

ABSTRACT
Lassa virus is a notorious human pathogen that infects many thousands of people each year in West Africa, causing severe viral hemorrhagic fevers and significant mortality. The surface glycoprotein of Lassa virus mediates receptor recognition through its GP1 subunit. Here we report the crystal structure of GP1 from Lassa virus, which is the first representative GP1 structure for Old World arenaviruses. We identify a unique triad of histidines that forms a binding site for LAMP1, a known lysosomal protein recently discovered to be a critical receptor for internalized Lassa virus at acidic pH. We demonstrate that mutation of this histidine triad, which is highly conserved among Old World arenaviruses, impairs LAMP1 recognition. Our biochemical and structural data further suggest that GP1 from Lassa virus may undergo irreversible conformational changes that could serve as an immunological decoy mechanism. Together with a variable region that we identify on the surface of GP1, those could be two distinct mechanisms that Lassa virus utilizes to avoid antibody-based immune response.
IMPORTANCE Structural data at atomic resolution for viral proteins is key for understanding their function at the molecular level and can facilitate novel avenues for combating viral infections. Here we used X-ray protein crystallography to decipher the crystal structure of the receptor-binding domain (GP1) from Lassa virus. This is a pathogenic virus that causes significant illness and mortality in West Africa. This structure reveals the overall architecture of GP1 domains from the group of viruses known as the Old World arenaviruses. Using this structural information, we elucidated the mechanisms for pH switch and binding of Lassa virus to LAMP1, a recently identified host receptor that is critical for successful infection. Lastly, our structural analysis suggests two novel immune evasion mechanisms that Lassa virus may utilize to escape antibody-based immune response.
INTRODUCTION
Lassa Virus (LASV) belongs to the Arenaviridae family of enveloped, negative-stranded RNA viruses (1). Arenaviruses are zoonotic viruses that are carried and spread to humans by rodents (2). Infection by some members of this family leads to severe viral hemorrhagic fevers (VHF) (2). LASV is the most predominant of the viruses causing VHF, with an estimated 300,000 annual cases in western Africa and high mortality rates (3). Arenaviruses are subdivided into two major subgroups, the “Old World” (OW) and the “New World” (NW) arenaviruses, which are endemic to Africa and South America, respectively (4).
Arenaviruses utilize various cell surface proteins as their cellular receptors for recognizing and attaching to target cells. NW arenaviruses that belong to clades A and B use transferrin receptor 1 (TfR1) (5, 6), whereas OW arenaviruses, as well as clade C NW arenaviruses, use α-dystroglycan (α-DG) (7–9). A trimeric class 1 viral glycoprotein complex (the spike complex) recognizes the cellular receptors and mediates membrane fusion upon exposure to low pH at the lysosome (10). The spike complex is expressed as a glycoprotein precursor that is cleaved into three segments by a signal peptidase and SKI-1/S1P protease (11). The functional spike complex consists of a receptor-binding subunit (GP1), a membrane-anchored fusion protein (GP2), and a unique structured signal peptide (SSP) (12). It was recently shown that successful infection requires LASV to switch from binding α-DG to binding a lysosomal protein termed LAMP1 in a pH-dependent manner (13).
No structural information is yet available for GP1 from LASV (GP1LASV) or any other OW arenaviruses. Currently, structures are available only for GP1 from the TfR1-tropic NW Machupo arenavirus (GP1MACV); crystallographic structures of GP1MACV were solved for the unbound protein (14) and for its complex with TfR1 (15). For GP1LASV, the overall architecture, molecular basis for receptor recognition, and mechanism of switching to LAMP1 are currently unknown. Here we provide the first crystal structure of the GP1 receptor-binding domain of an OW arenavirus. We have crystallized and solved the structure of GP1LASV to 2.6-Å resolution. We had to use an experimental phasing approach to solve the structure, emphasizing the great evolutionary distance between OW and NW arenaviruses. We compare the structures of GP1LASV and GP1MACV and highlight the structural diversification of the spike receptor-binding module. Our structural analysis reveals a variable region on the surface of GP1 that is likely to serve as an immunological decoy. We further used biochemical assays and structural analysis to identify the receptor-binding site on GP1LASV. We discovered a unique triad of histidines that forms the LAMP1-binding site in GP1LASV, thereby providing a molecular mechanism for the pH-dependent receptor switching. To verify our findings, we generated specific mutants of GP1LASV and showed the requirement of the histidine triad for interaction with LAMP1. Our structural analysis and biochemical data further suggest that GP1LASV undergoes irreversible conformational changes that may serve as a novel mechanism for evasion from protective immune responses against LASV.
MATERIALS AND METHODS
Materials.
ESF-921 protein-free cell culture medium, ESF-921 protein-free methionine-free cell culture medium, and ESF serum-free medium (ESF-SFM) were obtained from Expression Systems LLC. Spodoptera frugiperda (Sf9) and Trichoplusia ni High Five (Tni) insect cells that had been adapted to ESF-921 protein-free insect cell medium were obtained from Expression Systems LLC. HEK293ES suspension cells adapted to ESF-SFM were obtained from Expression Systems LLC. Disposable polycarbonate Erlenmeyer flasks were supplied by Corning. ProGreen baculovirus genomic vector DNA was obtained from ABvector. l(+)-selenomethionine (SeMet) was purchased from Carbosynth. Anti-Lamp1 antibody (CD107a) was obtained from Millipore, anti α-DG (IIH6) was obtained from the Developmental Studies Hybridoma Bank, and anti-human Fc horseradish peroxidase (HRP)-conjugated secondary antibody was from Jackson.
Construction of expression vectors.
