). With our current data set providing full-genome information for 96 clinical isolates, supplemented with 32 previously reported sequences, we have an ideal opportunity to further assess the occurrence of ORF-disrupting mutations in clinical HCMV isolates and provide a more detailed estimate of the mutation frequency in different genes.
An overview of all genes that contain ORF-disrupting mutations in one or more isolates is presented in Table 5, and more detailed information is provided in Table S2 in the supplemental material. In total, 26 of 170 genes (15%) have a disrupted ORF in at least one clinical isolate. Unsurprisingly, none of these genes are essential for growth on fibroblast cells, although UL30 was found to be growth augmenting (99, 100). Looking at the distribution of mutants over different clinical isolates (Fig. 4), only 28 of 124 isolates (23%) have the complete set of 170 intact genes, with the other isolates having 1 (33%), 2 (27%), 3 (13%), or 4 (3%) mutated genes. With these data, we show that only 1 out of 4 clinical isolates is genetically intact and that gene-disrupting mutations are extremely common in a defined set of nonessential genes. We cannot rule out the possibility that even more genes are mutated in our isolates, as discussed in Text S1 in the supplemental material. Some mutations affect only a small proportion of the ORF (e.g., in the US9 and US27 genes) (Table 5). These cases could be better described as variants than as mutants if the encoded gene products are not affected by these N- or C-terminal deletions, but functional experiments are needed to evaluate this.
TABLE 5.
Genes containing ORF-disrupting mutations in HCMV strains
| Gene | % of strains mutateda | Mutation type(s)b (no. of strains) | Median unaffected fraction of mutated ORFs (min–max) |
|---|---|---|---|
| UL9 | 34.6 (n = 127) | Sub in cod59 (5),c 23-nt del (4),c sub in cod6 (3),c 5-nt del (3),c sub in cod59 (2), 71-nt del (1),c sub in cod23 (1), sub in cod26 (1),c sub in cod40 (1),c sub in cod49 (1), sub in cod53 (1),c sub in cod59 (1), sub in cod63 (1), sub in cod71 (1),c sub in cod79 (1), sub in cod163 (1), 1-nt ins (1), 1-nt ins (1),c 1-nt ins (1), 178-nt del (1), 90-nt del (1), 51-nt del (1), 44-nt del (1), 40-nt del (1), 40-nt del (1), 29-nt del (1), 29-nt del (1),c 19-nt del (1), 19-nt del (1), 4-nt del (1), 2-nt del (1),c 1-nt del (1) | 27 (2–95) |
| RL5A | 20.3 | 11-nt del (14),c 2-nt del (4),c 17-nt del (3),c sub in cod12 (2),c sub in cod35 (1),c 44-nt del (1), 1-nt del (1) | 51 (6–58) |
| RL6 | 15.6 | 316-nt del (14),c 17-nt del (2), sub in cod57 (1),c 5-nt del (1), 2-nt del (1), 2-nt del (1) | 0 (0–66) |
| US9 | 15.0 (n = 127) | 35-nt del (18),c sub in cod227 (1)c | 94 (91–94) |
| UL1 | 10.2 | Sub in cod99 (6),c sub in cod86 (2), sub in cod147 (2), sub in cod42 (1), sub in cod45 (1),c 4-nt del (1) | 44 (8–68) |
| UL111A | 9.4 | Sub in cod36 (3),c 38-nt del (3),c sub in cod129 (2), sub in cod56 (1), 2-nt ins (1), 1-nt ins (1), 219-nt del (1)c | 32 (9–72) |
| UL150 | 6.3 | Sub in cod1 (1), 2-nt deletion (7)c | 1 (1–95) |
| UL40 | 5.5 | Sub in cod1 (7)c | 93 (93–93) |
| US7 | 5.5 (n = 127) | Sub in cod161 (1), sub in cod179 (1), 1-nt ins (1),c 112-nt del (1), 76-nt del (1),c 67-nt del (1), 47-nt del (1)c | 71 (3–93) |
| RL13 | 3.9 | Sub in cod150 (1), 2-nt ins (1), 279-nt del (1), 2-nt del (1), 1-nt del (1) | 50 (23–80) |
| UL128 | 2.4 (n = 126) | Sub in splice donor site (1), 1-nt ins (1), 14-nt del (1) | 32 (19–41) |
| UL133 | 2.3 | Sub in cod247 (1), 1-nt ins (1), 37-nt del (1) | 78 (61–97) |
| US13 | 2.3 | 24-nt del (3) | 97 (97–97) |
| RL12 | 1.6 | 5-nt del (1), 2-nt del (1) | 36 (27–45) |
| UL136 | 1.6 | Sub in cod210 (1), sub in cod227 (1) | 91 (87–94) |
| US27 | 1.6 | 18-nt del (1), 43-nt del (1)c | 97 (97–97) |
| RL1 | 0.8 | 84-nt del (1)c | 87 (87–87) |
| UL11 | 0.8 | 1-nt del (1) | 39 (39–39) |
| UL30 | 0.8 | 1-nt ins (1)c | 73 (73–73) |
| UL148 | 0.8 | Sub in cod104 (1) | 33 (33–33) |
| UL145 | 0.8 | 349-nt del (1) | 54 (54–54) |
| UL142 | 0.8 | 77-nt del (1)c | 73 (73–73) |
| UL150A | 0.8 | Sub in cod264 (1) | 97 (97–97) |
| IRS1 | 0.8 (n = 126) | Sub in cod836 (1) | 99 (99–99) |
| US6 | 0.8 (n = 127) | Sub in cod35 (1) | 19 (19–19) |
| US12 | 0.8 | 2-nt ins (1) | 44 (44–44) |
n = 128, unless stated otherwise (mutations that are certain to be artifacts of culture passage are omitted).
del, deletion; ins, insertion; sub, substitution; cod, codon (codon refers to the actual codon position in the specific strain that contains the mutation).
Mutations that were verified to be present in original clinical material (or strains sequenced directly from clinical material).
FIG 4.
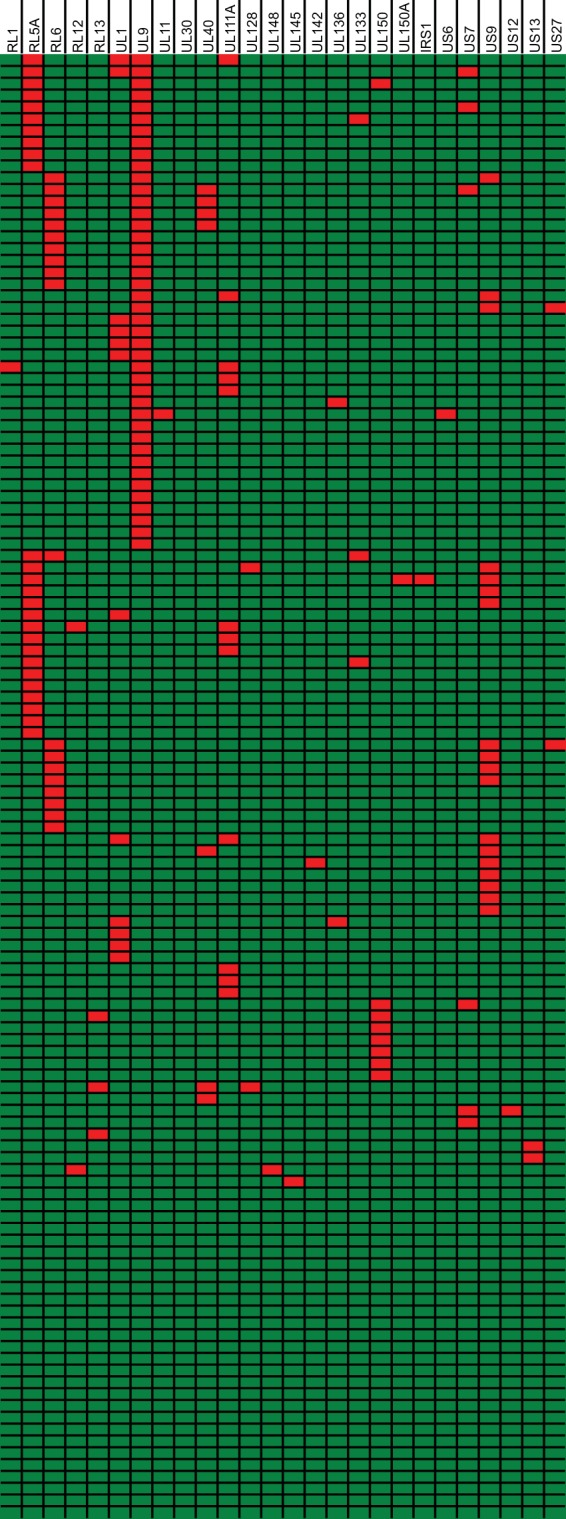
Distribution of ORF-disrupting mutations. Shown is a graphical representation of the distribution of ORF-disrupting mutations over 124 clinical isolates. Rows represent different isolates, and columns represent all 26 genes containing disruptive mutations. Disrupted genes are represented in red, and intact genes are represented in green.
