

ABSTRACT
As a herpesvirus, Epstein-Barr virus (EBV) establishes a latent infection that can periodically undergo reactivation, resulting in lytic replication and the production of new infectious virus. Latent membrane protein-1 (LMP1), the principal viral oncoprotein, is a latency-associated protein implicated in regulating viral reactivation and the maintenance of latency. We recently found that LMP1 hijacks the SUMO-conjugating enzyme Ubc9 via its C-terminal activating region-3 (CTAR3) and induces the sumoylation of cellular proteins. Because protein sumoylation can promote transcriptional repression, we hypothesized that LMP1-induced protein sumoylation induces the repression of EBV lytic promoters and helps maintain the viral genome in its latent state. We now show that with inhibition of LMP1-induced protein sumoylation, the latent state becomes less stable or leakier in EBV-transformed lymphoblastoid cell lines. The cells are also more sensitive to viral reactivation induced by irradiation, which results in the increased production and release of infectious virus, as well as increased susceptibility to ganciclovir treatment. We have identified a target of LMP1-mediated sumoylation that contributes to the maintenance of latency in this context: KRAB-associated protein-1 (KAP1). LMP1 CTAR3-mediated sumoylation regulates the function of KAP1. KAP1 also binds to EBV OriLyt and immediate early promoters in a CTAR3-dependent manner, and inhibition of sumoylation processes abrogates the binding of KAP1 to these promoters. These data provide an additional line of evidence that supports our findings that CTAR3 is a distinct functioning regulatory region of LMP1 and confirm that LMP1-induced sumoylation may help stabilize the maintenance of EBV latency.
IMPORTANCE Epstein-Barr virus (EBV) latent membrane protein-1 (LMP1) plays an important role in the maintenance of viral latency. Previously, we documented that LMP1 targets cellular proteins to be modified by a ubiquitin-like protein (SUMO). We have now identified a function for this LMP1-induced modification of cellular proteins in the maintenance of EBV latency. Because latently infected cells have to undergo viral reactivation in order to be vulnerable to antiviral drugs, these findings identify a new way to increase the rate of EBV reactivation, which increases cell susceptibility to antiviral therapies.
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous human gammaherpesvirus that causes persistent infection, generally asymptomatic, in over 90% of the world's population. Initially, the virus lytically infects oropharyngeal epithelial cells, producing virions containing linear genomes. The virus also quickly infects B lymphocytes, in which latent infection is established and persists in the form of episomes and subsets of viral latency genes are expressed. Periodically, latent virus can be reactivated and infectious virus is released in saliva (1). The processes that regulate the switch between latent and lytic infection have been studied for many years. One viral gene implicated in effecting this switch is latent membrane protein-1 (LMP1) (2–4), the principal oncoprotein of EBV.
LMP1, which is expressed in type II latency (Hodgkin's lymphoma and nasopharyngeal carcinoma [NPC]) and in type III latency (B-cell lymphomas in immunocompromised persons) (5–7), is an integral membrane signaling protein that mimics the tumor necrosis factor (TNF) receptor family members (such as CD40), with the exception that its activation is ligand independent and it is constitutively active (8). LMP1 consists of a short cytoplasmic N-terminal domain, six transmembrane domains, and a 200-amino-acid cytoplasmic C-terminal domain. The carboxyl terminus contains three C-terminal activating regions (CTARs; CTAR1 to CTAR3) (8, 9); most LMP1-mediated signal transduction events are mediated via the extensively characterized CTAR1 and CTAR2. Functions for CTAR3 are less well defined (10–13); however, we recently documented a novel function for CTAR3 in the dysregulation of sumoylation processes (14).
Protein sumoylation is a posttranslational modification characterized by the covalent, yet reversible, attachment of a small ubiquitin-like modifier (SUMO), a 12-kDa protein that shares 20% homology with ubiquitin (15), to a lysine residue of a target protein. It is a dynamic and reversible process that can regulate protein function by altering a protein's intracellular location, turnover, ability to interact with other proteins, or ability to interact with DNA (15–17). Protein sumoylation is involved in central cellular processes, and multiple oncogene and tumor suppressor proteins undergo sumoylation, altering their function (18–23). Furthermore, increases in protein sumoylation are a feature of a variety of types of cancer (24–27), and because cellular sumoylation processes are thought to be critical in regulating oncogenesis, elements of the sumoylation process have been proposed to be potential new targets for cancer therapies (26, 28).
Sumoylation processes have a role in the EBV life cycle (29–37). We documented that LMP1 CTAR3 physically interacts with the enzymatically active SUMO-conjugating enzyme Ubc9 during latent EBV infections (14), increases the sumoylation of cellular proteins, and contributes to basic features of the oncogenic phenotype produced by LMP1 (14). Our preliminary data have identified two additional mechanisms by which LMP1 regulates sumoylation processes: first, through the induction of sumo and, second, through the inhibition of desumoylating enzymes (unpublished data). Together, these findings led us to investigate aspects of the function of LMP1 in mediating the regulation of sumoylation processes during latent EBV infection.
One documented function of LMP1 is to help maintain viral latency (2–4). The conditional expression of LMP1 has been shown to inhibit EBV reactivation (2). LMP1-induced signaling also helps modulate the antiviral environment within latently infected cells (38–45), and our recent studies highlight a role for LMP1 CTAR3-induced sumoylation in modulating this antiviral environment (46). Sumoylation processes aid in transcriptional repression (47–49), and we have reported that LMP1 CTAR3-mediated sumoylation results in transcriptional repression (46). Therefore, we hypothesize that LMP1 induces, via CTAR3, the sumoylation of cellular proteins, which collectively promote transcriptional repression and help sustain the EBV latent state. Accordingly, inhibition of protein sumoylation might be expected to increase viral reactivation and result in increased cell susceptibility to drugs and other agents that inhibit viral replication.
We show here that LMP1-induced sumoylation contributes to the LMP1-mediated maintenance of EBV latency. Inhibition of LMP1-induced sumoylation results in the disruption of latency and viral reactivation as well as increased susceptibility to ganciclovir (GCV) treatment. In addition, we have identified a target of LMP1-induced sumoylation (KRAB-associated protein-1 [KAP1]) that contributes to the maintenance of EBV latency. KAP1 is a member of the tripartite motif family, is involved in transcriptional repression, and is thought to have antiviral activities (50–54). The endogenous sumoylation of KAP1, which occurred in an LMP1 CTAR3-dependent manner, was detected in EBV-transformed lymphoblastoid cell lines (LCLs). Further, we found that LMP1 CTAR3 and inhibition of cellular sumoylation processes affect KAP1 function in the context of chromatin association, a known requirement for KAP1-mediated transcriptional repression (53, 54). We also show that KAP1 binds to EBV OriLyt and immediate early promoters and inhibition of LMP1 CTAR3-induced protein sumoylation abrogates the binding of KAP1 to these promoters. These combined effects point to roles for LMP1-induced sumoylation, acting through CTAR3, in the maintenance of EBV latency.
MATERIALS AND METHODS
Cells.
Human embryonic kidney (HEK) 293 cells (293 cells) were maintained in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS). EBV-negative BL41 cells, BL41 cells infected with wild-type (WT) EBV, and BL41 cells infected with an EBV mutant (P3HR1 [55–57]), as well as Raji cells, were maintained in RPMI plus 10% FBS. EBV-expressing 293 cells and 293 cells expressing EBV with a CTAR3 deletion (dCTAR3) were a gift from Wolfgang Hammerschmidt (Munich, Germany) and maintained in RPMI with 10% FBS and hygromycin B (58). Paired EBV WT-transformed and EBV dCTAR3-transformed LCLs were made by the Lineberger Comprehensive Cancer Center Tissue Culture Facility and cultured in RPMI plus 10% FBS.
Plasmids and siRNA.
FLAG-LMP1 and FLAG-LMP1 dCTAR3 have been described previously (11, 59). Green fluorescent protein (GFP)-tagged KAP1 (GFP-KAP1) and hemagglutinin (HA)-tagged Ubc9 C93S (HA-Ubc9 C93S) were purchased from Addgene. Small interfering (siRNA) targeting Ubc9 or a scrambled sequence was purchased from Santa Cruz. The BZLF1-expressing plasmid was a gift from Wolfgang Hammerschmidt (58).
Immunoprecipitation.
293 cells were grown in 100-mm dishes and transfected with 8 μg of DNA using polyethyleneimine. At 48 h posttransfection, cells were lysed in boiling denaturing cell lysis buffer (20 mM Tris, pH 7.5, 70 mM β-mercaptoethanol [β-ME], 1× EDTA-free cOmplete protease inhibitor, 20 μM N-ethylmaleimide [NEM]). The lysates were boiled for 10 min, and 4 volumes of cell lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1× EDTA-free cOmplete protease inhibitor, 20 μM NEM) were added to the tubes. The lysates were centrifuged at 4,000 rpm for 5 min, and the supernatants were collected and incubated with 1 μg of antibody for 1 h at 4°C. Magnetic protein G beads (Life Technologies) were added to the samples, which were then incubated overnight at 4°C. The beads were washed four times with cell lysis buffer and resuspended in 4× sodium dodecyl sulfate (SDS; Sigma) loading buffer.