Lassa virus GP1-coding DNA was chemically synthesized (IDT). The gene was subcloned into the pACgp67b expression vector (BD Biosynthesis) to include an N-terminal His6 tag. LAMP1-Fc and GP1LASV-Fc fusion proteins were generated by cloning of the ectocellular domain of LAMP1 or Lassa virus GP1 along with a fragment encoding a portion of the human Fc (IgG1), using BamHI-KpnI restriction sites. The Fc fusion cassette was then digested with BamHI-NotI and ligated into a modified pHLsec plasmid for soluble protein expression, introducing a V5H8 signal peptide at the N′ terminus. Point mutations of histidine to tyrosine at residues 92, 93, and 230 of Lassa virus were introduced by PCR mutagenesis using Kapa HiFi DNA polymerase (Kapa Biosystems) according to QuikChange site-directed mutagenesis manual.
Protein expression and purification.
For expression of GP1LASV in insect cells, recombinant viruses were produced in Sf9 cells. Cells were cotransfected with 0.4 μg GP1LASV-pACgp67b and 0.25 μg linearized baculovirus genome (ABvector) using EscortIV transfection reagent (Sigma). Four days later, medium was collected from transfected cells (P0) and was used to infect fresh Sf9 cells (P1 to P2). Medium containing viruses was harvested 7 days later and was used to infect 3 liters of Tni insect cells. Culture medium containing the secreted soluble proteins was collected after 72 h of incubation, supplemented with 40 mM Tris-HCl (pH 8.0), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.02% (wt/vol) sodium azide, and incubated at 4°C overnight. Protein was concentrated and buffer exchanged from collected medium with TBS buffer (20 mM Tris-HCl [pH 8.0], 150 mM sodium chloride). For expression of SeMet-substituted GP1LASV protein, the protocol from Expression Systems was followed. Briefly, cells were split for two passages in methionine-free medium (ESF-921), infected with GP1LASV-encoding baculoviruses, and supplemented with 100 mg/liter SeMet at 8, 24, and 48 h posttransfection. Medium was collected at 72 h after infection and was treated as described above. For crystallization experiments, protein was purified using a HiTrap IMAC FF Ni2+ (GE Healthcare) affinity column and Superdex 75 10/300 size exclusion chromatography (GE Healthcare) and then concentrated to an optical density at 280 nm (OD280) of 17, in 20 mM Tris-HCl pH 8.0, 150 mM sodium chloride, 0.02% (wt/vol) sodium azide using an Amicon concentrator (Millipore). GP1LASV-Fc and GP1LASV mutants fused to Fc were expressed by transfection of the plasmid DNA into HEK293T cells in monolayers using linear polyethylenimine (PEI) (25 kDa; Polysciences). Medium was collected at 5 days posttransfection and assayed for levels of protein secretion by dot blotting using anti-human Fc. LAMP1-Fc was transfected using PEI into HEK293ES cells grown in suspension (Expression Systems), and 5 days later the medium was collected and protein was purified using a protein A affinity column (GE Healthcare).
Crystallization.
We used the method of vapor diffusion in sitting drops for crystallization screens. For crystallization experiments we used a Mosquito crystallization robot (TTP Labs) to set 60-, 120-, and 180-nl drops of protein with a 120-nl reservoir of commercially available crystallization screens. Initial crystallization hits for GP1LASV were identified using the PEGRx HT (Hampton) screen and then manually optimized. Thin rectangular layered crystals (space group P21, a = 56.36 Å, b = 93.08 Å, c = 111.95 Å, β = 92.8°; four molecules per asymmetric unit) were obtained upon mixing a protein solution at 17 mg/ml with 0.1 M bis(2-hydroxyethyl)-amino-tris(hydroxymethyl)-methane (pH 5.0), 0.2 M ammonium acetate (pH 7.0), 22% (wt/vol) polyethylene glycol 6000, and 5% (wt/vol) polyethylene glycol 200 at 25°C. Crystals were briefly soaked in mother liquor solution supplemented with 10% (wt/vol) polyethylene glycol 200 for cryopreservation before flash cooling in liquid nitrogen or were flash cooled without the addition of cryoprotectants.
Data collection, structure solution, and refinement.
X-ray diffraction data were collected at the European Synchrotron Radiation Facility (ESRF) beamlines BM14 using a MAR 225 charge-coupled device (CCD) detector and ID23-2 using a Pilatus 2M-F detector. Single-wavelength anomalous diffraction (SAD) data were collected to a resolution of 3.5 Å on BM14 at the selenium absorption edge (12.671 keV) using three different kappa angles. Images were indexed, integrated, and scaled using HKL2000 (16) to 3.8-Å resolution to maximize the anomalous signal. We used SHELXC/D (17) implemented in the HKL2MAP interface (18) to locate heavy atom sites. We then used AutoSol (19) and a subsequent run of AutoBuild (19) for phasing and initial model building at 3.8-Å resolution. The initial model included four partially traced monomers in the asymmetric unit (ASU). We selected the most complete monomer and manually omitted regions that did not agree well with the other partially traced polypeptide chains. This partial consensus structure was used as a search model for molecular replacement (MR) by Phaser (19), which located all four monomers in the ASU of a newly collected native and partially anisotropic data set that extended to 2.6-Å resolution. This data set was collected on ID23-2 and was processed using XDS (20). Subsequently, we used AutoBuild to extend the model almost to completion. The rest of the model was manually traced into electron density maps using Coot (21) and refined using Phenix Refine (19) in an iterative fashion.
Conservation analysis.
The structure of GP1LASV was submitted to the ConSurf server (http://consurf.tau.ac.il/) (22). A complete description of the conservation analysis by ConSurf is available through online documentation. Briefly, a multiple-sequence alignment of 58 Blast hits from UniRef90 (providing unique nonidentical sequences) (UniProt accession codes P08669, F1AM06, D7PQ63, D7PQ56, F1AM09, U5KQM4 Q4VZZ3, G3XEW0, F1AM21, A0A023J5A4P19240, F1AM15, C6ZK00, A0A023J4Z7, Q2A069, D4N7Z6, B7U9H2, P09991, D8L2A2, A0A023J501, Q27YE4, A4D941 Q4VZZ0, C3VVN0 Q77AV6, E2D674, C3VVM8 Q77AU9, K0IT71, U5KQ09 Q4VZY8, Q77AV1, Q4VZZ2, Q9WA84, Q9YPM4, Q995C5, Q9DK03, Q8B120, Q8AZ52, Q8B119, P03540, I3PX54 Q8B121, Q84168, Q4VZY9, and Q9QSP5) was used to construct a phylogenetic tree and to assign position-specific conservation scores. Scores were binned into a discrete scale of nine grades, showing the most conserved residues in purple, intermediate conservation in white, and the most variable residues in cyan.
