

ABSTRACT
Thermus thermophilus bacteriophage P23-77 is the type member of a new virus family of icosahedral, tailless, inner-membrane-containing double-stranded DNA (dsDNA) viruses infecting thermophilic bacteria and halophilic archaea. The viruses have a unique capsid architecture consisting of two major capsid proteins assembled in various building blocks. We analyzed the function of the minor capsid protein VP11, which is the third known capsid component in bacteriophage P23-77. Our findings show that VP11 is a dynamically elongated dimer with a predominantly α-helical secondary structure and high thermal stability. The high proportion of basic amino acids in the protein enables electrostatic interaction with negatively charged molecules, including nucleic acid and large unilamellar lipid vesicles (LUVs). The plausible biological function of VP11 is elucidated by demonstrating the interactions of VP11 with Thermus-derived LUVs and with the major capsid proteins by means of the dynamic-light-scattering technique. In particular, the major capsid protein VP17 was able to link VP11-complexed LUVs into larger particles, whereas the other P23-77 major capsid protein, VP16, was unable to link VP11-comlexed LUVs. Our results rule out a previously suggested penton function for VP11. Instead, the electrostatic membrane association of VP11 triggers the binding of the major capsid protein VP17, thus facilitating a controlled incorporation of the two different major protein species into the assembling capsid.
IMPORTANCE The study of thermophilic viruses with inner membranes provides valuable insights into the mechanisms used for stabilization and assembly of protein-lipid systems at high temperatures. Our results reveal a novel way by which an internal membrane and outer capsid shell are linked in a virus that uses two different major protein species for capsid assembly. We show that a positive protein charge is important in order to form electrostatic interactions with the lipid surface, thereby facilitating the incorporation of other capsid proteins on the membrane surface. This implies an alternative function for basic proteins present in the virions of other lipid-containing thermophilic viruses, whose proposed role in genome packaging is based on their capability to bind DNA. The unique minor capsid protein of bacteriophage P23-77 resembles in its characteristics the scaffolding proteins of tailed phages, though it constitutes a substantial part of the mature virion.
INTRODUCTION
Viruses from extremely hot environments need to develop special strategies to cope with high temperature. Hence, they provide a valuable source for the detection of novel enzymes for biotechnology and industrial processes. Their relative simple structure makes them an ideal model system for studying life at high temperatures. With respect to extreme thermophilic bacteria, the virus studied best on the structural level is P23-77, a strictly lytic phage infecting Thermus thermophilus ATCC 33923 (1, 2, 3). P23-77 is a representative of the novel Sphaerolipoviridae, comprising viruses infecting members of two domains of life, namely, extreme halophilic archaea of the family Halobacteriaceae and thermophilic bacteria of the family Thermaceae (4). All the family members have similar virion morphologies. The virus particles are spherical, tailless, and 50 to 80 nm in diameter. An inner lipid membrane is located between the capsid and the double-stranded DNA (dsDNA) genome, which is circular in the case of P23-77. The capsid is mainly built from two major capsid proteins (MCPs).
The recently solved structures of the P23-77 MCPs (5) revealed a structural relationship to other tailless icosahedral dsDNA viruses with internal membranes, such as the enterobacterial phage PRD1 (6), the marine bacteriophage PM2 (7), and the archaeal virus Sulfolobus turreted icosahedral virus (STIV) (8). Of these viruses, the assembly of bacteriophage PRD1 has been the most extensively examined (9). The morphogenesis starts with a lipid vesicle containing viral membrane proteins pinching off from the host cytoplasmic membrane as capsid-associated proteins assemble along it, resulting in the formation of an empty procapsid. The correct assembly is assisted by nonstructural viral assembly factors and the host GroEL-GroES chaperonin complex (10, 11, 12). The linear dsDNA genome is packaged through a special vertex by a viral packaging ATPase (13). DNA packaging into empty procapsids is the main mechanism for packaging linear genomes or unit length genome increments from concatemeric DNA (14, 15). Viruses with circular genomes, such as marine bacteriophages PM2 and P23-77, might follow a different packaging strategy without the involvement of an intermediate procapsid state (7, 16).
The internal membrane fulfills two functions in tailless icosahedral dsDNA viruses. First, it is involved in host infection. This is most evident in PRD1, where the membrane forms a tube through which the genome is injected into the host cell (17). Second, the internal membrane acts in concert with membrane proteins as a structural scaffold on which the capsid proteins assemble (12). Integral membrane proteins function as tape measures for the size of the membrane vesicle (7, 18, 19). The lipids are acquired selectively from the cytoplasmic membrane of the host during particle assembly in order to produce an appropriate membrane curvature (3, 20, 21, 22). The different lipid compositions of the outer and inner leaflets of the membrane enforce the association of the outermost DNA layer with the inner leaflet. The negative charge of the outer leaflet in turn promotes capsid assembly by electrostatic attraction to the positively charged bottoms of the capsomers (23, 24). Capsomers are formed either by a single MCP or by two MCPs, as in the cases of P23-77 and other sphaerolipoviruses.
Recently, a capsid model was developed for P23-77, in which capsomers are formed by various building blocks of the two major capsid proteins (5). The capsid model provides insights into the complex arrangement of the two MCPs in the capsid, suggesting a novel assembly mode not observed in other icosahedral viruses. The mechanism controlling the correct incorporation of the two different protein species, however, remained to be elucidated. Furthermore, knowledge of the proteins residing at the VP17-surrounded pentagonal vertices was elusive. The 5-fold axes of some icosahedral viruses are occupied by penton proteins that exhibit a highly conserved single beta-barrel core fold (7, 18, 25, 26). Among the 10 structural proteins identified in P23-77 virus particles, only three have been found to be associated with the capsid (3): the two MCPs, VP16 and VP17, and the 22-kDa minor capsid protein VP11. The minor capsid protein has no homologs in related viruses, prompting us to characterize it in detail. Experimental results indicate that the protein is a predominantly α-helical protein dimer. Thus, it does not exhibit the 5-fold symmetry typically associated with a penton protein. Moreover, it interacts with lipids and—in the presence of lipids—also with VP17, but not VP16. Based on these findings, we propose a role of VP11 as an assembly factor. A strong electrostatic association of the protein with the membrane triggers the binding of VP17, followed by VP16, to form the capsid. Thus, VP11 exhibits a scaffolding-protein-like function. However, it is retained in the virion.
MATERIALS AND METHODS
Plasmid construction.
The full-length 564-bp open reading frame 11 (ORF11) gene sequence was amplified by PCR with genomic P23-77 virus DNA as the template using primers creating an NdeI restriction site (underlined) at the 5′ terminus (5′AATAATTACATATGGTGATCCGCAGACAGCAGATCTG3′) and an XhoI site (underlined) at the 3′terminus (5′AACTGCAGCTCGAGTCATCGCCTCCTTTTGGGCTGGTA3′) of the gene. The purified PCR product was digested with NdeI and XhoI (Fermentas) and inserted into expression vector pET22b+ (Novagen), creating the VP11 expression plasmid pAP124. The integrity of the gene sequence was verified by DNA sequencing with an ABI Prism genetic analyzer 3100 (Life Technologies).
Protein expression and purification.
Recombinant VP11 was synthesized in Escherichia coli strain BL21(DE3) (Novagen) in LB medium supplemented with 150 μg/ml ampicillin. Cells were grown at 37°C to an optical density at 550 nm (OD550) of 0.7, and gene expression was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The cells were further incubated at 28°C for 20 h, after which they were washed in lysis buffer (50 mM Bicine, pH 8.5). The cell pellets were suspended in 1/50 of the original volume in ice-cold lysis buffer containing 1 mM Pefabloc SC protease inhibitor (Roche), disrupted with an EmulsiFlex-C3 (Avestin) homogenizer, and centrifuged (112,000 × g; 2 h; 5°C). The supernatant was briefly sonicated on ice to decrease the viscosity of the solution before filtering through 0.45-μm polyethersulfone filters (Sartorius Minisart) for subsequent chromatographic purification. All chromatography steps were performed at room temperature (RT) with an ÄKTAprime plus liquid chromatography system (GE Healthcare Bio-Science AB). The filtered crude extract was loaded onto a HiPrepSP HP 16/10 cation-exchange column (GE Healthcare Bio-Science AB) equilibrated in lysis buffer at a 1-ml/min flow rate (lysis buffer). After a washing step with 30% elution buffer (50 mM Bicine, pH 8.5, 2 M NaCl), VP11 was eluted from the column with a 400-ml linear gradient of 30% to 80% elution buffer at a 3-ml/min flow rate. Fractions were analyzed under denaturing conditions by 15% SDS-PAGE (27). A pool of fractions containing nearly pure VP11 was washed and concentrated to a final volume of 7 ml at 4°C with 50 mM Tris, pH 7.5, 1 M NaCl using a 10-kDa molecular weight cutoff (10 K) Amicon Ultra-15 filter device (Millipore). The filtrate was passed through a HiLoad 26/60 Superdex 200 prep-grade size exclusion chromatography (SEC) column (GE Healthcare Bio-Science AB) equilibrated with 50 mM Tris, pH 7.5, 1 M NaCl at a 1-ml/min flow rate. Protein fractions corresponding to distinct absorption peaks in the chromatogram were collected in separate pools and subjected to buffer exchange and concentration in 50 mM HEPES, pH 7.5, with a 10 K Amicon Ultra-15 filter device. Protein samples were stored at 8°C or −20°C.