Western blot analysis.
Samples were denatured in 4× SDS loading buffer and boiled for 10 min. The samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (GE). The membranes were blocked with 5% milk in Tris-buffered saline–Tween 20 (TBST) and incubated overnight at 4°C with primary antibodies. The membranes were washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The membranes were washed again, and bands were visualized with enhanced chemiluminescence reagent from GE.
Viral induction by ZTA.
EBV WT- and EBV dCTAR3-expressing 293 cells were grown and transfected with ZTA expression plasmids. Cells and supernatants were collected at 48 h after transfection. Total DNA was isolated from the cells and 100 μl supernatants with a Qiagen DNeasy blood and tissue kit. The remaining supernatants were added to Raji cells. Forty-eight hours later the percentages of GFP-positive Raji cells were determined by flow cytometry (60).
Viral induction by gamma irradiation.
EBV WT- and EBV dCTAR3-expressing 293 cells and paired EBV WT- and EBV dCTAR3-transformed LCLs were exposed to various doses of irradiation (0 to 10 Gy) (61, 62). Cells and supernatants were collected at 24 h after transfection. Total DNA was isolated from the cells and 100 μl supernatants with a Qiagen DNeasy blood and tissue kit. The remaining fluids were added to infect Raji cells. Forty-eight hours later the percentages of GFP-positive Raji cells were determined (60).
Real-time PCR.
Total RNA was harvested with the use of a Qiagen RNeasy Plus kit and reverse transcribed with an ABI Prism reverse transcription kit. DNA was harvested using a Qiagen DNeasy blood and tissue kit. DNA and cDNA were analyzed by real-time PCR with an ABI 7900HT real-time PCR system. The primers (sequences) used were EBV W-1 (5′-GCAGCCGCCCAGTCTCT-3′), EBV W-2 (5′-ACAGACAGTGCACAGGAGCCT-3′), GAPDH-1 (5′-TCATCAGCAATGCCTCCT-3′), GAPDH-2 (5′-AGGGGCCATCCACAGTCTTC-3′), OriLyt (flank left)-F (5′-GCGCAACAGTGCCACCAACC-3′), OriLyt (flank left)-R (5′-CAGGACCTGGCGGTAGTGCAG-3′), pBRLF1-F (5′-GGCTGACATGGATTACTGGTC-3′), pBRLF1-R (5′-TGATGCAGAGTCGCCTAATG-3′), pBZLF1-F (5′-GAGCCACAGGCATTGCTAA-3′), pBZLF1-R (5′-ACCAGCCTCCTCTGTGATGT-3′), pBALF5-F (5′-GATCGTGATAGCGTCTTCTGC-3′), pBALF5-R (5′-GCAACATGCCTCTGGTGA-3′), pBNRF1-F (5′-TGTACACCAACAGGTGTTGCCTTG-3′), pBNRF1-R (5′-ACCCCAAAGAGGGCAAAGCCTAC-3′), pBCRF1-F (5′-GGGAGGTACATGTCCCCCAGCATT-3′), pBNRF1-R (5′-CTGTGGACTGCAACACAACATTGCC-3′), Qp-F (5′-CTGTCACCACCTCCCTGATA-3′), Qp-R (5′-GAACACTCCCTCAGTGGTCA-3′), EFCAB7-F (5′-GCACCTCTTTCCTGCTGGTG-3′), EFCAB7-R (5′-AGAGCCCTTCTCCAAACGAT-3′), GABPB2-F (5′-GTGACGCACTTTTCCACCTC-3′), GABPB2-R (5′-GATTTCTGGCGGAGGAATCT-3′), ZNF-F (5′-GGCGAACTACAGTCGTGACA-3′), and ZNF-R (5′-CGCTATGCTGTTCACCTTCA-3′) (63–65). Bio-Rad Universal SYBR green Supermix (Bio-Rad) was used. Samples and experiments were run in triplicate.
Collecting chromatin fractions.
LCLs were grown and treated, and whole-cell lysates (WCLs) and chromatin fractions were collected. As described previously (66), cells were washed and resuspended in 200 μl of CSK buffer {10 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 0.1 mM ATP, 0.1% Triton X-100, cOmplete protease inhibitor}, and pellets were collected following centrifugation at 4,000 rpm for 5 min. The pellets were washed twice with 1 ml CSK buffer, and insoluble pellets were resuspended in 50 μl of 4× SDS sample buffer and boiled for 10 min. Western blot and slot blot assays were performed to examine total KAP1 expression in WCLs and chromatin fractions.
ChIP.
Chromatin immunoprecipitation (ChIP) assays were performed with an Active Motif ChIP-IT enzymatic kit (Active Motif) per the manufacturer's instruction and as previously described (67). LCLs were fixed with 37% formaldehyde (final concentration. 1%) for 10 min at 37°C; the reaction was stopped with cold 0.125 M glycine solution for 5 min at room temperature. The cells were then washed twice with phosphate-buffered saline and collected in 0.5 ml digestion buffer with 1× protease inhibitors. Chromatin was enzymatically sheared for 10 min at 37°C to obtain fragments with an average size of 200 to 1,000 bp. Sheared chromatin was incubated overnight at 4°C with protein G magnetic beads and KAP1 (Abcam) or control (Santa Cruz) antibodies. Immunoprecipitations were performed per the manufacturer's instructions; cross-linking was reversed by incubating immunoprecipitated complexes with 5 M NaCl and RNase A (final concentration, 25 μg/ml) for 2 h at 65°C, followed by proteinase K (final concentration, 50 mg/ml) treatment for 1 h at 37°C. Real-time PCR was performed to investigate KAP1 binding to lytic promoters.
Ganciclovir treatment.
EBV WT- and EBV dCTAR3-expressing 293 cells and paired EBV WT- and EBV dCTAR3-transformed LCLs were treated as indicated above, induced (5 Gy irradiation), and treated with ganciclovir (40 μM) (68). At 1 week after treatment, trypan blue exclusion assays were performed (69), and the fold change in cell death was determined.
Antibodies.
Anti-FLAG (M2) and antiactin (AG-15) antibodies were purchased from Sigma. Anti-SUMO-1 (clone D-11), anti-HA (clone F-7), anti-myc (clone 9E10), anti-histone H1 (clone AE-4), anti-EBV EA-R p85 (clone 6G7), and anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase; clone FL-335) antibodies were purchased from Santa Cruz. Anti-LMP1 antibodies (CS 1 to CS 4) were from Dako, and anti-KAP1 antibodies (clones ab22553 and ab10484) were from Abcam.
RESULTS
LMP1 CTAR3 is not essential for viral replication.
We have documented that LMP1 induces the sumoylation of cellular proteins by interacting with Ubc9 via CTAR3 (14), which led us to investigate possible functions of sumoylation during EBV latency. To investigate whether LMP1-induced sumoylation affects lytic EBV replication, 293 cells stably expressing WT EBV or EBV dCTAR3 (from Wolfgang Hammerschmidt) were transfected with EBV ZTA-expressing plasmids. The amounts of viral DNA (relative to genomic gapdh levels) were determined at 48 h posttransfection, and the fold changes in the amounts of viral DNA (relative to those in untreated EBV WT- and EBV dCTAR3-expressing 293 cells) were quantified (Fig. 1A). Supernatants were used to assay virus infectivity (Fig. 1B) (60). No differences in the production or release of infectious virus from either cell line were detected, which confirmed that LMP1 CTAR3 is not required for viral replication (13, 58). However, noninduced EBV dCTAR3-expressing 293 cells consistently contained significantly (P < 0.001) increased levels of viral DNA compared with untreated EBV WT-expressing 293 cells (Fig. 1C), which correlated with the increased release of new infectious virus (Fig. 1D). These data suggest that while LMP1 CTAR3 is not required for efficient viral replication, it may affect the LMP1-induced maintenance of latency.
FIG 1.

Deletion of CTAR3 did not affect viral replication but increased background levels of lytic replication. EBV WT- and EBV dCTAR3-expressing 293 cells were induced by transfection with ZTA expression plasmids or noninduced by transfection with the vector control, and cells and supernatants were collected at 48 h posttransfection. (A) Virus was harvested, and relative viral loads were quantitated by real-time PCR. The fold change in the amount of viral DNA (relative to that for noninduced EBV WT- and EBV dCTAR3-expressing 293 cells) was determined. (B) Raji cells were exposed to the remaining supernatants. The percentages of GFP-positive cells were determined by flow cytometry after 48 h. (C) For noninduced cells, the fold change in the amount of viral DNA (relative to that for noninduced EBV WT-expressing 293 cells) was determined. (D) Raji cells were exposed to the remaining supernatants. The percentages of GFP-positive cells were determined as described above. Results are shown as means ± standard deviations from experiments performed in triplicate.
LMP1 CTAR3 contributes to the maintenance of EBV latency.