Pulldown assays.
GP1LASV protein, produced and purified from insect cells (see above), was immobilized to N-hydroxysuccinimide (NHS)-activated beads (GE Healthcare) according to the manufacturer's protocol. As a control, NHS-activated beads were treated similarly to GP1LASV-coated beads, omitting the addition of a protein in the coupling step. Prior to pulldown, control/GP1LASV beads were adjusted to pH 8.0 or pH 5.0 by washing either with NETI buffer [50 mM Tris-HCl, 150 mM sodium chloride, 1 mM EDTA, 0.5% (vol/vol) octylphenoxypoly(ethyleneoxy)ethanol (branched) (IGEPAL), pH 8.0] or with NENI buffer (with 50 mM sodium acetate instead of Tris-HCl; pH 5.0), respectively. Whole-cell extracts from HEK293T cells were prepared in NETI buffer and were adjusted to pH 5.0 or pH 8.0 by addition of 20 mM Tris-HCl, 150 mM sodium chloride, and 0.5% (vol/vol) IGEPAL (pH 8.0) or of 20 mM sodium acetate, 150 mM sodium chloride, and 0.5% (vol/vol) IGEPAL (pH 5.0) (1:1 ratio). The cell lysate was clarified using centrifugation (18,800 × g for 10 min). Beads were incubated with lysates at 4°C for 2 h, followed by 3 washing steps using the above-mentioned buffers, after which beads were boiled in sample buffer and subjected to SDS-PAGE and immunoblot analysis. For pulldown assay using GP1LASV-Fc fusion proteins, proteins were incubated with protein A-Sepharose beads at 4°C for 1 h, followed by 3 wash steps in 20 mM Tris-HCl, 150 mM sodium chloride, and 0.5% (vol/vol) IGEPAL, pH 8.0. The beads were then incubated with whole-cell lysates as described above, after which proteins were eluted in 20 mM Tris-HCl–150 mM sodium chloride. Eluted protein samples were precipitated using cold acetone. Protein pellets were recovered in sample buffer and subjected to SDS-PAGE and immunoblot analysis.
Surface plasmon resonance (SPR) measurements.
The binding of wild-type (WT) GP1LASV-Fc and GP1LASV-Fc mutant fusion proteins to LAMP1 was compared using the ProteOn protein interaction array system (Bio-Rad). Purified LAMP1-Fc was immobilized at a coupling density of ∼1,500 resonance units (RU) on a GLH sensor chip (Bio-Rad) in 10 mM sodium acetate (pH 3.8) using primary amine coupling chemistry as described in the ProteOn manual. One of the flow cells on the sensor chip was mock coupled using buffer to serve as a blank. Experiments were performed at 25°C in 10 mM sodium acetate (pH 5.0), 0.5 M or 0.25 M sodium chloride, and 0.005% (vol/vol) Tween. Sensor chips were regenerated using TBS buffer. Purified GP1LASV-Fc (WT and mutants) and GP1JUNV-Fc were injected at a flow rate of 90 μl/min in a 2-fold dilution series (500 nM, 250 nM, 125 nM, 62.5 nM, and 31.25 nM).
ELISA.
Enzyme-linked immunosorbent assay (ELISA) was performed in 96-well plates coated with 1 μg/ml GP1LASV-Fc (WT and mutants) and GP1JUNV-Fc, diluted in phosphate-buffered saline (PBS). The plates were blocked for 1 h in 37°C with PBS containing 0.05% Tween (vol/vol) and 1% (wt/vol) bovine serum albumin (BSA) (blocking buffer). For detection, we used anti GP1LASV monoclonal antibody that was isolated from mice immunized with GP1LASV (our unpublished results). ELISA plates were overlaid and incubated for 1 h with hybridoma supernatants diluted 1:100 in blocking buffer. Subsequently, plates were incubated for 1 h with HRP-labeled anti-mouse IgG (1:5,000 in blocking buffer). Signal was detected using 3,3′,5,5′-tetramethylbenzidine (TMB) liquid substrate (Sigma-Aldrich) with an infinite 200-Pro Tecan plate reader.
CD analysis.
Stock solutions of 10 mg/ml GP1LASV-Fc (WT and mutants) in 1.3 mM Tris-HCl (pH 8.0)–10 mM sodium chloride were diluted 1:40 in 2 mM sodium acetate (pH 5.0) for recording circular dichroism (CD) spectra using a Chirascan-plus ACD spectrometer at 21.8°C.
Protein structure accession number.
The protein structure data have been submitted to the Protein Data Bank under accession number 4ZJF.
RESULTS
Determining the crystal structure of GP1LASV.