As our strains were minimally passaged in fibroblast cell culture, some mutations might be artifacts of culture adaptation. Therefore, the presence of mutations in the original clinical material was assessed by direct PCR sequencing of these samples, if available. All mutations that were confirmed with this procedure are shown in Table 5 and in Table S2 in the supplemental material. Importantly, all but one of the mutations that were characterized in the original clinical material could be confirmed to be unrelated to culture amplification. Mutations in the RL1, RL5A, RL6, UL1, UL9, UL30, UL40, UL111A, UL142, UL150, US7, US9, and US27 genes are thus indeed present in clinical isolates. Moreover, the occurrence of identical mutations in unrelated and geographically distinct isolates confirms their circulation in natural populations. The only mutation identified as an artifact of culture passage was a substitution in the first splice donor site of the UL128 gene in strain BE/11/2011; the specific case of mutations in the RL13 and UL128 genes is discussed in further detail in Text S1 in the supplemental material.
Mutated genes are highlighted on the genome map in Fig. 1 with a color code depicting mutation frequency. The RL11 gene family stands out, with 7 out of 14 members containing disruptive mutations (or 6 if RL13 is omitted). Furthermore, 4 out of 5 genes that are mutated in >10% of isolates are part of this family. RL11 genes share homology with the CR1 domain of the adenovirus E3 genes through their RL11 domain (101). The encoded proteins have similarities to the IgD family, and because of their hypervariability (see “HCMV displays the highest level of genetic diversity of all human herpesviruses,” above), it is believed they could have a function in modulating variable host proteins. Immunomodulatory capacities have recently been proposed for UL7 (102), UL11 (103), and RL12 and RL13 (73). While RL13 mutants are probably culture artifacts and RL12 and UL11 mutants are rare and unconfirmed in clinical material, mutations in RL5A, RL6, UL1, and UL9 are much more common and definitely circulate in the host. Functional data for these genes are limited, with UL1 being implicated as a tropism factor (104) and UL9 being implicated in growth temperance (100). While our analysis shows that almost 35% of strains have a mutation in UL9, a previous study that characterized UL9 sequences in unpassaged and moderately and highly passaged isolates from a diverse geographical background found only 2 mutations in 41 strains (5%) (GenBank accession numbers DQ847465 to DQ847505). It is unclear whether this discrepancy is due to geographical differences in UL9 mutations, the types of patients involved, or the body compartment that was sampled.
Several genes in the RL1 family (2/2), the US6 family (3/6), and the US12 family (2/10) are also affected. For the RL1 family genes RL1 and UL145, functional knowledge is lacking, and in both cases, only one strain contains a mutation. Members of the US6 family have established or tentative immunomodulatory functions, interfering with the major histocompatibility complex class I (MHC-I) antigen-processing pathway (71, 105). The role of US6 in inhibition of antigen peptide transport to MHC-I molecules has been well described (106). Studies on US7 and US9 function are much scarcer (107–109), but immunomodulatory functions have been predicted (71). The natural occurrence of mutants in US6, US7, and US9 may suggest some functional redundancy, as the US2, US3, US10, and US11 genes all target MHC-I antigen presentation (110). The US12 gene family encodes 10 seven-transmembrane proteins, with some members recently being associated with immunomodulation (111, 112) and cell tropism for epithelial and endothelial cells (113). Functions of the US12 and US13 genes have not yet been revealed. These different HCMV gene families exist as clusters of adjacent (apart from the RL1 family) and (distantly) related genes that probably originated from duplication of an ancestral gene (114). As recent experiments with poxviruses have shown, these so-called genomic accordions can rapidly expand under strong selective pressure and provide dsDNA viruses with low mutation rates an alternative mode to evolve more quickly under specific circumstances (115, 116). Subsequently, adaptive mutations in these new copies can accumulate at a much higher rate than in one isolated gene. Finally, when the selective environment changes, genes that did not acquire beneficial functions can be removed, and the accordion contracts. Especially for the RL11 and US6 gene families, where mutant genes were confirmed to be present in clinical isolates, this accordion contraction could be currently ongoing.
Several additional, isolated genes are mutated in the original clinical material. The fact that almost 10% of strains are affected in the viral interleukin-10 (IL-10)-encoding UL111A gene is striking. UL111A encodes separate transcripts during productive (cmvIL-10) and latent (LAcmvIL-10) infection, and both gene products are affected in all 12 mutant isolates (Fig. 5). cmvIL-10 binds and signals through the human IL-10 receptor and mimics its immunomodulatory properties. It has been shown to inhibit the production of proinflammatory cytokines and MHC-I and -II expression in monocytes and to stimulate monocyte differentiation to a phagocytic phenotype and B cell proliferation and differentiation. It is believed that these concerted cmvIL-10 actions have an important impact on the immune system's capacity to control HCMV replication (117). In this regard, cmvIL-10 has been shown to impair cytotrophoblast remodeling of the uterine vasculature, thereby possibly enhancing congenital disease (118). The latency-associated LAcmvIL-10 product cannot signal through human IL-10 receptors in the same fashion but was recently shown to upregulate the expression of cellular IL-10 and CCL8 (119). Another mutated gene with potential implications for the immunomodulatory capacities of isolates is UL40. The UL40 signal peptide is necessary for the cell surface expression of HCMV pUL18 and HLA-E molecules, both of which are natural killer (NK) cell ligands that can inhibit NK cell activation in the absence of normal MHC-I antigen presentation (120). In 5.5% of isolates, UL40 has a substitution in its original start codon, presumably leading to translation initiation from an alternative start codon located 15 nt downstream and truncating the signal peptide (see Table S2 in the supplemental material). Whereas this mutation was previously found only in strain 3157, we identified six more instances of signal peptide truncation in our isolates. By using UL40 from strain 3157 in comparison to wild-type UL40, it was recently demonstrated that this mutation did not affect UL40 translation or pUL18 surface expression but did inhibit the surface expression of HLA-E and thereby sensitized infected cells to NK cell lysis (120). For both the UL111A and UL40 genes, unrelated strains display identical deletions or substitutions, suggesting wide circulation of mutants in the population (Fig. 5; see also Table S2 in the supplemental material). Studying the impact of UL111A and UL40 mutants on the immune-evasive potential of strains and their implications for strain pathogenicity might be worthwhile, as initial findings for UL40 illustrate (120). The remaining genes containing disruptive mutations are discussed further in Text S1 in the supplemental material; mutants of these genes in clinical material were rare and/or unconfirmed.
FIG 5.

ORF-disrupting mutations in the UL111A gene. Shown is a nucleotide alignment of wild-type UL111A (strain Merlin) and all 12 mutants. Countries of isolation are listed for all strains with the international two-letter code (GB, Great Britain; BE, Belgium; CZ, Czech Republic; DE, Germany). Mutations (deletions, insertions, and substitutions) are highlighted in red, and the predicted stop codons are underlined, with untranslated sequences after stop codons being crossed out. Introns have a gray background, unless they are aberrantly translated because of the deletion of splice donor sites. LAcmvIL-10 transcripts are similar, but the second intron is not spliced, with translation proceeding into it.
HCMV evolution has been shaped by pervasive recombination.