To investigate the role of CTAR3 in the maintenance of latency, we changed the method of induction so that lower levels of viral reactivation would occur to better mimic endogenous viral reactivation. The induction of double-stranded DNA damage by gamma irradiation can promote EBV lytic reactivation (70–73) and is a common side effect of therapies for EBV-positive lymphomas. Exposure of WT EBV- and EBV dCTAR3-expressing 293 cells to graduated doses of gamma irradiation significantly (P < 0.001) increased the release of infectious virus in cells expressing LMP1 dCTAR3 compared with the amount released in WT EBV-expressing 293 cells (Fig. 2A). Quantitation of the relative levels of genomic viral DNA in induced EBV WT- and EBV dCTAR3-expressing 293 cells revealed that significantly (P < 0.001) more viral DNA was produced in cells containing the virus with the CTAR3 deletion (Fig. 2B). These results were confirmed in similar experiments performed in multiple paired lymphoblastoid cell lines established by immortalization of B cells with EBV WT or EBV dCTAR3 (Fig. 2C). Together, these data suggest that LMP1 dCTAR3 helps maintain the virus in its latent state.
FIG 2.
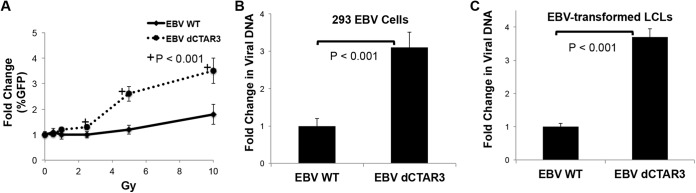
Deletion of CTAR3 increases cell susceptibility to viral reactivation by irradiation. (A) EBV WT- and EBV dCTAR3-expressing 293 cells were grown and exposed to various doses of irradiation, and supernatants were collected at 24 h postirradiation. The supernatants were added to Raji cells, and the percentage of GFP-positive cells was determined after 48 h. (B and C) EBV WT- and EBV dCTAR3-expressing 293 cells (293 EBV cells) (B) and paired EBV WT- and EBV dCTAR3-transformed LCLs (C) were exposed to 5 Gy of irradiation, and the fold changes in viral loads (relative to the viral load for noninduced cells) were determined. All results are shown as means ± standard deviations from experiments performed in triplicate.
LMP1-induced sumoylation helps maintain viral latency.
Because we previously identified a function for LMP1 CTAR3 in the induction of the sumoylation of cellular proteins (14), we examined whether protein sumoylation was a specific effector for regulating viral reactivation in our system. Recently, anacardic acid and ginkgolic acid were identified to block the formation of the SUMO-1/SUMO-activating enzyme E1 intermediate, which resulted in the decreased sumoylation of cellular proteins (74). Ubc9, the SUMO-conjugating enzyme, is another target for inhibiting sumoylation processes, and two mechanisms by which Ubc9 can be targeted is the exogenous expression of Ubc9 C93S, which renders Ubc9 enzymatically inactive and suppresses the function of endogenous Ubc9 (28, 75, 76), and the use of siRNA to inhibit Ubc9 expression (77–79). The effects of anacardic acid, ginkgolic acid, Ubc9 C93S, and Ubc9-specific siRNA (Ubc9 siRNA) on viral reactivation were tested.
The SUMO inhibitors, overexpression of Ubc9 C93S, and knockdown of Ubc9 (with Ubc9 siRNA) all blocked endogenous protein sumoylation in EBV-expressing 293 cells (Fig. 3A). Treatment of EBV WT-expressing 293 cells and EBV WT-transformed LCLs with SUMO inhibitors (ginkgolic acid and anacardic acid) revealed a significant (P < 0.001) increase in viral DNA levels when cells were treated with the SUMO inhibitor compared with those seen in control cells, which were treated with the vehicle control (water) and transfected with scrambled siRNA and an empty expression vector (Fig. 3B and C). No differences were observed when ginkgolic acid treatment was compared to anacardic acid treatment (data not shown), suggesting that these drugs have similar effects on viral reactivation. Transfection of cells with Ubc9 C93S and knockdown of Ubc9 with siRNA yielded results similar to those obtained by deletion of LMP1 CTAR3 and drug treatment (Fig. 3B), further supporting the hypothesis that LMP1-induced sumoylation contributes to the maintenance of EBV latency. Furthermore, noninduced cells that were treated with the SUMO inhibitors, transfected with Ubc9 C93S, and transfected with Ubc9 siRNA exhibited increased viral reactivation compared to mock-treated, noninduced cells (Fig. 3D). Together, these findings suggest that inhibiting protein sumoylation results in the impaired ability of the virus to be maintained in a latent state and that EBV-expressing cells are more susceptible to reactivation of lytic replication.
FIG 3.

Inhibition of LMP1-induced sumoylation increases lytic viral replication following irradiation. (A) EBV-expressing 293 cells were transfected with Ubc9 C93S, Ubc9 s9RNA, or control expression vectors. At 24 h posttransfection, cells were treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid). Whole-cell lysates were collected at 48 h posttransfection. Western blot analyses were performed for SUMO-1, Ubc9, and GAPDH (loading control). (B) EBV WT-expressing 293 cells were treated with a vehicle control and transfected with scrambled siRNA and an empty expression vector, treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid), transfected with Ubc9 C93S, or transfected with Ubc9 siRNA. (C and D) EBV WT-transformed LCLs were treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid) or the vehicle control and induced (C) or noninduced (D). Fold changes in viral loads were determined 24 h after irradiation. All results are shown as means ± standard deviations from experiments performed in triplicate.
Inhibition of LMP1-induced sumoylation makes cells more susceptible to antiviral treatment.
One proposed method for treating EBV-associated malignancies is the induction of EBV lytic reactivation in latently infected tumor cells (80–84), which would result in cell susceptibility to cytotoxic antiviral drugs (85). To investigate whether inhibition of LMP1-induced sumoylation results in increased cellular death in response to antiviral therapy, irradiated and mock-irradiated EBV WT-transformed LCLs, EBV dCTAR3-transformed LCLs, and EBV WT-transformed LCLs treated with SUMO inhibitors were treated with ganciclovir (GCV) or a vehicle control (water), and cell death was assayed by trypan blue exclusion (Fig. 4A). The results showed that GCV-induced LCL death was significantly (P < 0.001) increased in induced cells compared with noninduced cells, confirming that irradiation can induce viral reactivation. GCV-treated EBV dCTAR3-transformed LCLs and EBV WT-transformed, SUMO inhibitor-treated LCLs exhibited increased cell death compared with GCV-treated EBV WT LCLs, suggesting that viral reactivation increased in the absence of CTAR3 and inhibition of sumoylation processes. Furthermore, the death of LCLs transformed with EBV dCTAR3 or treated with a SUMO inhibitor was significantly (P < 0.001) greater than that of LCLs transformed with the EBV WT after mock irradiation, which verified that deletion of CTAR3 and inhibition of sumoylation processes result in a leakier hold on latency.
FIG 4.
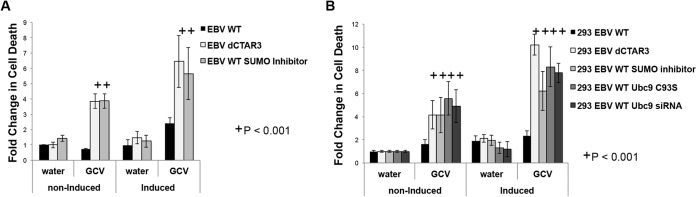
Inhibition of sumoylation increases cell susceptibility to cytotoxic antiviral drugs following irradiation. (A) EBV WT-transformed LCLs, EBV dCTAR3-transformed LCLs, and SUMO inhibitor (25 μM anacardic acid and 25 μM ginkgolic acid)-treated EBV WT-transformed LCLs were irradiated (5 Gy; induced) or noninduced. (B) EBV WT-expressing 293 cells, EBV dCTAR3-expressing 293 cells, and EBV WT-expressing 293 cells treated with water and transfected with scrambled siRNA and an empty expression vector, treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid), transfected with a Ubc9 C93S expression plasmid, or transfected with Ubc9 siRNA were induced or noninduced. At 24 h postirradiation, cells were treated with ganciclovir (10 mg/ml) or vehicle control (water). After 1 week, trypan blue exclusion assays were performed and fold changes in cell death were calculated. Results are shown as means ± standard deviations from experiments performed in triplicate.
To confirm the role of sumoylation processes in increased susceptibility to GCV treatment, similar experiments were performed in EBV WT-expressing 293 cells, EBV dCTAR3-expressing 293 cells, EBV WT-expressing 293 cells treated with a SUMO inhibitor, EBV WT-expressing 293 cells with exogenous expression of Ubc9 C93S, and EBV WT-expressing 293 cells transfected with Ubc9 siRNA (Fig. 4B). As a control, EBV WT-expressing 293 cells were treated with water and transfected with scrambled siRNA and an empty expression vector. Inhibition of sumoylation processes by deletion of CTAR3, treatment with ginkgolic acid and anacardic acid, overexpression of Ubc9 C93S, and knockdown of Ubc9 resulted in significantly (P < 0.001) increased susceptibility to GCV treatment. Together, these data imply that inhibition of LMP1 CTAR3-mediated protein sumoylation increases viral reactivation, which in turn results in increased susceptibility to antiviral drugs.
