

ABSTRACT
Respiratory syncytial virus (RSV) is the leading cause of acute respiratory tract viral infection in infants, causing bronchiolitis and pneumonia. The host antiviral response to RSV acts via retinoic acid-inducible gene I (RIG-I). We show here that RSV infection upregulates major histocompatibility complex class I (MHC-I) expression through the induction of NLRC5, a NOD-like, CARD domain-containing intracellular protein that has recently been identified as a class I MHC transactivator (CITA). RSV infection of A549 cells promotes upregulation of NLRC5 via beta interferon (IFN-β) production, since the NLRC5-inducing activity in a conditioned medium from RSV-infected A549 cells was removed by antibody to IFN-β, but not by antibody to IFN-γ. RSV infection resulted in RIG-I upregulation and induction of NLRC5 and MHC-I. Suppression of RIG-I induction significantly blocked NLRC5, as well as MHC-I, upregulation and diminished IRF3 activation. Importantly, Vero cells deficient in interferon production still upregulated MHC-I following introduction of the RSV genome by infection or transfection, further supporting a key role for RIG-I. A model is therefore proposed in which the host upregulates MHC-I expression during RSV infection directly via the induction of RIG-I and NLRC5 expression. Since elevated expression of MHC-I molecules can sensitize host cells to T lymphocyte-mediated cytotoxicity or immunopathologic damage, the results have significant implications for the modification of immunity in RSV disease.
IMPORTANCE Human respiratory syncytial virus (RSV) is the leading cause of bronchiolitis and pneumonia in infants and young children worldwide. Infection early in life is linked to persistent wheezing and allergic asthma in later life, possibly related to upregulation of major histocompatibility class I (MHC-I) on the cell surface, which facilitates cytotoxic T cell activation and antiviral immunity. Here, we show that RSV infection of lung epithelial cells induces expression of RIG-I, resulting in induction of a class I MHC transactivator, NLRC5, and subsequent upregulation of MHC-I. Suppression of RIG-I induction blocked RSV-induced NLRC5 expression and MHC-I upregulation. Increased MHC-I expression may exacerbate the RSV disease condition due to immunopathologic damage, linking the innate immune response to RSV disease.
INTRODUCTION
Respiratory syncytial virus (RSV) is the leading cause of lower respiratory tract infection in infants and young children, causing bronchiolitis and pneumonia in infants and young children worldwide. Due to the highly infectious nature of the virus, roughly two-thirds of children are infected by their first birthday, and this reaches essentially 100% by the age of 2 (1, 2). RSV infection is a leading cause of infant hospitalization due to bronchiolitis (2, 3). In the United States alone, an estimated 2.1 million children under 5 years of age with RSV infection require medical attention each year (4). Importantly, lower respiratory tract infection by RSV early in life is a risk factor for persistent wheezing and asthma in later life (5, 6). There are no RSV vaccines available to prevent childhood infection. These factors create an urgent need to understand the mechanisms of RSV disease, the molecular mechanisms associated with immunoregulation, and the downstream association between RSV infection and allergic asthma.
RSV belongs to the subfamily Pneumovirinae of the paramyxoviruses. A negative-sense, single-stranded RNA virus with a genome of approximately 15,000 nucleotides (7), the virus can infect a broad range of cells. In patients, however, infection is normally highly restricted to the superficial cells of the respiratory epithelium, the ciliated cells of the small bronchioles, and pneumocytes in the alveoli (8–10). Infection is initiated by cell surface binding via proteoglycans (11), followed by nucleolin-mediated fusion for RSV cell entry (9, 12) and infection. In response, the host initiates an early innate immune response at the site of infection. Receptors of innate immune recognition, like Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I), which are involved in detection of viral RNA, promote the activation of antiviral immunity and cytokine production, as well as the recruitment of proinflammatory cells (10, 13–16). This increased expression of inflammatory mediators, immune cell chemoattractants, and antigen-processing machinery is implicated in RSV-induced lung injury (13, 17–19). Indeed, several gene-based studies have linked the differences in outcomes after RSV infection to genes involved in immune responses, including those for interleukin 4 (IL-4), IL-6, and IL-8, as well as TLR4, in innate immunity (20). The importance of the T cell response in RSV disease is also supported by the observation that RSV infection of airway epithelial cells upregulates major histocompatibility complex class I (MHC-I) expression (21–23), although many viruses have the ability to downregulate MHC-I expression as a strategy for immune evasion (24–26). An apparent genetic susceptibility, based on the MHC class, to delayed sequelae of neonatal RSV infection is suggested by genome-wide association studies in murine models. The MHC haplotype and CD8+ T cell responses are linked as key determinants of the outcome in neonatal RSV infection (22). Despite this link, significant gaps in our knowledge remain regarding the mechanisms of MHC-I induction following RSV infection.
The expression of MHC-I molecules is transcriptionally regulated by transcription factors and a specific transactivator of MHC-I genes (CITA) (27, 28). The promoter regions consist of NF-κB binding regions, interferon (IFN)-stimulated response elements (ISRE), and an SXY module. The last is comprised of W/S, X1, X2, and Y boxes (27, 29) and is regulated by the binding of a multiprotein complex (30–34) to regulate the expression of MHC molecules (35–37). A recently identified NOD-, LRR-, and CARD-containing 5 (NLRC5) protein has been demonstrated to function as a specific cotransactivator of MHC-I genes (28, 38–41). NLRC5 is a multidomain protein expressed in lymphocytes, as well as in the lung (http://www.genecards.org/cgi-bin/carddisp.pl?gene=NLRC5). Viral infection, interferon stimulation, and similar challenges induce NLRC5 expression in a STAT1-dependent manner (28, 42). However, NLRC5 itself does not have a DNA-binding domain and therefore must cooperate with the multiprotein complex to regulate MHC-I gene expression (28, 39, 43, 44).