The concentration of the purified VP11 was determined by measuring the absorbance at 280 nm with an ND-1000 spectrophotometer (NanoDrop Technologies) using an extinction coefficient of 19,940 M−1 cm−1 and a theoretical molecular mass of 22,060 Da. Gel images were captured on a ChemiDoc gel documentation system (Bio-Rad). The verification of VP11 by peptide mass fingerprinting was carried out by the Proteomics Unit of the Institute of Biotechnology at the University of Helsinki.
DNA mobility shift assays.
The P23-77 genomic DNA was isolated from virus particles (0.5 mg/ml) by treatment with SDS and protease, followed by multiple extractions with phenol (28). Plasmid DNA (pUC19) was isolated using standard methods (29). P23-77 genomic DNA (20 μg/ml, digested into multiple fragments with restriction enzyme PstI) was incubated with increasing amounts of VP11 (0 to 150 μg/ml) in 30 μl 50 mM Tris, pH 7.5, for 10 min at 37°C. After 10 min centrifugation at 14,000 rpm, the RT supernatants were taken, and the pellets were resuspended in 30 μl 50 mM Tris, pH 7.5, supplemented with 1% SDS. The supernatant and pellet fractions were analyzed by 0.8% Tris-acetate-EDTA (TAE) agarose gel electrophoresis.
Immunodetection of VP11 in virus particles.
P23-77 virus particles were produced in T. thermophilus ATCC 33923 and purified by rate zonal sucrose gradient centrifugation (a linear 5 to 20% [wt/vol] gradient), followed by equilibrium centrifugation in 1.3 mg/ml CsCl2 as described previously (2). The purified virus particles were collected by differential centrifugation. The pellets were suspended in 20 mM Tris, pH 7.5, 5 mM MgCl2, 150 mM NaCl and stored at −20°C in SDS sample buffer containing either 1% or 5% β-mercaptoethanol. Polyclonal VP11 antibodies were generated in rabbit with native protein obtained from SEC purification (Storkbio Ltd.). Virus proteins were separated by SDS-15% PAGE and blotted on an Immobilon-P transfer membrane (EMD Millipore) at 4.0 mA/cm2 for 25 min using the TE-77 ECL Semi-Dry Transfer Unit (Amersham Biosciences) and a three-buffer system (30). The membrane was blocked overnight at 8°C in TEN (50 mM Tris, pH 7.4, 5 mM EDTA, 150 mM NaCl) plus 1% Tween 20. Immunolabeling was done using the Snap i.d. 2.0 protein detection system (EMD Millipore) according to the manufacturer's instructions with 1:30,000-diluted primary antibody against VP11 in TEN plus 1% Tween 20, followed by incubation with 0.26 μg/ml secondary antibody (anti-rabbit-horseradish peroxidase [HRP]; Dako) in TEN plus 0.1% Tween 20 for 10 min at RT on a platform shaker. The HRP detection was done with enhanced chemiluminescence (ECL) substrate (SuperSignalWest Pico chemiluminescence substrate; Thermo Scientific) according to the supplier's instructions. Immunoblot images were captured (20-min exposure time) on a ChemiDoc gel documentation system (Bio-Rad).
Estimation of the VP11 copy number per virus particle.
The copy number of VP11 per virus particle was estimated from Coomassie-stained 15% Tricine-SDS polyacrylamide gels (31) with the Quantity One program (Bio-Rad). The program detects each protein band on the gel individually and calculates its relative intensity. The disc value for background reduction was 10, and the width of lanes was 30 mm. The most abundant protein, VP16, was used as a reference band, and its measured intensity was stated as 1,080 protein copies. The measured intensities of protein bands were calibrated with the molecular masses of the proteins. The calibrated intensities of VP11 and VP17 were divided by the calibrated intensity of VP16, and the result was multiplied by 1,080 to obtain the copy numbers of VP11 and VP17.
CD measurements.
Circular dichroism (CD) spectra were recorded with quartz cuvettes of 1-cm path length (Hellma) using a J-715 spectropolarimeter (JASCO) equipped with a Peltier temperature controller and a circulating water bath. The protein concentration was 0.5 μM in 2 ml phosphate-buffered saline (PBS), pH 7.4. VP11 spectra were corrected by subtracting the buffer spectra, measured under the same conditions. Far-UV–CD spectra were recorded from 195 to 255 nm with the following parameters: 50-nm/min scan speed, 5-nm bandwidth, 1-s response, and three accumulations. Thermal scans were performed at 222 nm in the temperature range from 4°C to 85°C under the following conditions: 5-nm bandwidth, 8-s response, 2°C/min temperature slope, and an average spectrum produced from two independent consecutive scans.
DSC measurements.
VP11 was analyzed using a capillary VP-differential scanning calorimetry (DSC) instrument (MicroCal; Malvern Instruments Ltd.) with a protein concentration of 50 μM in 20 mM Tris, pH 7.5. The solution was degassed prior to measurement. Samples were heated from 20°C to 120°C at a scanning rate of 2°C/min. The feedback mode was set to low, and the filter period was 5 s. The melting temperature (Tm) and calorimetric heat change (ΔH) were obtained by subtracting the baseline and using the Levenberg-Marquardt nonlinear least-square method on the data using MicroCal Origin 7.0 software (Malvern Instruments Ltd.).
Production of Thermus large unilamellar vesicles (T-LUVs).
T. thermophilus cells were grown for 20 h at 70°C and 200 rpm in Thermus full medium containing 8 g polypeptone, 4 g yeast extract, and 2 g NaCl per liter. Fifteen milliliters of cell culture was pelleted by centrifugation, and lipids were extracted according to the method of Bligh and Dyer (32). The lipid extract obtained was vacuum dried and stored at −20°C.
For preparation of T-LUVs, dried lipid extract was resuspended in 800 μl 20 mM Tris, pH 7.5, and hydrated by six consecutive freeze-thaw cycles, during which the samples were sequentially incubated at 65°C for 10 min, vigorously vortexed, and frozen in liquid nitrogen. Homogeneous T-LUVs were prepared by extrusion of the lipid dispersion through polycarbonate membranes at 65°C using an Avanti Mini-Extruder (Avanti Polar Lipids) system according to the manufacturer's instructions with 10 repeated extrusions through a 400-nm membrane, followed by 15 repeated extrusions through a 100-nm membrane. The T-LUVs were stored at 8°C.
DLS and zeta potential measurements.
Dynamic-light-scattering (DLS) and zeta potential measurements were carried out using a Zetasizer Nano ZS instrument (Malvern) with the following settings: 0.9455-cP dispersant viscosity, 1.33 refractive index, 173° backscatter measurement angle, and 22°C. All measurements were performed in 20 mM Tris, pH 7.5, with freshly prepared proteins. The P23-77 major capsid proteins VP16 and VP17 were prepared as described previously (33). Myoglobin from equine skeletal muscle and bovine serum albumin (BSA) were obtained from Sigma-Aldrich. Protein stock solutions of 1 mg/ml in buffer were centrifuged before measurements at 13,000 rpm for 5 min. The protein concentrations were 1 mg/ml for molecular weight and zeta potential analysis and 0.3 mg/ml for the determination of the apparent average hydrodynamic diameter in combination with T-LUVs. T-LUVs were diluted for measurements as follows: 1:5 (zeta potential), 1:10 (T-LUV–protein interaction), or 1:500 (kinetics of T-LUV interactions with VP11 and VP17). The results were analyzed with Zetasizer software v.7.03 (Malvern).
RESULTS
VP11 sequence analysis.
Initial studies have shown that the VP11-encoding gene (ORF11) is exclusively found in P23-77 and has no homologs in the related sphaerolipovirus IN93 or associated proviruses (4, 34, 35). The only similarity we detected in data bank searches was to a protein sequence derived from Meiothermus timidus, indicating the presence of a P23-77-like provirus. ORF11 is located in the genome in an area with variable gene content, which comprises genes with predicted function in DNA packaging and virus exit (4, 34) (Fig. 1A). Notably, although there is no similarity on the amino acid sequence level to VP11, bacteriophage IN93 and provirus TP1 encode putative proteins with characteristics similar to those of VP11 (see below). The corresponding genes—ORF9 in the case of IN93 and ORF21 in the case of TP1—are located at genomic positions similar to that of ORF11. Those proteins in turn show similarities to virion-associated putative genome-packaging proteins of hyperthermophilic archeal viruses, such as Sulfolobus spindle-shaped virus 1 (SSV1) protein VP2 (36) and Sulfolobus turreted icosahedral virus 2 (STIV2) protein B72 (37), as well as to a predicted protein of Archaeoglobus veneficius SNP6 putative provirus (38).