An important role for recombination in HCMV evolution has long been suggested. Given the common occurrence of multiple infections in a single host, the potential for recombination is obvious (reviewed in reference 15). Recombination has been identified in individual gene sequences and by the small amount of gene linkage observed (98, 121–124). However, overall recombination on a full-genome scale has not yet been quantified.
Since standard phylogenetic trees cannot account for recombination and are incorrect if recombined sequences are included, we have analyzed phylogenetic relationships by constructing a split-decomposition network (Fig. 6A) (51). The large numbers of reticulate connections that are apparent at the root of the network indicate conflicting evolutionary signals such as those caused by recombination. By using the Phi-test for recombination, it was confirmed that recombination between isolates was detected (P = 0.0) (54). To further visualize recombination events, nine strains were selected from the network, and a BootScan analysis was performed based on a sequence alignment of these strains only (Fig. 6B). The constant shifting of phylogenetic relationships along the genome provides further evidence of numerous recombination events.
When split networks were constructed for HSV-1, VZV, and EBV, similar evidence for recombination was detected (P values are shown in Fig. 7). Previous studies have reported distinct geographic clusters for HSV-1, VZV, and EBV strains (48, 125–127). Split networks for these viruses indeed show distinct clusters (Fig. 7), while such clusters do not become apparent from our HCMV split network. However, apart from two Asian strains, all isolates were collected from European and North American patients. Interestingly, Asian strains JHC (128) and HAN are neighbors in the split network (highlighted in gray in Fig. 6A), but they do not cluster separately from European/North American strains. Additional full-genome sequences from Asian and African strains will be necessary to investigate the potential existence of separate geographic clusters of HCMV.
The amount of recombination was quantified by detecting individual recombination breakpoints with RDP3 (53). To allow comparison of recombination densities in HCMV, HSV-1, VZV, and EBV genomes, we selected a group of nine full-genome sequences for each virus species, i.e., the total number of genomes that was available for EBV. Strains were selected to properly reflect total diversity (Fig. 6A and 7). Of these four human herpesviruses, HCMV clearly has the highest recombination density (Table 6). There is a statistically significant, positive correlation between overall nucleotide diversity (Table 1) and recombination density (Table 6) for these four viruses (P = 0.017 and ρ = 0.98 by Pearson's product-moment correlation). In comparisons of clusterings of isolates in the split networks (Fig. 6A and 7), there is a tentative inverse relation between the existence of distinct clusters and the recombination breakpoint density. VZV has the lowest recombination density, and the divergence of clusters is very clear, with few recombinants between clusters. HSV-1 and EBV have intermediate recombination densities: distinct clusters are still recognizable, but their delineation is less pronounced because of the presence of intermediate recombinants. Finally, HCMV has a markedly higher recombination density, resulting in a star-like phylogeny with no apparent clustering. Hypothesizing that these clusters or “genome types” have evolved during human radiation across the planet, we may now see a fading of geographical genome types because of increased global travel and mixing of populations and viruses. This hypothesis is supported by findings that the geographic separation of VZV clades might be slowly fading because of recent immigration (129). Given their different recombination densities, distinct herpesviruses would currently be at different stages of this process. In comparisons of recombination in HSV-1 and VZV, it has been proposed that inherent disparities in biological characteristics might be at the root of recombination potential (130). Higher recombination rates in HSV-1 were attributed to more frequent reactivation, longer episodes of asymptomatic shedding, and subsequent increased occurrence of multiple infections. In the same fashion, its superior immunomodulatory capabilities and broad cell tropism might explain the even higher capacity for recombination in HCMV.
TABLE 6.
Comparative analysis of recombination in human herpesviruses
| Subfamily | Speciesa | No. of recombination events | No. of breakpoints | Recombination density (no. of breakpoints/kb) |
|---|---|---|---|---|
| Alphaherpesvirinae | HHV-1 (HSV-1) | 60 | 78 | 0.57 |
| HHV-3 (VZV) | 13 | 13 | 0.10 | |
| Betaherpesvirinae | HHV-5 (HCMV) | 314 | 392 | 1.33 |
| Gammaherpesvirinae | HHV-4 (EBV) | 61 | 69 | 0.40 |
While it is clear that recombination at the genome level permits the exchange of alleles at separate loci, we also wanted to evaluate the contribution of recombination to the generation of variation at the level of individual genes. Evidence for recombination in codon-aligned gene sequences was assessed by three different methods. The Phi-test for recombination simply determines whether there is statistically significant evidence for recombination, while GARD and RDP3 identify specific recombination breakpoints. An overview of results for all genes is presented in Table S6 in the supplemental material. Recombination was confirmed in 93 out of 170 genes (55%) by at least 2 out of 3 methods. The densities of breakpoints varied enormously between different genes, as illustrated in Fig. 1. Gene-specific recombination densities were grouped in different gene families, gene conservation groups, and gene functions, as described above for diversity (see second panel in Fig. 2 for gene families). There was a statistically significant difference between recombination densities in different gene families (P = 0.0031 by KWt) but not in different conservation categories (P = 0.13 by KWt). For the former, none of the pairwise comparisons were significant. Overall, gene families with higher levels of diversity display higher recombination densities, although there are clearly some exceptions in conserved genes, such as IRS1 and TRS1 of the US22 family (Fig. 2). When divided over different gene functions, there was still a statistically significant difference between recombination densities (P = 0.023 by KWt; no significant pairwise comparisons). The discrepancy between overall KWt results and the results of pairwise comparisons for recombination over different gene families and functions is caused by the rigorous correction for multiple testing in the latter. The numbers of genes assigned to gene families and functions vary widely, reducing the power of the tests. In summary, our data demonstrate the potential for recombination to generate diversity both at the level of the individual gene as well as over the complete genome. The high recombination rate in HCMV compared to that in other human herpesviruses could be one of the most important reasons for its higher level of strain diversity. Reciprocally, the increased diversity could also result in a higher sensitivity for detection of recombination events that are undetectable in more conserved herpesviruses.
Positively selected residues provide a genetic fingerprint of the evolutionary arms race between virus and host.
While intermolecular recombination is a very strong mechanism to increase nucleotide diversity, its ability to do so relies on preexisting variation through mutations caused by polymerase infidelity. Obviously, the accumulation of these mutations is dependent on the selective pressure that acts upon them. The persistent nature of HCMV and its constant interplay with host immune components highlight the potential selective pressure that might act on gene variants. To characterize the overall selective pressure acting on viral genes, we calculated the ratio of nonsynonymous substitutions per nonsynonymous site (dN) to synonymous substitutions per synonymous site (dS) for each gene alignment (Fig. 8; see also Table S4 in the supplemental material). Most HCMV genes seem to be under strong evolutionary constraints, as 96% of genes have a dN/dS ratio of <0.5 and 64% of genes have a ratio of <0.2. Recent studies of HSV-1 and MCMV also found a predominance of negative selection, but the proportion of genes with a dN/dS ratio of <0.1 is much higher for HCMV (38%, versus 7% for HSV-1 and 13% for MCMV) (48, 63). A few genes have dN/dS ratios closer to 1, indicating neutral selection or genetic drift. Most of these genes are poorly characterized, but the tegument protein ppUL25 was found to be a major target of anti-CMV antibodies (131). This strong negative/purifying selection is an indication of the excellent adaptation of HCMV to its human host, with most mutations having negative fitness effects and being quickly removed.
FIG 8.

The majority of HCMV genes are under strong purifying selection. The selection mode acting on genes is represented by calculation of dN/dS ratios. A ratio close to zero indicates strong negative/purifying selection, and a ratio close to 1 indicates neutral selection or genetic drift. A ratio significantly higher than 1 indicates positive/diversifying selection. Genes are binned in groups with similar dN/dS ratios in steps of 0.1.