We investigated MHC-I expression during RSV infection of A549 cells. Viral infection was always accompanied by upregulation of RIG-I and, subsequently, production of interferons. Notably, NLRC5 induction followed beta interferon (IFN-β) induction and RIG-I upregulation, since suppression of RIG-I ablated both NLRC5 and MHC-I gene expression. The results implicate NLRC5 as a key effector of MHC upregulation downstream of interferon during RSV infection of human airway epithelial cells.
MATERIALS AND METHODS
Cells and virus.
The A549 human lung adenocarcinoma epithelial cell line was purchased from the ATCC, Manassas, VA (CCL-185). The Vero cell line (African green monkey kidney epithelial cells) originated from the ATCC and was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose that was supplemented with 2 mM l-glutamine, nonessential amino acids, sodium pyruvate, and 10% heat-inactivated fetal bovine serum (FBS) (all from Life Technologies). Newborn calf serum was used instead for Vero cells. All cells were cultured at 37°C in a humidified incubator with 5% CO2.
Human RSV type A (strain A2) was used in previous studies, and the titer of the virus was determined by following a previously reported protocol (45, 46). The resulting stock solution was stored at −70°C. Because RSV retains a close association with the host cell membrane and has a tendency to aggregate during centrifugation procedures, the stock of crude supernatant was used after a titer was determined.
To prepare genomic RNA for transfection studies, RSV was purified by ultracentrifugation by following a previously reported protocol (47). The purified virus was then treated with 500 μg/ml of proteinase K and 0.3% SDS at 56°C for 20 min (48). After removal of proteins by phenol-chloroform extraction, viral RNA was recovered by ethanol precipitation and used for transfection studies.
Antibodies and reagents.
Recombinant human IFN-β, IFN-γ, IL-28B, and IL-29 proteins were purchased from SinoBiologicals Inc. (Beijing, China). Lipofectamine 2000 was purchased from Life Technologies. p3x-FLAG-NLRC5 was kindly provided by Jean de Silva Correia. Antibodies used for immunoblotting studies of RSV F protein (sc-101362) and MHC-I (sc-32235) were purchased from Santa Cruz Biotechnology Inc. Antibody against p-IRF3 (4947) was purchased from Cell Signaling Technology. Monoclonal antibody to RIG-I was purchased from Alexis/Enzo Life Sciences (Farmingdale, NY). Antibodies against IFN-β (BS6631), IFN-γ (BS3486), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (AP0063) were purchased from Bioworld Technology Inc. Allophycocyanin (APC)-conjugated HLA-ABC (number 555571) and fluorescein isothiocyanate (FITC)-conjugated HLA-DR, -DP, and -DQ (number 555573) antibody kits for fluorescence-activated cell sorter (FACS) studies of MHC-I and MHC-II expression were purchased from BD Biosciences.
Preparation and validation of a monoclonal antibody against NLRC5.
To prepare a monoclonal antibody for the detection of endogenous NLRC5, a recombinant protein corresponding to the C-terminal 258 amino acid residues of human NLRC5 (NM_032206) was prepared in Escherichia coli and used to immunize BALB/c mice at AB-Mart (Shanghai, China). Eight individual clones were identified and subjected to further screening for the ability to recognize NLRC5. Clone 7 was used in this project, since it recognized FLAG-tagged NLRC5 expressed in HEK293T cells. The clone also selectively detected a set of protein bands at approximately 230 kDa from IFN-γ-treated A549 cells. Animal experiments were carried out with the authorization of Nanjing University, Jiangsu, China, in accordance with the China law for animal protection.
Preparation of conditioned medium.
Conditioned medium from RSV-infected A549 cells (CM-RSV A549) or Vero cells (CM-RSV Vero) was prepared by infecting A549 or Vero cells, respectively, with RSV at a multiplicity of infection (MOI) of 1.0 PFU/cell in DMEM containing 2% FBS. The medium was collected at approximately 48 h postinfection (p.i.), centrifuged at 3,000 × g for 30 min to remove cellular debris, and then stored at −70°C. To inactivate RSV in the CM-RSV, ice-cold supernatant was UV irradiated for 15 min at 1,200 mJ/cm2 using a UV Stratalinker 2400 (Stratagene).
Western blotting assay.
Cells plated in 6-well culture plates were harvested in lysis buffer containing 1% NP-40, 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), and a cocktail of protease inhibitors (Roche). After removal of cellular debris, soluble proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). The proteins were detected by incubation with a primary antibody, followed by horseradish peroxidase-conjugated secondary antibody, and the ECL reagent kit (Pierce). The images were captured using the Clinx (Shanghai, China) ChemiScope imaging system.
We used ImageJ software (NIH) to semiquantitatively measure changes in the ratio of MHC-I to GAPDH by boxing each band of a representative image with the rectangular selection tool and calculating the total area of the band in pixels. The total area of the MHC-I bands was normalized to the total area of the GAPDH band and plotted, using 1 for the MHC/GAPDH ratio in the scrambled control (SC).
Flow cytometry assay.
Monolayers of A549 cells were infected with RSV under the indicated conditions or remained uninfected. The cells were rinsed twice with diluted trypsin-EDTA (1× 0.25% trypsin-EDTA diluted with PBS at a ratio of 1:5). After removal of the trypsin-EDTA, the cells were left at room temperature for 3 to 5 min to detach the cells with residual trypsin-EDTA. The cells were then collected, washed twice with complete medium, and then resuspended in ice-cold PBS. MHC-I and MHC-II staining was carried out using APC- or FITC-conjugated antibody staining kits. The data were collected on a FACSCalibur and analyzed using FCS Express v3 software (De Novo Software, Glendale, CA).