KAP1 is a target of LMP1 CTAR3-induced sumoylation.
To identify a cellular target of LMP1-induced sumoylation that might be implicated in the progression of lytic replication, we first focused on a members of the tripartite motif family (KAP1). KAP1 can function as a transcriptional repressor (50, 86), and the modification of KAP1 by SUMO at lysine 554, 575, 676, 750, 779, and/or 804 aids its repressive functions (54, 87–92). Sumoylated KAP1 has been reported to bind to and repress Kaposi's sarcoma-associated herpesvirus (KSHV) lytic promoters (93); however, its function during the EBV life cycle remains unexamined. These findings led us to propose that LMP1-mediated sumoylation of KAP1 helps in the maintenance of EBV latency by inhibiting lytic replication.
The endogenous sumoylation of KAP1 during latent EBV infection was examined using the paired BL41 cell lines that were either EBV negative, latently infected with EBV WT, or latently infected with the EBV P3HR1 mutant (Fig. 5A) (39, 56, 57). The results showed that increased levels of sumoylated KAP1 were detected in EBV WT-expressing BL41 cells compared with EBV-negative BL41 cells, which suggested that the sumoylation of KAP1 is increased during EBV latency. In addition, cells latently infected with the P3HR1 mutant, which lacks EBNA2, resulting in undetectable levels of LMP1 expression (39, 56, 57), showed lower levels of sumoylated KAP1 than EBV WT-expressing BL41 cells. Densitometric analysis of repeat experiments confirmed these findings (Fig. 5B), which led us to propose that the LMP1-mediated regulation of sumoylation processes significantly contributes to the sumoylation of KAP1 during EBV latency.
FIG 5.

LMP1 expression correlates with endogenous KAP1 sumoylation in a CTAR3-dependent manner. (A) Denaturing immunoprecipitations (IP) were performed on BL41 EBV-negative, BL41 EBV-positive, and BL41 P3HR1 mutant-infected cells with SUMO-1-specific antibodies or IgG control antibodies. Western blot (WB) analyses were used to detect KAP1 covalently modified by SUMO-1. (B) Densitometric analysis of repeat experiments was performed. Results are shown as means ± standard deviations from experiments performed in triplicate. pos, positive; neg, negative. (C) Western blot analyses were used to detect KAP1 covalently modified by SUMO-1 in paired EBV WT- and EBV dCTAR3-transformed LCLs with SUMO-1-specific antibodies or IgG control antibodies. (D) Densitometric analysis of repeat experiments was performed. Results are shown as means ± standard deviations from experiments performed in triplicate. (E) 293 cells were transfected with GFP-KAP1 and either FLAG-LMP1, FLAG-LMP1 dCTAR3, or vector control expression constructs. At 48 h posttransfection, whole-cell lysates were collected and immunoblotting assays were performed to detect LMP1 (FLAG) and KAP1 (GFP) levels. GAPDH was used as a loading control.
We previously documented a function for LMP1 CTAR3 in the induction of the sumoylation of cellular proteins (14), and results from paired EBV WT- and EBV dCTAR3-transformed LCLs showed that LMP1 CTAR3 is required for the sumoylation of KAP1 during viral latency (Fig. 5C). Sumoylated KAP1 was detected in all LCLs (Fig. 5A); however, the levels of sumoylated KAP1 were significantly (P < 0.001) increased in all EBV WT-expressing cells compared with those in EBV dCTAR3-expressing cells (Fig. 5D), which suggests that the sumoylation of KAP1 occurred in a CTAR3-dependent manner.
The importance of LMP1 and LMP1 CTAR3 in the observed increased sumoylation of KAP1 was confirmed by overexpression experiments in 293 cells (Fig. 5E). Taken together, these findings document that KAP1 is sumoylated during latent EBV infection and EBV-induced sumoylation of KAP1 is highly dependent on LMP1, specifically, CTAR3. Because we proposed that the LMP1 CTAR3-mediated sumoylation of KAP1 contributes to the maintenance of EBV latency, next we investigated the role of LMP1 CTAR3-induced sumoylation on a known function of KAP1.
The association of KAP1 with chromatin increases with LMP1-induced sumoylation.
While KAP1 lacks a DNA-binding domain, it associates with KRAB-containing zinc finger proteins (KRAB-ZFPs), which in turn bind gene promoter regions (53, 86, 87). The sumoylation of KAP1 is required for its repressive function at these promoter regions (87, 90–92), so the effect of LMP1-induced sumoylation on the ability of KAP1 to associate with cellular chromatin was examined. The results showed that LMP1-expressing cells contained increased detectable levels of sumoylated KAP1 (Fig. 6A, WCL), which confirms that sumoylation of KAP1 increases in the presence of LMP1. Following separation of the chromatin and supernatant fractions, sumoylated KAP1 was detected only in the chromatin fractions of LMP1-expressing cells (Fig. 6A, chrom), while control-expressing cells contained almost undetectable KAP1 levels. In EBV WT- and EBV dCTAR3-transformed LCLs and SUMO inhibitor-treated EBV WT-transformed LCLs (Fig. 6B), densitometric analysis of repeat experiments showed a significant (P < 0.001) 40 to 60% decrease in the amount of chromatin-associated KAP1 in cells expressing LMP1 dCTAR3 compared with that in cells expressing the LMP1 WT. Similar results were observed when EBV WT-transformed LCLs were treated with a SUMO inhibitor for 24 h before harvesting. Furthermore, similar experiments were performed in 293 cells where LMP1-mediated dysregulation of sumoylation processes was targeted by deletion of CTAR3, treatment with SUMO inhibitors, overexpression of Ubc9 C93S, and knockdown of Ubc9 (Fig. 6C). These findings suggest that LMP1 induced the sumoylation of KAP1, which increased the association of KAP1 with cellular chromatin.
FIG 6.

LMP1 CTAR3-induced sumoylation regulates KAP1 function. (A) Whole-cell lysates, chromatin-enriched extracts (chrom), and the corresponding supernatants (sup) collected from 293 cells transfected with LMP1 expression constructs or control expression constructs; (B) paired EBV WT- and EBV dCTAR3-transformed LCLs and EBV WT-transformed LCLs treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid); (C) 293 cells transfected with a vector control expression construct and scrambled siRNA, LMP1 dCTAR3 expression constructs, LMP1 expression constructs, LMP1 expression constructs treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid), LMP1 and Ubc9 C93S expression constructs, and LMP1 expression constructs treated with Ubc9 siRNA. (A) Western blot analyses were performed, and KAP1 and GAPDH levels were detected. (B and C) Densitometric analysis of repeat immunoblots and slot blot analyses were performed, and the KAP1 levels in chromatin fractions relative to the levels in whole-cell lysates were determined. Fold changes in KAP1 chromatin association were determined. (D and E) Paired EBV WT- and EBV dCTAR3-transformed LCLs and EBV WT-transformed LCLs treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid) (D) and EBV WT-expressing 293 cells, EBV dCTAR3-expressing 293 cells, and EBV WT-expressing 293 cells treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid), transfected with Ubc9 C93S-expression constructs, or transfected with Ubc9 siRNA (E) were grown, and chromatin immunoprecipitations were performed with KAP1-specific antibodies or control IgG antibodies. Real-time PCR analyses were performed to examine the KAP1 association with the GABPB2, EFCAB7, and ZNF gene promoters. The fold change in DNA binding (relative to that for the input controls) was determined. Results are shown as means ± standard deviations from experiments performed in triplicate.
ChIP analyses were performed to confirm that KAP1 was associating with known cellular promoters, specifically, zinc finger (ZNF) genes, GABPB2, and EFCAB7 (65). Input controls were positive for these three promoters, and the fold change in DNA binding (relative to that for the input controls) was determined. In both EBV-transformed LCLs and EBV-expressing 293 cells, inhibition of LMP1 CTAR3-induced protein sumoylation significantly (P < 0.001) inhibited the association of KAP1 with the GABPB2 and EFCAB7 promoters (Fig. 6D and E). While KAP1 binding to the ZNF genes was not detected in the LCLs, KAP1 did associate with ZNF genes in a CTAR3-dependent manner in EBV-expressing 293 cells (Fig. 6E). Inhibition of cellular sumoylation processes abrogated this interaction.
Together, these results confirm that LMP1 expression significantly (P < 0.001) increases the association of KAP1 with cellular chromatin and select cellular promoters. Inhibition of LMP1-induced sumoylation processes abrogated this increase. These findings demonstrate that LMP1-mediated sumoylation increases the association of KAP1 with chromatin, which leads us to propose that LMP1 may inhibit lytic gene expression through the association of sumoylated KAP1 with viral chromatin.
KAP1 binds to EBV OriLyt and the immediate early promoters.