Notwithstanding the strong purifying selection acting on most HCMV genes, individual residues might be experiencing a different selective regimen. To identify positive/diversifying and negative/purifying selection at the codon level, we made use of four separate algorithms (SLAC, FEL, MEME, and FUBAR) included in the HyPhy package for phylogenetic hypothesis testing (56) (see Table S7 in the supplemental material). Since these methods have different sensitivities and specificities, we retained only sites that were independently confirmed by at least two out of four methods. Of 68,287 codons analyzed, 431 (0.6%) showed evidence of positive selection (in 105 genes), and 7,731 (11.3%) showed evidence of negative selection (in 169 genes), again demonstrating the predominance of negative selection acting on HCMV genes. However, there were clear discrepancies in the distribution of positively selected residues, as shown in Fig. 1. There was a statistically significant difference between gene families (P = 0.0054 by KWt) and gene conservation groups (P = 0.00055 by KWt); genes with higher levels of variability generally display higher percentages of positively selected codons (statistically significant only for cytomegalovirus-specific versus core genes [P = 0.0033 by WRSt]). Clearly, there are multiple exceptions, most notably the MHC family (UL18 and UL142) and the RL1 (RL1 family), US7 (US6 family), IRS1 and TRS1 (US22 family), and US14 and US18 (US12 family) genes, which are subjected to higher levels of positive selection than would be expected from their diversity (Fig. 2, third panel). Their products function in the evasion of both adaptive immune responses (UL18, UL142, and US18) and innate antiviral mechanisms (IRS1 and TRS1), or their functions are not yet characterized. Hence, it could be suggested that these functionally uncharacterized genes might also interact with host antiviral mechanisms. Also, for negative selection, there was a significant difference between gene families (P = 0.00023 by KWt) and gene conservation groups (P = 0.015 by KWt), but this was unrelated to gene diversity (Fig. 2, bottom). No statistically significant differences in selection between gene functions were observed (P = 0.16 for positive selection; P = 0.32 for negative selection [determined by KWt]).
Of the genes experiencing the highest percentages of positive selection, there is a clear predominance of genes modulating host immune and antiviral pathways (Fig. 1; see also Table S7 in the supplemental material). The UL147 gene encodes an α-chemokine (132), UL142 interferes with NK cell activation (133), IRS1 inhibits the protein kinase R antiviral pathway (134), and UL20 is poorly characterized but has also been implicated in immune evasion (135). The role of the UL40 signal peptide in inhibiting NK cells is discussed above (see “Wild-type HCMV strains contain ORF-disrupting mutations in a wide range of nonessential genes”). Interestingly, 6 out of 8 residues under positive selection in pUL40 are located within this signal peptide, and more specifically, 4 out of 8 are located within the nonamer peptide that is processed to bind the viral MHC-I mimic pUL18 and HLA-E. For pUL18, a crystal structure of its interaction with human β-2-microglobulin (MHC-I light chain), an actin peptide, and the inhibitory LIR-1 receptor was reported (136). We have visualized pUL18 residues under positive selection on the pUL18 structure (Fig. 9). All positively selected residues are located on the surface of pUL18, with L265 interacting with β-2-microglobulin and D248, V250, and E252 being situated adjacent to LIR-1 binding sites. Although the overall evolutionary mode of HCMV is strongly shaped by constraints and purifying selection, these examples illustrate that specific protein residues can experience diversifying selective pressure through their interactions with the host.
FIG 9.
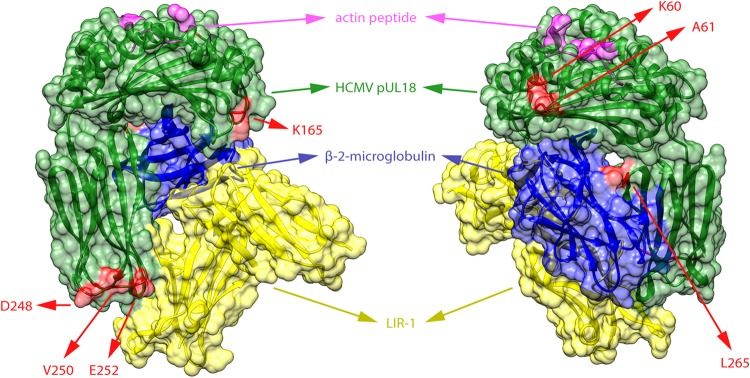
Residues under diversifying selection in pUL18. Codons under positive/diversifying selection in the UL18 gene were determined with the SLAC, FEL, MEME, and FUBAR algorithms of the HyPhy package. Sites that showed significant evidence of positive selection by at least two of four methods are represented in red on the protein structure of pUL18 (green). The structure shows a complex of pUL18 (a viral MHC-I homolog), human β-2-microglobulin (blue) (a MHC-I light chain), and an actin peptide (pink) bound to the inhibitory immunoglobulin receptor LIR-1 (yellow) (136). The three-dimensional structure is visualized from two opposite angles. All selected residues are located at the surface of pUL18.
Concluding remarks.
In this study, we have applied our previously described workflow (36) to the characterization of HCMV genomes of 96 distinct clinical isolates from Belgian and Czech patients. Based on a comparative analysis of 124 full-genome sequences, HCMV interstrain nucleotide diversity, π, was estimated to be 0.021, significantly higher than the diversity in other human herpesviruses. Nevertheless, overall levels of purifying selection were very high, reflecting the remarkable adaptation of HCMV to its human host. Given the proofreading capacity of the HCMV DNA polymerase, additional strategies are necessary to generate diversity apart from replication error. Because of the wide range of immune-evasive functions, multiple infections are common, permitting the virus to recombine extensively. Furthermore, gene duplications can effectively enlarge mutational space. Finally, interactions with host components generate positive selective pressure at specific loci, which may help the virus avoid immune recognition.
We have demonstrated the widespread occurrence of gene-disrupting mutations in wild-type HCMV strains, unrelated to culture passage. While some of these mutants might be evidence of ongoing contraction of gene family duplications, others could have implications for the immunomodulatory and, ultimately, the pathogenic potential of the isolates involved. In particular, a closer look at mutants of the UL40 and UL111A genes is warranted. We are currently retrieving clinical data regarding our patient population to explore associations of gene variants and mutants with disease outcome.
Our data set is completely derived from European patients. Currently, only two complete genome sequences from patients outside Europe and North America are publicly available. The availability of more complete genomes, especially from African and Asian patients, will reveal whether there are geographical discrepancies in gene diversity and mutational patterns. Additionally, the majority of our isolates (74/96) were derived from urine samples. Our preliminary results show identical mutations in different body compartments for several genes (including nasopharyngeal isolates, blood, amniotic fluid, and bronchoalveolar lavage fluid), but we cannot exclude the possibility that specific mutations can influence the tissue tropism of the viral strain. In the future, it will be interesting to see whether there are differences in the occurrence of disruptive mutations depending on the body compartment that is sampled.
To our knowledge, this study is the most comprehensive analysis of genetic variability and evolution in HCMV to date, providing both conceptual insights into diversity generation and a large source of sequence information of outstanding value for functional experiments with this important human pathogen.
Supplementary Material
ACKNOWLEDGMENTS
Steven Sijmons and Piet Maes were supported by the Research Foundation Flanders (FWO [Fonds voor Wetenschappelijk Onderzoek, Vlaanderen]).
The FWO was not involved in experimental design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
We thank all colleagues of the Laboratory of Clinical Virology for helpful comments and insightful discussions. We are also indebted to the laboratory technicians of the molecular diagnostics unit (CEMOL) at University Hospitals Leuven and Marc De Foor (Iris-Lab, Brussels, Belgium) for cell culture inoculation of patient samples and to Carl Van Hove (Janssen R&D) for preparing sequencing libraries and conducting sequencing experiments. Finally, we acknowledge the efforts and contributions of authors of previously reported sequences included in our analyses.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00578-15.
