

ABSTRACT
Herpesviruses have evolved a unique mechanism for nucleocytoplasmic transport of nascent nucleocapsids: the nucleocapsids bud through the inner nuclear membrane (INM; primary envelopment), and the enveloped nucleocapsids then fuse with the outer nuclear membrane (de-envelopment). Little is known about the molecular mechanism of herpesviral de-envelopment. We show here that the knockdown of both CD98 heavy chain (CD98hc) and its binding partner β1 integrin induced membranous structures containing enveloped herpes simplex virus 1 (HSV-1) virions that are invaginations of the INM into the nucleoplasm and induced aberrant accumulation of enveloped virions in the perinuclear space and in the invagination structures. These effects were similar to those of the previously reported mutation(s) in HSV-1 proteins gB, gH, UL31, and/or Us3, which were shown here to form a complex(es) with CD98hc in HSV-1-infected cells. These results suggested that cellular proteins CD98hc and β1 integrin synergistically or independently regulated HSV-1 de-envelopment, probably by interacting directly and/or indirectly with these HSV-1 proteins.
IMPORTANCE Certain cellular and viral macromolecular complexes, such as Drosophila large ribonucleoprotein complexes and herpesvirus nucleocapsids, utilize a unique vesicle-mediated nucleocytoplasmic transport: the complexes acquire primary envelopes by budding through the inner nuclear membrane into the space between the inner and outer nuclear membranes (primary envelopment), and the enveloped complexes then fuse with the outer nuclear membrane to release de-enveloped complexes into the cytoplasm (de-envelopment). However, there is a lack of information on the molecular mechanism of de-envelopment fusion. We report here that HSV-1 recruited cellular fusion regulatory proteins CD98hc and β1 integrin to the nuclear membrane for viral de-envelopment fusion. This is the first report of cellular proteins required for efficient de-envelopment of macromolecular complexes during their nuclear egress.
INTRODUCTION
Herpesviruses are enveloped double-stranded DNA viruses that replicate their genomes and package the nascent progeny viral genomes into capsids in the nucleus, but these nascent viruses acquire their final envelopes in the cytoplasm (1, 2). Therefore, herpesvirus nucleocapsids must traverse the inner nuclear membrane (INM) and outer nuclear membrane (ONM) for viral morphogenesis. Since herpesvirus nucleocapsids are too large to cross the INM and ONM through nuclear pores, the viruses evolved a unique nuclear egress mechanism: progeny nucleocapsids acquire primary envelopes by budding through the INM into the perinuclear space between the INM and ONM (primary envelopment) and enveloped nucleocapsids then fuse with the ONM to release de-enveloped nucleocapsids into the cytoplasm (de-envelopment) (1, 2). Although this type of vesicle-mediated nucleocytoplasmic transport has not been reported previously, other than for herpesvirus nuclear egress, it has recently been reported that Drosophila cellular ribonucleoprotein (RNP) complexes utilize a similar mechanism for their nucleocytoplasmic transport in neurons (3). This suggested that vesicle-mediated nucleocytoplasmic transport may be a general cellular process for export of large macromolecular complexes from the nucleus, mediated by specific cellular proteins. However, although vesicle-mediated nucleocytoplasmic transport of nucleocapsids is readily detectable in herpesvirus-infected cells, it has not been reported for other cellular macromolecular complexes in normal cells, except for the Drosophila RNP complexes in neurons described above (3). Therefore, specific signaling(s) may be required to initiate and/or carry out vesicle-mediated nucleocytoplasmic transport, and herpesvirus infection may efficiently take over the signaling(s), probably by viral proteins that interact with the cellular proteins that regulate this process. In agreement with this hypothesis, herpesviruses have been reported to recruit host cell protein kinase C (PKC) isoforms to the nuclear membrane for phosphorylation and dissolution of the nuclear lamina (1, 2, 4, 5). This has been suggested to facilitate herpesvirus nucleocapsid access to the INM in the first step of nucleocytoplasmic transport, primary envelopment, using a heterodimeric complex, designated the nuclear egress complex (NEC), of two herpesvirus proteins that are conserved throughout the Herpesviridae family (1, 2, 6).
Herpes simplex virus 1 (HSV-1) is one of the best-characterized members of the Herpesviridae family and an important human pathogen causing a variety of diseases, such as mucocutaneous diseases, keratitis, skin diseases, and encephalitis (7). The HSV-1 NEC, which consists of UL31 and UL34 proteins or their homologs in other herpesviruses, has been reported to play a critical role in primary envelopment by mediating modification of the nuclear lamina as described above (4, 5, 8, 9), recruiting nucleocapsids into primary envelopes (10, 11) and budding these primary enveloped virions through the INM (12–14). In contrast, little is known about the next step of herpesviral nuclear egress, de-envelopment. It has been reported that HSV-1 de-envelopment appeared to be reduced by mutations in several viral proteins. Mutations that abrogate either the expression or catalytic activity of HSV-1 serine/threonine protein kinase Us3, the expression of both HSV-1 envelope glycoprotein B (gB) and gH, or the phosphorylation of UL31 have been reported to induce membranous structures containing primary enveloped virions that are invaginations of the INM into the nucleoplasm and to induce the aberrant accumulation of primary enveloped virions in the perinuclear space and in the induced invagination structures (15–18). These observations suggested that gB, gH, UL31, and Us3 were required for efficient HSV-1 de-envelopment during HSV-1 nuclear egress. Although these observations suggested that these viral proteins are involved in regulation of HSV-1 de-envelopment, the precise mechanisms by which these HSV-1 and cellular proteins act in this process remain largely unknown. In particular, there is a lack of information on how fusion between primary viral envelopes and the ONM is regulated in HSV-1 de-envelopment. To identify a cellular protein(s) involved in HSV-1 de-envelopment, we screened for cellular proteins that interact with gB, a possible HSV-1 regulator for this process, in HSV-1-infected cells by tandem affinity purification coupled with mass spectrometry-based proteomics technology. Of the putative gB-interacting cellular proteins that were identified, we focused on CD98 heavy chain (CD98hc; also known as SLC3A2 or 4F2hc) as a novel gB-interacting protein.
CD98hc, a type II membrane glycoprotein, is expressed on the cell surface of a wide variety of cell types in cell cultures and is ubiquitously expressed in vivo (19–22). CD98hc has two distinct functions. CD98hc acts as an amino acid transporter on the cell surface by associating with one of several light chains through its extracellular domain (22) and also regulates integrin-signaling that is involved in cell adhesion and migration by associating with β1 and β3 integrins through its cytoplasmic and transmembrane domains (23–26). Interestingly, it has been shown that stimulation of CD98hc with specific monoclonal antibodies enhanced or inhibited cell fusion induced by enveloped viruses, including Newcastle disease virus (NDV), human parainfluenza virus type 2 (HPIV-2), and human immunodeficiency virus (HIV) (27–30), and that this regulation required β1 integrin (28), suggesting that CD98hc and β1 integrin may be regulators of membrane fusion mediated by enveloped viruses.
In this study, we have shown that, in HSV-1-infected cells, CD98hc formed a complex(es) with gB, gH, UL31, UL34, and Us3, most of which were reported to be required for efficient HSV-1 de-envelopment (15–18) as described above. We also showed that CD98hc formed a complex with β1 integrin in HSV-1-infected cells, and both of these cellular proteins accumulated at the nuclear membrane, where CD98hc colocalized with gB, gH, UL31, UL34, and β1 integrin. Furthermore, both CD98hc and β1 integrin knockdown induced aberrant accumulation of primary enveloped virions in the perinuclear space and in the membranous invagination structures adjacent to the nuclear membrane that were induced in CD98hc- and β1 integrin-depleted cells. The effects of CD98hc and β1 integrin knockdown on HSV-1 de-envelopment appeared to be similar to those of the mutation(s) in HSV-1 gB, gH, UL31, and/or Us3 reported previously (15–18). Taken together with the known role of CD98hc and β1 integrin in enveloped virus-induced membrane fusion (27–30) described above, these results supported the hypothesis that CD98hc and β1 integrin synergistically or independently regulated HSV-1 de-envelopment, probably by interacting directly and/or indirectly with viral gB, gH, UL31, UL34, and Us3.
MATERIALS AND METHODS
Cells and viruses.
HEp-2, Vero, and 293T cells were described previously (31, 32). sh-Luc-HEp-2 cells expressing shRNA against firefly luciferase were described previously (33). The HSV-1 wild-type strain HSV-1(F), recombinant virus YK711 (MEF-gB) encoding gB fused to an MEF tag with myc and Flag epitopes and a TEV protease cleavage site (MEF-gB), recombinant virus YK538 (MEF-UL34) encoding MEF-UL34, and influenza virus [A/WSN/33(H1N1)] have been described previously (34, 35).