Interestingly, sumoylation of KAP1 is reported to help maintain KSHV latency by repressing KSHV lytic promoters (93). When KAP1 is desumoylated, it no longer associates with viral promoters and lytic viral replication is initiated (93). To examine whether LMP1-induced sumoylation of KAP1 serves a similar repressive function on EBV lytic promoters, ChIP analyses were performed to determine the association of KAP1 with OriLyt, Qp, and the ZTA, RTA, BALF5, BNRF1, and BCRF1 promoters. Input controls were positive for all promoters examined, and the fold change in DNA binding (relative to that for the input controls) was determined. The results showed that KAP1 associated with OriLyt as well as the ZTA and RTA promoters in EBV WT-transformed LCLs (Fig. 7A) from our sets of paired LCLs. However, significantly (P < 0.001) less KAP1 associated with these promoters in paired LCLs when sumoylation processes were inhibited (in EBV dCTAR3-transformed LCLs and EBV WT-transformed LCLs treated with a SUMO inhibitor). Similar results were observed in EBV-expressing 293 cells where LMP1-induced sumoylation was inhibited by deletion of CTAR3, treatment with the SUMO inhibitor, and transfection of Ubc9 C93S or Ubc9 siRNA (Fig. 7B). In addition, the data revealed that neither KAP1 nor the IgG control interacted with anything bound to select early gene promoters (BALF5), late gene promoters (BNRF1), or latency-associated promoters (Qp) (Fig. 7A and B). No significant differences between KAP1 and IgG control antibody binding to the BCRF1 promoter were observed, which suggests that KAP1 does not bind to the BCRF1 promoter. These results demonstrate that LMP1 CTAR3 promotes the binding of KAP1 to EBV OriLyt and the immediate early promoters and inhibition of cellular sumoylation processes abrogates this response. We propose that the binding of KAP1 at these sites promotes the transcriptional repression that contributes to the maintenance of viral latency.
FIG 7.

KAP1 binds to the EBV OriLyt and the immediate early promoters. Paired EBV WT-transformed LCLs, EBV dCTAR3-transformed LCLs, and EBV WT-transformed LCLs treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid) (A) and EBV WT-expressing 293 cells, EBV dCTAR3-expressing 293 cells, and EBV WT-expressing 293 cells treated with the vehicle control (water) and transfected with scrambled siRNA and an empty expression vector and treated with SUMO inhibitors (25 μM anacardic acid and 25 μM ginkgolic acid), transfected with Ubc9 C93S expression constructs, or transfected with Ubc9 siRNA (B) were grown, and chromatin immunoprecipitations were performed with KAP1-specific antibodies or control IgG antibodies. Real-time PCR analyses were done for OriLyt, the ZTA promoter, the RTA promoter, the BALF5 promoter, the BNRF1 promoter, the BCRF1 promoter, and Qp. The fold change in DNA binding (relative to input controls) was determined. Results are shown as means ± standard deviations from experiments performed in triplicate.
Together, these findings provide additional evidence that CTAR3 is a distinct regulatory region of LMP1 and identify a function and mechanism of action for LMP1-induced sumoylation, acting through CTAR3, in the maintenance of EBV latency.
DISCUSSION
Earlier, we identified a novel function for LMP1 CTAR3 in the regulation of cellular sumoylation processes during EBV latency (14). Our new findings now point to a role for CTAR3 and the induction of sumoylation by LMP1 in the maintenance of EBV latency. Additionally, we found that KAP1 is a cellular target of LMP1 CTAR3-induced sumoylation that can contribute to LMP1 CTAR3-mediated maintenance of EBV latency. These data are the first to identify a role for KAP1 in the maintenance of EBV latency. Together, these findings provide further support for the functions of LMP1 CTAR3, specifically, in the dysregulation of cellular sumoylation processes. They also highlight the importance of LMP1-induced sumoylation in the EBV life cycle.
The finding that noninduced cells expressing LMP1 lacking CTAR3 exhibited increased levels of viral DNA led us to study the role of CTAR3 in the maintenance of EBV latency. However, while transfection of BZLF1 expression plasmids is an efficient inducer of lytic reactivation in EBV-expressing 293 cells, we found that it was not ideal for these studies due to high transfection efficiencies and high levels of viral reactivation. Instead, we used induction of double-stranded DNA damage by irradiation (61, 62, 71–73), which proved to induce lower levels of viral reactivation and facilitated examination of the endogenous reactivation of EBV in LCLs. The finding that LCLs became susceptible to GCV treatment when LMP1-induced sumoylation was inhibited and cells were irradiated demonstrates that viral reactivation occurred and progressed to lytic replication. Furthermore, the finding that nonirradiated EBV dCTAR3-transformed LCLs were susceptible to GCV treatment provides additional support to our claim that these cells are leakier than their EBV WT-transformed LCL counterparts.
In the course of these and our previous studies (14), we used four different methods to inhibit LMP1-induced sumoylation: (i) deletion of CTAR3, (ii) overexpression of enzymatically inactive Ubc9, (iii) treatment of cells with a combination of anacardic acid and ginkgolic acid, and (iv) siRNA-mediated knockdown of Ubc9. Due to the increased sumoylation observed in multiple malignancies, cellular sumoylation processes are proposed targets for anticancer therapies (26, 28). Ubc9 can specifically be targeted with siRNA (77–79), and Ubc9 function can be suppressed by expression of a dominant negative Ubc9 mutant (Ubc9 C93S) (75, 76). Inhibition of LMP1-induced protein sumoylation by any of the four tested methods partially abolished the inhibition of lytic replication in EBV-positive cells, which resulted in increased levels of viral DNA in noninduced and induced cells. In all experiments, inhibition of cellular sumoylation processes produced results similar to those observed with deletion of CTAR3, which suggests that any of the other tested methods of inhibiting sumoylation would be potential mechanisms by which LMP1-induced sumoylation could be inhibited in vivo during EBV latency. Two novel sumoylation inhibitors, spectomycin B1 and davidiin, have recently been identified (94, 95). Davidiin, an ellagitannin, functions in a similar manner as ginkgolic acid and anacardic acid and inhibits the formation of the SUMO-activating enzyme–SUMO-1 intermediate (95). Spectomycin B1 binds to Ubc9 and inhibits the SUMO-Ubc9 intermediate (94). Because our studies targeted the same steps of the sumoylation process, we propose that treatment of EBV-positive cells with either davidiin or spectomycin B1 would abrogate the LMP1 sumoylation-dependent maintenance of EBV latency, promoting viral reactivation.
We now document that KAP1 sumoylation, a well-studied process (54, 88–92), occurs in an LMP1 CTAR3-dependent manner. While KAP1 sumoylation has been documented during latent KSHV infection (93), this is the first report to show endogenous sumoylation of KAP1 during latent EBV infection. The mechanism by which KAP1 is sumoylated during EBV latency remains unknown, but there are multiple possible manners by which the sumoylation of KAP1 may occur. KAP1 undergoes autosumoylation following its binding to Ubc9 (90). We documented that LMP1 CTAR3 interacts with Ubc9 (14), so following the interaction of LMP1 with Ubc9, Ubc9 may bind KAP1, which results in the direct SUMO conjugation of KAP1 (90). Supporting this idea is the finding that KAP1 serves as a SUMO E3 ligase for interferon regulatory factor 7 (IRF7) (96), which was the first identified target of LMP1 CTAR3-induced sumoylation (46). We previously showed that the LMP1-induced sumoylation of IRF7 is dependent on cooperation between LMP1 CTAR2 and LMP1 CTAR3 (46). We proposed that sumoylation of IRF7 was due to LMP1 bringing together IRF7, which indirectly interacts with CTAR2 (38, 42), and Ubc9, which interacts with CTAR3 (14). Therefore, the possibility exists that KAP1 is also associated with the LMP1/Ubc9/IRF7 complex, resulting in KAP1 sumoylation as well as the action of KAP1 as a SUMO ligase for IRF7. Finally, it is possible that the sumoylation of KAP1 is a result of its interactions with downstream targets of LMP1. For instance, KAP1 also interacts with NF-κB and STAT3 (97), which are known targets of SUMO-1 (98–101) and downstream targets of LMP1-induced signaling (8, 38, 102, 103), and the ability of KAP1 to interact with known sumoylated proteins may promote the sumoylation of KAP1 itself. Deciphering the mechanism by which LMP1 mediates the sumoylation of KAP1 will be part of our future studies into the function of LMP1 CTAR3 and LMP1-induced protein sumoylation.
KAP1 is a well-characterized transcriptional corepressor (86) that binds to thousands of sites in the human genome via KRAB-ZFPs (53, 86, 87) when it is sumoylated (53). Here we show KAP1 associates with cellular chromatin and select cellular promoters (65) in an LMP1 CTAR3-induced sumoylation-dependent manner. These findings suggest that one mechanism by which LMP1 can inhibit gene expression is via the sumoylation of KAP1, which promotes KAP1 chromatin association and transcriptional repression.
In addition to associating with cellular chromatin, we now document that KAP1 also associates with viral DNA. KAP1 binds to EBV OriLyt and the immediate early promoters ZTA and RTA but not select early, late, and latency-associated promoters. The binding of KAP1 to these promoters occurs in a sumoylation-dependent manner in both LCLs and EBV-expressing 293 cells. Due to the documented abundance of repressive histone methylation (63), the specific piece of OriLyt that was analyzed in these studies was OriLyt Flank (63), which comprises auxiliary components of OriLyt that contribute to and enhance the activity of OriLyt (104). The ability of KAP1 to bind to the core components of OriLyt remains to be examined; however, we propose that multiple KAP1-binding sites which contribute to SUMO-dependent repression of lytic reactivation are present. In addition, the wide array of functions attributed to KAP1 (53) suggests that investigation of the effects of LMP1 CTAR3 in DNA repair responses, as well as in the maintenance of EBV genomic integrity, may yield interesting results.