REFERENCES
- 1.Cannon MJ, Schmid DS, Hyde TB. 2010. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 20:202–213. doi: 10.1002/rmv.655. [DOI] [PubMed] [Google Scholar]
- 2.Sinclair J, Reeves M. 2014. The intimate relationship between human cytomegalovirus and the dendritic cell lineage. Front Microbiol 5:389. doi: 10.3389/fmicb.2014.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Invest 121:1673–1680. doi: 10.1172/JCI45449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R, National Vaccine Advisory Committee . 2004. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin Infect Dis 39:233–239. doi: 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 6.Krause PR, Bialek SR, Boppana SB, Griffiths PD, Laughlin CA, Ljungman P, Mocarski ES, Pass RF, Read JS, Schleiss MR, Plotkin SA. 2013. Priorities for CMV vaccine development. Vaccine 32:4–10. doi: 10.1016/j.vaccine.2013.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J Gen Virol 85:1301–1312. doi: 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 8.Murphy E, Shenk T. 2008. Human cytomegalovirus genome. Curr Top Microbiol Immunol 325:1–19. doi: 10.1007/978-3-540-77349-8_1. [DOI] [PubMed] [Google Scholar]
- 9.Waner JL, Weller TH. 1978. Analysis of antigenic diversity among human cytomegaloviruses by kinetic neutralization tests with high-titered rabbit antisera. Infect Immun 21:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drew WL, Sweet ES, Miner RC, Mocarski ES. 1984. Multiple infections by cytomegalovirus in patients with acquired immunodeficiency syndrome: documentation by Southern blot hybridization. J Infect Dis 150:952–953. doi: 10.1093/infdis/150.6.952. [DOI] [PubMed] [Google Scholar]
- 11.Kilpatrick BA, Huang ES, Pagano JS. 1976. Analysis of cytomegalovirus genomes with restriction endonucleases Hin D III and EcoR-1. J Virol 18:1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pignatelli S, Dal Monte P, Rossini G, Landini MP. 2004. Genetic polymorphisms among human cytomegalovirus (HCMV) wild-type strains. Rev Med Virol 14:383–410. doi: 10.1002/rmv.438. [DOI] [PubMed] [Google Scholar]
- 14.Puchhammer-Stockl E, Gorzer I. 2006. Cytomegalovirus and Epstein-Barr virus subtypes—the search for clinical significance. J Clin Virol 36:239–248. doi: 10.1016/j.jcv.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Puchhammer-Stockl E, Gorzer I. 2011. Human cytomegalovirus: an enormous variety of strains and their possible clinical significance in the human host. Future Virol 6:259–271. doi: 10.2217/fvl.10.87. [DOI] [Google Scholar]
- 16.Shepp DH, Match ME, Ashraf AB, Lipson SM, Millan C, Pergolizzi R. 1996. Cytomegalovirus glycoprotein B groups associated with retinitis in AIDS. J Infect Dis 174:184–187. doi: 10.1093/infdis/174.1.184. [DOI] [PubMed] [Google Scholar]
- 17.Torok-Storb B, Boeckh M, Hoy C, Leisenring W, Myerson D, Gooley T. 1997. Association of specific cytomegalovirus genotypes with death from myelosuppression after marrow transplantation. Blood 90:2097–2102. [PubMed] [Google Scholar]
- 18.Correia-Silva JF, Resende RG, Arao TC, Abreu MH, Teixeira MM, Bittencourt H, Silva TA, Gomez RS. 2011. HCMV gB genotype and its association with cytokine levels in hematopoietic stem cell transplantation. Oral Dis 17:530–537. doi: 10.1111/j.1601-0825.2011.01801.x. [DOI] [PubMed] [Google Scholar]
- 19.Emery VC, Manuel O, Asberg A, Pang X, Kumar D, Hartmann A, Preiksaitis JK, Pescovitz MD, Rollag H, Jardine AG, Gahlemann CG, Humar A. 2012. Differential decay kinetics of human cytomegalovirus glycoprotein B genotypes following antiviral chemotherapy. J Clin Virol 54:56–60. doi: 10.1016/j.jcv.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossini G, Pignatelli S, Dal Monte P, Camozzi D, Lazzarotto T, Gabrielli L, Gatto MR, Landini MP. 2005. Monitoring for human cytomegalovirus infection in solid organ transplant recipients through antigenemia and glycoprotein N (gN) variants: evidence of correlation and potential prognostic value of gN genotypes. Microbes Infect 7:890–896. doi: 10.1016/j.micinf.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Pignatelli S, Lazzarotto T, Gatto MR, Dal Monte P, Landini MP, Faldella G, Lanari M. 2010. Cytomegalovirus gN genotypes distribution among congenitally infected newborns and their relationship with symptoms at birth and sequelae. Clin Infect Dis 51:33–41. doi: 10.1086/653423. [DOI] [PubMed] [Google Scholar]
- 22.Paradowska E, Jablonska A, Studzinska M, Suski P, Kasztelewicz B, Zawilinska B, Wisniewska-Ligier M, Dzierzanowska-Fangrat K, Wozniakowska-Gesicka T, Czech-Kowalska J, Lipka B, Kornacka M, Pawlik D, Tomasik T, Kosz-Vnenchak M, Lesnikowski ZJ. 2013. Distribution of cytomegalovirus gN variants and associated clinical sequelae in infants. J Clin Virol 58:271–275. doi: 10.1016/j.jcv.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Paradowska E, Jablonska A, Studzinska M, Kasztelewicz B, Zawilinska B, Wisniewska-Ligier M, Dzierzanowska-Fangrat K, Wozniakowska-Gesicka T, Kosz-Vnenchak M, Lesnikowski ZJ. 2014. Cytomegalovirus glycoprotein H genotype distribution and the relationship with hearing loss in children. J Med Virol 86:1421–1427. doi: 10.1002/jmv.23906. [DOI] [PubMed] [Google Scholar]
- 24.Arav-Boger R, Willoughby RE, Pass RF, Zong JC, Jang WJ, Alcendor D, Hayward GS. 2002. Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J Infect Dis 186:1057–1064. doi: 10.1086/344238. [DOI] [PubMed] [Google Scholar]
- 25.Arav-Boger R, Battaglia CA, Lazzarotto T, Gabrielli L, Zong JC, Hayward GS, Diener-West M, Landini MP. 2006. Cytomegalovirus (CMV)-encoded UL144 (truncated tumor necrosis factor receptor) and outcome of congenital CMV infection. J Infect Dis 194:464–473. doi: 10.1086/505427. [DOI] [PubMed] [Google Scholar]
- 26.Waters A, Hassan J, De Gascun C, Kissoon G, Knowles S, Molloy E, Connell J, Hall WW. 2010. Human cytomegalovirus UL144 is associated with viremia and infant development sequelae in congenital infection. J Clin Microbiol 48:3956–3962. doi: 10.1128/JCM.01133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paradowska E, Jablonska A, Plociennikowska A, Studzinska M, Suski P, Wisniewska-Ligier M, Dzierzanowska-Fangrat K, Kasztelewicz B, Wozniakowska-Gesicka T, Lesnikowski ZJ. 2014. Cytomegalovirus alpha-chemokine genotypes are associated with clinical manifestations in children with congenital or postnatal infections. Virology 462–463:207–217. doi: 10.1016/j.virol.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 28.Arav-Boger R, Boger YS, Foster CB, Boger Z. 2008. The use of artificial neural networks in prediction of congenital CMV outcome from sequence data. Bioinform Biol Insights 2:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He R, Ruan Q, Qi Y, Ma YP, Huang YJ, Sun ZR, Ji YH. 2006. Sequence variability of human cytomegalovirus UL146 and UL147 genes in low-passage clinical isolates. Intervirology 49:215–223. doi: 10.1159/000091468. [DOI] [PubMed] [Google Scholar]
- 30.Heo J, Petheram S, Demmler G, Murph JR, Adler SP, Bale J, Sparer TE. 2008. Polymorphisms within human cytomegalovirus chemokine (UL146/UL147) and cytokine receptor genes (UL144) are not predictive of sequelae in congenitally infected children. Virology 378:86–96. doi: 10.1016/j.virol.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pati SK, Pinninti S, Novak Z, Chowdhury N, Patro RK, Fowler K, Ross S, Boppana S, NIDCD CHIMES Study Investigators . 2013. Genotypic diversity and mixed infection in newborn disease and hearing loss in congenital cytomegalovirus infection. Pediatr Infect Dis J 32:1050–1054. doi: 10.1097/INF.0b013e31829bb0b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nijman J, Mandemaker FS, Verboon-Maciolek MA, Aitken SC, van Loon AM, de Vries LS, Schuurman R. 2014. Genotype distribution, viral load and clinical characteristics of infants with postnatal or congenital cytomegalovirus infection. PLoS One 9:e108018. doi: 10.1371/journal.pone.0108018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vales-Gomez M, Shiroishi M, Maenaka K, Reyburn HT. 2005. Genetic variability of the major histocompatibility complex class I homologue encoded by human cytomegalovirus leads to differential binding to the inhibitory receptor ILT2. J Virol 79:2251–2260. doi: 10.1128/JVI.79.4.2251-2260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerboni C, Achour A, Warnmark A, Mousavi-Jazi M, Sandalova T, Hsu ML, Cosman D, Karre K, Carbone E. 2006. Spontaneous mutations in the human CMV HLA class I homologue UL18 affect its binding to the inhibitory receptor LIR-1/ILT2/CD85j. Eur J Immunol 36:732–741. doi: 10.1002/eji.200425220. [DOI] [PubMed] [Google Scholar]