FIG 1.

VP11 distribution and sequence analysis. (A) (Top) Localization of ORF11 in the P23-77 genome. The circular genome is shown linearized for simpler representation. Predicted transcriptional units and genes encoding structural proteins are indicated. (Bottom) Corresponding genome regions in related virus IN93 and proviruses (4). ORFs encoding gene products with similar predicted functions are depicted in identical colors. ORFs without predicted functions are shown in black (shared by all members), gray (shared by at least two members), and white (present in only one member). (B) Secondary-structure and disorder prediction was according to the PSIPRED and DISOPRED 3 programs (41, 43). Alpha-helical regions are depicted as red bars. Beta sheets are marked with blue arrows and areas with predicted disorder with dashed lines.
Computational analysis (39, 40, 41, 42, 43, 44) classified VP11 as a highly basic protein (pI = 10.71) with a high content of α-helices and without transmembrane domains (Fig. 1B). VP11 was predicted to be highly structured, with disordered regions mainly restricted to a few residues at the N and C termini, which according to the DISOPRED 3 program (43) are predicted to be involved in protein-protein interactions. The N- and C-terminal disordered regions are highly positively charged (arginine and lysine residues), suggesting that electrostatic repulsions render them structurally disordered.
Expression and purification of recombinant VP11.
Large amounts of soluble recombinant protein were produced in E. coli. As expected for a highly basic protein, VP11 bound to a cation-exchange (CIX) column. VP11 was eluted by a slow rise in the NaCl concentration in the elution buffer, which removed most of the impurities. The elution took place at an NaCl concentration of 0.75 M. The eluted protein fractions were pooled and further purified by SEC in Tris buffer containing 1 M NaCl. The protein eluates mainly contained a 25-kDa protein species, identified as VP11 by peptide mass fingerprinting, and a small amount of an ∼50-kDa protein species, corresponding to a VP11 dimer, as demonstrated below. The OD260/OD280 ratio of VP11, obtained from UV-visible (Vis) spectra of the purified protein samples, was 0.57, suggesting the protein preparations were free of DNA contamination.
VP11 oligomerization and copy number.
The 50-kDa band visible in denaturing protein gels of recombinant protein preparations corresponds to the size of a VP11 dimer (Fig. 2A, I). Proteins forming strong disulfide bonds have been shown to remain in a multimeric state even after boiling in standard SDS sample buffer containing 1% SDS and 1% β-mercaptoethanol but could be dissociated with a higher reducing agent concentration (45). Therefore, a sample of VP11 was boiled in SDS sample buffer containing a high (5%) concentration of the reducing agent β-mercaptoethanol. Under these conditions, the ∼50-kDa band—corresponding to the putative VP11 dimer—vanished completely from the gel, leaving only the ∼25-kDa monomeric VP11 band (Fig. 2A, II). The addition of 0.1 M dithiothreitol (DTT) had a similar effect, whereas elevating the SDS concentration to 5% did not affect the relative ratio of the two bands (data not shown).
FIG 2.
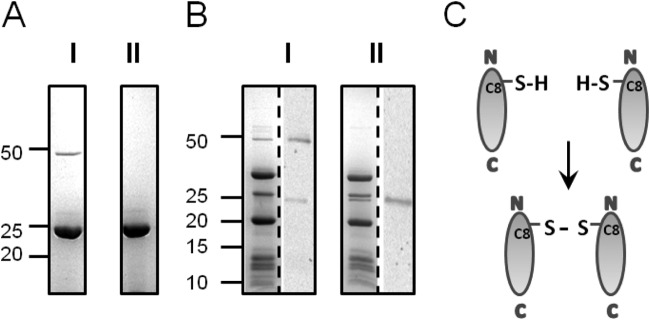
VP11 dimer formation. (A) Coomassie blue-stained SDS-15% polyacrylamide gel of purified recombinant VP11 boiled in SDS sample buffer with 1% β-mercaptoethanol (I) or 5% β-mercaptoethanol (II). (B) P23-77 virus particles prepared with 1% β-mercaptoethanol (I) and 5% β-mercaptoethanol (II) as for panel A. The protein pattern of a Coomassie-stained gel is shown on the left side of each gel, and Western blots incubated with VP11 polyclonal antibody are shown on the right side. (A and B) Protein sizes are indicated in kilodaltons according to the All Blue Precision Plus protein standard (Bio-Rad). (C) Schematic presentation of intermolecular disulfide bond formation between cysteine residues at the N terminus of VP11.
Next, we analyzed the oligomeric state of native VP11 derived from virus particles. Freshly prepared virions boiled in 1% β-mercaptoethanol showed a 50-kDa band in SDS-PAGE, similar to recombinant VP11 samples (Fig. 2B, I). A polyclonal antibody against purified recombinant VP11 detected the 50-kDa protein band in Western blot analysis and—to a lesser extent—also a 25-kDa protein band that was not visible in the corresponding SDS gel. On the other hand, SDS gels of particles boiled with 5% β-mercaptoethanol lacked the 50-kDa band yet showed an additional band at ∼25 kDa (Fig. 2B, II). The VP11 antibody recognized only the 25-kDa protein band in the Western blot. Moreover, the intensity of the signal was equal to the total intensity of both signals in the Western blot obtained from samples treated with 1% β-mercaptoethanol (Fig. 2B, I). These findings rule out cross-reactivity of the VP11 antibody and clearly demonstrate that the 50-kDa band corresponds to a VP11 dimer. The protein dimerizes through an intermolecular disulfide bridge between single cysteine residues at position 8 on the N terminus (Fig. 2C). The dimer formation is not an artifact of recombinant expression but also occurs in virus particles. Recombinant VP11 was purified mostly in monomeric form, while the dimeric form dominated in virus particles. Higher multimerization states were not detected in our experiments either in SEC or in gradient centrifugation experiments (data not shown).
The copy number of VP11 in the capsid was estimated by measuring the intensities of Coomassie-stained protein bands in Tricine-SDS polyacrylamide gels. According to the P23-77 virus capsid model (5), there are 18 copies of 19-kDa VP16 and 9 copies of 32-kDa VP17 per asymmetric unit. The capsid consists of 60 asymmetric units, thus accounting for 1,080 copies of VP16 and 540 copies of VP17 in total. The band intensities were 36.2% for VP16, 29.7% for VP17, and 5.7% for VP11. Taking into account the molecular masses of the capsid proteins and setting the VP16 band intensity as a copy number of 1,080, the estimated copy numbers were 527 for VP17 and 147 for VP11. The value for VP17 is in the range of the theoretical value (540 copies). The value for VP11 suggests a distribution of either 2 or 3 copies per asymmetric unit, with an absolute copy number of 120 or 180 per capsid. The results are based on the assumption that Coomassie stain binds equally to the proteins studied here.
VP11 is a heat-stable α-helical protein with refolding capacity.
Secondary-structure prediction proposed a large content of α-helices for VP11 (Fig. 1B). The theoretical assumption was confirmed by far-UV–CD spectroscopy (Fig. 3A): The spectrum showed two negative dichroic minima at 208 nm and—less prominent—at 222 nm, characteristic of α-helical proteins. Below 208 nm, the signal increased toward a positive range. The spectrum below 205 nm could not be recorded due to high absorption of buffer substances, especially at high temperatures. A gradual increase of the temperature led to a successive, but not complete, loss of helicity, indicating a partially folded state of VP11 even at 85°C.
FIG 3.

VP11 secondary structure and heat stability. (A) UV CD spectra at various temperatures. The CD spectra were recorded in PBS buffer in the far-UV region (200 to 255 nm) in the 4°C to 85°C temperature range. The spectra are represented as molar ellipticity obtained by normalizing the measured CD spectra with the mean molar residue concentration of VP11. (B) Thermal scan of VP11. The temperature was increased from 4°C to 85°C with a 2°C per min increment (unfolding reaction [black line]) and decreased back to 4°C under the same conditions (refolding reaction [gray line]). (C) CD spectra of folded VP11 measured prior to the unfolding reaction (black line) and after refolding (gray line). (D) Unfolding profile of VP11 protein measured with a capillary VP-DSC instrument. The difference of heat capacity (Cp) between the sample and the reference cell was recorded in the 40°C to 120°C range (black line). Curve fitting was done using the non-2-state curve-fitting model (gray line).