Plasmids.
pPEP-gB, pPEP-gH, pPEP-gL, and pPEP-gD, expression plasmids for gB, gH, gL, and gD of HSV-1 KOS strain, respectively, were described previously (36). pcDNA-MEF-CD98hc, an expression plasmid for CD98hc fused to the MEF tag, was constructed by amplifying the CD98hc open reading frame (ORF) by PCR from cDNA synthesized from the total RNA of HEp-2 cells, as described previously (33), and cloning it into pcDNA-MEF (37). pSSCH-CD98hc, used to generate a stable cell line expressing shRNA against the 3′ untranslated region (3′-UTR) of CD98hc mRNA, was constructed as follows. Oligonucleotides 5′-TTTGTCTCATTCTGGTTCTACTGGGCTTCCTGTCACCCAGTAGAACCAGAATCAGACTTTTTTG-3′ and 5′-AATTCAAAAAAGTCTGATTCTGGTTCTACTGGGTGACAGGAAGCCCAGTAGAACCAGAATCAGA-3′ were annealed and cloned into the BbsI and EcoRI sites of pmU6 (35). The BamHI-SalI fragment of the resultant plasmid, containing the U6 promoter and the sequence encoding shRNA against the 3′-UTR of CD98hc, was cloned into the BamHI and SalI sites of pSSCH (35), a derivative of the retrovirus vector pMX containing a hygromycin B resistance gene, to produce pSSCH-CD98hc. Plasmid pSSCH-β1 integrin encoding shRNA against β1 integrin mRNA was constructed by the same procedure as pSSCH-pCD98hc, except using the oligonucleotides 5′-TTTGTAGCAATTTCCATAGTCACAGCTTCCTGTCACTGTGACTATGGAAATTGCTACTTTTTTG-3′ and 5′-AATTCAAAAAAGTAGCAATTTCCATAGTCACAGTGACAGGAAGCTGTGACTATGGAAATTGCTA-3′. pMXs-CD98hc, a retrovirus vector expressing CD98hc, was constructed by cloning the entire CD98hc coding sequence, amplified by PCR from pcDNA-MEF-CD98hc, into pMXs-puro (38). pMXs-β1 integrin expressing β1 integrin was constructed by cloning the entire β1 integrin coding sequence, amplified by PCR from cDNA synthesized from total RNA of HEp-2 cells generated previously (33), into pMXs-puro. pBS-CD98hc was constructed by cloning the EcoRV-XbaI fragment of pcDNA-MEF-CD98hc containing the entire CD98hc coding sequence into pBluescript II KS(+) (Stratagene). pBS-CD98hc-Kan was generated by amplifying the domain of pEPkan-S (39) carrying the I-SceI site and the kanamycin resistance gene by PCR from pEPkan-S and cloning it into the MluI site of pBS-CD98hc. pBS-CD98hc-Kan was used to generate recombinant virus YK715 (MEF-CD98hc) as described below.
Identification of proteins that interact with gB.
HEp-2 cells were infected with YK711 (MEF-gB) at a multiplicity of infection (MOI) of 5, harvested at 24 h postinfection, and lysed in 0.1% NP-40 buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF, 0.1% NP-40) containing a protease inhibitor cocktail (Nacalai Tesque). After centrifugation, the supernatants were immunoprecipitated with an anti-Myc monoclonal antibody, and the immunoprecipitates were incubated with AcTEV protease (Invitrogen). After another centrifugation, the supernatants were immunoprecipitated with an anti-Flag monoclonal antibody, and the immunoprecipitates were washed three times with wash buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF). Flag elution buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5 mg of Flag peptide/ml) was added, and the immunoprecipitates were rotated for 2 h at 4°C. The eluted protein solution was treated with trypsin and analyzed by nano-liquid chromatography tandem mass spectrometry (MS/MS) as described previously (40). For this analysis, we used Q-STAR Elite (AB Sciex) coupled with Dina (KYA Technologies). Proteins were identified by analyzing the MS and the MS/MS signals against the 68,711 protein sequences in the RefSeq human protein database (National Center for Biotechnology Information) and the 74 virus protein sequences based on the complete genome sequence of human herpesvirus 1 strain F (GenBank accession number GU734771) using the Mascot algorithm (v2.4.1; Matrix Science) with the following parameters: variable modifications, oxidation (Met), protein N-terminal acetylation, pyroglutamination (Gln); maximum missed cleavages, 2; peptide mass tolerance, 100 ppm; and MS/MS tolerance, 0.5 Da. Protein identification was based on the criterion of having at least one MS/MS data signal with a Mascot score greater than the threshold (P < 0.05).
Generation of recombinant retroviruses and establishment of cell lines stably expressing shRNA against CD98hc and β1 integrin.
Recombinant retroviruses were generated as described previously (35). sh-CD98hc-HEp-2 and sh-β1 integrin-HEp-2 cells were isolated from HEp-2 cells infected with retrovirus-containing supernatants of Plat-GP cells that had been transfected with pSSCH-CD98hc or pSSCH-β1 integrin, respectively, and selected with 50 μg of hygromycin B/ml.
Establishment of sh-CD98hc-HEp-2 and sh-β1 integrin-HEp-2 cells exogenously expressing CD98hc or β1 integrin, respectively.
sh-CD98hc-HEp-2 and sh-β1 integrin-HEp-2 cells were transduced by infection with retrovirus-containing supernatants of Plat-GP cells that had been transfected with pMXs-CD98hc or pMXs-β1 integrin, respectively, and selected with 2 μg of puromycin/ml, which led to the isolation of sh-CD98hc/CD98hc-HEp-2 and sh-β1 integrin/β1 integrin-HEp2 cells.
Assay for cell viability.
The viability of sh-Luc-HEp-2, shCD98hc-HEp-2, shCD98hc/CD98hc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells was assayed using a Cell Counting Kit-8 (Dojindo) according to the manufacturer's instructions.
Mutagenesis of HSV-1 genomes and generation of recombinant HSV-1.
To generate YK715 (MEF-CD98hc) carrying an expression cassette consisting of the Egr-1 promoter, the entire human CD98hc coding sequence, and bidirectional polyadenylation signals of the HSV-1 UL21 and UL22 genes, a two-step Red-mutagenesis procedure was carried out. For this procedure, we used the primers 5′-GTGAGCTCGGATCCGCCACCATGGACTACAAGGACGACGATGACAAAGATATGGAGCTACAGCCTCCTGA-3′ and 5′-TCGATAAGCTTGATATCGAATTCCTGCAGCCCGGGGGATCCACTAGTTTATCAGGCCGCGTAGGGGAAGC-3′, pBS-CD98hc, and Escherichia coli GS1783 carrying mutant HSV-1 bacterial artificial chromosome, which included the Egr-1 promoter, an MEF tag, and bidirectional polyadenylation signals of the HSV-1 UL21 and UL22 genes in the intergenic region between UL50 and UL51 (33), as described previously (41).
Antibodies.
Commercial mouse monoclonal antibodies to Flag (M2; Sigma), Myc (PL14; MBL), gB (H1817; Virusys), gD (DL6; Santa Cruz Biotechnology), gH (52-S; American Type Culture Collection), gC (H1A022; Virusys), gE (9H3; Abcam), pan-cadherin (CH-19; Sigma), lamin A/C (636; Santa Cruz Biotechnology), α5 integrin (1/CD49e; BD Biosciences), αV integrin (21/CD51; BD Biosciences), and α-tubulin (DM1A; Sigma), commercial rabbit polyclonal antibodies to CD98hc (H-300; Santa Cruz Biotechnology) and VP23 (CAC-CT-HSV-UL18; CosmoBio), and commercial goat polyclonal antibody to β1 integrin (N20; Santa Cruz Biotechnology) were used. The rabbit polyclonal antibodies to Us3, UL34, and UL31 and mouse polyclonal antibody to UL31 were as described previously (34, 42). Rabbit polyclonal antibody to UL31 was used for immunoblotting, and mouse polyclonal antibody to UL31 was used for immunofluorescence.
Immunoblotting.
Immunoblotting analyses were performed as described previously (43).
Immunoprecipitation.
Infected and transfected cells were lysed in NP-40 buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% Nonidet P-40) containing a protease inhibitor cocktail (Nacalai Tesque). The supernatants obtained after centrifugation of the cell lysates were precleared by incubation with protein A-Sepharose beads at 4°C for 30 min and then reacted with the indicated antibodies at 4°C for 2 h. Protein A-Sepharose beads were added to the supernatants, and the reaction continued for another 1 h. Immunoprecipitates were collected by a brief centrifugation, washed extensively with NP-40 buffer, and analyzed by immunoblotting with the indicated antibodies.
Immunofluorescence.
Cells mock infected or infected with the indicated viruses on 35-mm-diameter glass-bottom dishes (Matsunami) were fixed at the indicated times after infection with 4% formaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.1% Triton X-100 in PBS, blocked with 10% human serum (Sigma) in PBS, reacted with the indicated antibodies, reacted with goat anti-mouse IgG-Alexa Fluor 546, goat anti-mouse IgG-Alexa Fluor 488, goat anti-rabbit IgG-Alexa Fluor 488, and/or donkey anti-goat IgG-Alexa Fluor 546 (Invitrogen) and examined with a Zeiss LSM5 laser scanning confocal microscope. All images shown are of a single focal plane.
Electron microscopic analysis.
sh-Luc-HEp-2, shCD98hc-HEp-2, shCD98hc/CD98hc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells infected with wild-type HSV-1(F) at an MOI of 10 for 24 h were examined by ultrathin-section electron microscopy as described previously (42).
RESULTS
Identification of cell proteins that interacted with gB.
To identify host cell proteins that interact with gB, we used tandem affinity purification coupled with MS-based proteomics to analyze lysates of HEp-2 cells infected with recombinant virus HSV-1 YK711 (MEF-gB) encoding MEF-gB (35). These experiments identified one viral and 17 cellular proteins that coimmunoprecipitated with MEF-gB (see Table S1 and S2 in the supplemental material). Of these proteins, we focused on CD98hc here because of its putative role as a regulator of enveloped virus membrane fusion.
Interaction of CD98hc with gB, gH, UL31, UL34, Us3, and β1 integrin.
To confirm and extend the interaction data obtained with MS-based proteomics screening, HEp-2 cells infected with wild-type HSV-1(F) at an MOI of 5 were lysed at 24 h postinfection and immunoprecipitated with anti-gB, anti-gC, or anti-Myc antibody, and the immunoprecipitates were analyzed by immunoblotting with anti-CD98hc antibody. As shown in Fig. 1, anti-gB antibody, but not anti-Myc or anti-gC antibody, coprecipitated endogenous CD98hc and gB from lysates of wild-type HSV-1(F)-infected cells.
FIG 1.