- 35.Sijmons S, Van Ranst M, Maes P. 2014. Genomic and functional characteristics of human cytomegalovirus revealed by next-generation sequencing. Viruses 6:1049–1072. doi: 10.3390/v6031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sijmons S, Thys K, Corthout M, Van Damme E, Van Loock M, Bollen S, Baguet S, Aerssens J, Van Ranst M, Maes P. 2014. A method enabling high-throughput sequencing of human cytomegalovirus complete genomes from clinical isolates. PLoS One 9:e95501. doi: 10.1371/journal.pone.0095501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 38.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prichard MN, Penfold ME, Duke GM, Spaete RR, Kemble GW. 2001. A review of genetic differences between limited and extensively passaged human cytomegalovirus strains. Rev Med Virol 11:191–200. doi: 10.1002/rmv.315. [DOI] [PubMed] [Google Scholar]
- 41.Bradley AJ, Lurain NS, Ghazal P, Trivedi U, Cunningham C, Baluchova K, Gatherer D, Wilkinson GW, Dargan DJ, Davison AJ. 2009. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J Gen Virol 90:2375–2380. doi: 10.1099/vir.0.013250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wernersson R, Pedersen AG. 2003. RevTrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res 31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otto TD, Dillon GP, Degrave WS, Berriman M. 2011. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res 39:e57. doi: 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang G, Raghavan B, Kotur M, Cheatham J, Sedmak D, Cook C, Waldman J, Trgovcich J. 2007. Antisense transcription in the human cytomegalovirus transcriptome. J Virol 81:11267–11281. doi: 10.1128/JVI.00007-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc Natl Acad Sci U S A 108:19755–19760. doi: 10.1073/pnas.1115861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szpara ML, Gatherer D, Ochoa A, Greenbaum B, Dolan A, Bowden RJ, Enquist LW, Legendre M, Davison AJ. 2014. Evolution and diversity in human herpes simplex virus genomes. J Virol 88:1209–1227. doi: 10.1128/JVI.01987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benson G. 1999. Tandem Repeats Finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 51.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 52.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- 56.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 57.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kosakovsky Pond SL, Frost SD. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 59.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205. doi: 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 62.Cunningham C, Gatherer D, Hilfrich B, Baluchova K, Dargan DJ, Thomson M, Griffiths PD, Wilkinson GW, Schulz TF, Davison AJ. 2010. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J Gen Virol 91:605–615. doi: 10.1099/vir.0.015891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith LM, McWhorter AR, Shellam GR, Redwood AJ. 2013. The genome of murine cytomegalovirus is shaped by purifying selection and extensive recombination. Virology 435:258–268. doi: 10.1016/j.virol.2012.08.041. [DOI] [PubMed] [Google Scholar]
- 64.Renzette N, Bhattacharjee B, Jensen JD, Gibson L, Kowalik TF. 2011. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog 7:e1001344. doi: 10.1371/journal.ppat.1001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanton R, Westmoreland D, Fox JD, Davison AJ, Wilkinson GW. 2005. Stability of human cytomegalovirus genotypes in persistently infected renal transplant recipients. J Med Virol 75:42–46. doi: 10.1002/jmv.20235. [DOI] [PubMed] [Google Scholar]
- 66.Bradley AJ, Kovacs IJ, Gatherer D, Dargan DJ, Alkharsah KR, Chan PK, Carman WF, Dedicoat M, Emery VC, Geddes CC, Gerna G, Ben-Ismaeil B, Kaye S, McGregor A, Moss PA, Pusztai R, Rawlinson WD, Scott GM, Wilkinson GW, Schulz TF, Davison AJ. 2008. Genotypic analysis of two hypervariable human cytomegalovirus genes. J Med Virol 80:1615–1623. doi: 10.1002/jmv.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lurain NS, Fox AM, Lichy HM, Bhorade SM, Ware CF, Huang DD, Kwan SP, Garrity ER, Chou S. 2006. Analysis of the human cytomegalovirus genomic region from UL146 through UL147A reveals sequence hypervariability, genotypic stability, and overlapping transcripts. Virol J 3:4. doi: 10.1186/1743-422X-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorzer I, Guelly C, Trajanoski S, Puchhammer-Stockl E. 2010. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J Virol 84:7195–7203. doi: 10.1128/JVI.00475-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murthy S, Hayward GS, Wheelan S, Forman MS, Ahn JH, Pass RF, Arav-Boger R. 2011. Detection of a single identical cytomegalovirus (CMV) strain in recently seroconverted young women. PLoS One 6:e15949. doi: 10.1371/journal.pone.0015949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J. 2000. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J Mol Evol 50:56–68. http://link.springer.com/article/10.1007%2Fs002399910007. [DOI] [PubMed] [Google Scholar]
- 71.Van Damme E, Van Loock M. 2014. Functional annotation of human cytomegalovirus gene products: an update. Front Microbiol 5:218. doi: 10.3389/fmicb.2014.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, Davison AJ, Wilkinson GW. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cortese M, Calo S, D'Aurizio R, Lilja A, Pacchiani N, Merola M. 2012. Recombinant human cytomegalovirus (HCMV) RL13 binds human immunoglobulin G Fc. PLoS One 7:e50166. doi: 10.1371/journal.pone.0050166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hannan AJ. 2012. Tandem repeat polymorphisms: mediators of genetic plasticity, modulators of biological diversity and dynamic sources of disease susceptibility. Adv Exp Med Biol 769:1–9. doi: 10.1007/978-1-4614-5434-2_1. [DOI] [PubMed] [Google Scholar]
- 75.Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. 2010. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet 44:445–477. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- 76.Brouwer JR, Willemsen R, Oostra BA. 2009. Microsatellite repeat instability and neurological disease. Bioessays 31:71–83. doi: 10.1002/bies.080122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duke GM, Osorio JE, Palmenberg AC. 1990. Attenuation of Mengo virus through genetic engineering of the 5′ noncoding poly(C) tract. Nature 343:474–476. doi: 10.1038/343474a0. [DOI] [PubMed] [Google Scholar]
- 78.Hahn H, Palmenberg AC. 1995. Encephalomyocarditis viruses with short poly(C) tracts are more virulent than their mengovirus counterparts. J Virol 69:2697–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perdue ML, Garcia M, Senne D, Fraire M. 1997. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res 49:173–186. doi: 10.1016/S0168-1702(97)01468-8. [DOI] [PubMed] [Google Scholar]