For the study of NLRC5 in MHC-I expression, A549 cells were transfected with p3x-FLAG-NLRC5 or an empty vector as a control using Lipofectamine 2000. Thirty-six hours after transfection, cells were harvested and analyzed for NLRC5 expression by Western blotting or for MHC-I expression by FACS.
Gene-silencing study.
Small interfering RNA (siRNA) duplexes targeting NLRC5 (siNLRC5) or RIG-I (siRIG-I) expression were purchased from GenePharma (Shanghai, China) and were used to transfect A549 cells using Lipofectamine 2000 according to the manufacturer's instructions. A scrambled oligonucleotide pair (NLRC5 number 1 and RIG-I number 1) was used as a control (SC). The sequences of the oligonucleotides for RIG-I and NLRC5 are as follows: RIG-I number 1, 5′-GCAUAUUGACUGGACGUGGCATT; RIG-I number 2, 5′-GGUGGAGGAUAUUGCAACUTT; NLRC5 number 1, 5′-CAGGGUUCUCUCCCUGUUAGA; and NLRC5 number 2, 5′-CUGCUGAUCUUUGAUGGGCUA.
Twenty-four hours after transfection, the cells were infected with RSV for another 36 h. The cells were then harvested and analyzed for NLRC5 and MHC-I expression by Western blotting and by quantitative PCR (qPCR) for the NLRC5 and HLA-A, -B, and -C genes.
Reverse transcription (RT)-PCR and quantitative real-time PCR.
RNA from A549 cells was isolated using TRIzol reagent (Life Technologies). Quantitative real-time PCR was performed on the Bio-Rad C1000 real-time PCR system using SYBR green Master Mix reagent (Bio-Rad). GAPDH was used as an internal control for normalization. The data were analyzed using the 2−ΔΔCT formula, and the control sample value was normalized to 1. The sequences of all the primers used in this study are as follows (forward and reverse): NLRC5, 5′-CTCAGCCAGGAGCACGTAG and 5′-CCAGCAGGTTTGCTGAGAGA; HLA-A, 5′-CCGTCCAGAGGATGTATGGC and 5′-CCAGGTAGGCTCTCAACTG; HLA-B: 5′-AACCGTCCTCCTGCTGCTCTC and 5′-CTGTGTGTTCCGGTCCCAATAC; HLA-C, 5′-GGATGTCTGGCTGCGACCTG and 5′-TCTTTGGGGGTTCTGCGCGC; IFN-β, 5′-CTTGGATCCTACAAAGAAGC and 5′-CATCTCATAGATGGTCAATGC; RIG-I, 5′-TGATTGCCACCTCAGTTGCT and 5′-ACTGCTTCGTCCCATGTCTG; STAT1, 5′-GGCACGCACACAAAAGTGAT and 5′-TTGGAGATCACCACAACGGG; and GAPDH, 5′-ATGACATCAAGAAGGTGGTG and 5′-CATACCAGGAAATGAGCTTG.
Immunodepletion of IFN-β from UV-irradiated CM-RSV.
UV-irradiated CM-RSV (100 μl) was incubated at 4°C for 60 min with an antibody against IFN-β at a final concentration of 10 or 30 μg/ml or an antibody against IFN-γ at 30 μg/ml. After removal of the antibody with protein A-Sepharose beads (Pierce), the supernatants were mixed with complete DMEM (5% [vol/vol]) and used to treat A549 cells.
RESULTS
RSV infection upregulates MHC-I and NLRC5 in A549 airway epithelial cells.
Previous studies have shown that RSV infection promotes MHC-I expression (21, 23). To delineate a mechanism of MHC-I upregulation during RSV infection, we first performed FACS analysis to determine whether RSV infection upregulated MHC complexes. To this end, A549 cells were infected with RSV at an MOI of 1.0 for 36 h. MHC-I and MHC-II expression in RSV-infected and uninfected controls were stained with anti-MHC-I and anti-MHC-II antibodies followed by FACS analysis. Consistent with the report by Garofalo et al. (21), RSV infection selectively induced MHC-I expression, and no induction of MHC-II was detected in RSV-infected A549 cells (Fig. 1A). The upregulation of MHC-I molecules was verified by qPCR for the heavy-chain molecules. As shown in Fig. 1B, elevated expression of HLA-A, HLA-B, and HLA-C was detected in RSV-infected cells compared with that in uninfected controls.
FIG 1.

Determination of MHC-I and MHC-II expression in RSV-infected A549 airway epithelial cells. (A) Surface expression of MHC-I and MHC-II by flow cytometry study. The A549 cells of a human alveolar basal epithelial cell line were infected with RSV at an MOI of 1.0 for 36 h. Surface expression of MHC-I or MHC-II was stained for with commercial antibody kits and determined by FACS. Values on the x axes are fluorescence intensities of APC-MHC I and FITC-MHC II, respectively. Blue lines indicate results for the idiotype controls. The experiment was performed independently 3 times. (B) Determination of MHC-I molecules by quantitative PCR. A549 cells were left uninfected (Con) or infected with various amounts of RSV for 24 h. Expression of the HLA-A, HLA-B, and HLA-C genes was determined by qPCR. The results are representative of three independent experiments with duplicate samples. The data are presented as means and standard deviations (SD) of duplicate samples. An unpaired t test was performed for statistical analysis. NS, no significance; **, P ≤ 0.01.