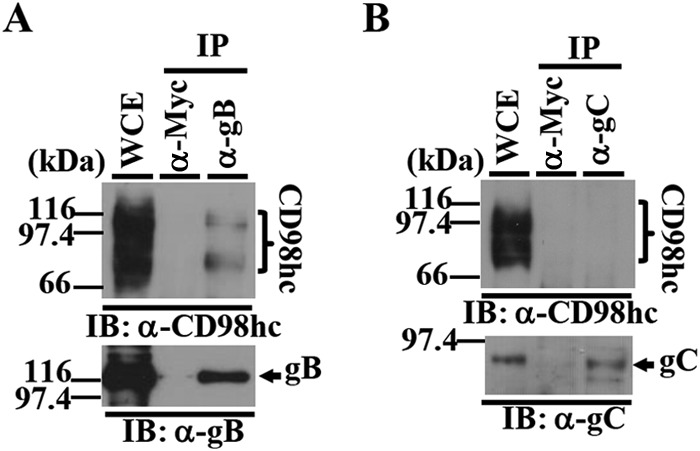
Coimmunoprecipitation of CD98hc with HSV-1 gB in HSV-1-infected cells. (A and B) HEp-2 cells infected with wild-type HSV-1(F) at an MOI of 5 (3 × 107 PFU/ml) for 24 h were harvested, immunoprecipitated (IP) with anti-Myc (α-Myc), anti-gB (α-gB), or anti-gC (α-gC) antibody, and analyzed by immunoblotting (IB) with anti-CD98hc (α-CD98hc) (A and B), anti-gB (A), or anti-gC (B) antibody. Molecular mass markers (in kilodaltons) are shown on the left. WCE, whole-cell extract.
Recombinant virus YK715 (MEF-CD98hc) expressing MEF-tagged CD98hc was then generated and shown to have a growth curve in Vero cells infected at an MOI of 5 almost identical to that of wild-type HSV-1(F) (Fig. 2A and B). HEp-2 cells were then infected with YK715 (MEF-CD98hc) or wild-type HSV-1(F) at an MOI of 5, lysed at 24 h postinfection, and immunoprecipitated with anti-Myc antibody, and the immunoprecipitates were analyzed by immunoblotting with antibodies to various HSV-1 and cellular proteins (Fig. 2C). As shown in Fig. 2C, anti-Myc antibody coprecipitated gB, gH, UL31, UL34, Us3, and β1 integrin with MEF-CD98hc from the lysates of cells infected with YK715 (MEF-CD98hc) but did not coprecipitate gC, gE, and VP23. In contrast, anti-Myc antibody did not coprecipitate any of these viral and cellular proteins from lysates of wild-type HSV-1(F)-infected cells (Fig. 2C). These results indicated that CD98hc specifically formed a complex(es) with gB, gH, UL31, UL34, Us3, and β1 integrin in HSV-1-infected cells. The complex formation of CD98hc and β1 integrin demonstrated here was in agreement with previous reports that CD98hc directly binds to β1 integrin and regulates integrin signaling (23–26).
FIG 2.
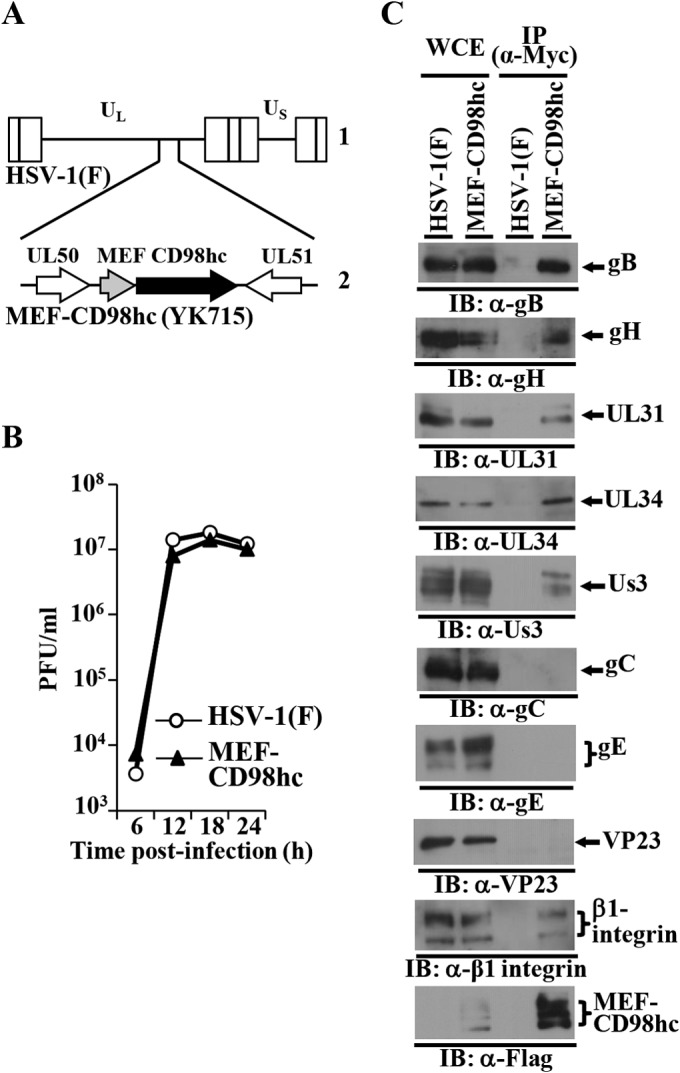
Coimmunoprecipitation of CD98hc with HSV-1 gB, gH, UL31, UL34, and Us3, and β1 integrin in HSV-1-infected cells. (A) Schematic of the genome of wild-type HSV-1(F) (line 1) and of the MEF-CD98hc domain of recombinant virus YK715 (MEF-CD98hc) (line 2). (B) Characterization of YK715 (MEF-CD98hc). Vero cells were infected with wild-type HSV-1(F) or YK715 (MEF-CD98hc) at an MOI of 5 (5 × 106 PFU/ml), and the total virus from cell culture supernatants and infected cells was harvested and assayed on Vero cells. (C) HEp-2 cells infected with wild-type HSV-1(F) or YK715 (MEF-CD98hc) at an MOI of 5 (3 × 107 PFU/ml) for 24 h were harvested, immunoprecipitated with anti-Myc antibody, and analyzed by immunoblotting with the indicated antibodies.
293T cells were then cotransfected with pcDNA-MEF-CD98hc expressing MEF-CD98hc and either (i) pPEP-gB expressing gB, (ii) pPEP-gH and pPEP-gL expressing HSV-1 envelope glycoproteins gH and gL, respectively, or (iii) pPEP-gD expressing HSV-1 envelope glycoprotein gD. The cells were lysed at 48 h posttransfection and immunoprecipitated with anti-Flag antibody, and the immunoprecipitates were analyzed by immunoblotting with antibodies to Flag, gB, gH, and gD. As shown in Fig. 3, anti-Flag antibody coprecipitated gB with MEF-CD98hc from lysates of cells cotransfected with MEF-CD98hc and gB, and gH with MEF-CD98hc from lysates of cells cotransfected with MEF-CD98hc, and both gH and gL but did not coprecipitate gD from lysates of cells cotransfected with MEF-CD98hc and gD. These results confirmed the complex formation of CD98hc with gB and gH and also indicated that gB and gH can interact with CD98hc in the absence of other HSV-1 proteins and independently of each other.
FIG 3.
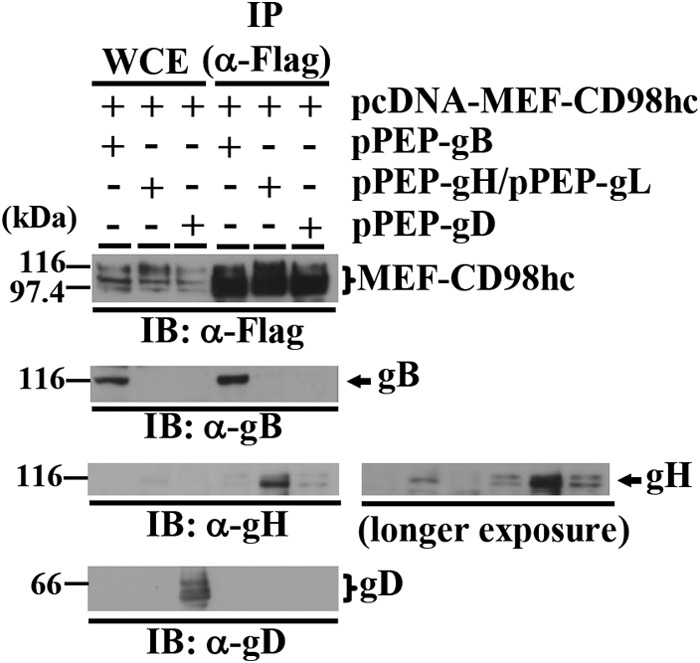
Interaction of CD98hc with gB and gH. 293T cells were cotransfected with pcDNA-MEF-CD98hc and either pPEP-gB, pPEPgH, and pPEP-gL, or pPEP-gD. At 2 days posttransfection, the cells were harvested, immunoprecipitated with anti-Flag antibody, and analyzed by immunoblotting with the indicated antibodies. Molecular mass markers (in kilodaltons) are shown on the left. A longer exposure is also shown for gH.
HSV-1 infection caused the accumulation of CD98hc at the nuclear membrane.
To investigate the subcellular localization of CD98hc, gB, gH, UL31, and UL34 in HSV-1-infected cells, HEp-2 cells were mock infected or infected with wild-type HSV-1(F) or YK538 (MEF-UL34) encoding MEF-tagged UL34 (MEF-UL34) for 24 h, and the localization of CD98hc, gB, gH, UL31, and MEF-UL34 in the infected cells was analyzed by immunofluorescence microscopy. As reported previously (19), CD98hc was detected predominantly at the plasma membrane in mock-infected cells (Fig. 4A). Interestingly, in HSV-1-infected cells, although some CD98hc was still at the plasma membrane, CD98hc was predominantly at the nuclear rim, colocalized with lamins A and C (Fig. 4A), which are INM markers. This accumulation of CD98hc was also observed at 16 h postinfection (data not shown). In contrast, HSV-1 infection had no effect on localization of cadherin, a transmembrane cell adhesion protein, at the plasma membrane (Fig. 4B). In HSV-1-infected cells, CD98hc colocalized with gB, gH, UL31, and MEF-UL34 at the nuclear membrane (Fig. 4C to F). These results indicated that HSV-1 infection specifically induced the accumulation of CD98hc at the nuclear membrane.