We performed thermal-CD scans to analyze the thermal stability of VP11 (Fig. 3B). The CD signal lacked a sigmoidal profile characteristic of cooperative protein unfolding. Instead, the signal decreased continuously between 4°C and 85°C without a clear transition, a behavior characteristic of proteins with a high content of disordered regions (46). The predicted content of unstructured regions for VP11 is rather low, and the far-UV–CD signal profile contradicts the presence of large disordered regions. Thus, dynamic transitions between α-helical structures and disordered regions could explain this type of CD behavior. The partial unfolding was completely reversed when the sample was cooled from 85°C to 4°C under the same conditions. The CD spectrum measured after the heating-cooling cycle was almost identical, indicating a nearly complete reversal of the partial unfolding (Fig. 3C). The result suggests a partially loose α-helical structure for VP11.
We used DSC to analyze the unfolding and refolding of VP11 at even higher temperatures. For this purpose, the protein was gradually heated from 20°C to 120°C, followed by cooling to 20°C under the same conditions. The DSC profile (Fig. 3D) revealed a single transition peak with a Tm of 84.4°C. A second scan after cooling (refolding reaction) did not show any transition peak, indicating irreversible denaturation of the protein. It thus seems that VP11 can withstand partial unfolding, caused by a short exposure to temperatures around the melting point, without losing the capability to refold. Further heating, on the other hand, causes transition to conformations that become structurally trapped when cooled. Such a phenomenon has been observed in several other proteins (47, 48). The estimated melting point of approximately 85°C for the minor capsid protein is in line with the melting temperatures of the major capsid proteins (49), demonstrating the extreme heat stability of all P23-77 virus capsid-associated proteins characterized so far.
VP11 binds DNA in a nonspecific manner.
Next, we were interested in studying the capability of VP11 to interact with various macromolecules, such as DNA, lipids, and proteins. We performed mobility shift assays to analyze the DNA-binding capability of purified VP11. During the incubation of DNA with increasing amounts of VP11, we observed precipitation of the VP11-DNA complex, which prevented the DNA from migrating into the gel. Therefore, DNA-protein mixtures were centrifuged, and the pellet fractions were treated with SDS to release the protein from the DNA and to enable the migration of fragments in the electric field (Fig. 4). At protein concentrations lower than the 20-μg/ml DNA concentration used in the experiments, all the DNA was found in the supernatant and the DNA migrated as without protein. At equal concentrations (20 μg/ml), the DNA was partitioned between the supernatant and pellet fractions, with a larger fraction in the pellet. At protein concentrations higher than 20 μg/ml, all the DNA was found in the pellet fraction. The intensities of all the bands vanished to the same extent, regardless of whether virus DNA (Fig. 4), plasmid DNA, linearized DNA molecules, or DNA molecules digested into multiple fragments were used as the template (data not shown). The results indicate a predominantly electrostatic binding of VP11 to DNA without DNA sequence specificity. The positively charged VP11 neutralizes the negatively charged DNA, thus disabling electrophoretic movement. However, at a low protein/DNA mass ratio, the amount of protein should be too small for complete neutralization of the DNA, and one would expect a shift of bands in agarose gels. This was not observed. Instead, the bands vanished completely from the gel at a protein/DNA mass ratio of approximately 1:1, and precipitation of the protein-DNA complex occurred. VP11 was found to bind DNA very tightly, since a high concentration of SDS (1%) was necessary to resolve the complex.
FIG 4.
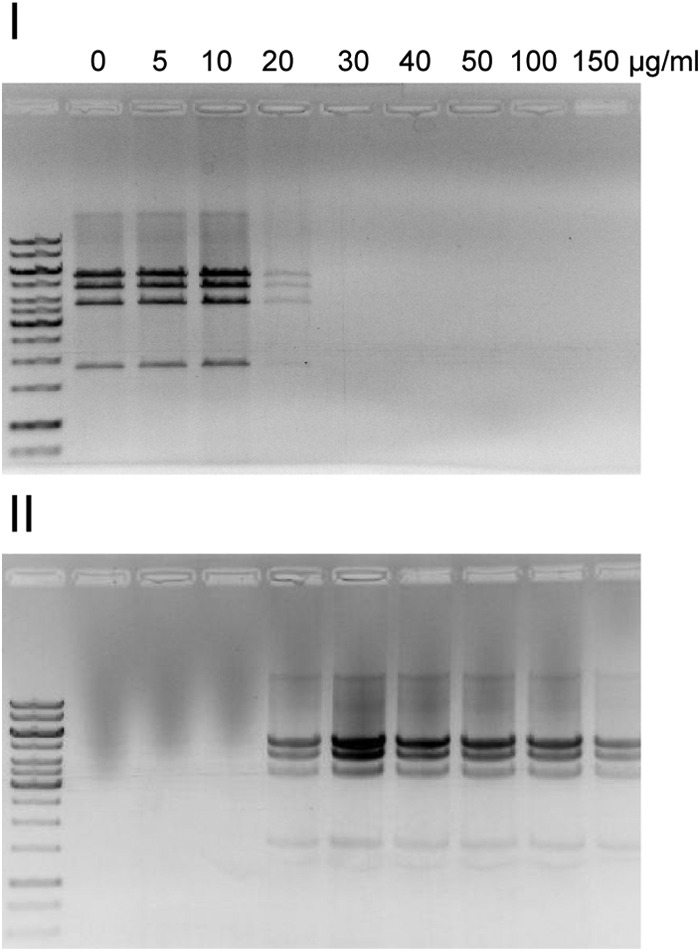
VP11 interaction with DNA. For DNA mobility shift assays, 20 μg/ml of PstI-digested genomic P23-77 DNA was incubated with increasing concentrations of purified VP11 for 10 min at 37°C. After centrifugation, the supernatant was taken and the pellet containing the precipitated DNA-protein complex was resuspended in buffer containing 1% SDS, which released the DNA from the protein. The supernatant (I) and corresponding pellet (II) fractions were analyzed on a 1% agarose gel. The marker in the far-left lanes was a GeneRuler 1-kb DNA ladder (Thermo Scientific).
VP11 associates with T-LUVs.
It has been shown that the major capsid proteins could be released from the P23-77 virion in denaturing agents, such as urea and guanidine hydrochloride (3). Lowering the pH to 6.0 also dissociated the minor capsid protein VP11, in addition to the major capsid proteins. This finding, along with a predicted lack of transmembrane helices, suggests localization of VP11 underneath the capsid shell peripheral to the membrane. Thus, we used DLS to investigate the interaction of VP11 with T-LUVs.
The lipid composition of P23-77 differs from that of the host. The viral membrane contains neutral lipids and phosphoglycolipids, whereas glycolipids, a substantial component of the host lipid membrane, are nearly completely omitted (3). However, we assumed the lipid composition of T-LUVs to be similar enough to functionally mimic the viral membrane. Lipids were isolated from the host strain T. thermophilus ATCC 33923, and T-LUVs were prepared by extrusion through 100-nm-pore-diameter membranes. Vesicles were highly stable and did not show any sign of fusion or aggregation during storage for at least 2 weeks. The hydrodynamic diameter of the T-LUVs was approximately 120 nm. The hydrodynamic diameter of VP11 was 8.4 ± 1.1 nm. Thus, the measured size refers to a molecular mass of 27.4 kDa in the case of a linear protein or 96.7 kDa in the case of a globular protein. Therefore, the DLS analysis suggests a rather linear shape for VP11. The zeta potential of T-LUVs was −59.5 ± 3.96 mV, a value characteristic of negatively charged phospholipids, such as phosphatidylglycerol, indicating a negative surface charge for Thermus-derived lipid vesicles. As expected for a positively charged protein, the zeta potential of pure VP11 was 21.8 ± 2.54 mV. Addition of VP11 to T-LUVs shifted the zeta potential to 34.1 ± 1.43 mV, indicating the capability of the protein to adsorb to Thermus lipid vesicles. On the other hand, protein-protein interactions of capsid-associated proteins were not observed in DLS measurements, either between VP11 and the major capsid proteins VP16 and VP17 or between the MCPs alone (data not shown).
Rapid lipid aggregation induced by VP11 together with the large major capsid protein VP17.