A construct spanning residues 75 to 237 of LASV GP1 (Fig. 1A) expressed in insect cells (here referred to as GP1LASV) yielded sufficient quantities of soluble, monodispersed protein as evaluated by size exclusion chromatography and SDS-PAGE (Fig. 1B). Using this protein, we obtained several crystallization hits, all at acidic pH. X-ray diffraction analysis after crystal optimization revealed that GP1LASV crystallized in a monoclinic space group (P1211) with several monomers in the asymmetric unit (ASU) as estimated from the unit-cell dimensions (Table 1). First, we collected a complete data set to 2.8-Å resolution at the European Synchrotron Radiation Facility (ESRF). Molecular replacement (MR) approaches using the known crystal structure of GP1MACV as a search model or as a template for homology modeling-based MR failed to provide solutions. We therefore produced GP1LASV labeled with selenomethionine and collected a complete data set at the selenium absorption edge from a single crystal for SAD phasing. Using the SAD approach, we were able to solve the structure and to partially build four monomers in the ASU. This solution was subsequently used as search model for MR with a newly collected native data set that extended to 2.6-Å resolution. Final model consists of four polypeptide chains that were refined to 20.9% and 24.1% for Rwork and Rfree, respectively. The four monomers in the ASU are packed in the crystal with two distinct chemical environments: one for chains A and C and another for chains B and D (Fig. 1C). On average, chain C exhibits higher thermal factors than the other chains in the ASU and had less-defined electron density (Fig. 1C). The four monomers in the ASU almost perfectly align with each other, showing only minor deviations. Residues 77 to 237 in chains A and C and residues 81 to 237 in chains B and D are ordered and were included in the final model (Fig. 1A). Electron density for N-linked glycans was visible at four sites on GP1LASV. On chain C, only two glycans linked to Asn109 and Asn224 were modeled. On chains A, B, and D, two additional glycans at Asn119 and Asn167 were modeled. Based on the quality of the electron density, the first one or two N-acetylglucosamine units were included for each glycan (Fig. 1C).
FIG 1.

Production and overview of the crystal structure of GP1LASV. (A) Schematic diagram showing the Lassa virus glycoprotein complex (GPC) precursor, the GP1 construct that was crystallized, and the ordered residues that were modeled into electron density maps for the 4 copies of GP1LASV of the asymmetric unit in the crystal. (B) Chromatogram of size exclusion chromatography used as the last purification step for GP1LASV. The arrow indicates the monomeric peak of GP1LASV, and Coomassie blue-stained SDS-PAGE analysis shows purified GP1LASV. (C) The 4 polypeptide chains that were modeled in the asymmetric unit are represented as tubes using different colors. The width of the tube is proportional to the B factor of the atoms. (D) Ribbon diagram of GP1LASV colored as a rainbow, starting from the N terminus in blue to the C terminus in red. Secondary structure elements are labeled according to type (α for α-helices and β for β-strands) and order of appearance in the primary structure. D designates disulfide bridges. The image on the right is rotated 90° around the y axis with respect to the image on the left. (E) Ribbon diagram showing a superposition of GP1MACV (PDB code 2WFO) on the structure of GP1LASV that was calculated using the central Cα atoms of β-strands 2, 7, and 4 from GP1LASV and β-strands 3, 7, and 6 from GP1MACV. GP1LASV is shown in magenta, except for the C and N termini in red and the extended β-hairpin V region in orange. GP1MACV is colored green, except for the C and N termini in blue and the loop connecting α-helix 2 and β-strand 7 (GP1MACV numbering) in cyan. (F) Multiple-sequence alignment of GP1s from Lassa virus, lymphocytic choriomeningitis virus (LCMV), Dandenong virus, Ippy virus, and Morogoro virus OW arenaviruses, showing the secondary structure elements as observed in the crystal structure of GP1LASV. The numbering of the amino acids is based on the sequence of the Lassa virus glycoprotein precursor. Fully conserved residues are highlighted with a red background, and partially conserved residues are shown in red. A blue-to-white scale bar below the sequences indicates solvent accessibility (exposed to buried). Numbers below the sequences designate the locations of the disulfide bridges. The symbol ‘Ψ’ marks glycosylated asparagine residues. This graphical representation was generated by ESPript (25) (http://espript.ibcp.fr).
TABLE 1.
Data collection and refinement statistics
| Parameter | Valuea for: |
|
|---|---|---|
| Native protein | SeMet protein | |
| Data collection | ||
| Wavelength (Å) | 0.953 | 0.978 |
| Space group | P1211 | P1211 |
| Cell dimensions | ||
| a, b, c (Å) | 49.6, 86.5, 109.5 | 60.6, 93.0, 110.7 |
| α, β, γ (°) | 90.0, 92.8, 90.0 | 90.0, 93.9, 90.0 |
| Resolution (Å)b | 49.6–2.6 (2.7–2.6) | 50.0–3.8 (3.9–3.8) |
| Rmeas (%) | 10.2 (139.0) | 10.6 (20.9) |
| CC1/2 | 99.9 (74.6) | 99.8 (99.5) |
| I/sI | 14.3 (1.6) | 35.1 (18.8) |
| Completeness (%)c | 99.1 (94.2) | 100.0 (100.0) |
| Multiplicity | 6.8 | 24.6 |
| Reflections | 195,511 | 301,648 |
| Unique reflections | 28,667 | 12,240 |
| Refinement | ||
| Resolution (Å) | 49.6–2.6 | |
| No. of reflections | 24,011 | |
| Rwork/Rfreed | 20.9/24.1 | |
| No. of atoms | ||
| Protein | 5,359 | |
| Ligand/ion | 280 | |
| Water | 85 | |
| B factors | ||
| Protein | 58.2 | |
| Ligand/ion | 72.6 | |
| Water | 49.4 | |
| Ramachandran | ||
| Favored (%) | 96.5 | |
| Allowed (%) | 3.5 | |
| Outlier (%) | 0.0 | |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.004 | |
| Bond angles (°) | 0.862 | |
Values in parentheses are for the highest-resolution shell.
For the native data set, resolution limits are 3.1 Å, 2.6 Å, and 2.6 Å along a*, b*, and c*, respectively.
After truncating the native data to 3.1 Å along a*, overall completeness drops to 83.5%. The overall completeness in the resolution range of 49.6 to 3.1 is 100%.
8.35% of unique reflections were removed as a test set for the Rfree calculation.
Molecular architecture of GP1LASV.