FIG 4.
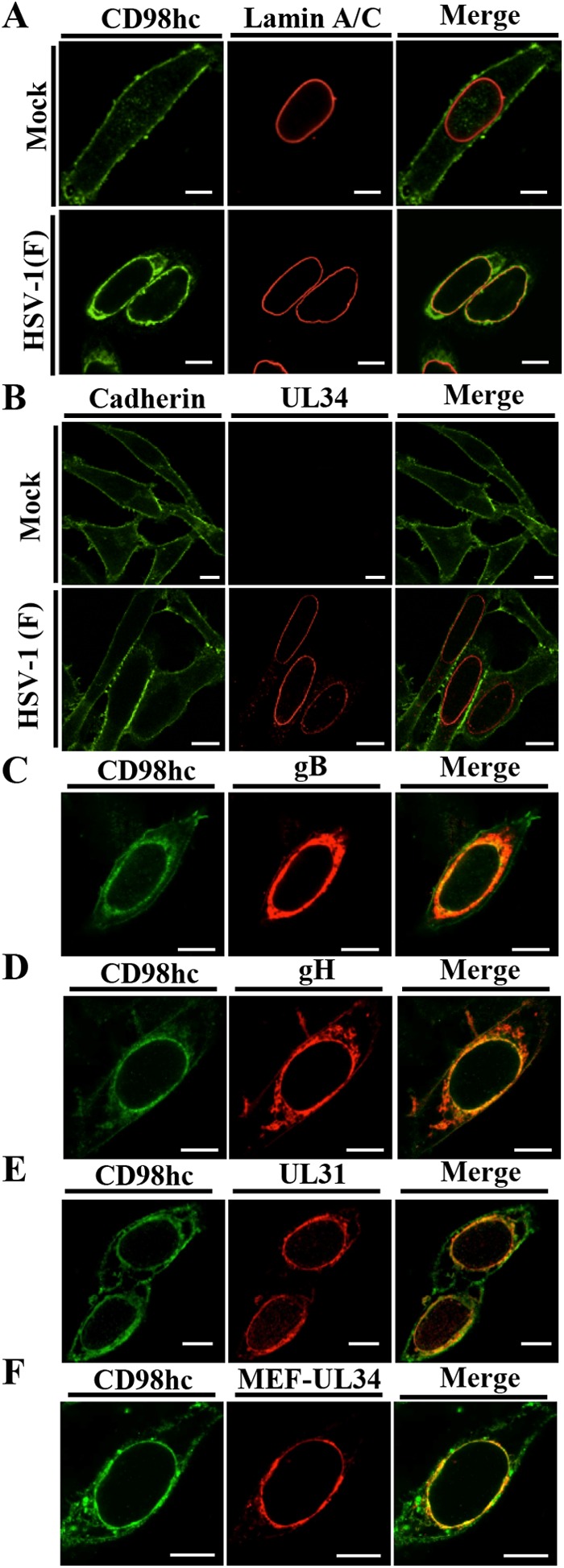
Effect of HSV-1 infection on localization of CD98hc. HEp-2 cells were mock infected (A and B) or infected with wild-type HSV-1(F) (A to E) or YK538 (MEF-UL34) (F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Bars, 10 μm.
Effect of CD98hc knockdown on localization of UL31 and UL34 in HSV-1-infected cells.
The data above, showing both CD98hc coimmunoprecipitation with gB, gH, UL34, UL31, and Us3, and CD98hc accumulation at the nuclear membrane in HSV-1-infection cells, led us to investigate the effect of CD98hc on HSV-1 nuclear egress, since this event takes place at the nuclear membrane (1, 2) and has been reported to be regulated by most of the CD98hc interacting HSV-1 proteins identified above, including gB, gH, UL31, and Us3 (15–18). Therefore, we generated HEp-2 cell lines stably expressing shRNA against the 3′-UTR of CD98hc mRNA (sh-CD98hc-HEp-2) to knock down CD98hc expression, and a control cell line (sh-Luc-HEp-2) expressing shRNA against firefly luciferase mRNA. In addition, to examine whether the phenotype(s) observed in sh-CD98hc-HEp-2 cells was due to a nonspecific effect(s) of the shRNA, we generated sh-CD98hc/CD98hc-HEp2 cells in which CD98hc was expressed exogenously by transduction of sh-CD98hc-HEp-2 cells with a retrovirus vector expressing CD98hc. As shown in Fig. 5A, the expression of endogenous CD98hc in sh-CD98hc-HEp-2 cells was significantly less than in sh-Luc-HEp-2 cells, but the expression of endogenous CD98hc in sh-CD98hc/CD98hc-HEp-2 cells was comparable to that in sh-Luc-HEp-2 cells. The viability of sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells was similar, suggesting that the reduced expression of CD98hc in sh-CD98hc-HEp-2 cells had no effect on HEp-2 cell viability (Fig. 5B).
FIG 5.
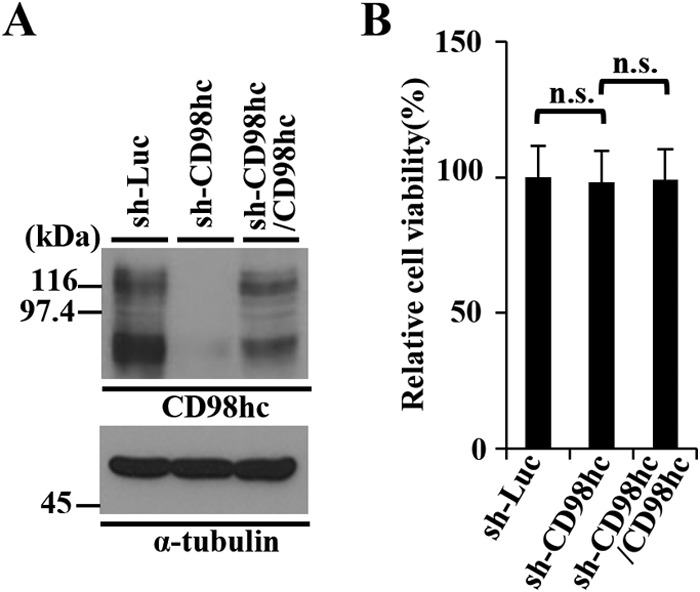
Characterization of sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells. (A) The expression of CD98hc in sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells was analyzed by immunoblotting with anti-CD98hc (top) and anti-α-tubulin (bottom) antibodies. Molecular mass markers (in kilodaltons) are shown on the left. (B) The cell viability of sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells was assayed 24 h after 2 × 104 cells were seeded on 96-well plates. Each value is the mean ± the standard error of the results of three independent triplicate experiments and is expressed relative to the mean for sh-Luc-HEp-2 cells, which was normalized to 100%. Statistical analysis was performed by one-way analysis of variance (ANOVA) and Tukey's test. n.s., not statistically significant.
We then examined the effect(s) of CD98hc on UL31 and UL34, which are critical HSV-1 nuclear egress factors, in HSV-1-infected cells by comparing the localization of these viral proteins in sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells infected with wild-type HSV-1(F). In agreement with previous reports (6), UL31 and UL34 localized at the nuclear membrane with a uniform distribution in most sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells (95 and 92%, respectively) infected with wild-type HSV-1(F) (Fig. 6). However, in a significant fraction (26%) of HSV-1(F)-infected sh-CD98hc-HEp-2 cells, UL31 and UL34 localized both at the nuclear membrane and in aberrant punctate structures adjacent to the nuclear membrane (Fig. 6). These results indicated that CD98hc was required for the proper localization of UL31 and UL34 at the nuclear membrane in a fraction of HSV-1-infected cells.
FIG 6.
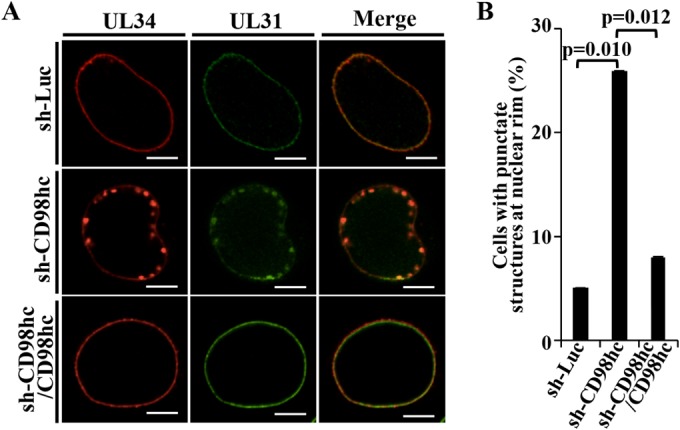
Effect of CD98hc on localization of UL31 and UL34 in HSV-1-infected cells. (A) sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy. Bars, 10 μm. (B) sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy as described for panel A. The percentage of cells with aberrant punctate structures at the nuclear rim was determined. Each value is the mean ± the standard error of the results of three independent experiments. Statistical analysis was performed by one-way ANOVA with Turkey's test. We note that the standard errors are too small to be displayed.
CD98hc was required for efficient HSV-1 de-envelopment.