Next, we used DLS to analyze the interaction of all three capsid-associated proteins in the presence of lipids by estimating the apparent mean hydrodynamic diameter (Fig. 5A). Neither the addition of the major capsid proteins VP16 and VP17 alone nor in combination changed the diameter of the T-LUVs (120 nm), whereas the addition of VP11 immediately led to a significant increase in size (∼200 nm). Thereafter, the particle size did not change during the observed period of 16 h. On the other hand, whenever VP11 was added to T-LUVs in combination with the major capsid protein VP17, particle size increased immediately to over 600 nm, and a visible precipitate formed in the cuvette. Aggregation and sedimentation of particles increased during the course of the measurement. A determination of the exact hydrodynamic size was therefore impossible at the later time of the experiment. In contrast, incubation of T-LUVs in combination with VP11 and the small major capsid protein VP16 increased the size of the lipid vesicles to 200 nm, similarly to the addition of VP11 alone. The same was true for the incubation of T-LUVs in combination with VP11 and horse skeletal muscle myoglobin (data not shown). Incubation of VP11 with BSA, another unrelated protein used as a control, caused heavy aggregation even in the absence of T-LUVs. BSA is negatively charged at neutral pH, and electrostatic interactions lead to the observed protein aggregation. The results indicate that the interaction between T-LUV-associated VP11 and VP17 is not based simply on electrostatic attraction, since incubation of VP11 with VP17 in the absence of T-LUVs did not cause aggregation (see above). Addition of myoglobin (neutrally charged at pH 7.5) or BSA alone to T-LUVs did not change the size of the lipid vesicles.
FIG 5.
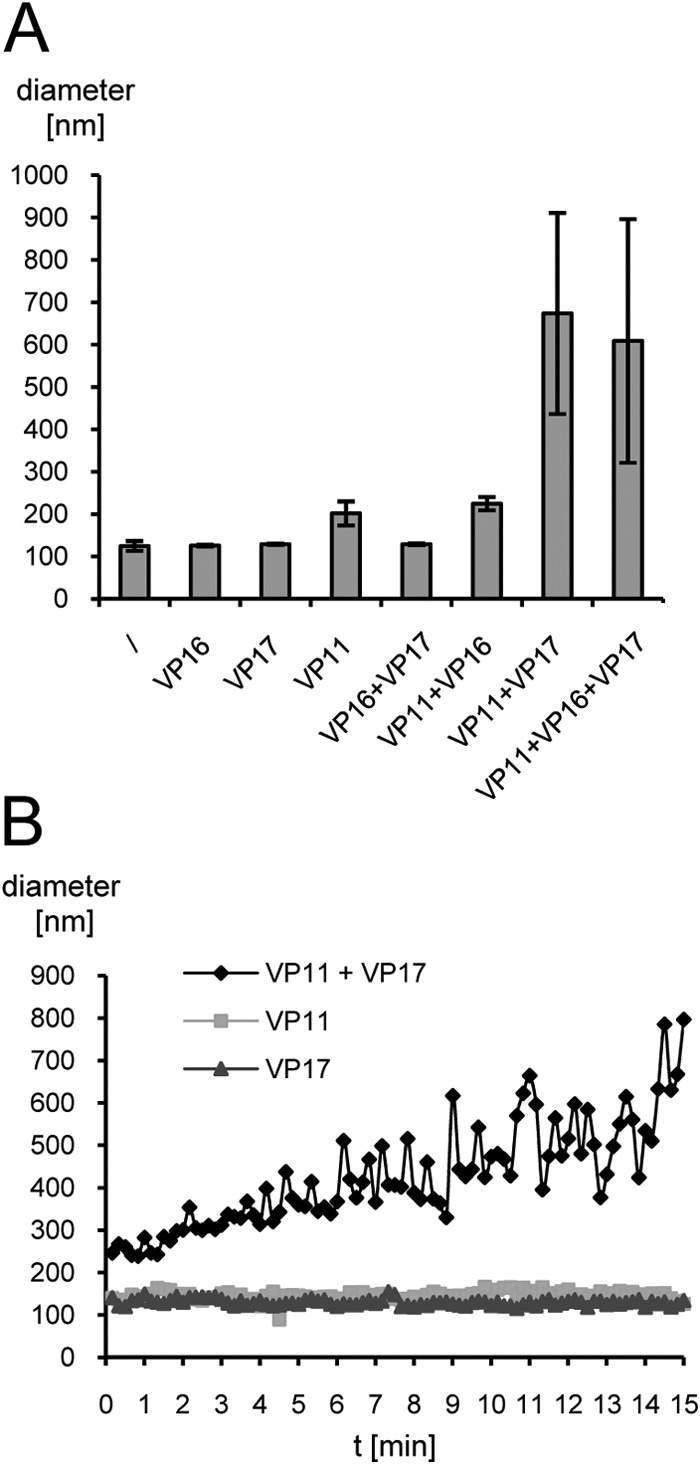
Interaction of T-LUVs with P23-77 capsid-associated proteins. (A) T-LUVs (diluted 1:10) were mixed with either buffer (\) or various combinations of the minor capsid protein VP11, the small major capsid protein VP16, and the large major capsid protein VP17 (0.3 mg/ml each) in 20 mM Tris, pH 7.5. The bars show the mean apparent hydrodynamic diameters of T-LUVs from three independent measurements, each performed 10 times for 10 s each time. The error bars indicate standard deviations. (B) Kinetics of the T-LUV interaction with VP11 and VP17. T-LUVs (diluted 1:500) were incubated with 0.3 mg/ml of VP11, VP17, or a combination of the two in 20 mM Tris, pH 7.5, for 15 min. The mean apparent hydrodynamic diameter was estimated every 10 s, with 10 runs per data point. Particle size estimates are presented as an average diameter (based on volume distribution).
In order to analyze the kinetics of the interaction between T-LUVs, VP11, and VP17, lipid vesicles were diluted to the point where no immediate aggregation was observed (1:500) and incubated with the same concentration of protein as previously (Fig. 5B). Addition of either VP11 or VP17 alone did not change the diameter of the vesicles significantly (∼130 nm) during 15 min. The addition of both proteins, on the other hand, doubled the size of the lipid vesicles at subminute time scales. The diameter of the particles increased continuously during the measurement to ∼800 nm, yet after approximately 8 min, the accuracy of the determination became compromised by large and heterogeneous particles resulting from vesicle-protein aggregation and sedimentation. Interestingly, the addition of VP11 did not increase the size of T-LUVs, as observed with 50 times more concentrated vesicles in the initial interaction assays (Fig. 5A).
DISCUSSION
VP11 forms homodimers.
VP11 dimerizes via an intermolecular disulfide bridge between single cysteine residues at the N terminus of the protein (Fig. 2). It is the dominant form in freshly prepared virus particles, indicating the dimer as the native form of VP11 in the virion. Viruses assemble in the cytoplasm of the host cell, a reducing environment that usually precludes formation of disulfide bonds. Consequently, VP11 was purified from E. coli as a monomer. Interestingly, intracellular disulfide bond formation is a common phenomenon in thermophilic organisms, where it serves as a mechanism of protein stabilization at high temperatures (50, 51, 52). Intracellular disulfide bond formation is mediated by the protein disulfide oxidoreductase (53), an enzyme specifically found in thermophiles, including the P23-77 host T. thermophilus (51). Therefore, the presence of a VP11 dimer in the virus particle is considered biologically relevant. Intra- or intermolecular disulfide bridges enhance the thermal stability of viral proteins (54, 55). The intermolecular disulfide bridge in VP11 cannot be disrupted, even by boiling in 1% β-mercaptoethanol reducing agent, providing rigidity to the capsid at high temperatures. This is in line with the estimated melting point of around 85°C. VP11, as well as the major capsid proteins (33), exhibits high thermal stability, reflecting adaptation to the natural habitat of the host, T. thermophilus, which is able to grow at 80°C and higher (56). On the other hand, VP11 seems to possess a flexible structure, as indicated by the continuous destabilization of α-helices with increasing temperatures in CD thermal scans. Taking the CD and DLS results together, VP11 is suggested to be a predominantly α-helical protein with a dynamic, elongated shape. This might allow structural adaptation of the virus to varying temperatures in the natural environment.
VP11 binds to DNA without sequence specificity.
VP11 strongly associates with DNA (Fig. 4). However, a putative function of VP11 in DNA replication or genome packaging remains unclear due to its capability to bind any type of negatively charged molecule. VP11 could be separated from the membrane and DNA in virion dissociation experiments (3) and is most likely localized on the membrane outer surface (see below). Thus, the interaction of VP11 with DNA might be an in vitro artifact. However, at this stage, we cannot rule out a dual role for VP11, functioning in genome condensation as well as in assisting capsid assembly, as outlined below.
The gene encoding VP11 is located in the P23-77 genome in an area with variable gene content, reflecting adaptation to the host. At a similar genomic position, the related virus IN93 carries a gene encoding a highly basic, mainly α-helical protein with similarity to structural proteins of SSV1 and STIV2 (36, 37) (Fig. 1). The SSV1 structural protein VP2 was assigned to play a role in genome packaging based on the observation that it binds tightly to SSV1 DNA (36). Remarkably, it is dispensable for the viability of the virus and therefore might not be a genome-packaging protein (57). All of the above-mentioned viruses have a lipid moiety in their virions, which is an internal membrane in the cases of P23-77, IN93, and STIV and a putative outer envelope in the case of SSV1. On the basis of our results, one could likewise assume a membrane-associated function for positively charged virion components in those thermophilic viruses. Hence, IN93 and P23-77 have acquired proteins with similar characteristics from different sources to carry out similar functions in the virus.