- 80.Bates PA, DeLuca NA. 1998. The polyserine tract of herpes simplex virus ICP4 is required for normal viral gene expression and growth in murine trigeminal ganglia. J Virol 72:7115–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pfister LA, Letvin NL, Koralnik IJ. 2001. JC virus regulatory region tandem repeats in plasma and central nervous system isolates correlate with poor clinical outcome in patients with progressive multifocal leukoencephalopathy. J Virol 75:5672–5676. doi: 10.1128/JVI.75.12.5672-5676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaufer BB, Jarosinski KW, Osterrieder N. 2011. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J Exp Med 208:605–615. doi: 10.1084/jem.20101402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis CL, Field D, Metzgar D, Saiz R, Morin PA, Smith IL, Spector SA, Wills C. 1999. Numerous length polymorphisms at short tandem repeats in human cytomegalovirus. J Virol 73:6265–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walker A, Petheram SJ, Ballard L, Murph JR, Demmler GJ, Bale JF Jr. 2001. Characterization of human cytomegalovirus strains by analysis of short tandem repeat polymorphisms. J Clin Microbiol 39:2219–2226. doi: 10.1128/JCM.39.6.2219-2226.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Picone O, Ville Y, Costa JM, Rouzioux C, Leruez-Ville M. 2005. Human cytomegalovirus (HCMV) short tandem repeats analysis in congenital infection. J Clin Virol 32:254–256. doi: 10.1016/j.jcv.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 86.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog 7:e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vinces MD, Legendre M, Caldara M, Hagihara M, Verstrepen KJ. 2009. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324:1213–1216. doi: 10.1126/science.1170097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martin P, Makepeace K, Hill SA, Hood DW, Moxon ER. 2005. Microsatellite instability regulates transcription factor binding and gene expression. Proc Natl Acad Sci U S A 102:3800–3804. doi: 10.1073/pnas.0406805102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kawakami K, Salonga D, Park JM, Danenberg KD, Uetake H, Brabender J, Omura K, Watanabe G, Danenberg PV. 2001. Different lengths of a polymorphic repeat sequence in the thymidylate synthase gene affect translational efficiency but not its gene expression. Clin Cancer Res 7:4096–4101. [PubMed] [Google Scholar]
- 90.Tiscornia G, Mahadevan MS. 2000. Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol Cell 5:959–967. doi: 10.1016/S1097-2765(00)80261-0. [DOI] [PubMed] [Google Scholar]
- 91.King DG, Soller M, Kashi Y. 1997. Evolutionary tuning knobs. Endeavour 21:36–40. doi: 10.1016/S0160-9327(97)01005-3. [DOI] [Google Scholar]
- 92.Faux NG, Bottomley SP, Lesk AM, Irving JA, Morrison JR, de la Banda MG, Whisstock JC. 2005. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res 15:537–551. doi: 10.1101/gr.3096505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rupp B, Ruzsics Z, Buser C, Adler B, Walther P, Koszinowski UH. 2007. Random screening for dominant-negative mutants of the cytomegalovirus nuclear egress protein M50. J Virol 81:5508–5517. doi: 10.1128/JVI.02796-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. 2014. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88:249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, Coen DM. 2015. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J Virol 89:523–534. doi: 10.1128/JVI.02426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dargan DJ, Douglas E, Cunningham C, Jamieson F, Stanton RJ, Baluchova K, McSharry BP, Tomasec P, Emery VC, Percivalle E, Sarasini A, Gerna G, Wilkinson GW, Davison AJ. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–1546. doi: 10.1099/vir.0.018994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aoki T, Hirono I, Kurokawa K, Fukuda H, Nahary R, Eldar A, Davison AJ, Waltzek TB, Bercovier H, Hedrick RP. 2007. Genome sequences of three koi herpesvirus isolates representing the expanding distribution of an emerging disease threatening koi and common carp worldwide. J Virol 81:5058–5065. doi: 10.1128/JVI.00146-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sekulin K, Gorzer I, Heiss-Czedik D, Puchhammer-Stockl E. 2007. Analysis of the variability of CMV strains in the RL11D domain of the RL11 multigene family. Virus Genes 35:577–583. doi: 10.1007/s11262-007-0158-0. [DOI] [PubMed] [Google Scholar]
- 99.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davison AJ, Akter P, Cunningham C, Dolan A, Addison C, Dargan DJ, Hassan-Walker AF, Emery VC, Griffiths PD, Wilkinson GW. 2003. Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J Gen Virol 84:657–663. doi: 10.1099/vir.0.18856-0. [DOI] [PubMed] [Google Scholar]
- 102.Engel P, Perez-Carmona N, Alba MM, Robertson K, Ghazal P, Angulo A. 2011. Human cytomegalovirus UL7, a homologue of the SLAM-family receptor CD229, impairs cytokine production. Immunol Cell Biol 89:753–766. doi: 10.1038/icb.2011.55. [DOI] [PubMed] [Google Scholar]
- 103.Gabaev I, Steinbruck L, Pokoyski C, Pich A, Stanton RJ, Schwinzer R, Schulz TF, Jacobs R, Messerle M, Kay-Fedorov PC. 2011. The human cytomegalovirus UL11 protein interacts with the receptor tyrosine phosphatase CD45, resulting in functional paralysis of T cells. PLoS Pathog 7:e1002432. doi: 10.1371/journal.ppat.1002432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shikhagaie M, Merce-Maldonado E, Isern E, Muntasell A, Alba MM, Lopez-Botet M, Hengel H, Angulo A. 2012. The human cytomegalovirus-specific UL1 gene encodes a late-phase glycoprotein incorporated in the virion envelope. J Virol 86:4091–4101. doi: 10.1128/JVI.06291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Powers C, DeFilippis V, Malouli D, Fruh K. 2008. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 325:333–359. doi: 10.1007/978-3-540-77349-8_19. [DOI] [PubMed] [Google Scholar]
- 106.Dugan GE, Hewitt EW. 2008. Structural and functional dissection of the human cytomegalovirus immune evasion protein US6. J Virol 82:3271–3282. doi: 10.1128/JVI.01705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maidji E, Tugizov S, Abenes G, Jones T, Pereira L. 1998. A novel human cytomegalovirus glycoprotein, gpUS9, which promotes cell-to-cell spread in polarized epithelial cells, colocalizes with the cytoskeletal proteins E-cadherin and F-actin. J Virol 72:5717–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huber MT, Tomazin R, Wisner T, Boname J, Johnson DC. 2002. Human cytomegalovirus US7, US8, US9, and US10 are cytoplasmic glycoproteins, not found at cell surfaces, and US9 does not mediate cell-to-cell spread. J Virol 76:5748–5758. doi: 10.1128/JVI.76.11.5748-5758.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mandic L, Miller MS, Coulter C, Munshaw B, Hertel L. 2009. Human cytomegalovirus US9 protein contains an N-terminal signal sequence and a C-terminal mitochondrial localization domain, and does not alter cellular sensitivity to apoptosis. J Gen Virol 90:1172–1182. doi: 10.1099/vir.0.008466-0. [DOI] [PubMed] [Google Scholar]
- 110.Noriega V, Redmann V, Gardner T, Tortorella D. 2012. Diverse immune evasion strategies by human cytomegalovirus. Immunol Res 54:140–151. doi: 10.1007/s12026-012-8304-8. [DOI] [PubMed] [Google Scholar]
- 111.Fielding CA, Aicheler R, Stanton RJ, Wang EC, Han S, Seirafian S, Davies J, McSharry BP, Weekes MP, Antrobus PR, Prod'homme V, Blanchet FP, Sugrue D, Cuff S, Roberts D, Davison AJ, Lehner PJ, Wilkinson GW, Tomasec P. 2014. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog 10:e1004058. doi: 10.1371/journal.ppat.1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gurczynski SJ, Das S, Pellett PE. 2014. Deletion of the human cytomegalovirus US17 gene increases the ratio of genomes per infectious unit and alters regulation of immune and endoplasmic reticulum stress response genes at early and late times after infection. J Virol 88:2168–2182. doi: 10.1128/JVI.02704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bronzini M, Luganini A, Dell'Oste V, De Andrea M, Landolfo S, Gribaudo G. 2012. The US16 gene of human cytomegalovirus is required for efficient viral infection of endothelial and epithelial cells. J Virol 86:6875–6888. doi: 10.1128/JVI.06310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Davison AJ. 2011. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann N Y Acad Sci 1230:E37–E49. doi: 10.1111/j.1749-6632.2011.06358.x. [DOI] [PubMed] [Google Scholar]