We next investigated viral morphogenesis in sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells infected with wild-type HSV-1(F) by electron microscopy. As shown in Fig. 7, and Table 1, many (37.4%) of the enveloped HSV-1 virions in infected sh-CD98hc-HEp-2 cells were in membranous invagination structures in the nucleoplasm adjacent to the nuclear membrane, but no enveloped virions were in these invaginations in infected sh-Luc-HEp-2, and few (5.5%) were in these invaginations in infected sh-CD98hc/CD98hc-HEp-2 cells. In infected sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells, 9.5 and 14.3%, respectively, of enveloped HSV-1 virions were in the invaginations or the perinuclear space (Fig. 7B and Table 1). However, in sh-CD98hc-HEp-2 cells, 51.2% of enveloped HSV-1 virions were in the invaginations and perinuclear space, which was 5.4- and 3.6-fold more than in sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells, respectively (Fig. 7B and Table 1). In sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells, 62.5 and 55.6%, respectively, of enveloped HSV-1 virions were on the cell surface, but only 17.2% of enveloped virions were on the cell surface in sh-CD98hc-HEp-2 cells (Table 1). Therefore, although most enveloped HSV-1 virions were in the invaginations and perinuclear space in sh-CD98hc-HEp-2 cells, most were on the surfaces of sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells. The fraction of virus particles that were nuclear capsids in sh-CD98hc-HEp-2 cells (44.7%) was similar to that in sh-Luc-HEp-2 (45.6%) and sh-CD98hc/CD98hc-HEp-2 (48.5%) cells (Table 1). These results indicated that CD98hc knockdown induced membranous invaginations containing primary enveloped virions adjacent to the nuclear membrane and induced aberrant accumulation of primary envelope virions in these invagination structures and in the perinuclear space.
FIG 7.

Effect of CD98hc on HSV-1 nuclear egress. (A) sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 10 (107 PFU/ml), fixed at 24 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. Arrowheads indicate invagination structures containing primary enveloped virions. Nu, nucleus; Cyt, cytoplasm; Nm, nuclear membrane. Bars, 500 nm. (B) sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 10 (107 PFU/ml), fixed at 24 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy as described for panel A. The percentage of enveloped perinuclear virions was determined.
TABLE 1.
Effect of CD98hc knockdown on distribution of enveloped HSV-1(F) virus particles in infected HEp-2 cells
| Knockdown cell | No. of invaginations | % of virus particles in morphogenetic stagea |
Total no. (particles/cells) | |||
|---|---|---|---|---|---|---|
| EV in the intranuclear vesicles | EV in the perinuclear space | EV in the cytoplasm | Extracellular EV | |||
| sh-Luc-HEp-2 | 0 | 0 (0) | 9.5 (70) | 28.0 (206) | 62.5 (460) | 736/9 |
| sh-CD98hc-HEp-2 | 15 | 37.4 (274) | 13.8 (101) | 31.6 (231) | 17.2 (126) | 732/10 |
| sh-CD98hc/CD98hc-HEp-2 | 5 | 5.5 (39) | 8.8 (62) | 30.1 (213) | 55.6 (393) | 707/10 |
Numbers in parentheses are the numbers of virus particles. EV, enveloped virions.
Effect of β1 integrin depletion on localization of UL31 and UL34 in HSV-1-infected cells and on HSV-1 de-envelopment.
Since, as we showed above, CD98hc formed a complex with β1 integrin in HSV-1-infected cells (Fig. 2C) and since anti-CD98hc antibody-dependent enhancement of HIV-mediated cell fusion has been reported to require β1 integrin (28), we also examined whether β1 integrin, like CD98hc, accumulated at the nuclear membrane in HSV-1-infected cells. As shown in Fig. 8, β1 integrin was diffusely localized in the cytoplasm and at the plasma membrane of mock-infected cells as reported previously (44), but was mainly localized at the nuclear rim and colocalized with CD98hc and gB in HSV-1-infected cells. These results indicated that HSV-1 infection induced the accumulation of β1 integrin at the nuclear membrane. We noted that HSV-1 infection did not downregulate the accumulation of CD98hc and β1 integrin proteins in infected cells (data not shown).
FIG 8.
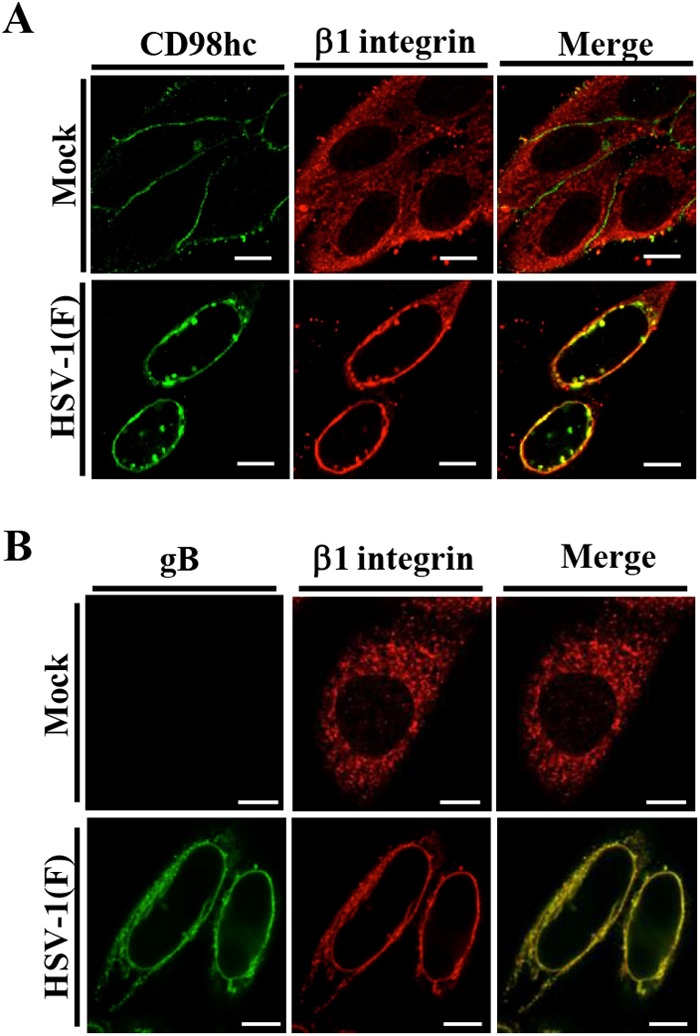
Effect of HSV-1 infection on localization of β1 integrin. (A and B) HEp-2 cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Bars, 10 μm.
We then investigated the effect of β1 integrin on HSV-1 nuclear egress in HSV-1-infected cells. For this study, we generated HEp-2 cell lines stably expressing shRNA against the 3′-UTR of β1 integrin mRNA (sh-β1 integrin-HEp-2) to knock down β1 integrin expression and sh-β1 integrin/β1 integrin-HEp-2 cells in which β1 integrin was expressed exogenously by transduction of sh-β1 integrin-HEp-2 cells with a retrovirus vector expressing β1 integrin. As shown in Fig. 9A, the expression of endogenous β1 integrin in sh-β1 integrin-HEp-2 cells was significantly less than in sh-Luc-HEp-2 cells, but the expression of endogenous β1 integrin in sh-β1 integrin/β1 integrin-HEp-2 cells was comparable to that in sh-Luc-HEp-2 cells. The viability of sh-β1 integrin-HEp-2 cells tended to be slightly lower than that of sh-Luc-HEp-2 and sh-β1 integrin/β1 integrin-HEp-2 cells (Fig. 9B), as reported elsewhere (45). As shown in Fig. 10 and 11, and Table 2, the HSV-1 nuclear egress phenotype in β1 integrin knockdown cells was almost identical to that in CD98hc knockdown cells, including induction of (i) aberrant localization of UL31 and UL34 at the nuclear membrane in a fraction of HSV-1-infected cells, (ii) membranous invagination structures containing primary enveloped virions adjacent to the nuclear membrane, and (iii) aberrant accumulation of primary enveloped virions in the perinuclear space and in the induced invagination structures.
FIG 9.
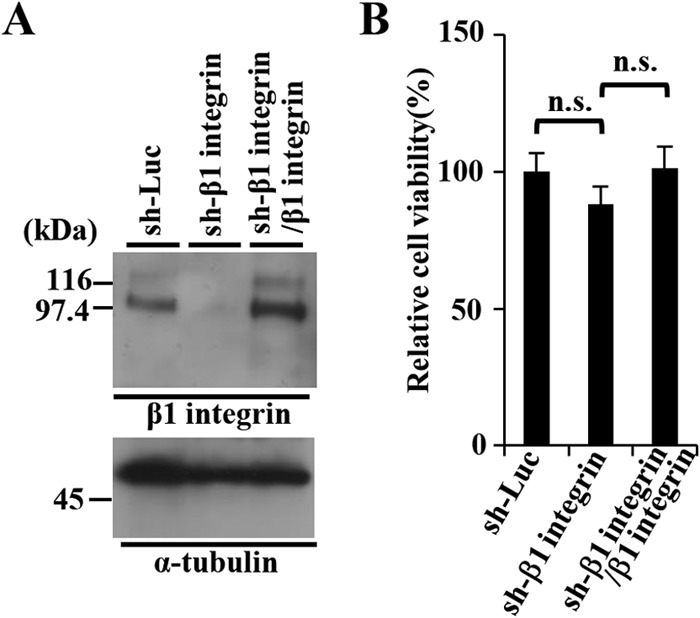
Characterization of sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells. (A) Expression of β1 integrin in sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells was analyzed by immunoblotting with anti-β1 integrin (top) and anti-α-tubulin (bottom) antibodies. Molecular mass markers (in kilodaltons) are shown on the left. (B) Cell viability of sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells assayed 24 h after 2 × 104 cells were seeded on 96-well plates. Each value is the mean ± the standard error of the results of three independent triplicate experiments and is expressed relative to the mean for sh-Luc-HEp-2 cells, which was normalized to 100%. Statistical analysis was performed by one-way ANOVA and Tukey's test. n.s., not statistically significant.
FIG 10.