NLRC5 was recently identified as a cotransactivator for MHC-I expression, and we next investigated whether RSV infection promoted the expression of NLRC5. The induction of NLRC5 was first demonstrated using RT-PCR. As shown in Fig. 2A, RSV infection induced NLRC5 expression in A549 cells. In addition, we also detected increased expression of IFN-β, RIG-I, and STAT1, genes of the innate immune response. No expression of IFN-γ was detected even after 40 cycles of amplification. To closely investigate the role of NLRC5 induction in viral infection, we prepared a panel of monoclonal antibodies that could be used for immunoblotting studies. Clone 7 recognized both overexpressed and endogenously expressed NLRC5 (Fig. 2B) and was used in this study. A549 cells expressed NLRC5 at low levels (Fig. 2C). RSV infection induced NLRC5 expression in a dose- and time-dependent manner. The induction of NLRC5 was specific, since pretreatment of A549 cells with two different siRNAs targeting NLRC5 expression (siNLRC5 number 1 and number 2) significantly suppressed NLRC5 expression (Fig. 2D). Although siRNA treatment suppressed NLRC5 expression, the treatment did not significantly affect RSV infection, since F protein expression was detected at comparable levels in the samples (Fig. 2D).
FIG 2.

RSV infection promotes NLRC5 expression. (A) Gene induction determined by RT-PCR. A549 cells were infected with RSV at different MOIs (0.1, 0.3, and 1.0) for 24 h. Total RNA was isolated for the detection of NLRC5, IFN-β, RIG-I, and STAT1 by RT-PCR. GAPDH expression was used as an internal control. (B) Demonstration of a monoclonal antibody that recognizes both overexpressed and IFN-γ-induced NLRC5. A recombinant protein corresponding to the C-terminal 258 amino acid residues of human NLRC5 was prepared in E. coli and used to immunize BALB/c mice to generate a monoclonal antibody that could be used for immunoblotting (IB) studies. Clone 7 recognized two protein bands at approximately 230 kDa from p3x-FLAG-NLRC5-transfected 293T cells. It also recognized a pair of protein bands at approximately 230 kDa from IFN-γ-treated A549 cells. A band at approximately 130 kDa was a nonspecific band (ns). (C) Dose-response and time course studies of NLRC5 induction by RSV infection. A549 cells were infected with RSV at various MOIs, as indicated, for 24 h or at an MOI of 1 for the indicated times. NLRC5 induction was detected using clone 7. RSV F protein expression was used as an indication of infection, while GAPDH was used as a loading control. (D) Pretreatment of A549 cells with siNLRC5 suppresses NLRC5 induction. A549 cells were left untreated or treated with two different siRNAs targeting NLRC5 (siNLRC5 number 1 and number 2) or an SC. The cells were then infected with RSV at an MOI of 1 and used for detection of NLRC5 induction and F protein expression. GAPDH expression was used as a loading control.
Correlation between NLRC5 expression and MHC-I upregulation during RSV infection.
As a CITA, NLRC5 regulates MHC class I gene expression. We next investigated whether NLRC5 upregulation was involved in MHC-I induction during RSV infection. The effect of NLRC5 on MHC-I expression was first demonstrated by transfection of A549 cells with a plasmid for FLAG-NLRC5 expression (Fig. 3A, inset). Surface expression of MHC-I in NLRC5- and control vector-transfected A549 cells was detected by FACS analysis 36 h after transfection. As shown in Fig. 3A, overexpression of NLRC5 resulted in increased detection of MHC-I. The induction of MHC-I was also demonstrated by RNA levels. As shown in Fig. 3B, the levels of HLA-A, HLA-B, and HLA-C mRNAs in FLAG-NLRC5-transfected cells were significantly elevated compared to those in vector-transfected controls, indicating overexpression of NLRC5 promoted MHC-I expression in A549 cells.
FIG 3.
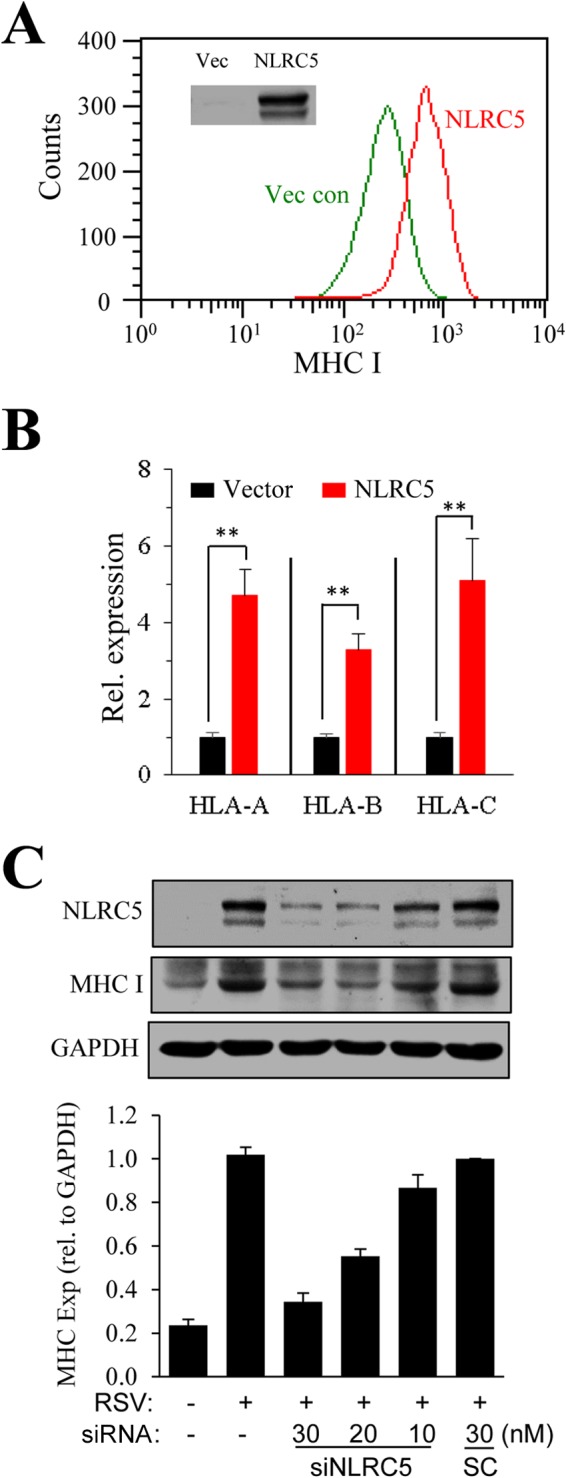
Correlation of NLRC5 expression and MHC-I induction. (A) Overexpression of NLRC5 enhances MHC-I expression. A549 cells were transiently transfected with an empty vector (Vec con) or with p3x-FLAG-NLRC5 for 36 h. Surface expression of MHC-I was determined by FACS. The inset shows NLRC5 expression determined by Western blotting. The experiment was performed twice independently. (B) In parallel experiments, gene expression of MHC-I molecules in the samples was determined by qPCR. The results are representative of three independent experiments. The data are presented as means and SD of duplicate samples. **, P ≤ 0.01. (C) Suppression of NLRC5 expression by siRNA inhibits MHC-I induction. A549 cells were treated by transfection with an SC or with siNLRC5 number 1 at various concentrations (10, 20, and 30 nM) for 24 h. The cells were left uninfected or infected with RSV for another 36 h. NLRC5 and MHC-I expression (Exp) was determined by immunoblotting. The experiment was performed three times independently. The fold change in the ratio of MHC-I to GAPDH was measured in the three blots using ImageJ software and plotted by normalizing to the ratio in the SC, set as 1.