- 115.Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150:831–841. doi: 10.1016/j.cell.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brennan G, Kitzman JO, Rothenburg S, Shendure J, Geballe AP. 2014. Adaptive gene amplification as an intermediate step in the expansion of virus host range. PLoS Pathog 10:e1004002. doi: 10.1371/journal.ppat.1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McSharry BP, Avdic S, Slobedman B. 2012. Human cytomegalovirus encoded homologs of cytokines, chemokines and their receptors: roles in immunomodulation. Viruses 4:2448–2470. doi: 10.3390/v4112448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamamoto-Tabata T, McDonagh S, Chang HT, Fisher S, Pereira L. 2004. Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol 78:2831–2840. doi: 10.1128/JVI.78.6.2831-2840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Poole E, Avdic S, Hodkinson J, Jackson S, Wills M, Slobedman B, Sinclair J. 2014. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J Virol 88:13947–13955. doi: 10.1128/JVI.02424-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Prod'homme V, Tomasec P, Cunningham C, Lemberg MK, Stanton RJ, McSharry BP, Wang EC, Cuff S, Martoglio B, Davison AJ, Braud VM, Wilkinson GW. 2012. Human cytomegalovirus UL40 signal peptide regulates cell surface expression of the NK cell ligands HLA-E and gpUL18. J Immunol 188:2794–2804. doi: 10.4049/jimmunol.1102068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chou SW. 1989. Reactivation and recombination of multiple cytomegalovirus strains from individual organ donors. J Infect Dis 160:11–15. doi: 10.1093/infdis/160.1.11. [DOI] [PubMed] [Google Scholar]
- 122.Haberland M, Meyer-Konig U, Hufert FT. 1999. Variation within the glycoprotein B gene of human cytomegalovirus is due to homologous recombination. J Gen Virol 80(Part 6):1495–1500. [DOI] [PubMed] [Google Scholar]
- 123.Rasmussen L, Geissler A, Winters M. 2003. Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J Infect Dis 187:809–819. doi: 10.1086/367900. [DOI] [PubMed] [Google Scholar]
- 124.Faure-Della Corte M, Samot J, Garrigue I, Magnin N, Reigadas S, Couzi L, Dromer C, Velly JF, Dechanet-Merville J, Fleury HJ, Lafon ME. 2010. Variability and recombination of clinical human cytomegalovirus strains from transplantation recipients. J Clin Virol 47:161–169. doi: 10.1016/j.jcv.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 125.Kolb AW, Ane C, Brandt CR. 2013. Using HSV-1 genome phylogenetics to track past human migrations. PLoS One 8:e76267. doi: 10.1371/journal.pone.0076267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Quinlivan M, Hawrami K, Barrett-Muir W, Aaby P, Arvin A, Chow VT, John TJ, Matondo P, Peiris M, Poulsen A, Siqueira M, Takahashi M, Talukder Y, Yamanishi K, Leedham-Green M, Scott FT, Thomas SL, Breuer J. 2002. The molecular epidemiology of varicella-zoster virus: evidence for geographic segregation. J Infect Dis 186:888–894. doi: 10.1086/344228. [DOI] [PubMed] [Google Scholar]
- 127.Santpere G, Darre F, Blanco S, Alcami A, Villoslada P, Mar Alba M, Navarro A. 2014. Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1,000 Genomes Project. Genome Biol Evol 6:846–860. doi: 10.1093/gbe/evu054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jung GS, Kim YY, Kim JI, Ji GY, Jeon JS, Yoon HW, Lee GC, Ahn JH, Lee KM, Lee CH. 2011. Full genome sequencing and analysis of human cytomegalovirus strain JHC isolated from a Korean patient. Virus Res 156:113–120. doi: 10.1016/j.virusres.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 129.Sauerbrei A, Wutzler P. 2007. Different genotype pattern of varicella-zoster virus obtained from patients with varicella and zoster in Germany. J Med Virol 79:1025–1031. doi: 10.1002/jmv.20879. [DOI] [PubMed] [Google Scholar]
- 130.Norberg P, Tyler S, Severini A, Whitley R, Liljeqvist JA, Bergstrom T. 2011. A genome-wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus. PLoS One 6:e22527. doi: 10.1371/journal.pone.0022527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lazzarotto T, Varani S, Gabrielli L, Pignatelli S, Landini MP. 2001. The tegument protein ppUL25 of human cytomegalovirus (CMV) is a major target antigen for the anti-CMV antibody response. J Gen Virol 82:335–338. [DOI] [PubMed] [Google Scholar]
- 132.Penfold ME, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ. 1999. Cytomegalovirus encodes a potent alpha chemokine. Proc Natl Acad Sci U S A 96:9839–9844. doi: 10.1073/pnas.96.17.9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ashiru O, Bennett NJ, Boyle LH, Thomas M, Trowsdale J, Wills MR. 2009. NKG2D ligand MICA is retained in the cis-Golgi apparatus by human cytomegalovirus protein UL142. J Virol 83:12345–12354. doi: 10.1128/JVI.01175-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jelcic I, Reichel J, Schlude C, Treutler E, Sinzger C, Steinle A. 2011. The polymorphic HCMV glycoprotein UL20 is targeted for lysosomal degradation by multiple cytoplasmic dileucine motifs. Traffic 12:1444–1456. doi: 10.1111/j.1600-0854.2011.01236.x. [DOI] [PubMed] [Google Scholar]
- 136.Yang Z, Bjorkman PJ. 2008. Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci U S A 105:10095–10100. doi: 10.1073/pnas.0804551105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Qi Y, Mao ZQ, Ruan Q, He R, Ma YP, Sun ZR, Ji YH, Huang Y. 2006. Human cytomegalovirus (HCMV) UL139 open reading frame: sequence variants are clustered into three major genotypes. J Med Virol 78:517–522. doi: 10.1002/jmv.20571. [DOI] [PubMed] [Google Scholar]
- 138.Rasmussen L, Geissler A, Cowan C, Chase A, Winters M. 2002. The genes encoding the gCIII complex of human cytomegalovirus exist in highly diverse combinations in clinical isolates. J Virol 76:10841–10848. doi: 10.1128/JVI.76.21.10841-10848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hitomi S, Kozuka-Hata H, Chen Z, Sugano S, Yamaguchi N, Watanabe S. 1997. Human cytomegalovirus open reading frame UL11 encodes a highly polymorphic protein expressed on the infected cell surface. Arch Virol 142:1407–1427. doi: 10.1007/s007050050169. [DOI] [PubMed] [Google Scholar]
- 140.Pignatelli S, Dal Monte P, Rossini G, Chou S, Gojobori T, Hanada K, Guo JJ, Rawlinson W, Britt W, Mach M, Landini MP. 2003. Human cytomegalovirus glycoprotein N (gpUL73-gN) genomic variants: identification of a novel subgroup, geographical distribution and evidence of positive selective pressure. J Gen Virol 84:647–655. doi: 10.1099/vir.0.18704-0. [DOI] [PubMed] [Google Scholar]
- 141.Lurain NS, Kapell KS, Huang DD, Short JA, Paintsil J, Winkfield E, Benedict CA, Ware CF, Bremer JW. 1999. Human cytomegalovirus UL144 open reading frame: sequence hypervariability in low-passage clinical isolates. J Virol 73:10040–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bar M, Shannon-Lowe C, Geballe AP. 2001. Differentiation of human cytomegalovirus genotypes in immunocompromised patients on the basis of UL4 gene polymorphisms. J Infect Dis 183:218–225. doi: 10.1086/317939. [DOI] [PubMed] [Google Scholar]
- 143.Hayajneh WA, Contopoulos-Ioannidis DG, Lesperance MM, Venegas AM, Colberg-Poley AM. 2001. The carboxyl terminus of the human cytomegalovirus UL37 immediate-early glycoprotein is conserved in primary strains and is important for transactivation. J Gen Virol 82:1569–1579. [DOI] [PubMed] [Google Scholar]
- 144.Ji YH, Rong Sun Z, Ruan Q, Guo JJ, He R, Qi Y, Ma YP, Mao ZQ, Huang YJ. 2006. Polymorphisms of human cytomegalovirus UL148A, UL148B, UL148C, UL148D genes in clinical strains. J Clin Virol 37:252–257. doi: 10.1016/j.jcv.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 145.Ji YH, Sun ZR, Ruan Q, He R, Qi Y, Ma YP, Huang YJ. 2006. High variability of human cytomegalovirus UL150 open reading frame in low-passaged clinical isolates. Chin Med Sci J 21:69–74. [PubMed] [Google Scholar]
- 146.Deckers M, Hofmann J, Kreuzer KA, Reinhard H, Edubio A, Hengel H, Voigt S, Ehlers B. 2009. High genotypic diversity and a novel variant of human cytomegalovirus revealed by combined UL33/UL55 genotyping with broad-range PCR. Virol J 6:210. doi: 10.1186/1743-422X-6-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.