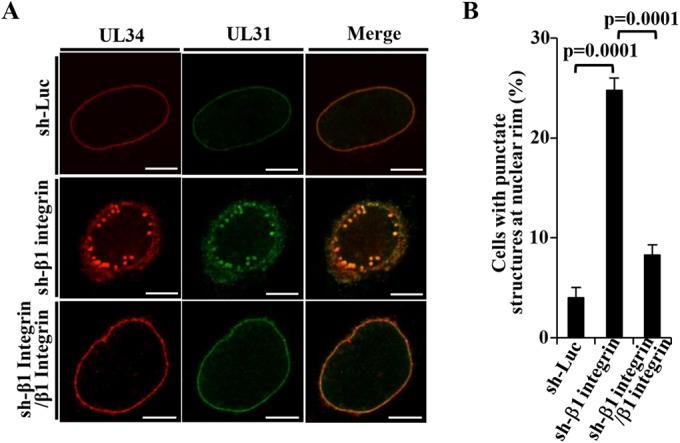
Effect of β1 integrin on localization of UL31 and UL34 in HSV-1-infected cells. (A) sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy. Bars, 10 μm. (B) sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy as described for panel A. The percentage of cells with aberrant punctate structures at the nuclear rim was determined. Each value is the mean ± the standard error of the results of three independent experiments. Statistical analysis was performed by one-way ANOVA and Tukey's test.
FIG 11.

Effect of β1 integrin on HSV-1 nuclear egress. (A) sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 10 (107 PFU/ml), fixed at 24 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. Arrowheads indicate invagination structures containing primary enveloped virions. Nu, nucleus; Cyt, cytoplasm; NM, nuclear membrane. Bars, 500 nm. (B) sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 10 (107 PFU/ml), fixed at 24 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy as described for panel A. The percentage of enveloped perinuclear virions was determined.
TABLE 2.
Effect of β1 integrin knockdown on distribution of enveloped HSV-1(F) virus particles in infected HEp-2 cells
| Knockdown cell | No. of invaginations | % of virus particles in morphogenetic stagea |
Total no. (particles/cells) | |||
|---|---|---|---|---|---|---|
| EV in the intranuclear vesicles | EV in the perinuclear space | EV in the cytoplasm | Extracellular EV | |||
| sh-Luc-HEp-2 | 6 | 10.4 (76) | 1.4 (10) | 16.9 (122) | 71.3 (516) | 724/9 |
| sh-β1 integrin-HEp-2 | 20 | 35.8 (282) | 15.5 (122) | 12.6 (99) | 36.2 (285) | 788/9 |
| sh-β1 integrin/β1 integrin-HEp-2 | 4 | 3.3 (26) | 9.0 (71) | 11.3 (89) | 80.3 (633) | 819/9 |
Numbers in parentheses are the numbers of virus particles. EV, enveloped virions.
Effect of CD98hc and β1 integrin on HSV-1 replication.
To investigate the effects of CD98hc and β1 integrin in HSV-1 replication, sh-Luc-HEp-2, sh-CD98hc-HEp-2, sh-CD98hc/CD98hc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 0.05 or 5. The titers of total intracellular and extracellular progeny virus production and of extracellular progeny virus production were then assayed at various times postinfection. As shown in Fig. 12A, in cells infected at an MOI of 0.05, wild-type HSV-1(F) replicated in sh-CD98hc-HEp-2 cells less efficiently than in sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells. The total progeny virus titer in sh-CD98hc-HEp-2 cells at 72 h postinfection was significantly less than in sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells (8.3- and 5.6-fold, respectively) (Fig. 12B). In agreement with these results, the extracellular progeny virus titer of wild-type HSV-1(F) in sh-CD98hc-HEp-2 cells infected at an MOI of 0.05 was reduced compared to that in sh-Luc-HEp-2 or sh-CD98hc/CD98hc-HEp-2 cells, and the titer of extracellular progeny virus in sh-CD98hc-HEp-2 cells at 72 h postinfection was significantly less than in sh-Luc-HEp-2 and sh-CD98hc/CD98hc-HEp-2 cells (16- and 8.1-fold, respectively) (Fig. 12C and D). Similarly, CD98hc knockdown significantly reduced both total intracellular and extracellular progeny virus production and extracellular progeny virus production in HEp-2 cells infected at an MOI of 5 (Fig. 12E to H). Furthermore, CD98hc knockdown had little effect on influenza virus replication in HEp-2 cells (Fig. 13A). These results indicated that proper expression of CD98hc was specifically required for efficient HSV-1 replication.
FIG 12.

Effect of CD98hc and on HSV-1 replication. (A to H) sh-Luc-HEp-2, sh-CD98hc-HEp-2, and sh-CD98hc/CD98hc-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 0.05 (5 × 104 PFU/ml). At the indicated times postinfection, total virus from cell culture supernatants and infected cells (A, B, E, and F) and extracellular progeny virus from cell culture supernatants (C, D, G, and H) was harvested and assayed. Virus titers in panels A and C at 72 h postinfection and in panels E and G at 36 h postinfection are shown in panels B, D, F, and H, respectively. Each value is the mean ± the standard error of the results of three independent experiments. Statistical analysis was performed by one-way ANOVA and Tukey's test.
FIG 14.
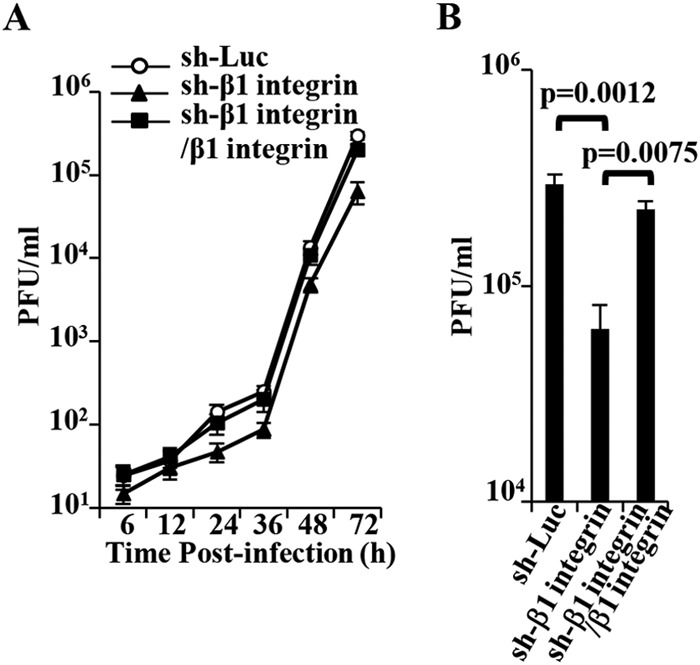
Effect of CD98hc and β1 integrin on HSV-1 replication. (A) sh-Luc-HEp-2, sh-β1 integrin-HEp-2, and sh-β1 integrin/β1 integrin-HEp-2 cells were infected with wild-type HSV-1(F) at an MOI of 0.05 (5 × 104 PFU/ml). At the indicated times postinfection, extracellular virus from cell culture supernatants were harvested and assayed. Each value is the mean ± the standard error of the results of three independent experiments. (B) Virus titers in panel A at 72 h postinfection. Each value is the mean ± the standard error of the results of three independent experiments. Statistical analysis was performed by one-way ANOVA and Tukey's test.
In agreement with the effect of CD98hc knockdown on HSV-1 extracellular progeny virus production and replication of influenza virus as described above, wild-type HSV-1(F) extracellular progeny virus production in sh-β1 integrin-HEp-2 cells infected at an MOI of 0.05 was significantly lower (4.7- and 3.6-fold, respectively) than in sh-Luc-HEp-2 and sh-β1 integrin/β1 integrin-HEp-2 cells (Fig. 14A and B). β1 integrin knockdown had little effect on influenza virus replication in HEp-2 cells (Fig. 13B). In contrast, the growth curve of total intracellular and extracellular progeny virus production in sh-β1 integrin-HEp-2 cells infected with wild-type HSV-1(F) at an MOI of 5 or 0.05 and the growth curve of extracellular progeny virus production of wild-type HSV-1(F) in sh-β1 integrin-HEp-2 cells infected at an MOI of 5 were similar to those in sh-Luc-HEp-2 cells (data not shown). These results indicated that β1 integrin was required for efficient extracellular viral production in cells infected at an MOI of 0.05.
FIG 13.
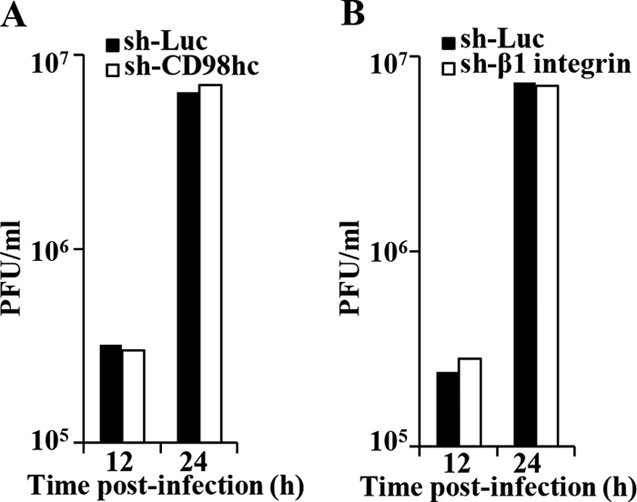
Effect of CD98hc and β1 integrin on influenza virus replication. (A) sh-Luc-HEp-2 and sh-CD98hc-HEp-2 cells were infected with influenza virus at an MOI of 0.01 (5 × 104 PFU/ml). At the indicated times postinfection, extracellular virus from cell culture supernatants was harvested and assayed. (B) sh-Luc-HEp-2 and sh-β1 integrin-HEp-2 cells were infected with influenza virus at an MOI of 0.01. At the indicated times postinfection, extracellular virus from cell culture supernatants was harvested and assayed.
Effect of HSV-1 infection on αV and α5.