We then asked whether suppression of NLRC5 expression by siRNA treatment would block MHC-I induction during RSV infection. A549 cells were transfected with various amounts of siNLRC5 number 1 prior to RSV infection. The expression of NLRC5 and MHC-I was determined by immunoblotting studies (Fig. 3C). Treatment with siNLRC5, but not an SC, significantly suppressed NLRC5 expression. MHC-I induction was also significantly reduced in these samples. The results, taken together, indicated that NLRC5 upregulation was a contributing factor for MHC-I induction during RSV infection.
RSV promotes NLRC5 expression through IFN-β secretion in A549 cells.
Several factors, including interferons and double-stranded RNA, are known to induce NLRC5 expression. We therefore investigated whether RSV infection promoted NLRC5 and MHC-I expression through a secreted factor(s) or by direct sensing of viral RNA. We first tested whether a conditioned medium from RSV-infected A549 cells contained NLRC5-inducing activity by following a widely used protocol (49). A conditioned medium from RSV-infected cells (CM-RSV) was harvested. The medium was used without treatment or treated by heat deactivation or by exposure to UV light. The samples were tested for their activity in inducing NLRC5 expression. As shown in Fig. 4A, the untreated medium (−UV) had both infectivity and NLRC5-inducing ability. Heat treatment (HK) abolished both infectivity and NLRC5-inducing activity. In contrast, UV irradiation (+UV) retained NLRC5-inducing ability, even though the treatment completely destroyed infectivity, as was confirmed by the absence of F protein expression in the sample (Fig. 4A). When we titrated for NLRC5-inducing activity in the medium, we found that the activity was relatively strong. Conditioned medium at as low as 2% promoted NLRC5 expression (Fig. 4B).
FIG 4.
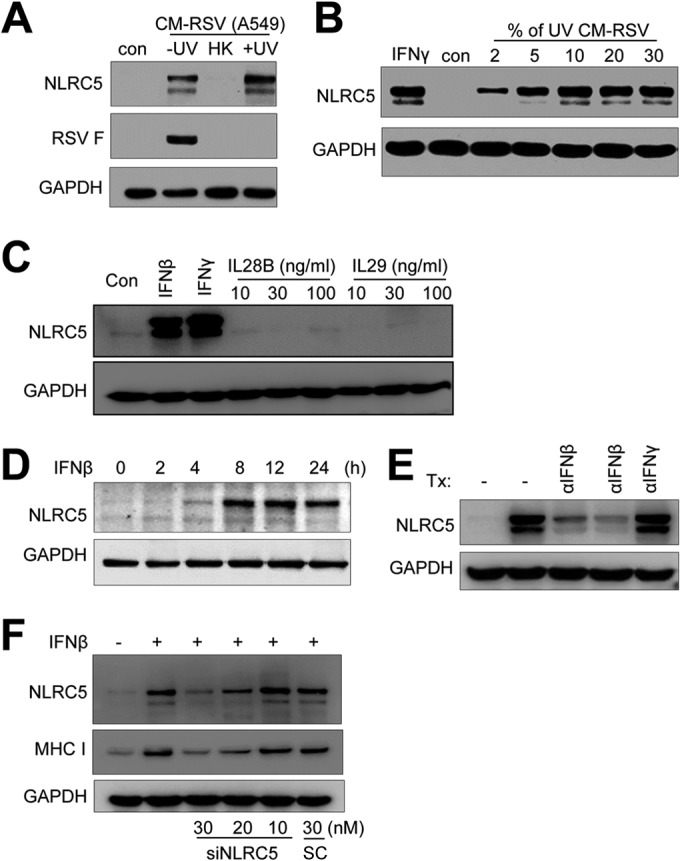
RSV promotes NLRC5 expression in A549 cells through IFN-β secretion. (A and B) Conditioned medium contains NLRC5-inducing activity. (A) A conditioned medium from RSV-infected A549 cells (CM-RSV A549) was UV irradiated to deactivate RSV (+UV), heat inactivated (HK), or left untreated (−UV). The medium was then mixed with fresh DMEM at a 10% ratio and used to treat A549 cells. (B) Dose response of NLRC5 induction to the conditioned medium. UV-irradiated conditioned medium was mixed with fresh DMEM at various ratios (percent [vol/vol]) and used to treat A549 cells. NLRC5 induction was determined by Western blotting. The expression of viral F protein was determined to show the effect of UV irradiation or heat deactivation on infectious virions. The experiment was performed two times independently. (C) Induction of NLRC5 by recombinant interferons. A549 cells were treated with IFN-β (100 U/ml), IFN-γ (100 U/ml), and IL-28B or IL-29 (10, 30, and 100 ng/ml) for 24 h. Cells were harvested, and NLRC5 induction was detected by Western blotting. GAPDH expression was used as an internal control. (D) Time course of NLRC5 induction by recombinant IFN-β. A549 cells were treated with recombinant IFN-β at 100 U for the indicated times. NLRC5 induction was determined by immunoblotting. GAPDH was the loading control. (E) Immunodepletion of IFN-β removes NLRC5-inducing activity. Aliquots (100 μl) of UV-irradiated conditioned medium were left untreated or incubated at 4°C with anti-IFN-β (10 μg/ml and 30 μg/ml) or anti-IFN-γ (30 μg/ml) for 60 min. The immunocomplexes were removed by protein A-Sepharose beads. The supernatants were mixed with fresh DMEM at 5% (vol/vol) and tested on A549 cells for NLRC5-inducing activity. The results are representative of two independent experiments. GAPDH was used as a loading control. Tx, treatment. (F) Suppression of NLRC5 expression inhibits NLRC5 and MHC-I induction by IFN-β. A549 cells were transfected with scrambled control or with siNLRC5 number 1 (10, 20, or 30 nM) for 24 h. The cells were then treated with 100 U of IFN-β for 24 h. NLRC5 and MHC-I expression was determined by Western blotting. GAPDH expression was the loading control.