The crystal structure of GP1LASV reveals a central β-sheet sandwiched by the N and C termini on one side and an array of α-helices and loops on the other (Fig. 1D). GP1LASV shares the same overall fold as was previously observed in the crystal structure of GP1MACV (14), but major structural differences in the form of rearrangements, additions, and deletions of secondary structure elements can be observed (Fig. 1E). The most structurally conserved region is the central part of the core β-sheet. Three β-strands from each structure were aligned, emphasizing the differences between GP1LASV and GP1MACV in other regions of the protein (Fig. 1E). For example, α-helices 1 and 3 of GP1LASV are almost perpendicular to the corresponding α-helices (numbered 1 and 2) in GP1MACV. In addition, the longer α-helix 2 in GP1MACV corresponds to two shorter α-helices (numbered 2 and 3) with different orientations in GP1LASV. A loop that connects α-helix 3 and β-strand 7 is much longer than the corresponding loop at GP1MACV and forms a novel β-hairpin-like structure (Fig. 1E). This β-hairpin is stabilized by a disulfide bridge between β-strand 6 and α-helix 2. Interestingly, multiple-sequence alignment of representative OW arenaviruses reveals that β-strand 5 is highly variable both in length and in composition (Fig. 1F). We thus term this region the variable (V) region. Another significant difference between the two GP1 structures is a complete rearrangement of the termini. The first β-strand at the N terminus of GP1MACV and the helical segment at the C terminus are completely reoriented and adopt loop-like structures on the surface of the central β-sheet of GP1LASV (Fig. 1E).
Structural analysis indicates a putative receptor-binding site.
Superimposing GP1LASV on the GP1MACV/TfR1 structure (15) (Fig. 2A, left) reveals that the comparable region of the TfR1-binding site in GP1LASV is occupied by the N and C termini of the protein (Fig. 2A, right). Thus, the termini of GP1LASV may pose a steric barrier that will preclude receptor binding in a fashion similar to that for GP1MACV. As was also observed in the case of GP1MACV (14), the glycans on GP1LASV cluster on one side of the protein (Fig. 2B), roughly defining the exposed surface of GP1 in the context of the trimer that should harbor the receptor-binding site. To identify possible alternative sites for receptor binding, we analyzed the structure of GP1LASV using the ConSurf server (22) (Fig. 2C). This analysis revealed a conserved continuous patch that forms on β-strand 1 and additional residues from both the N and C termini (Fig. 2D). Central residues of this patch are His92 and His93 (from β-strand 1) and His230 (near the C terminus), which together form an unusual cluster of three histidines (Fig. 2D). Next to this histidine triad and part of the conserved patch is an exposed disulfide bridge between Cys86 and Cys231 (Fig. 2D). Additional residues that contribute to this patch are Ser91, Asn89, and Tyr217 (Fig. 2D). Interestingly, the chemical nature of this patch is mostly hydrophilic. Considering that histidine side chains have a pKa of around 6, endocytosis of LASV from the outside of the cell (pH ∼7.4) to the lysosome (pH ∼5.0) may dramatically change the electrostatic potential of this conserved patch. Specifically, full or partial protonation of the histidine triad will create a strong positive charge on this conserved region of GP1.
FIG 2.

Putative receptor-binding site on the surface of GP1LASV. (A) The structure of GP1MACV (green surface) in complex with TfR1 (orange ribbon) (PDB code 3KAS) (left image) compared with that of GP1LASV (magenta surface) that was superimposed on GP1MACV (omitted from the figure) in complex with TfR1 (right). This superimposition reveals a potential clash with the terminal region of GP1LASV (red) and excludes this region as a binding site. (B) Ribbon representation of GP1LASV showing N-linked glycans as spheres. The cyan and light gray distinguish the two halves of the molecule that are either lacking or bearing N-linked glycans, respectively. The four asparagine residues that were found to be glycosylated are listed. (C) Molecular surface of GP1LASV that is colored by a relative conservation score based on multiple-sequence alignment of 58 unique sequences, calculated using the ConSurf server (22). Relatively variable and conserved residues are shown in cyan and dark purple, respectively. Glycans are shown as sticks. (D) Detailed view of the conserved patch on the surface of GP1LASV using the same color coding as in panel C. Individual amino acids that contribute to this patch are shown as sticks. An adjacent glycan is shown as spheres.
Recombinant GP1LASV interacts with LAMP1.
Notably, we have crystallized GP1LASV in a reservoir that contained a buffer at pH 5.0 (see Materials and Methods). Therefore, this GP1LASV structure most likely represents the positively charged state of the conserved patch as will appear at the lysosome. Moreover, it has been shown recently that LASV switches from α-DG to LAMP1 in a pH-dependent manner when it reaches the lysosome (13). We were thus interested in whether the GP1LASV that we have crystallized could interact with LAMP1. To test this idea, we decorated activated resin with GP1LASV, performed pulldown assays from cell lysates, and probed for LAMP1 using Western blot analysis (Fig. 3). At pH 5.0, but not at pH 8.0, GP1LASV was able to pull down endogenous LAMP1 from HEK293 cells, indicating that the LAMP1-binding site was properly formed at the lower pH. Interestingly, we were not able to pull down α-DG at pH 8.0 (Fig. 3) or at pH 5.0 (data not shown) using the same assay. In contrast, the full trimeric glycoprotein spike complex was previously shown to pull down α-DG at alkaline and slightly acidic pHs (13). We thus conclude that our GP1LASV is in a LAMP1-compatible state that could not revert to an α-DG-compatible state using a simple pH switch.
FIG 3.