Recently, it was documented that DNA-damaging agents, including hydrogen peroxide, induce EBV reactivation (70). Hydrogen peroxide and the induction of reactive oxygen species are known to result in lower levels of overall protein sumoylation due to their effects on the Ubc9-SUMO interaction (105). Therefore, both reports add direct support for our hypothesis that LMP1-induced sumoylation affects the maintenance of EBV latency.
In summary, we propose that during latent EBV infection, when LMP1 is expressed (type II and type III EBV latency), sumoylation processes are dysregulated, resulting in increased protein sumoylation. KAP1 is sumoylated, resulting in transcriptional repression, including the repression of lytic viral promoters. Inhibition of LMP1-induced sumoylation by deletion of LMP1 CTAR3, treatment with anacardic acid and ginkgolic acid, expression of enzymatically inactive Ubc9, knockdown of Ubc9, or potentially even treatment with hydrogen peroxide results in the loss of KAP1 sumoylation and a leakier switch from latent infection to lytic replication. The result is the increased ability to induce lytic replication. Thus, because lytic replication is necessary for antiviral drugs to be activated and exert their cytotoxic effects, these findings identify methods by which latent EBV infections can be disrupted and made susceptible to such approaches.
ACKNOWLEDGMENTS
G.L.B. was supported by a grant from NCI (CA160786). J.S.P. was supported by grant CA163217 and supplemental funding from NCI to the UNC Lineberger Comprehensive Cancer Center and CFAR. C.R.M. was supported by grant RSG-13-229-01 from ACS.
REFERENCES
- 1.Morgan DG, Niederman JC, Miller G, Smith HW, Dowaliby JM. 1979. Site of Epstein-Barr virus replication in the oropharynx. Lancet ii:1154–1157. [DOI] [PubMed] [Google Scholar]
- 2.Adler B, Schaadt E, Kempkes B, Zimber-Strobl U, Baier B, Bornkamm GW. 2002. Control of Epstein-Barr virus reactivation by activated CD40 and viral latent membrane protein 1. Proc Natl Acad Sci U S A 99:437–442. doi: 10.1073/pnas.221439999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahsan N, Kanda T, Nagashima K, Takada K. 2005. Epstein-Barr virus transforming protein LMP1 plays a critical role in virus production. J Virol 79:4415–4424. doi: 10.1128/JVI.79.7.4415-4424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prince S, Keating S, Fielding C, Brennan P, Floettmann E, Rowe M. 2003. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J Virol 77:5000–5007. doi: 10.1128/JVI.77.8.5000-5007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pagano J. 2009. EBV diseases, p 217–240. Springer, New York, NY. [Google Scholar]
- 6.Pagano JS. 2010. Molecular pathobiology of EBV infection, p 409–451. John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 7.Pagano JS, Blaser M, Buendia MA, Damania B, Khalili K, Raab-Traub N, Roizman B. 2004. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol 14:453–471. doi: 10.1016/j.semcancer.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Li HP, Chang YS. 2003. Epstein-Barr virus latent membrane protein 1: structure and functions. J Biomed Sci 10:490–504. doi: 10.1007/BF02256110. [DOI] [PubMed] [Google Scholar]
- 9.Kieser A. 2008. Abstr 13th Biennial Conf Int Assoc Res Epstein-Barr Virus Associated Dis, Guangzhou, China, abstr 27. [Google Scholar]
- 10.Brennan P, Floettmann JE, Mehl A, Jones M, Rowe M. 2001. Mechanism of action of a novel latent membrane protein-1 dominant negative. J Biol Chem 276:1195–1203. doi: 10.1074/jbc.M005461200. [DOI] [PubMed] [Google Scholar]
- 11.Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C, Zeidler R, Scheffer B, Ueffing M, Hammerschmidt W. 1999. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J 18:3064–3073. doi: 10.1093/emboj/18.11.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higuchi M, Kieff E, Izumi KM. 2002. The Epstein-Barr virus latent membrane protein 1 putative Janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J Virol 76:455–459. doi: 10.1128/JVI.76.1.455-459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumi KM, Cahir McFarland ED, Riley EA, Rizzo D, Chen Y, Kieff E. 1999. The residues between the two transformation effector sites of Epstein-Barr virus latent membrane protein 1 are not critical for B-lymphocyte growth transformation. J Virol 73:9908–9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentz GL, Whitehurst CB, Pagano JS. 2011. Epstein-Barr virus latent membrane protein 1 (LMP1) C-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J Virol 85:10144–10153. doi: 10.1128/JVI.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerscher O, Felberbaum R, Hochstrasser M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 16.Kroetz MB. 2005. SUMO: a ubiquitin-like protein modifier. Yale J Biol Med 78:197–201. [PMC free article] [PubMed] [Google Scholar]
- 17.Kerscher O. 2007. SUMO junction—what's your function? New insights through SUMO-interacting motifs. EMBO Rep 8:550–555. doi: 10.1038/sj.embor.7400980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bies J, Markus J, Wolff L. 2002. Covalent attachment of the SUMO-1 protein to the negative regulatory domain of the c-Myb transcription factor modifies its stability and transactivation capacity. J Biol Chem 277:8999–9009. doi: 10.1074/jbc.M110453200. [DOI] [PubMed] [Google Scholar]
- 19.Buschmann T, Fuchs SY, Lee CG, Pan ZQ, Ronai Z. 2000. SUMO-1 modification of Mdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinate p53. Cell 101:753–762. doi: 10.1016/S0092-8674(00)80887-9. [DOI] [PubMed] [Google Scholar]
- 20.Ghioni P, D'Alessandra Y, Mansueto G, Jaffray E, Hay RT, La Mantia G, Guerrini L. 2005. The protein stability and transcriptional activity of p63 alpha are regulated by SUMO-1 conjugation. Cell Cycle 4:183–190. doi: 10.4161/cc.4.1.1359. [DOI] [PubMed] [Google Scholar]
- 21.Huang YP, Wu GJ, Guo ZM, Osada M, Fomenkov T, Park HL, Trink B, Sidransky D, Fomenkov A, Ratovitski EA. 2004. Altered sumoylation of p63 alpha contributes to the split-hand/foot malformation phenotype. Cell Cycle 3:1587–1596. doi: 10.4161/cc.3.12.1290. [DOI] [PubMed] [Google Scholar]
- 22.Muller S, Matunis MJ, Dejean A. 1998. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J 17:61–70. doi: 10.1093/emboj/17.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt D, Muller S. 2002. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A 99:2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alarcon-Vargas D, Ronai Z. 2002. SUMO in cancer—wrestlers wanted. Cancer Biol Ther 1:237–242. doi: 10.4161/cbt.74. [DOI] [PubMed] [Google Scholar]
- 25.Katayama A, Ogino T, Bandoh N, Takahara M, Kishibe K, Nonaka S, Harabuchi Y. 2007. Overexpression of small ubiquitin-related modifier-1 and sumoylated Mdm2 in oral squamous cell carcinoma: possible involvement in tumor proliferation and prognosis. Int J Oncol 31:517–524. doi: 10.3892/ijo.31.3.517. [DOI] [PubMed] [Google Scholar]
- 26.Duan X, Trent JO, Ye H. 2009. Targeting the SUMO E2 conjugating enzyme Ubc9 interaction for anti-cancer drug design. Anticancer Agents Med Chem 9:51–54. doi: 10.2174/187152009787047716. [DOI] [PubMed] [Google Scholar]
- 27.Stewart MJ, Smoak K, Blum MA, Sherry B. 2005. Basal and reovirus-induced beta interferon (IFN-beta) and IFN-beta-stimulated gene expression are cell type specific in the cardiac protective response. J Virol 79:2979–2987. doi: 10.1128/JVI.79.5.2979-2987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mo YY, Moschos SJ. 2005. Targeting Ubc9 for cancer therapy. Expert Opin Ther Targets 9:1203–1216. doi: 10.1517/14728222.9.6.1203. [DOI] [PubMed] [Google Scholar]
- 29.Lin J, Johannsen E, Robertson E, Kieff E. 2002. Epstein-Barr virus nuclear antigen 3C putative repression domain mediates coactivation of the LMP1 promoter with EBNA-2. J Virol 76:232–242. doi: 10.1128/JVI.76.1.232-242.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosendorff A, Illanes D, David G, Lin J, Kieff E, Johannsen E. 2004. EBNA3C coactivation with EBNA2 requires a SUMO homology domain. J Virol 78:367–377. doi: 10.1128/JVI.78.1.367-377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hille A, Badu-Antwi A, Holzer D, Grasser FA. 2002. Lysine residues of Epstein-Barr virus-encoded nuclear antigen 2 do not confer secondary modifications via ubiquitin or SUMO-like proteins but modulate transcriptional activation. J Gen Virol 83:1037–1042. [DOI] [PubMed] [Google Scholar]