A549 cells are known to produce interferons in response to viral infection (21, 50), factors that are known to induce NLRC5 expression. We therefore first tested whether recombinant interferons had the ability to induce NLRC5 in A549 cells using IFN-γ as a positive control. Treatment of A549 cells with IFN-β or IFN-γ, but not IL-28B or IL-29, members of the IFN-λ group (51), strongly induced NLRC5 expression (Fig. 4C). The effect of IFN-β on NLRC5 induction was rapid and robust, since elevated NLRC5 induction was detected at 4 h and persisted throughout the experiment (Fig. 4D), suggesting IFN-β production was potentially a factor in NLRC5 induction. We therefore treated the UV-irradiated medium with an antibody against IFN-β or IFN-γ to determine whether immunodepletion of IFN-β would remove NLRC5-inducing activity in the medium. As shown in Fig. 4E, depletion of IFN-β, but not IFN-γ, eliminated NLRC5-inducing activity, suggesting IFN-β production was responsible for NLRC5 and MHC-I induction during RSV infection of A549 cells. The conclusion was substantiated by the results from gene-silencing studies. A549 cells were treated with siNLRC5 number 1 at various concentrations or with a scrambled siRNA at 30 nM, followed by treatment with recombinant IFN-β. The induction of NLRC5 and MHC-I expression in the samples was determined by immunoblotting studies. As shown in Fig. 4F, IFN-β treatment induced both NLRC5 and MHC-I expression. Suppression of NLRC5 expression significantly inhibited MHC-I induction, indicating that IFN-β acted through NLRC5 to induce MHC-I expression during RSV infection of A549 cells.
Role of RIG-I in NLRC5 expression during RSV infection.
RIG-I, a pattern recognition receptor (PRR), has been demonstrated to sense viral RNA to mediate the early response to RSV infection in airway epithelial cells (14). We next investigated whether RIG-I expression played a role in NLRC5 induction. To this end, A549 cells were transfected with a preparation of RSV genomic RNA. NLRC5 expression was evaluated at 24 h posttransfection. As shown in Fig. 5A, transfection with RSV RNA resulted in induction of NLRC5 protein, indicating that the host had the ability to sense RSV RNA for NLRC5 induction.
FIG 5.
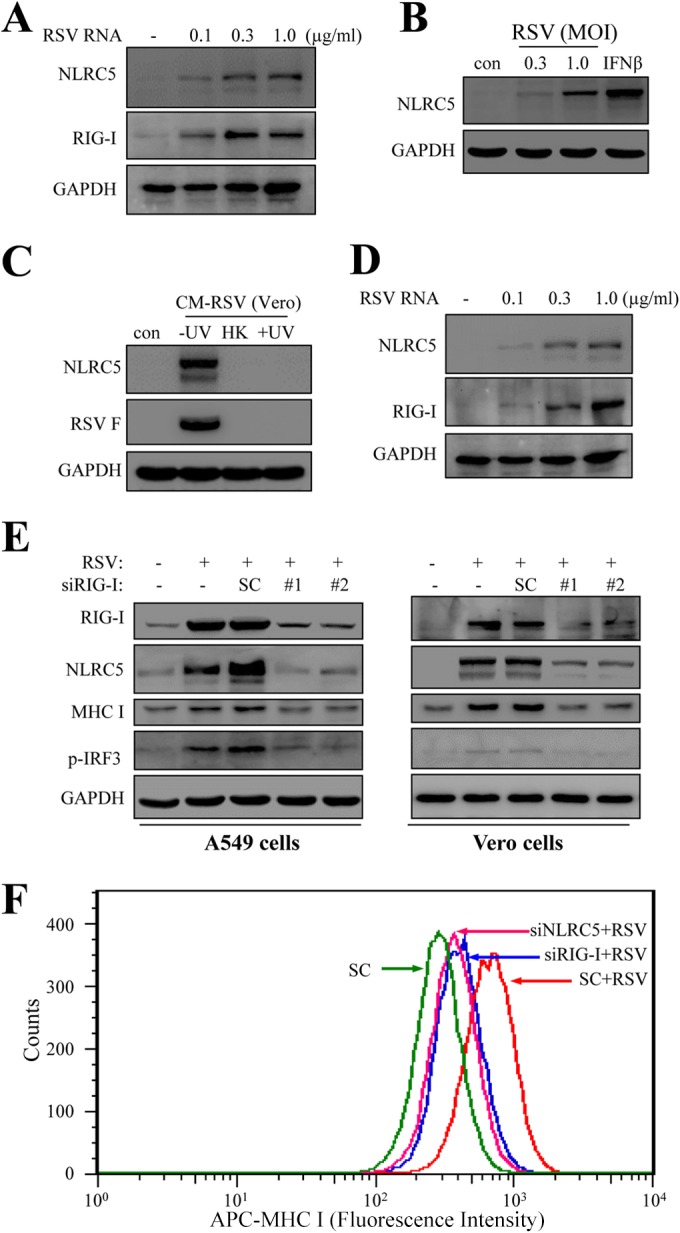
Role of RIG-I in NLRC5 induction. (A) Induction of NLRC5 and RIG-I in A549 cells by RSV RNA. A549 cells were transfected with various amounts of RSV RNA for 24 h. NLRC5 and RIG-I induction was detected by Western blotting. GAPDH expression was used as a loading control. The experiment was performed 2 times independently. (B) Induction of NLRC5 in type I IFN-defective Vero cells by RSV infection. Vero cells were infected with RSV at two different MOIs for 36 h or treated with IFN-β (100 U/ml). NLRC5 expression was detected by immunoblotting. RSV infection or IFN-β stimulation promoted NLRC5 expression, even though the cells are defective in type I interferon production (54, 55). The experiment was performed 2 times independently. (C) UV irradiation destroys NLRC5-inducing activity in a conditioned medium from RSV-infected Vero cells. CM-RSV Vero was processed as described in the legend to Fig. 4A. The medium was then tested on A549 cells for NLRC5-inducing activity by immunoblotting. A nonirradiated (−UV) sample contained infectious virions (F protein expression) and promoted NLRC5 expression. UV irradiation (+UV) destroyed infectious virions (absence of F protein). The sample also lost NLRC5-inducing activity. The experiment was performed 3 times independently. (D) Induction of NLRC5 by RSV RNA in Vero cells. Vero cells were transfected with various amounts of viral RNA for 24 h. NLRC5 and RIG-I induction was detected by Western blotting. GAPDH expression was used as a loading control. The experiment was performed 2 times independently. (E) Suppression of RIG-I expression blocks NLRC5 and MHC-I expression and IRF3 phosphorylation