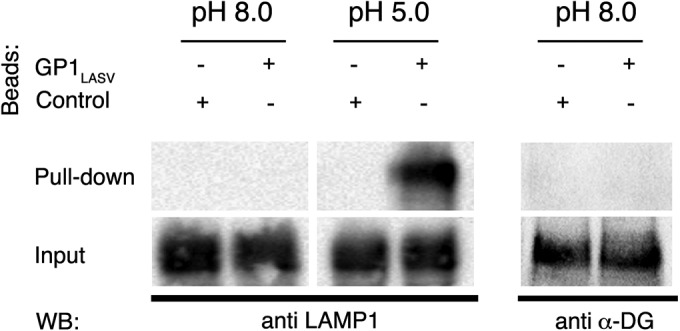
GP1LASV can pull down endogenous LAMP1. Purified GP1LASV was immobilized on agarose beads, incubated with a soluble phase of a cell lysate at pH 8.0 and pH 5.0, pulled down, and washed. Western blot analysis of the pulled-down fraction was performed using anti-LAMP1 antibody (left) or anti-α-DG (right, at pH 8.0).
The conserved histidine triad forms part of the LAMP1-binding site.
The pH-dependent LAMP1 binding suggests that pH probes like histidine could play a role in forming the binding site. We thus postulated that the conserved histidine triad could make part of this binding site. To investigate this notion, we mutated each histidine of the conserved histidine triad (Fig. 4A) to tyrosine and tested the binding to LAMP1. Importantly, structural analysis suggests that each histidine could be replaced by a tyrosine without causing steric clashes with neighboring residues, and in all three cases the hydroxyl group of the tyrosine is predicted to extend into the solvent. Wild-type GP1LASV and the three mutated variants were expressed in HEK293 cells as chimeras having an Fc portion of an antibody fused to their C termini. All Fc fusion variants were readily expressed and secreted into the media, indicating that the proteins were properly folded. The Fc fusion products assemble into dimers through the Fc portion, enabling avidity for LAMP1 capture. Supernatants were loaded onto protein A beads, washed, and incubated at pH 5.0 with a soluble fraction of detergent-lysed HEK293 cells. The pulled-down fractions were analyzed using anti-LAMP1 antibody. Compared to WT, all three histidine mutations significantly reduced the ability of GP1 to capture LAMP1 (Fig. 4B). To further analyze the role of the histidine residues in forming the LAMP1-binding site, we cloned the luminal ectodomain (residues 29 to 350) of human LAMP1, fused it to Fc (similar to a previously reported construct [13]), and expressed and purified it from HEK293 cells (Fig. 4C). We also produced and purified in large scale GP1LASV-Fc, GP1LASVH92Y-Fc, GP1LASVH93Y-Fc, and GP1LASVH230Y-Fc. Purified LAMP1-Fc was immobilized on an SPR sensor chip, and the binding of GP1LASV-Fc was compared to the binding of GP1LASVH92Y-Fc, GP1LASVH93Y-Fc, and GP1LASVH230Y-Fc by flowing a dilution series of each of the proteins in a pH 5.0 buffer (Fig. 4D). As a control, we injected a concentration series of GP1JUNV-Fc and analyzed its interaction with immobilized LAMP1 (Fig. 4D). Compared to the binding of GP1LASV-Fc, GP1LASVH92Y-Fc showed a reduced response with an apparent lower affinity (Fig. 4D). The GP1LASVH93Y-Fc and GP1LASVH230Y-Fc proteins almost completely lost their capacity to interact with LAMP1. GP1JUNV-Fc was also inert to LAMP1. Importantly, avidity due to the dimeric nature of the Fc fusion constructs enhances the interaction with the immobilized LAMP1, resulting in an apparent higher affinity. To validate that the mutations in the histidine triad did not change the overall fold of the GP1 domain, we performed ELISA by immobilizing the Fc fusion proteins and used an anti-GP1LASV monoclonal antibody (our unpublished results) for detecting them (Fig. 4E). All mutants were recognized to a level similar to that for GP1LASV-Fc (Fig. 4E). GP1JUNV-Fc served as a control for the ELISA (Fig. 4E). Since the ELISA cannot be performed in acidic pH, we recorded circular dichroism (CD) spectra from GP1LASV-Fc, GP1LASVH92Y-Fc, GP1LASVH93Y-Fc, and GP1LASVH230Y-Fc at pH 5.0 (Fig. 4F). The spectra are almost identical, indicating that mutating the histidine triad did not affect the overall fold of GP1.
FIG 4.

The conserved histidine triad is important for LAMP1 binding. (A) Surface representation of GP1LASV showing the contribution of His92, His93, and His230 to the formation of the putative LAMP1-binding site. (B) The ability of the GP1LASV-Fc chimera to pull down endogenous LAMP1 via protein A-conjugated beads is compared to those of GP1-Fc variants harboring a His-to-Tyr mutation in each of the three conserved histidines. LAMP1 was detected using anti-LAMP1 antibody. All three mutations significantly reduced the amount of LAMP1 that was precipitated. As a control, the total load of GP1-Fc conjugate was verified using anti human-Fc antibody. A schematic diagram illustrates the fusion proteins used in this assay; green circles represent GP1, and blue rectangles illustrate the Fc portions that promote dimerization. (C) Coomassie blue-stained SDS-PAGE showing purified Fc fusion proteins of GP1LASV-WT and mutants, GP1JUNV, and LAMP1 that were used in the SPR and ELISAs. A schematic diagram shows the chimeric LAMP1 luminal ectodomain fused to Fc. (D) Sensograms showing surface plasmon resonance analysis of a 2-fold dilution series (500 nM, 250 nM, 125 nM, 62.5 nM, 31.25 nM, and 0 nM) for GP1LASV-Fc, GP1LASVH92Y-Fc, GP1LASVH93Y-Fc, and GP1LASVH230Y-Fc that were injected over immobilized LAMP1-Fc at pH 5.0 (orange curves). A concentration series of GP1JUNV-Fc was used as a control, and the response curve for the highest concentration used (500 nM) is included in black to the sensogram of GP1LASV-Fc. Measurements were repeated twice in buffers containing 0.5 M and 0.25 M sodium chloride. Results from the high-ionic-strength buffer are presented. (E) Bar graph indicating the relative response of an anti-GP1LASV monoclonal antibody against the WT, various GP1LASV-Fc mutants, and GP1JUNV-Fc in ELISA. Values are the means from two separate experiments, each containing three duplicates. Error bars indicate the spread of the values. (F) CD spectra of GP1LASV-Fc, GP1LASVH92Y-Fc, GP1LASVH93Y-Fc, and GP1LASVH230Y-Fc, shown in red, blue, green, and orange, respectively.