- 32.Adamson AL. 2005. Effects of SUMO-1 upon Epstein-Barr virus BZLF1 function and BMRF1 expression. Biochem Biophys Res Commun 336:22–28. doi: 10.1016/j.bbrc.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 33.Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol 75:2388–2399. doi: 10.1128/JVI.75.5.2388-2399.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang LK, Lee YH, Cheng TS, Hong YR, Lu PJ, Wang JJ, Wang WH, Kuo CW, Li SS, Liu ST. 2004. Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J Biol Chem 279:38803–38812. doi: 10.1074/jbc.M405470200. [DOI] [PubMed] [Google Scholar]
- 35.Chang LK, Liu ST, Kuo CW, Wang WH, Chuang JY, Bianchi E, Hong YR. 2008. Enhancement of transactivation activity of Rta of Epstein-Barr virus by RanBPM. J Mol Biol 379:231–242. doi: 10.1016/j.jmb.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Hagemeier SR, Dickerson SJ, Meng Q, Yu X, Mertz JE, Kenney SC. 2010. Sumoylation of the Epstein-Barr virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase. J Virol 84:4383–4394. doi: 10.1128/JVI.02369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu ST, Wang WH, Hong YR, Chuang JY, Lu PJ, Chang LK. 2006. Sumoylation of Rta of Epstein-Barr virus is preferentially enhanced by PIASxbeta. Virus Res 119:163–170. doi: 10.1016/j.virusres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 38.Ning S, Campos AD, Darnay BG, Bentz GL, Pagano JS. 2008. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol Cell Biol 28:6536–6546. doi: 10.1128/MCB.00785-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ning S, Hahn AM, Huye LE, Pagano JS. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J Virol 77:9359–9368. doi: 10.1128/JVI.77.17.9359-9368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ning S, Huye LE, Pagano JS. 2005. Interferon regulatory factor 5 represses expression of the Epstein-Barr virus oncoprotein LMP1: braking of the IRF7/LMP1 regulatory circuit. J Virol 79:11671–11676. doi: 10.1128/JVI.79.18.11671-11676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ning S, Huye LE, Pagano JS. 2005. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J Biol Chem 280:12262–12270. doi: 10.1074/jbc.M404260200. [DOI] [PubMed] [Google Scholar]
- 42.Ning S, Pagano JS. 2010. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J Virol 84:6130–6138. doi: 10.1128/JVI.00364-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ning S, Pagano JS, Barber GN. 2011. IRF7: activation, regulation, modification and function. Genes Immun 12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Pagano JS. 2001. Interferon regulatory factor 7: a key cellular mediator of LMP-1 in EBV latency and transformation. Semin Cancer Biol 11:445–453. doi: 10.1006/scbi.2001.0411. [DOI] [PubMed] [Google Scholar]
- 45.Zhang L, Wu L, Hong K, Pagano JS. 2001. Intracellular signaling molecules activated by Epstein-Barr virus for induction of interferon regulatory factor 7. J Virol 75:12393–12401. doi: 10.1128/JVI.75.24.12393-12401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bentz GL, Shackelford J, Pagano JS. 2012. Epstein-Barr virus latent membrane protein 1 regulates the function of interferon regulatory factor 7 by inducing its sumoylation. J Virol 86:12251–12261. doi: 10.1128/JVI.01407-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anckar J, Sistonen L. 2007. SUMO: getting it on. Biochem Soc Trans 35:1409–1413. doi: 10.1042/BST0351409. [DOI] [PubMed] [Google Scholar]
- 48.Andreou AM, Tavernarakis N. 2009. SUMOylation and cell signalling. Biotechnol J 4:1740–1752. doi: 10.1002/biot.200900219. [DOI] [PubMed] [Google Scholar]
- 49.Bossis G, Melchior F. 2006. SUMO: regulating the regulator. Cell Div 1:13. doi: 10.1186/1747-1028-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cammas F, Khetchoumian K, Chambon P, Losson R. 2012. TRIM involvement in transcriptional regulation. Adv Exp Med Biol 770:59–76. doi: 10.1007/978-1-4614-5398-7_5. [DOI] [PubMed] [Google Scholar]
- 51.Micale L, Chaignat E, Fusco C, Reymond A, Merla G. 2012. The tripartite motif: structure and function. Adv Exp Med Biol 770:11–25. doi: 10.1007/978-1-4614-5398-7_2. [DOI] [PubMed] [Google Scholar]
- 52.Ozato K, Shin DM, Chang TH, Morse HC III. 2008. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iyengar S, Farnham PJ. 2011. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem 286:26267–26276. doi: 10.1074/jbc.R111.252569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kepkay R, Attwood KM, Ziv Y, Shiloh Y, Dellaire G. 2011. KAP1 depletion increases PML nuclear body number in concert with ultrastructural changes in chromatin. Cell Cycle 10:308–322. doi: 10.4161/cc.10.2.14551. [DOI] [PubMed] [Google Scholar]
- 55.Calender A, Billaud M, Aubry JP, Banchereau J, Vuillaume M, Lenoir GM. 1987. Epstein-Barr virus (EBV) induces expression of B-cell activation markers on in vitro infection of EBV-negative B-lymphoma cells. Proc Natl Acad Sci U S A 84:8060–8064. doi: 10.1073/pnas.84.22.8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calender A, Cordier M, Billaud M, Lenoir GM. 1990. Modulation of cellular gene expression in B lymphoma cells following in vitro infection by Epstein-Barr virus (EBV). Int J Cancer 46:658–663. doi: 10.1002/ijc.2910460418. [DOI] [PubMed] [Google Scholar]
- 57.Cherney BW, Sgadari C, Kanegane C, Wang F, Tosato G. 1998. Expression of the Epstein-Barr virus protein LMP1 mediates tumor regression in vivo. Blood 91:2491–2500. [PubMed] [Google Scholar]
- 58.Dirmeier U, Neuhierl B, Kilger E, Reisbach G, Sandberg ML, Hammerschmidt W. 2003. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by Epstein-Barr virus. Cancer Res 63:2982–2989. [PubMed] [Google Scholar]
- 59.Miller WE, Mosialos G, Kieff E, Raab-Traub N. 1997. Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. J Virol 71:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gershburg E, Raffa S, Torrisi MR, Pagano JS. 2007. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J Virol 81:5407–5412. doi: 10.1128/JVI.02398-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O'Nions J, Turner A, Craig R, Allday MJ. 2006. Epstein-Barr virus selectively deregulates DNA damage responses in normal B cells but has no detectable effect on regulation of the tumor suppressor p53. J Virol 80:12408–12413. doi: 10.1128/JVI.01363-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olive PL, Banath JP. 1993. Detection of DNA double-strand breaks through the cell-cycle after exposure to X-rays, bleomycin, etoposide and (125)Idurd. Int J Radiat Biol 64:349–358. doi: 10.1080/09553009314551531. [DOI] [PubMed] [Google Scholar]
- 63.Ramasubramanyan S, Osborn K, Flower K, Sinclair AJ. 2012. Dynamic chromatin environment of key lytic cycle regulatory regions of the Epstein-Barr virus genome. J Virol 86:1809–1819. doi: 10.1128/JVI.06334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitehurst CB, Ning S, Bentz GL, Dufour F, Gershburg E, Shackelford J, Langelier Y, Pagano JS. 2009. The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J Virol 83:4345–4353. doi: 10.1128/JVI.02195-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iyengar S, Ivanov AV, Jin VX, Rauscher FJ III, Farnham PJ. 2011. Functional analysis of KAP1 genomic recruitment. Mol Cell Biol 31:1833–1847. doi: 10.1128/MCB.01331-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Izumi M, Yatagai F, Hanaoka F. 2001. Cell cycle-dependent proteolysis and phosphorylation of human Mcm10. J Biol Chem 276:48526–48531. [DOI] [PubMed] [Google Scholar]
- 67.Bheda A, Yue W, Gullapalli A, Shackelford J, Pagano JS. 2011. PU.1-dependent regulation of UCH L1 expression in B-lymphoma cells. Leuk Lymphoma 52:1336–1347. doi: 10.3109/10428194.2011.562571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sides MD, Block GJ, Shan B, Esteves KC, Lin Z, Flemington EK, Lasky JA. 2011. Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein-Barr positive epithelial cells. Virology 416:86–97. doi: 10.1016/j.virol.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bentz GL, Yurochko AD. 2008. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and beta1 and beta3 integrins. Proc Natl Acad Sci U S A 105:5531–5536. doi: 10.1073/pnas.0800037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hagemeier SR, Barlow EA, Meng Q, Kenney SC. 2012. The cellular ataxia telangiectasia-mutated kinase promotes Epstein-Barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J Virol 86:13360–13370. doi: 10.1128/JVI.01850-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. 2002. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res 62:1920–1926. [PubMed] [Google Scholar]