in both A549 and Vero cells. A549 cells or Vero cells were left untreated or transfected with an SC or two different siRNAs (siRIG-I number 1 and number 2) for 24 h. The cells were then infected with RSV. RIG-I expression and protein induction, as well as IRF3 phosphorylation, were detected by Western blotting. (F) Effect of NLRC5 or RIG-I knockdown on the induction of MHC-I in RSV-infected A549 cells. A549 cells were transfected with an SC or the indicated siRNA for 24 h. The cells were then infected with RSV for 36 h and used for the detection of MHC-I induction using FACS. Uninfected SC was used as a control.
We showed that A549 upregulated NLRC5 expression through IFN-β secretion. The host initiates an innate immune response to virus infection to promote antiviral gene expression, including interferon production (52, 53). To dissect the effect of IFN-β production and direct detection of viral RNA on NLRC5 expression, we then transfected Vero cells with viral RNA. Vero cells are derived from a green monkey kidney cell line that has an impaired pathway for type I IFN production (54, 55) and therefore can be used to exclude the effect of secreted IFN-β in NLRC5 upregulation. As shown in Fig. 5B, Vero cells responded to IFN-β stimulation. RSV infection also promoted NLRC5 expression. Unlike A549 cells, which produced a factor to induce NLRC5 expression, RSV-infected Vero cells did not produce NLRC5-inducing factors, since after UV irradiation, a conditioned medium from RSV-infected Vero cells (CM-RSV Vero) lost NLRC5-inducing activity (Fig. 5C, lane 4). When tested for NLRC5 induction by direct transfection of RSV RNA, the cells responded to viral RNA by upregulating NLRC5 expression (Fig. 5D). The data indicated that the host senses viral RNA to initiate an innate immune response for NLRC5 upregulation.
The essential role of RIG-I expression was demonstrated with gene silencing of RIG-I expression. As shown in Fig. 5E, treatment of A549 or Vero cells with siRNA targeting RIG-I blocked RIG-I induction by RSV infection. As expected, suppression of RIG-I expression blocked the induction of both NLRC5 and MHC-I in A549 cells and Vero cells (Fig. 5E). In addition, suppression of RIG-I expression also blocked RSV-induced IRF3 phosphorylation, a critical player in IFN response. The importance of RIG-I and NLRC5 in RSV-induced MHC-I expression was finally demonstrated with reduced surface expression of MHC-I in A549 cells after knockdown of NLRC5 or RIG-I. As shown in Fig. 5F, suppression of NLRC5 or RIG-I expression markedly blocked MHC-I induction by RSV infection. These results demonstrated that the host senses viral RNA to upregulate NLRC5 and MHC-I expression.
DISCUSSION
RSV infection of lung epithelial cells leads to increased expression of MHC-I. In this report, we examined the regulation of MHC-I induction in RSV-infected airway epithelial cells. We found that the host upregulates RIG-I expression by direct detection of viral RNA, leading to IRF3 activation and IFN-β secretion, which in turn upregulates NLRC5 expression in A549 cells. In addition, detection of viral RNA also leads to NLRC5 expression independent of IFN-β production, as was demonstrated using Vero cells, a cell line that is defective in IFN-β production due to a genetic lesion in the IFN-β locus (54, 56). RSV infection promotes significant production of IFN-λ (57–59). Treatment of A549 cells with recombinant IL-28B or IL-29, however, did not significantly induce NLRC5 expression, excluding IFN-λ as a player in MHC-I induction in A549 cells. Although IFN-γ treatment promotes NLRC5 expression in both macrophages (60) and A549 cells (Fig. 2B and 4C), both immunodepletion studies and studies using Vero cells showed that NLRC5 induction relied on viral RNA sensing and IFN-β secretion. Our data therefore implicate RIG-I and NLRC5 as critical players in MHC-I induction in RSV-infected epithelial cells (Fig. 6).