DISCUSSION
The structure of GP1LASV and the LAMP1-binding site.
Solving the first crystal structure of GP1 from an OW arenavirus allowed us to compare it with the only other known structure of an arenavirus GP1, from the NW MACV arenavirus. This comparison revealed highly diversified structures (Fig. 1D), reflecting the evolutionary distance of those viruses. Nevertheless, GP1LASV and GP1MACV share the same overall basic architecture, with a central β-sheet and two distinct halves: a glycosylated half that contains the receptor-binding site and is made mostly by the central β-sheet and surrounding loops and a half that contains mostly helices and probably faces the trimer axis.
Conservation analysis indicated a conserved patch of GP1LASV (Fig. 2B) that is distant from the known TfR1-binding site of GP1MACV (15). This patch contains a unique cluster of three histidines on the surface of GP1LASV (Fig. 2C) that are important for LAMP1 binding (Fig. 4B and D). Mutating His92 resulted in a reduction in the apparent affinity (Fig. 4D). This kind of reduction is expected when a mutation at protein-protein interaction interface is introduced and has some cost in terms of binding energy. Mutating His93 and His230, on the other hand, resulted in a much more deleterious effect on binding (Fig. 4D). The binding energy that is provided by His93 and His230 might be higher than that provided by His92, or mutating His93 or His230 may result in some conformational changes that indirectly affect binding. Such changes, however, must be local, as the histidine mutants are well folded and secreted from cells (Fig. 4C), are recognized to similar extents by a monoclonal antibody (Fig. 4E), and are almost indistinguishable by CD analysis (Fig. 4F). While performing the SPR analysis, we noticed that the ionic strength is inversely and strongly correlated with the binding response of GP1LASV-Fc to LAMP1 (250 nM versus 500 nM NaCl) (data not shown). This observation implies that polar interactions, as predicted for the histidine triad, are important for binding. Altogether, we can conclude that the histidine triad is a structural element that directly interacts with LAMP1 and is perhaps further needed to stabilize a LAMP1-“compatible” conformation. The high conservation of residues that make the LAMP1-binding site in other OW arenaviruses (Fig. 1E) implies that LAMP1 switching may be common to other OW arenaviruses, although LCMV was shown not to require LAMP1 for infecting cells (13). Identifying the histidine triad as a central component of the LAMP1-binding site provides a molecular mechanism for the pH dependency of LASV switching to LAMP1.
Possible conformational changes of GP1LASV.
GP1LASV can pull down LAMP1 but not α-DG (Fig. 3), implying that the α-DG-binding site is not properly formed on GP1LASV and hence not faithfully represented in our crystal structure. Different explanations may account for this observation. One possibility is that the α-DG-binding site spans two GP1 protomers in the context of the trimeric spike complex. We cannot completely rule out this possibility, but we consider it to be less probable. Such an explanation suggests that at least half of the binding site is presented on each GP1 protomer and that a complete site could transiently form by the GP1LASV-Fc constructs or by GP1 conjugated to beads due to the relative flexibility between the GP1s, a scenario that should lead to some detectable binding. Yet, both GP1LASV-Fc (data not shown) and GP1LASV beads (Fig. 3) failed to pull down α-DG. Another possible explanation, which we favor, is irreversible conformational changes that GP1 undergoes preventing it from binding α-DG. Such putative conformational changes could be induced by dissociation of GP1 from the trimer (or producing it alone in our case), a change in the pH, or a combination of both. We do not have direct evidence of conformational changes in GP1LASV, and perhaps such evidence will not be available until the structure of an intact trimeric spike complex is available. Nevertheless, our structural analysis provides insights that support this notion. One of the significant differences between GP1MACV and GP1LASV is the position of the termini (Fig. 1D). This structural difference could reflect either the great evolutionary distance between LASV and MACV or a novel conformational state that is available to GP1LASV and was trapped in our structure.
By superimposing the two structures, a reasonable hypothesis can be made that the termini of GP1LASV could rearrange to adopt a conformation similar to that seen in GP1MACV, by swinging around an axis formed between two pivot points (Fig. 5A). The molecular surface underneath the termini that could potentially be exposed by such structural rearrangements is fairly hydrophilic (Fig. 5B). A close inspection of the putative molecular surface reveals a buried glutamic acid (Glu104) (Fig. 5C) that is also highly conserved (Fig. 1E). At neutral pH, Glu104 is likely to be charged. Considering the current observed conformation of GP1LASV, there is no obvious countercharged partner that could compensate and stabilize a negatively charged Glu104 at the protein core. At low pH, on the other hand, Glu104 may be either protonated or compensated by the close proximity of the positively charged histidine triad, although not making any obvious salt bridge in our structure. This observation indirectly supports the possibility that at neutral pH and when the spike complex is intact, this region of the protein is solvent exposed. Thus, the α-DG-binding site may be partially concealed by the termini and hence unavailable.
FIG 5.

Proposed conformational changes for GP1LASV. (A) Ribbon representation of GP1LASV colored in magenta, showing the terminal regions in red. The terminal regions from GP1MACV extracted from the superposition on GP1LASV are shown in blue and represent a potential alternative conformation for the GP1LASV termini. Arrows at the end of the N-terminal region and the beginning of the C-terminal region define the potential pivot points that would allow the terminal region of GP1LASV to swing and change conformation. (B) The molecular surface of GP1LASV was calculated after omitting the terminal regions to show the chemical nature of the buried surface underneath it. The termini are shown as red ribbons. Oxygen and nitrogen atoms are colored red and blue, respectively, to highlight hydrophilic regions. (C) A close look at the buried area, highlighting some of the hydrophilic residues. Glu104, in the vicinity of the histidine triad, is almost fully buried but would be exposed if the termini adopted a conformation similar to that seen in GP1MACV.