To investigate whether CD98hc and β1 integrin formed a complex with αV and α5 integrins, which were reported to interact with β1 integrin and/or gH/gL (46, 47), in HSV-1-infected cells, HEp-2 cells were infected with YK715 (MEF-CD98hc) or wild-type HSV-1(F) at an MOI of 5, lysed at 24 h postinfection, and immunoprecipitated with anti-Myc antibody, and the immunoprecipitates were analyzed by immunoblotting with antibodies to αV, α5, and β1 integrins. As shown in Fig. 15A, anti-Myc antibody coprecipitated β1 integrin as described in Fig. 2C but did not coprecipitate αV and α5 integrins. In contrast, anti-Myc antibody did not coprecipitate any of these integrins from lysate of wild-type HSV-1(F)-infected cells (Fig. 15A). We also examined the subcellular localization of αV and α5 integrins in wild-type HSV-1(F)-infected HEp-2 cells by immunofluorescence microscopy. As shown in Fig. 15B and C, αV and α5 integrins were diffusely localized in the cytoplasm and at the plasma membrane of mock-infected cells as reported previously (48). In agreement with Fig. 4, CD98hc was detected predominantly at the plasma membrane in mock-infected cells and at the nuclear rim in HSV-1(F)-infected cells (Fig. 15B and C). In contrast, unlike CD98hc and β1 integrin, HSV-1(F) infection had little effect on the localization of αV and α5 integrins, and the accumulation of these integrins at the nuclear membrane was not observed in HSV-1(F)-infected cells (Fig. 15B and C). These results suggested that a complex of CD98hc and β1 integrin did not further form a complex with αV and α5 integrins in HSV-1-infected cells and that αV and α5 integrins, which can interact with β1 integrin and/or gH/gL, did not accumulate at the nuclear membrane in HSV-1-infected cells, unlike CD98hc and β1 integrin.
FIG 15.
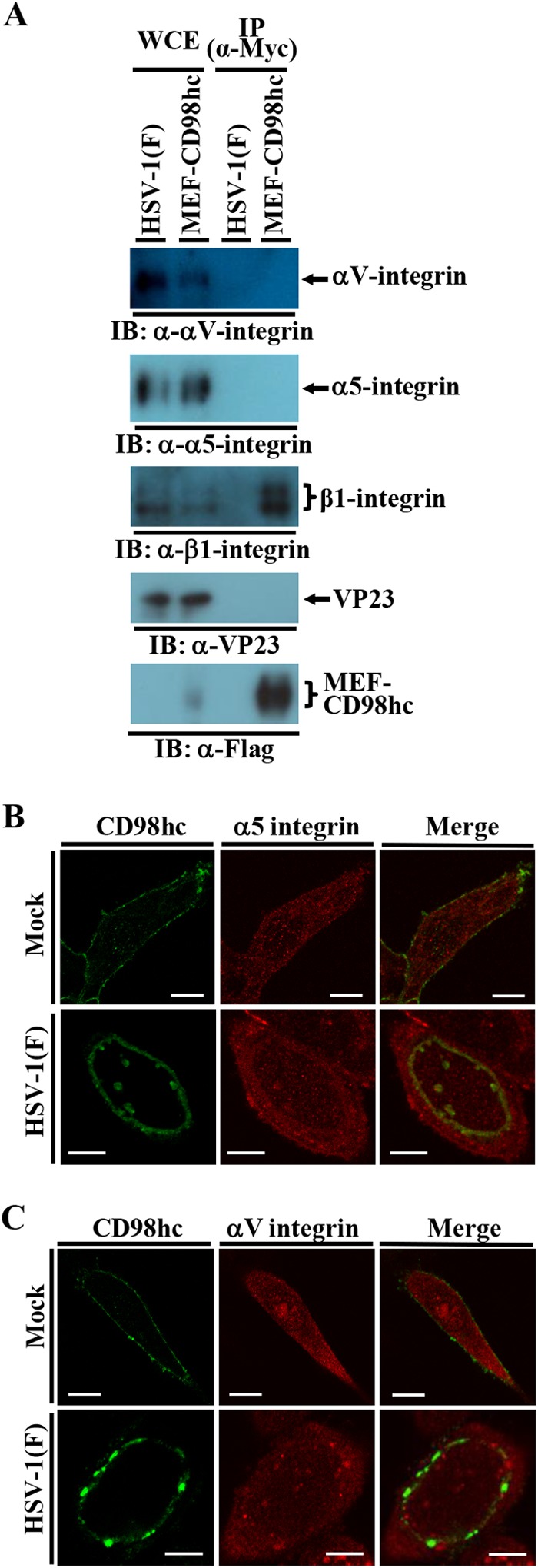
Effect of HSV-1 infection on αV and α5 integrins. (A) HEp-2 cells infected with wild-type HSV-1(F) or YK715 (MEF-CD98hc) at an MOI of 5 (3 × 107 PFU/ml) for 24 h were harvested, immunoprecipitated with anti-Myc antibody, and analyzed by immunoblotting with the indicated antibodies. (B and C) HEp-2 cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5 (5 × 106 PFU/ml), fixed at 24 h postinfection, permeabilized, stained with the indicated antibodies, and examined by confocal microscopy. Bars, 10 μm.
DISCUSSION
A CD98hc regulatory function in membrane fusion was suggested by the observations that some monoclonal anti-CD98hc antibodies were shown to enhance NDV- and HIV-mediated cell-cell fusion and to inhibit HPIV-2-mediated cell-cell fusion. This antibody-regulated cell-cell fusion required fusogenic envelope glycoproteins and/or their cofactors, including the F and HN proteins of NDV and HPIV-2, and HIV gp120 (27–30). In addition, CD98hc appeared to regulate membrane fusion in the absence of any viral envelope glycoproteins, based on the observations that anti-CD98hc monoclonal antibodies induced the fusion of human peripheral blood monocytes (49) and the depletion of CD98hc inhibited the fusion of cytotrophoblast-derived BeWo cells mediated by forskolin (50). The HIV-mediated and monocyte cell fusion induced by anti-CD98hc antibody was shown to be inhibited by antibody to β1 integrin, a binding partner of CD98hc, suggesting that the CD98hc function in membrane fusion involved β1 integrin. This CD98hc/β1 integrin function suggested that CD98hc/β1 integrin may also regulate de-envelopment fusion between HSV-1 envelopes and the ONM during HSV-1 nuclear egress. In agreement with this hypothesis, we have shown here that CD98hc formed a complex with β1 integrin in HSV-1-infected cells and HSV-1 infection induced the accumulation of CD98hc and β1 integrin at the nuclear membrane, as found for HSV-1 recruitment of cellular PKC to the nuclear rim as a regulator for primary envelopment during herpesvirus nuclear egress (5). In addition, we showed that both CD98hc and β1 integrin knockdown reduced extracellular progeny virus production in cells infected at an MOI of 0.05, led to aberrant localization of HSV-1 NEC, UL31, and UL34 at the nuclear membrane, induced membranous invagination structures containing primary enveloped virions adjacent to the nuclear membrane, and produced aberrant accumulation of primary enveloped virions in the perinuclear space and in the induced invagination structures. Although we cannot eliminate the possibility that CD98hc and β1 integrin played a role(s) of in extracellular progeny HSV-1 production other than in HSV-1 de-envelopment, the regulatory role of CD98hc and β1 integrin in HSV-1 de-envelopment detected here may contribute to efficient progeny virus production. We need to note that there is an additional possibility that CD98hc and β1 integrin independently function in HSV-1 de-envelopment. As described above, CD98hc has been reported to interact with several light chains of CD98 and β3 integrin (22, 26), whereas β1 integrin was shown to interact with several α integrins, including α1, α3, α5, α6, α8, α10, and αV integrins (47). CD98hc and β1 integrin might independently regulate HSV-1 de-envelopment by interacting with their binding partners. We showed that α5 and αV integrins did not accumulate at the nuclear membrane, unlike CD98hc and β1 integrin, and were not coimmunoprecipitated with CD98hc and β1 integrin in HSV-1-infected cells. These results might support the possibility that CD98hc/β1 integrin regulates HSV-1 de-envelopment. However, we cannot completely eliminate the additional possibility. β1 integrin and CD98hc might independently function in HSV-1 de-envelopment by interacting with the other α integrins and with the known CD98hc binding partners, respectively. Further studies to clarify whether CD98hc and β1 integrin regulate HSV-1 de-envelopment synergistically or independently will be needed and interesting. These studies are under investigation in these laboratories.
We have shown here that CD98hc formed a complex(es) with gB, gH, UL31, UL34, and Us3: most of these HSV-1 proteins (gB, gH, UL31, and Us3) have been reported to be important for HSV-1 de-envelopment in HSV-1-infected cells (15–18). The phenotypes of HSV-1 viruses carrying a mutation(s) in gB, gH, UL31, and/or Us3 in de-envelopment fusion reported previously (15–18) appeared to be similar to that of the HSV-1 de-envelopment fusion in infected CD98hc and β1 integrin knockdown cells shown in the present study. Thus, HSV-1 with a mutation in Us3 abrogating either Us3 expression or catalytic activity, HSV-1 with a mutation in UL31 abrogating Us3 phosphorylation of UL31, HSV-1 with mutations in gB and gH abrogating both gB and gH expression, and HSV-1 with mutations in gB and gH abrogating Us3 phosphorylation of gB and expression of gH were shown to (i) mislocalize UL31 and UL34 at the nuclear membrane, (ii) induce membranous invagination structures containing primary enveloped virions adjacent to the nuclear membrane, and/or (iii) accumulate aberrant primary enveloped virions in the perinuclear space and in the induced invagination structures (17, 18, 51). In particular, in previous reports, ca. 75 to 85% of enveloped progeny virions accumulated in the perinuclear space and in the induced invaginations in HaCaT and Vero cells infected with an HSV-1 mutant lacking both gB and gH or with an HSV-1 Us3 mutant, respectively, in contrast to the 38 to 74% of enveloped progeny virions that were on the cell surface in wild-type HSV-1-infected cells (16, 17). Similar to those results, we found that 51% of enveloped progeny virions accumulated in the perinuclear space and induced invaginations in wild-type HSV-1-infected, CD98hc-depleted or β1 integrin-depleted HEp-2 cells, in contrast to 63 to 71% of enveloped progeny virions that were on the cell surface of infected control HEp-2 cells. Therefore, we propose a model in which CD98hc interacts with β1 integrin and this complex promotes the fusion process in HSV-1 de-envelopment by forming a complex(es) with the HSV-1 regulatory proteins, including gB, gH, UL31, UL34, and Us3.