VP11 serves as a linker between the internal membrane and the capsid shell.
Our results rule out localization of VP11 at the 5-fold vertex base. Pentons of related viruses have a core fold consisting of an eight-stranded beta-barrel (7, 18, 25, 26). In contrast, CD measurements in the far-UV region revealed a high content of alpha-helices (Fig. 3), excluding the presence of a beta-barrel core fold in VP11. Furthermore, VP11 is a dimer instead of a pentamer, and its estimated copy number is too high for a penton protein. An icosahedral capsid contains 12 vertices, a total of 60 copies of a penton protein. The predicted copy number of 147 for VP11, estimated by comparison of band intensities in polyacrylamide gels, exceeds this number by factor of 2.5. Even by taking into account unequal incorporation of Coomassie stain, the difference is striking and makes it unlikely that VP11 is the penton protein. The predicted copy number for VP17 (527) matched the theoretical value (540) quite well, demonstrating that reliable results could be produced by this method. VP11 dimers might be located underneath the vertices, linking the penton base to the membrane, which is in line with the estimated copy number per capsid and the observed lipid interaction in DLS measurements. However, we regard this possibility as unlikely. It is evident from the fitting of the MCPs to the P23-77 cryo-electron microscopy density (2) that the P23-77 5 folds contain a penton structure not comprised of VP16 or VP17 but instead of another protein species (5). This is similar to the capsid architecture of structurally related viruses, including PRD1 (18), PM2 (7), and STIV (26). Assuming that VP11 is localized underneath the 5-fold vertices, it would most likely require the penton protein as an interaction partner, yet our results indicate an interaction with lipids and the major capsid protein VP17. The capsids of other, distantly related viruses are linked to the internal membrane by α-helical domains present in their major capsid proteins (18, 58) or integral membrane proteins (7). The mainly α-helical secondary structure and fairly linear shape of VP11, in addition to its relatively high copy number, lack of predicted transmembrane domains, and observed interaction with both lipids and VP17, strongly suggest localization on the outer membrane surface across the virion.
The VP11-lipid interaction seems to be mostly based on electrostatic attraction of positively charged protein surface domains to anionic phospholipids, as shown for several protein species (59). Although zeta potential measurements clearly indicate adsorption to Thermus lipid vesicles, aggregation of the protein-lipid complex was not observed, in contrast to experiments where VP11 was studied in the presence of other negatively charged partners, such as DNA and BSA (see above). Even though the addition of VP11 doubled the size of the lipid vesicles immediately, no further increase in size was observed. We have no explanation for this effect so far. A size increase was not observed with 50 times more diluted T-LUVs in the kinetics experiment (Fig. 5). In this sense, VP11 differs from other basic proteins, such as lysozyme, which cause rapid aggregation with lipid vesicles (60, 61, 62). Rather, it functions as a trigger for the binding of the large major capsid protein VP17, which in turn results in a rapid aggregation of the protein-membrane complex. The VP11-mediated interaction is specific, since VP16 and myoglobin did not induce aggregation when added to T-LUVs together with VP11. So far, we have not been able to detect an interaction between VP11 and VP17 in the absence of lipids, either in DLS measurements or with other methods, such as gradient centrifugation and yeast two-hybrid assays. The results presented here strongly suggest that the association of the minor capsid protein VP11 with the lipid membrane is the key to the binding of the large major capsid protein VP17. Thus, VP11 might function as an assembly factor, comparable to scaffolding proteins (see below).
The minor capsid protein resembles scaffolding proteins.
In its characteristics, VP11 resembles scaffolding proteins of tailed bacteriophages. Scaffolding proteins are chaperones required for the correct formation of procapsids, an intermediate state during the assembly of tailed dsDNA bacteriophages. They are removed before or during genome packaging (63). In that sense, VP11 is not a scaffolding protein per definitionem because it is a significant component of the mature virion. However, the similarities are striking: α-helical structure, elongated shape, structural flexibility, and protein binding via electrostatic interactions (64). The scaffolding proteins of podoviruses P22 and Phi29 are predominantly α-helical proteins with elongated shapes (65, 66, 67). A similar estimated structure was obtained for VP11 from far-CD spectra and hydrodynamic-diameter estimation. Positively charged amino acid residues on the surface of the C-terminal helix of the P22 scaffolding protein are essential for binding to the major capsid protein via electrostatic interaction (68), as electrostatic forces account for the interaction of VP11 with lipids. In Phi29, the unstructured C terminus of the scaffolding protein interacts with the coat protein (69). Short unstructured stretches are also present at the N and C termini of VP11. They may provide interaction sites with the large major capsid protein VP17. VP17 is proposed to bind to the internal membrane via hydrophobic residues at the N terminus connected by a proline-rich linker to the rest of the protein (5). Proline-rich regions are often involved in protein-protein interactions (70), suggesting the proline linker of VP17 is a putative interaction site for VP11.
In the membrane-containing bacteriophage PRD1, the nonstructural protein P10 covers the membranes of newly synthesized virus particles. The protein is supposed to act as a scaffold for the assembly of the capsid proteins (12, 15). As with other scaffolding proteins, it is removed during capsid assembly, whereas VP11 remains associated with the capsid. However, the striking similarity to scaffolding proteins indicates an assisting role of VP11 in capsid assembly, facilitating the binding of capsid components. In contrast to the above-described viruses, P23-77 viral particles are exposed to extremely high temperatures. This affords additional factors to enhance the rigidity of the capsid. As a stable linker between the capsid shell and the membrane, VP11 might represent such a factor.
The role of VP11 in capsid assembly.
In P23-77, as well as in other icosahedral, tailless dsDNA viruses with an inner membrane, a transmembrane channel beneath the vertices forms the only visible connection between the capsid and the internal membrane in cryo-electron microscopy images (2, 26, 71, 72). This finding supported the idea of the vertices as starting points for the capsid shell assembly (5, 26). The tight anchoring of the vertex complex to the membrane might serve as a nucleation point for capsid growth, which is thought to be triggered by VP11, based on our results. The strong electrostatic association of VP11 with the membrane, achieved through a large content of negatively charged phospholipids in the membrane outer leaflet, promotes binding of the major capsid protein VP17. Protein-protein interaction is probably stabilized by conformational or electrostatic modulation in VP11, VP11 dimerization, and interaction of the VP17 N terminus with the membrane. Binding of the major capsid protein allows subsequent binding of VP16 dimers to VP17, finalizing the assembly of the P23-77 virion (Fig. 6).
FIG 6.
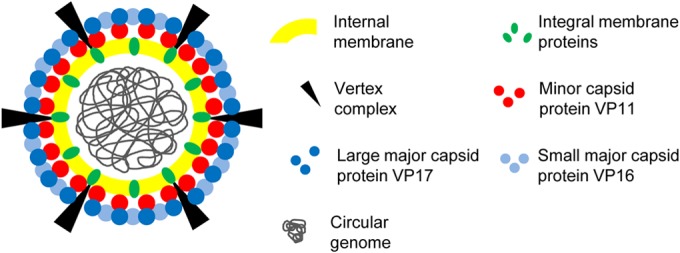
Model of the P23-77 virion. Lipids and integral membrane proteins surround the circular dsDNA genome. A yet-unidentified vertex complex interacts with the integral membrane protein(s), forming a channel connecting the vertex to the membrane. The minor capsid protein VP11 links the capsid to the internal membrane. Its electrostatic interaction with the lipid surface facilitates the interaction with the major capsid protein VP17. VP17 in turn binds the small major capsid protein VP16.
The complex capsid architecture of viruses with two major capsid proteins requires factors that facilitate correct insertion of the various building blocks into the growing capsid. By interacting specifically with the large major capsid protein VP17, but not with the small major capsid protein VP16, VP11 likely constitutes such a factor. It represents a novel type of minor capsid protein of prokaryotic icosahedral dsDNA viruses. However, the presence of a highly basic protein in the virions of several thermophilic viruses implies the presence of similar assembly factors in viruses from high-temperature environments.
ACKNOWLEDGMENTS
This work was funded by the Acadamy of Finland Centre of Excellence Programme, CoE in Biological Interactions 2012-2017 (252411; responsible leader for the grant, Johanna Mappes), and Academy of Finland personal grants to J.K.H.B. (251106) J.A.I. (134061), and V.P.H. (273192).
We thank Johanna Mappes for supporting the project. We thank Petri Papponen, Alli Liukkonen, and Elina Dadu for technical help. The use of the facilities and expertise of the Protein Technologies core facility, a member of Biocenter Finland, is gratefully acknowledged.