Immune evasion mechanisms of LASV.
During infection, antibody responses against LASV as well as other OW arenaviruses are not thought to facilitate clearance, as high viremia and high antibody titers often coexist (23). Our structural data suggest two possible mechanisms for LASV immune evasion: presentation of a surface variable region and conformational changes of GP1 upon shedding. Structure-based sequence alignment allowed us to identify a variable region made of β-strand 5 of GP1LASV (Fig. 1E). This variable region is likely to be highly exposed in the context of the functional trimeric spike complex and is probably under immunological pressure that drives the observed diversification. The variability of this region suggests that it is not a crucial element for the function of the trimeric spike. Thus, antibodies that target this region may have a less deleterious effect on LASV than antibodies that target other functional sites of GP1. Therefore, the variable region may serve as an immunological decoy. Moreover, GP1 shedding was detected during acute LASV infection, a process that was suggested to serve immunological decoying (24). If GP1 indeed changes conformation to conceal the α-DG-binding site upon dissociation from the spike complex, this ratifies the role of shed GP1 as an immunological decoy. Soluble GP1 in this scenario will present a nonfunctional altered conformation, which is likely to elicit an ineffective neutralizing-antibody response.
ACKNOWLEDGMENTS
We thank Babu A. Manjasetty (EMBL) for providing critical support on the beamline and EMBL-DBT for providing access to the BM14 beamline at the ESRF. We thank Yosef Scolnik for his kind help in recording CD spectra. The α-DG (IIH6) antibody developed by Kevin P. Campbell was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA.
Ron Diskin is incumbent of the Tauro career development chair in biomedical research. This research was supported by a research grant from the Enoch Foundation, a research grant from the Abramson Family Center for Young Scientists, a research grant from Rudolfine Steindling, and the I-CORE Program of the Planning and Budgeting Committee and The Israel Science Foundation (grant no. 1775/12).
REFERENCES
- 1.Oldstone MB. 2002. Arenaviruses. I. The epidemiology molecular and cell biology of arenaviruses. Introduction. Curr Top Microbiol Immunol 262:v–xii. [PubMed] [Google Scholar]
- 2.Geisbert TW, Jahrling PB. 2004. Exotic emerging viral diseases: progress and challenges. Nat Med 10:S110–121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- 3.McCormick JB, Fisher-Hoch SP. 2002. Lassa fever. Curr Top Microbiol Immunol 262:75–109. [DOI] [PubMed] [Google Scholar]
- 4.Nunberg JH, York J. 2012. The curious case of arenavirus entry, and its inhibition. Viruses 4:83–101. doi: 10.3390/v4010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, Li W, Nagel J, Schmidt PJ, Nunberg JH, Andrews NC, Farzan M, Choe H. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zong M, Fofana I, Choe H. 2014. Human and host species transferrin receptor 1 use by North American arenaviruses. J Virol 88:9418–9428. doi: 10.1128/JVI.01112-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Nichol ST, Compans RW, Campbell KP, Oldstone MB. 1998. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- 8.Kunz S, Rojek JM, Perez M, Spiropoulou CF, Oldstone MB. 2005. Characterization of the interaction of lassa fever virus with its cellular receptor alpha-dystroglycan. J Virol 79:5979–5987. doi: 10.1128/JVI.79.10.5979-5987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spiropoulou CF, Kunz S, Rollin PE, Campbell KP, Oldstone MB. 2002. New World arenavirus clade C, but not clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J Virol 76:5140–5146. doi: 10.1128/JVI.76.10.5140-5146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eschli B, Quirin K, Wepf A, Weber J, Zinkernagel R, Hengartner H. 2006. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J Virol 80:5897–5907. doi: 10.1128/JVI.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasquato A, Burri DJ, Traba EG, Hanna-El-Daher L, Seidah NG, Kunz S. 2011. Arenavirus envelope glycoproteins mimic autoprocessing sites of the cellular proprotein convertase subtilisin kexin isozyme-1/site-1 protease. Virology 417:18–26. doi: 10.1016/j.virol.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 12.Burri DJ, da Palma JR, Kunz S, Pasquato A. 2012. Envelope glycoprotein of arenaviruses. Viruses 4:2162–2181. doi: 10.3390/v4102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jae LT, Raaben M, Herbert AS, Kuehne AI, Wirchnianski AS, Soh TK, Stubbs SH, Janssen H, Damme M, Saftig P, Whelan SP, Dye JM, Brummelkamp TR. 2014. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowden TA, Crispin M, Graham SC, Harvey DJ, Grimes JM, Jones EY, Stuart DI. 2009. Unusual molecular architecture of the machupo virus attachment glycoprotein. J Virol 83:8259–8265. doi: 10.1128/JVI.00761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abraham J, Corbett KD, Farzan M, Choe H, Harrison SC. 2010. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat Struct Mol Biol 17:438–444. doi: 10.1038/nsmb.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr A Found Crystallogr 64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 18.Pape T, Schneider TR. 2004. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J Appl Crystallogr 37:843–844. doi: 10.1107/S0021889804018047. [DOI] [Google Scholar]
- 19.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. 2005. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher-Hoch SP, McCormick JB. 2001. Towards a human Lassa fever vaccine. Rev Med Virol 11:331–341. doi: 10.1002/rmv.329. [DOI] [PubMed] [Google Scholar]
- 24.Branco LM, Grove JN, Moses LM, Goba A, Fullah M, Momoh M, Schoepp RJ, Bausch DG, Garry RF. 2010. Shedding of soluble glycoprotein 1 detected during acute Lassa virus infection in human subjects. Virol J 7:306. doi: 10.1186/1743-422X-7-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]