It is noteworthy that an HSV-1 mutant lacking both gB and gH has been reported to exhibit a significant defect in viral de-envelopment (17), as also described above, but HSV-1 mutants lacking either gB or gH showed only a small reduction in de-envelopment (17). This suggested that gB and gH acted in a redundant fashion to mediate HSV-1 de-envelopment fusion. We have shown here that CD98hc coimmunoprecipitated with gB or gH in cells transiently overexpressing CD98hc and either gB or gH, respectively. This result indicated that gB and gH interacted with CD98hc independently of each other and was in agreement with the previous report (17) on HSV-1 mutants lacking gB and/or gH. The fusion components for viral de-envelopment, which are probably induced at the nuclear membrane in HSV-1-infected cells, should include a host cell fusogenic protein(s) and perhaps cellular cofactor(s) such as CD98hc and β1 integrin, whose crystal structures have no structural homology with any known fusion proteins (52, 53). This is suggested because (i) HSV-1 gB, the only viral fusogenic protein that has structural homology to a vesicular stomatitis virus fusion protein, glycoprotein G, plays only a minor role in viral de-envelopment fusion (17), as described above, and (ii) nuclear egress of the Drosophila cellular RNP complex may only require cellular fusion components at the nuclear membrane (3). CD98hc and β1 integrin may provide experimental tools for identifying the cellular fusogenic protein(s).
In conclusion, we identified CD98hc and β1 integrin, which are accumulated at the nuclear membrane by HSV-1 infection, as novel cellular regulators for HSV-1 de-envelopment fusion during viral nuclear egress. To our knowledge, this is the first report of cellular proteins required for efficient herpesvirus de-envelopment. Further studies, including identification of cellular fusogenic protein(s) mediating HSV-1 de-envelopment fusion during viral nuclear egress and elucidation of the mechanism for accumulation of CD98hc and β1 integrin at the nuclear membrane in HSV-1-infected cells, are needed to understand this unique vesicle-mediated nucleocytoplasmic transport process. Both CD98hc and β1 integrin have been reported to be involved in various membrane fusion events, including enveloped virus-mediated cell fusion, the entry of various enveloped viruses involving fusion between the cell membrane and the virus envelope, monocyte cell fusion, and trophoblast cell fusion (27, 28, 30, 49, 50, 54). Therefore, this and further studies may also provide insight into the mechanism(s) of membrane fusion events, as well as herpesvirus de-envelopment fusion and perhaps the cellular pathways for nucleocytoplasmic transport of macromolecular complexes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants for Scientific Research from the Japan Society for the Promotion of Science, a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases, and a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports, and Technology (MEXT) of Japan, as well as grants from the Takeda Science Foundation, the Tokyo Biochemical Research Foundation, and the Asahi Glass Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00741-15.
REFERENCES
- 1.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 2.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 3.Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V. 2012. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149:832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 5.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott/The Williams & Wilkins Co, Philadelphia, PA. [Google Scholar]
- 8.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81:6459–6470. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J Virol 82:8094–8104. doi: 10.1128/JVI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108:14276–14281. doi: 10.1073/pnas.1108564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J. 2012. Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol 86:594–598. doi: 10.1128/JVI.05988-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78:399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura E. 1999. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem 274:3009–3016. doi: 10.1074/jbc.274.5.3009. [DOI] [PubMed] [Google Scholar]
- 20.Verrey F, Jack DL, Paulsen IT, Saier MH Jr, Pfeiffer R. 1999. New glycoprotein-associated amino acid transporters. J Membr Biol 172:181–192. doi: 10.1007/s002329900595. [DOI] [PubMed] [Google Scholar]
- 21.Chillaron J, Roca R, Valencia A, Zorzano A, Palacin M. 2001. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Physiol Renal Physiol 281:F995–F1018. [DOI] [PubMed] [Google Scholar]
- 22.Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. 2004. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch Eur J Physiol 447:532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 23.Fenczik CA, Sethi T, Ramos JW, Hughes PE, Ginsberg MH. 1997. Complementation of dominant suppression implicates CD98 in integrin activation. Nature 390:81–85. doi: 10.1038/36349. [DOI] [PubMed] [Google Scholar]
- 24.Zent R, Fenczik CA, Calderwood DA, Liu S, Dellos M, Ginsberg MH. 2000. Class- and splice variant-specific association of CD98 with integrin beta cytoplasmic domains. J Biol Chem 275:5059–5064. doi: 10.1074/jbc.275.7.5059. [DOI] [PubMed] [Google Scholar]
- 25.Feral CC, Nishiya N, Fenczik CA, Stuhlmann H, Slepak M, Ginsberg MH. 2005. CD98hc (SLC3A2) mediates integrin signaling. Proc Natl Acad Sci U S A 102:355–360. doi: 10.1073/pnas.0404852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prager GW, Feral CC, Kim C, Han J, Ginsberg MH. 2007. CD98hc (SLC3A2) interaction with the integrin beta subunit cytoplasmic domain mediates adhesive signaling. J Biol Chem 282:24477–24484. doi: 10.1074/jbc.M702877200. [DOI] [PubMed] [Google Scholar]
- 27.Ito Y, Komada H, Kusagawa S, Tsurudome M, Matsumura H, Kawano M, Ohta H, Nishio M. 1992. Fusion regulation proteins on the cell surface: isolation and characterization of monoclonal antibodies which enhance giant polykaryocyte formation in Newcastle disease virus-infected cell lines of human origin. J Virol 66:5999–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohta H, Tsurudome M, Matsumura H, Koga Y, Morikawa S, Kawano M, Kusugawa S, Komada H, Nishio M, Ito Y. 1994. Molecular and biological characterization of fusion regulatory proteins (FRPs): anti-FRP MAbs induced HIV-mediated cell fusion via an integrin system. EMBO J 13:2044–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohgimoto S, Tabata N, Suga S, Nishio M, Ohta H, Tsurudome M, Komada H, Kawano M, Watanabe N, Ito Y. 1995. Molecular characterization of fusion regulatory protein-1 (FRP-1) that induces multinucleated giant cell formation of monocytes and HIV gp160-mediated cell fusion. FRP-1 and 4F2/CD98 are identical molecules. J Immunol 155:3585–3592. [PubMed] [Google Scholar]
- 30.Okamoto K, Tsurudome M, Ohgimoto S, Kawano M, Nishio M, Komada H, Ito M, Sakakura Y, Ito Y. 1997. An anti-fusion regulatory protein-1 monoclonal antibody suppresses human parainfluenza virus type 2-induced cell fusion. J Gen Virol 78(Pt 1):83–89. [DOI] [PubMed] [Google Scholar]
- 31.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor α. J Virol 83:4520–4527. doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato A, Hirohata Y, Arii J, Kawaguchi Y. 2014. Phosphorylation of herpes simplex virus 1 dUTPase upregulated viral dUTPase activity to compensate for low cellular dUTPase activity for efficient viral replication. J Virol 88:7776–7785. doi: 10.1128/JVI.00603-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 36.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B, Jazag A, Ohta M, Imamura J, Ikenoue T, Ijichi H, Kawabe T, Isobe T, Omata M. 2006. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 25:633–642. [DOI] [PubMed] [Google Scholar]
- 38.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 40.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J Virol 87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tharmalingam S, Daulat AM, Antflick JE, Ahmed SM, Nemeth EF, Angers S, Conigrave AD, Hampson DR. 2011. Calcium-sensing receptor modulates cell adhesion and migration via integrins. J Biol Chem 286:40922–40933. doi: 10.1074/jbc.M111.265454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang R, Li J, Lyte K, Yashpal NK, Fellows F, Goodyer CG. 2005. Role for beta1 integrin and its associated alpha3, alpha5, and alpha6 subunits in development of the human fetal pancreas. Diabetes 54:2080–2089. doi: 10.2337/diabetes.54.7.2080. [DOI] [PubMed] [Google Scholar]
- 46.Gianni T, Salvioli S, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2013. αvβ6- and αvβ8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog 9:e1003806. doi: 10.1371/journal.ppat.1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takada Y, Ye X, Simon S. 2007. The integrins. Genome Biol 8:215. doi: 10.1186/gb-2007-8-5-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH. 2009. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials 30:1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higuchi S, Tabata N, Tajima M, Ito M, Tsurudome M, Sudo A, Uchida A, Ito Y. 1998. Induction of human osteoclast-like cells by treatment of blood monocytes with anti-fusion regulatory protein-1/CD98 monoclonal antibodies. J Bone Miner Res 13:44–49. doi: 10.1359/jbmr.1998.13.1.44. [DOI] [PubMed] [Google Scholar]
- 50.Kudo Y, Boyd CA. 2004. RNA interference-induced reduction in CD98 expression suppresses cell fusion during syncytialization of human placental BeWo cells. FEBS Lett 577:473–477. doi: 10.1016/j.febslet.2004.10.047. [DOI] [PubMed] [Google Scholar]
- 51.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fort J, de la Ballina LR, Burghardt HE, Ferrer-Costa C, Turnay J, Ferrer-Orta C, Uson I, Zorzano A, Fernandez-Recio J, Orozco M, Lizarbe MA, Fita I, Palacin M. 2007. The structure of human 4F2hc ectodomain provides a model for homodimerization and electrostatic interaction with plasma membrane. J Biol Chem 282:31444–31452. doi: 10.1074/jbc.M704524200. [DOI] [PubMed] [Google Scholar]
- 53.Nagae M, Re S, Mihara E, Nogi T, Sugita Y, Takagi J. 2012. Crystal structure of alpha5beta1 integrin ectodomain: atomic details of the fibronectin receptor. J Cell Biol 197:131–140. doi: 10.1083/jcb.201111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stewart PL, Nemerow GR. 2007. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol 15:500–507. doi: 10.1016/j.tim.2007.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.