REFERENCES
- 1.Yu MX, Slater MR, Ackermann HW. 2006. Isolation and characterization of Thermus bacteriophages. Arch Virol 151:663–679. doi: 10.1007/s00705-005-0667-x. [DOI] [PubMed] [Google Scholar]
- 2.Jaatinen ST, Happonen LJ, Laurinmäki P, Butcher SJ, Bamford DH. 2008. Biochemical and structural characterisation of membrane-containing icosahedral dsDNA bacteriophages infecting thermophilic Thermus thermophilus. Virology 379:10–19. doi: 10.1016/j.virol.2008.06.023. [DOI] [PubMed] [Google Scholar]
- 3.Jalasvuori M, Jaatinen ST, Laurinavičius S, Ahola-Iivarinen E, Kalkkinen N, Bamford DH, Bamford JK. 2009. The closest relatives of icosahedral viruses of thermophilic bacteria are among viruses and plasmids of the halophilic archaea. J Virol 83:9388–9397. doi: 10.1128/JVI.00869-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawlowski A, Rissanen I, Bamford JK, Krupovic M, Jalasvuori M. 2014. Gammasphaerolipovirus, a newly proposed bacteriophage genus, unifies viruses of halophilic archaea and thermophilic bacteria within the novel family Sphaerolipoviridae. Arch Virol 159:1541–1554. doi: 10.1007/s00705-013-1970-6. [DOI] [PubMed] [Google Scholar]
- 5.Rissanen I, Grimes JM, Pawlowski A, Mäntynen S, Harlos K, Bamford JK, Stuart DI. 2013. Bacteriophage P23-77 capsid protein structures reveal the archetype of an ancient branch from a major virus lineage. Structure 21:718–726. doi: 10.1016/j.str.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson SD, Bamford JK, Bamford DH, Burnett RM. 1999. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures, Cell 98:825–833. [DOI] [PubMed] [Google Scholar]
- 7.Abrescia NG, Grimes JM, Kivelä HM, Assenberg R, Sutton GC, Butcher SJ, Stuart DI. 2008. Insights into virus evolution and membrane biogenesis from the structure of the marine lipid-containing bacteriophage PM2. Mol Cell 31:749–761. doi: 10.1016/j.molcel.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 8.Khayat R, Tang L, Larson ET, Lawrence CM, Young M, Johnson JE. 2005. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc Natl Acad Sci U S A 102:18944–18949. doi: 10.1073/pnas.0506383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butcher SJ, Manole V, Karhu NJ. 2012. Lipid-containing viruses: bacteriophage PRD1 assembly, p 365–377. In Rossmann MG, Rao VB (ed), Viral molecular machines. Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- 10.Hänninen AL, Bamford DH, Bamford JK. 1997. Assembly of membrane-containing bacteriophage PRD1 is dependent on GroEL and GroES. Virology 227:207–210. doi: 10.1006/viro.1996.8308. [DOI] [PubMed] [Google Scholar]
- 11.Caldentey J, Hänninen AL, Holopainen JM, Bamford JK, Kinnunen PK, Bamford DH. 1999. Purification and characterization of the assembly factor P17 of the lipid-containing bacteriophage PRD1. Euro J Biochem 260:549–558. [DOI] [PubMed] [Google Scholar]
- 12.Rydman PS, Bamford JK, Bamford DH. 2001. A minor capsid protein P30 is essential for bacteriophage PRD1 capsid assembly. J Mol Biol 313:785–795. doi: 10.1006/jmbi.2001.5068. [DOI] [PubMed] [Google Scholar]
- 13.Strömsten NJ, Bamford DH, Bamford JK. 2005. In vitro DNA packaging of PRD1: a common mechanism for internal-membrane viruses. J Mol Biol 348:617–629. doi: 10.1016/j.jmb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Earnshaw WC, Casjens SR. 1980. DNA packaging by the double-stranded DNA bacteriophages. Cell 21:319–331. doi: 10.1016/0092-8674(80)90468-7. [DOI] [PubMed] [Google Scholar]
- 15.Hong C, Oksanen HM, Liu X, Jakana J, Bamford DH, Chiu W. 2014. A structural model of the genome packaging process in a membrane-containing double stranded DNA virus. PLoS Biol 12:e1002024. doi: 10.1371/journal.pbio.1002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huiskonen JT, Kivelä HM, Bamford DH, Butcher SJ. 2004. The PM2 virion has a novel organization with an internal membrane and pentameric receptor binding spikes. Nat Struct Mol Biol 11:850–856. doi: 10.1038/nsmb807. [DOI] [PubMed] [Google Scholar]
- 17.Grahn AM, Daugelavičius R, Bamford DH. 2002. Sequential model of phage PRD1 DNA delivery: active involvement of the viral membrane. Mol Microbiol 46:1199–1209. doi: 10.1046/j.1365-2958.2002.03250.x. [DOI] [PubMed] [Google Scholar]
- 18.Abrescia NG, Cockburn JJ, Grimes JM, Sutton GC, Diprose JM, Butcher SJ, Fuller SD, San Martin C, Burnett RM, Stuart DI, Bamford DH, Bamford JK. 2004. Insights into assembly from structural analysis of bacteriophage PRD1. Nature 432:68–74. doi: 10.1038/nature03056. [DOI] [PubMed] [Google Scholar]
- 19.Fu CY, Wang K, Gan L, Lanman J, Khayat R, Young MJ, Johnson JE. 2010. In vivo assembly of an archaeal virus studied with whole-cell electron cryotomography. Structure 18:1579–1586. doi: 10.1016/j.str.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braunstein SN, Franklin RM. 1971. Structure and synthesis of a lipid-containing bacteriophage. V. Phospholipids of the host BAL-31 and of the bacteriophage PM2. Virology 43:685–695. [DOI] [PubMed] [Google Scholar]
- 21.Laurinavičius S, Käkelä R, Somerharju P, Bamford DH. 2004. Phospholipid molecular species profiles of tectiviruses infecting Gram-negative and Gram-positive hosts. Virology 322:328–336. doi: 10.1016/j.virol.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Bamford DH, Ravantti JJ, Rönnholm G, Laurinavičius S, Kukkaro P, Dyall-Smith M, Somerharju P, Kalkkinen N, Bamford JK. 2005. Constituents of SH1, a novel lipid-containing virus infecting the halophilic euryarchaeon Haloarcula hispanica. J Virol 79:9097–9107. doi: 10.1128/JVI.79.14.9097-9107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cockburn JJ, Abrescia NG, Grimes JM, Sutton GC, Diprose JM, Benevides JM, Stuart DI. 2004. Membrane structure and interactions with protein and DNA in bacteriophage PRD1. Nature 432:122–125. doi: 10.1038/nature03053. [DOI] [PubMed] [Google Scholar]
- 24.Schäfer R, Hinnen R, Franklin RM. 1974. Structure and synthesis of a lipid-containing bacteriophage. Eur J Biochem 50:15–27. doi: 10.1111/j.1432-1033.1974.tb03868.x. [DOI] [PubMed] [Google Scholar]
- 25.Zubieta C, Schoehn G, Chroboczek J, Cusack S. 2005. The structure of the human adenovirus 2 penton. Mol Cell 17:121–135. doi: 10.1016/j.molcel.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 26.Veesler D, Ng TS, Sendamarai AK, Eilers BJ, Lawrence CM, Lok SM, Fu CY. 2013. Atomic structure of the 75 MDa extremophile Sulfolobus turreted icosahedral virus determined by cryoEM and X-ray crystallography. Proc Natl Acad Sci U S A 110:5504–5509. doi: 10.1073/pnas.1300601110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olkkonen VM, Bamford DH. 1989. Quantitation of the adsorption and penetration stages of bacteriophage ϕ6 infection. Virology 171:229–238. doi: 10.1016/0042-6822(89)90530-8. [DOI] [PubMed] [Google Scholar]
- 28.Bamford JK, Bamford DH. 1991. Large-scale purification of membrane-containing bacteriophage PRD1 and its subviral particles. Virology 181:348–352. doi: 10.1016/0042-6822(91)90501-2. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 30.EMD Millipore. 2012. Protein blotting handbook, 6th ed EMD Millipore, Billerica, MA. [Google Scholar]
- 31.Schägger H, Von Jagow G. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 32.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 33.Rissanen I, Pawlowski A, Harlos K, Grimes JM, Stuart DI, Bamford JK. 2012. Crystallization and preliminary crystallographic analysis of the major capsid proteins VP16 and VP17 of bacteriophage P23-77. Acta Crystallogr Sect F Struct Biol Cryst Commun 68:580–583. doi: 10.1107/S1744309112010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsushita I, Yanase H. 2009. The genomic structure of Thermus bacteriophage ϕIN93. J Biochem 146:775–785. doi: 10.1093/jb/mvp125. [DOI] [PubMed] [Google Scholar]
- 35.Jalasvuori M, Pawlowski A, Bamford JK. 2010. A unique group of virus-related genome-integrating elements found solely in the bacterial family Thermaceae and the archaeal family Halobacteriaceae. J Bacteriol 192:3231–3234. doi: 10.1128/JB.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiter WD, Palm P, Henschen A, Lottspeich F, Zillig W, Grampp B. 1987. Identification and characterization of the genes encoding three structural proteins of the Sulfolobus virus-like particle SSV1. Mol Gen Genet 206:144–153. [Google Scholar]