- 72.Westphal EM, Blackstock W, Feng W, Israel B, Kenney SC. 2000. Activation of lytic Epstein-Barr virus (EBV) infection by radiation and sodium butyrate in vitro and in vivo: a potential method for treating EBV-positive malignancies. Cancer Res 60:5781–5788. [PubMed] [Google Scholar]
- 73.Liu MT, Chen YR, Chen SC, Hu CY, Lin CS, Chang YT, Wang WB, Chen JY. 2004. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 23:2531–2539. doi: 10.1038/sj.onc.1207375. [DOI] [PubMed] [Google Scholar]
- 74.Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, Kimura K, Sodeoka M, Yoshida M. 2009. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol 16:133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 75.Giorgino F, de Robertis O, Laviola L, Montrone C, Perrini S, McCowen KC, Smith RJ. 2000. The sentrin-conjugating enzyme mUbc9 interacts with GLUT4 and GLUT1 glucose transporters and regulates transporter levels in skeletal muscle cells. Proc Natl Acad Sci U S A 97:1125–1130. doi: 10.1073/pnas.97.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mo YY, Yu Y, Shen Z, Beck WT. 2002. Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J Biol Chem 277:2958–2964. doi: 10.1074/jbc.M108263200. [DOI] [PubMed] [Google Scholar]
- 77.Kurihara I, Shibata H, Kobayashi S, Suda N, Ikeda Y, Yokota K, Murai A, Saito I, Rainey WE, Saruta T. 2005. Ubc9 and protein inhibitor of activated STAT 1 activate chicken ovalbumin upstream promoter-transcription factor I-mediated human CYP11B2 gene transcription. J Biol Chem 280:6721–6730. doi: 10.1074/jbc.M411820200. [DOI] [PubMed] [Google Scholar]
- 78.Lin X, Liang M, Liang YY, Brunicardi FC, Feng XH. 2003. SUMO-1/Ubc9 promotes nuclear accumulation and metabolic stability of tumor suppressor Smad4. J Biol Chem 278:31043–31048. doi: 10.1074/jbc.C300112200. [DOI] [PubMed] [Google Scholar]
- 79.Wang QE, Zhu Q, Wani G, El-Mahdy MA, Li J, Wani AA. 2005. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res 33:4023–4034. doi: 10.1093/nar/gki684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Daibata M, Bandobashi K, Kuroda M, Imai S, Miyoshi I, Taguchi H. 2005. Induction of lytic Epstein-Barr virus (EBV) infection by synergistic action of rituximab and dexamethasone renders EBV-positive lymphoma cells more susceptible to ganciclovir cytotoxicity in vitro and in vivo. J Virol 79:5875–5879. doi: 10.1128/JVI.79.9.5875-5879.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feng WH, Cohen JI, Fischer S, Li L, Sneller M, Goldbach-Mansky R, Raab-Traub N, Delecluse HJ, Kenney SC. 2004. Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J Natl Cancer Inst 96:1691–1702. doi: 10.1093/jnci/djh313. [DOI] [PubMed] [Google Scholar]
- 82.Feng WH, Hong G, Delecluse HJ, Kenney SC. 2004. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. J Virol 78:1893–1902. doi: 10.1128/JVI.78.4.1893-1902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Feng WH, Kenney SC. 2006. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res 66:8762–8769. doi: 10.1158/0008-5472.CAN-06-1006. [DOI] [PubMed] [Google Scholar]
- 84.Sides MD, Sosulski ML, Luo F, Lin Z, Flemington EK, Lasky JA. 2013. Co-treatment with arsenic trioxide and ganciclovir reduces tumor volume in a murine xenograft model of nasopharyngeal carcinoma. Virol J 10:152. doi: 10.1186/1743-422X-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, Kenney SC. 2010. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J Virol 84:4534–4542. doi: 10.1128/JVI.02487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Urrutia R. 2003. KRAB-containing zinc-finger repressor proteins. Genome Biol 4:231. doi: 10.1186/gb-2003-4-10-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cheng CT, Kuo CY, Ann DK. 2014. KAPtain in charge of multiple missions: emerging roles of KAP1. World J Biol Chem 5:308–320. doi: 10.4331/wjbc.v5.i3.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Campagna M, Herranz D, Garcia MA, Marcos-Villar L, Gonzalez-Santamaria J, Gallego P, Gutierrez S, Collado M, Serrano M, Esteban M, Rivas C. 2011. SIRT1 stabilizes PML promoting its sumoylation. Cell Death Differ 18:72–79. doi: 10.1038/cdd.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wolyniec K, Shortt J, de Stanchina E, Levav-Cohen Y, Alsheich-Bartok O, Louria-Hayon I, Corneille V, Kumar B, Woods SJ, Opat S, Johnstone RW, Scott CL, Segal D, Pandolfi PP, Fox S, Strasser A, Jiang YH, Lowe SW, Haupt S, Haupt Y. 2012. E6AP ubiquitin ligase regulates PML-induced senescence in Myc-driven lymphomagenesis. Blood 120:822–832. doi: 10.1182/blood-2011-10-387647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ III. 2007. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28:823–837. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee YK, Thomas SN, Yang AJ, Ann DK. 2007. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J Biol Chem 282:1595–1606. doi: 10.1074/jbc.M606306200. [DOI] [PubMed] [Google Scholar]
- 92.Mascle XH, Germain-Desprez D, Huynh P, Estephan P, Aubry M. 2007. Sumoylation of the transcriptional intermediary factor 1beta (TIF1beta), the co-repressor of the KRAB multifinger proteins, is required for its transcriptional activity and is modulated by the KRAB domain. J Biol Chem 282:10190–10202. doi: 10.1074/jbc.M611429200. [DOI] [PubMed] [Google Scholar]
- 93.Chang PC, Fitzgerald LD, Van Geelen A, Izumiya Y, Ellison TJ, Wang DH, Ann DK, Luciw PA, Kung HJ. 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res 69:5681–5689. doi: 10.1158/0008-5472.CAN-08-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hirohama M, Kumar A, Fukuda I, Matsuoka S, Igarashi Y, Saitoh H, Takagi M, Shin-ya K, Honda K, Kondoh Y, Saito T, Nakao Y, Osada H, Zhang KY, Yoshida M, Ito A. 2013. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem Biol 8:2635–2642. doi: 10.1021/cb400630z. [DOI] [PubMed] [Google Scholar]
- 95.Takemoto M, Kawamura Y, Hirohama M, Yamaguchi Y, Handa H, Saitoh H, Nakao Y, Kawada M, Khalid K, Koshino H, Kimura K, Ito A, Yoshida M. 2014. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. J Antibiot 67:335–338. doi: 10.1038/ja.2013.142. [DOI] [PubMed] [Google Scholar]
- 96.Liang QM, Deng HY, Li XJ, Wu XF, Tang QY, Chang TH, Peng HZ, Rauscher FJ, Ozato K, Zhu FX. 2011. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J Immunol 187:4754–4763. doi: 10.4049/jimmunol.1101704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kamitani S, Togi S, Ikeda O, Nakasuji M, Sakauchi A, Sekine Y, Muromoto R, Oritani K, Matsuda T. 2011. Kruppel-associated box-associated protein 1 negatively regulates TNF-alpha-induced NF-kappa B transcriptional activity by influencing the interactions among STAT3, p300, and NF-kappa B/p65. J Immunol 187:2476–2483. doi: 10.4049/jimmunol.1003243. [DOI] [PubMed] [Google Scholar]
- 98.Lang V, Rodriguez MS. 2008. Innate link between NF-kappaB activity and ubiquitin-like modifiers. Biochem Soc Trans 36:853–857. doi: 10.1042/BST0360853. [DOI] [PubMed] [Google Scholar]
- 99.Mabb AM, Miyamoto S. 2007. SUMO and NF-kappaB ties. Cell Mol Life Sci 64:1979–1996. doi: 10.1007/s00018-007-7005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guo B, Yang SH, Witty J, Sharrocks AD. 2007. Signalling pathways and the regulation of SUMO modification. Biochem Soc Trans 35:1414–1418. doi: 10.1042/BST0351414. [DOI] [PubMed] [Google Scholar]
- 101.Rui HL, Fan E, Zhou HM, Xu Z, Zhang Y, Lin SC. 2002. SUMO-1 modification of the C-terminal KVEKVD of axin is required for JNK activation but has no effect on Wnt signaling. J Biol Chem 277:42981–42986. doi: 10.1074/jbc.M208099200. [DOI] [PubMed] [Google Scholar]
- 102.Luftig M, Prinarakis E, Yasui T, Tsichritzis T, Cahir-McFarland E, Inoue J, Nakano H, Mak TW, Yeh WC, Li X, Akira S, Suzuki N, Suzuki S, Mosialos G, Kieff E. 2003. Epstein-Barr virus latent membrane protein 1 activation of NF-kappaB through IRAK1 and TRAF6. Proc Natl Acad Sci U S A 100:15595–15600. doi: 10.1073/pnas.2136756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huye LE, Ning S, Kelliher M, Pagano JS. 2007. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol Cell Biol 27:2910–2918. doi: 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hammerschmidt W, Sugden B. 1988. Identification and characterization of OriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55:427–433. doi: 10.1016/0092-8674(88)90028-1. [DOI] [PubMed] [Google Scholar]
- 105.Bossis G, Melchior F. 2006. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell 21:349–357. doi: 10.1016/j.molcel.2005.12.019. [DOI] [PubMed] [Google Scholar]