- 37.Happonen LJ, Redder P, Peng X, Reigstad LJ, Prangishvili D, Butcher SJ. 2010. Familial relationships in hyperthermo- and acidophilic archaeal viruses. J Virol 84:4747–4754. doi: 10.1128/JVI.02156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makarova KS, Wolf YI, Forterre P, Prangishvili D, Krupovic M, Koonin EV. 2014. Dark matter in archaeal genomes: a rich source of novel mobile elements defense systems and secretory complexes. Extremophiles 18:877–893. doi: 10.1007/s00792-014-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cserzö M, Wallin E, Simon I, von Heijne G, Elofsson A. 1997. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng 10:673–676. doi: 10.1093/protein/10.6.673. [DOI] [PubMed] [Google Scholar]
- 40.Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A 95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 42.Pasquier C, Promponas VJ, Palaios GA, Hamodrakas JS, Hamodrakas SJ. 1999. A novel method for predicting transmembrane segments in proteins based on a statistical analysis of the SwissProt database: the PRED-TMR algorithm. Protein Eng 12:381–385. doi: 10.1093/protein/12.5.381. [DOI] [PubMed] [Google Scholar]
- 43.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 44.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607. In Walker JM. (ed), The proteomics protocols handbook. Humana Press, Totowa, NJ. [Google Scholar]
- 45.Grigorian AL, Bustamante JJ, Hernandez P, Martinez AO, Haro LS. 2005. Extraordinarily stable disulfide-linked homodimer of human growth hormone. Protein Sci 14:902–913. doi: 10.1110/ps.041048805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perdomo D, Baron B, Rojo-Domínguez A, Raynal B, England P, Guillén N. 2013. The α-helical regions of KERP1 are important in Entamoeba histolytica adherence to human cells. Sci Rep 3:1171. doi: 10.1038/srep01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flora K, Brennan JD, Baker GA, Doody MA, Bright FV. 1998. Unfolding of acrylodan-labeled human serum albumin probed by steady-state and time-resolved fluorescence methods. Biophys J 75:1084–1096. doi: 10.1016/S0006-3495(98)77598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson CM. 2013. Differential scanning calorimetry as a tool for protein folding and stability. Arch Biochem Biophys 531:100–109. doi: 10.1016/j.abb.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 49.Rissanen I. 2014. An ancient virus type from extreme environments. Ph.D. thesis University of Jyväskylä, Jyväskylä, Finland. [Google Scholar]
- 50.Mallick P, Boutz DR, Eisenberg D, Yeates TO. 2002. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc Natl Acad Sci U S A 99:9679–9684. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beeby MD, O'Connor B, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. 2005. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS Biol 3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorda J, Yeates TO. 2011. Widespread disulfide bonding in proteins from thermophilic archaea. Archaea 2011:409156. doi: 10.1155/2011/409156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pedone E, Ren B, Ladenstein R, Rossi M, Bartolucci S. 2004. Functional properties of the protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus. Eur J Biochem 271:3437–3448. doi: 10.1111/j.0014-2956.2004.04282.x. [DOI] [PubMed] [Google Scholar]
- 54.Larson ET, Eilers B, Menon S, Reiter D, Ortmann A, Young MJ, Lawrence CM. 2007. A winged-helix protein from Sulfolobus turreted icosahedral virus points toward stabilizing disulfide bonds in the intracellular proteins of a hyperthermophilic virus. Virology 368:249–261. doi: 10.1016/j.virol.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 55.Menon SK, Maaty WS, Corn GJ, Kwok SC, Eilers BJ, Kraft P, Lawrence CM. 2008. Cysteine usage in Sulfolobus spindle-shaped virus 1 and extension to hyperthermophilic viruses in general. Virology 376:270–278. doi: 10.1016/j.virol.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 56.Manaia CM, Hoste B, Carmen Gutierrez M, Gillis M, Ventosa A, Kersters K, Da Costa MS. 1995. Halotolerant Thermus strains from marine and terrestrial hot springs belong to Thermus thermophilus (ex Oshima and Imahori 1974) nom. rev. emend. Syst Appl Microbiol 17:526–532. doi: 10.1016/S0723-2020(11)80072-X. [DOI] [Google Scholar]
- 57.Iverson E, Stedman K. 2012. A genetic study of SSV1, the prototypical fusellovirus. Front Microbiol 3:200. doi: 10.3389/fmicb.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khayat R, Fu CY, Ortmann AC, Young MJ, Johnson JE. 2010. The architecture and chemical stability of the archaeal Sulfolobus turreted icosahedral virus. J Virol 84:9575–9583. doi: 10.1128/JVI.00708-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mulgrew-Nesbitt A, Diraviyam K, Wang J, Singh S, Murray P, Li Z, Murray D. 2006. The role of electrostatics in protein-membrane interactions. Biochim Biophys Acta 1761:812–826. doi: 10.1016/j.bbalip.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 60.Diraviyam K, Murray D. 2006. Computational analysis of the membrane association of group IIA secreted phospholipases A2: a differential role for electrostatics. Biochemistry 45:2584–2598. doi: 10.1021/bi051901t. [DOI] [PubMed] [Google Scholar]
- 61.Gorbenko GP, Ioffe VM, Kinnunen PK. 2007. Binding of lysozyme to phospholipid bilayers: evidence for protein aggregation upon membrane association. Biophys J 93:140–153. doi: 10.1529/biophysj.106.102749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coutinho A, Loura L, Fedorov A, Prieto M. 2008. Pinched multilamellar structure of aggregates of lysozyme and phosphatidylserine-containing membranes revealed by FRET. Biophys J 95:4726–4736. doi: 10.1529/biophysj.108.134379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prevelige PE, Fane BA. 2012. Building the machines: scaffolding protein functions during bacteriophage morphogenesis, p 325–350. In Rossmann MG, Rao VB (ed), Viral molecular machines. Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- 64.Mateu MG. 2013. Assembly stability and dynamics of virus capsids. Arch Biochem Biophys 531:65–79. doi: 10.1016/j.abb.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 65.Fuller MT, King J. 1982. Assembly in vitro of bacteriophage P22 procapsids from purified coat and scaffolding subunits. J Mol Biol 156:633–665. doi: 10.1016/0022-2836(82)90270-4. [DOI] [PubMed] [Google Scholar]
- 66.Tuma R, Parker MH, Weigele P, Sampson L, Sun Y, Krishna NR, Casjens S, Thomas GJ Jr, Prevelige PE Jr. 1998. A helical coat protein recognition domain of the bacteriophage P22 scaffolding protein. J Mol Biol 281:81–94. [DOI] [PubMed] [Google Scholar]
- 67.Morais MC, Kanamaru S, Badasso MO, Koti JS, Owen BA, McMurray CT, Anderson DL, Rossmann MG. 2003. Bacteriophage phi29 scaffolding protein gp7 before and after prohead assembly. Nat Struct Biol 10:572–576. doi: 10.1038/nsb939. [DOI] [PubMed] [Google Scholar]
- 68.Cortines JR, Weigele PR, Gilcrease EB, Casjens SR, Teschke CM. 2011. Decoding bacteriophage P22 assembly: identification of two charged residues in scaffolding protein responsible for coat protein interaction. Virology 421:1–11. doi: 10.1016/j.virol.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu CY, Morais MC, Battisti AJ, Rossmann MG, Prevelige PE Jr. 2007. Molecular dissection of ϕ29 scaffolding protein function in an in vitro assembly system. J Mol Biol 366:1161–1173. doi: 10.1016/j.jmb.2006.11.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williamson MP. 1994. The structure and function of proline-rich regions in proteins. Biochem J 297:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rice G, Tang L, Stedman K, Roberto F, Spuhler J, Gillitzer E, Young M. 2004. The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc Natl Acad Sci U S A 101:7716–7720. doi: 10.1073/pnas.0401773101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jäälinoja HT, Roine E, Laurinmäki P, Kivelä HM, Bamford DH, Butcher SJ. 2008. Structure and host-cell interaction of SH1, a membrane-containing halophilic euryarchaeal virus. Proc Natl Acad Sci U S A 105:8008–8013. doi: 10.1073/pnas.0801758105. [DOI] [PMC free article] [PubMed] [Google Scholar]