FIG 6.

Model of MHC-I upregulation during RSV infection of airway epithelial cells. The host senses invading RSV with RIG-I and initiates an antiviral response, including upregulation of antiviral genes and secretion of IFN-β, which in turn promotes NLRC5 expression and MHC-I upregulation. The “p” in red circles denotes phosphorylated proteins.
The host senses invading virus with PRRs. Members of the PRRs, including TLRs, RIG-I-like receptors (RLRs), and NOD-like receptors (NLRs), contribute to viral detection, leading to induction of cytokines, chemokines, and type I interferons and subsequent eradication of the virus (61). Viral infection and interferon stimulation promote NLRC5 expression. Although NLRC5 was originally identified as a regulator of antiviral immunity (62, 63), the mechanisms underlying this role began to become clearer when it was reported to interact with RIG-I to induce the antiviral response against influenza virus (50). RIG-I is a key factor in recognizing RNA viruses, and while RSV infection has not been performed in RIG-I−/− mice, they are defective in both interferon production and immunity to a host of other RNA viruses (64). In addition, we show that RSV promotes NLRC5 expression and subsequent MHC-I upregulation through RIG-I induction. The identification of factors that promote MHC-I accumulation during viral infection is a challenging but interesting proposition, since viruses frequently suppress MHC expression for immune evasion (24–26, 65, 66).
The host response to RSV infection in the lung contributes to its pathology via significant increases in inflammation and mucus production (67). Therefore, a delicate balance involving control of infection by virus-specific cytotoxic T lymphocytes (CTL) while mitigating initiation of general inflammation mediating airway pathology may represent an optimal solution (68–70). Aside from exacerbating the immediate response, it is not unreasonable that upregulation of MHC-I sensitizes the host to allergic asthma later in life. Indeed, RSV infection is ubiquitous, and it remains unclear whether immunologic aspects of initial infection contribute to these sequelae later in life. It would be interesting to determine whether patients with RSV infection have increased NLRC5 and RIG-I expression. Nonetheless, the demonstration of MHC-I upregulation and the delineation of a mechanism regulating MHC-I expression during RSV infection of epithelial cells provide a link for T cell response in RSV disease.
ACKNOWLEDGMENTS
This work was financially supported by grants from the NSFC (81421091 and 81371772). X.G. was the recipient of a predoctoral scholarship from the Jiangsu Innovation Program.
We thank Dwayne Stupack and Ying Xia for English editing.
REFERENCES
- 1.Zhou H, Thompson WW, Viboud CG, Ringholz CM, Cheng PY, Steiner C, Abedi GR, Anderson LJ, Brammer L, Shay DK. 2012. Hospitalizations associated with influenza and respiratory syncytial virus in the United States, 1993-2008. Clin Infect Dis 54:1427–1436. doi: 10.1093/cid/cis211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tregoning JS, Schwarze J. 2010. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev 23:74–98. doi: 10.1128/CMR.00032-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourgeois FT, Valim C, McAdam AJ, Mandl KD. 2009. Relative impact of influenza and respiratory syncytial virus in young children. Pediatrics 124:e1072–e1080. doi: 10.1542/peds.2008-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N Engl J Med 360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomsen SF, van der Sluis S, Stensballe LG, Posthuma D, Skytthe A, Kyvik KO, Duffy DL, Backer V, Bisgaard H. 2009. Exploring the association between severe respiratory syncytial virus infection and asthma: a registry-based twin study. Am J Respir Crit Care Med 179:1091–1097. doi: 10.1164/rccm.200809-1471OC. [DOI] [PubMed] [Google Scholar]
- 6.Openshaw PJ, Dean GS, Culley FJ. 2003. Links between respiratory syncytial virus bronchiolitis and childhood asthma: clinical and research approaches. Pediatr Infect Dis J 22:S58–S64. doi: 10.1097/01.inf.0000053887.26571.eb. [DOI] [PubMed] [Google Scholar]
- 7.Collins PL, Crowe JEJ. 2007. Respiratory syncytial virus and metapneumovirus, p 1601–1646. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. 2007. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol 20:108–119. doi: 10.1038/modpathol.3800725. [DOI] [PubMed] [Google Scholar]
- 9.Shakeri A, Mastrangelo P, Griffin JK, Moraes TJ, Hegele RG. 2015. Respiratory syncytial virus receptor expression in the mouse and viral tropism. Histol Histopathol 30:401–411. doi: 10.14670/HH-30.401. [DOI] [PubMed] [Google Scholar]
- 10.Collins PL, Graham BS. 2008. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 82:2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallak LK, Kwilas SA, Peeples ME. 2007. Interaction between respiratory syncytial virus and glycosaminoglycans, including heparan sulfate. Methods Mol Biol 379:15–34. doi: 10.1007/978-1-59745-393-6_2. [DOI] [PubMed] [Google Scholar]
- 12.Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. 2011. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med 17:1132–1135. doi: 10.1038/nm.2444. [DOI] [PubMed] [Google Scholar]
- 13.Bueno SM, Gonzalez PA, Riedel CA, Carreno LJ, Vasquez AE, Kalergis AM. 2011. Local cytokine response upon respiratory syncytial virus infection. Immunol Lett 136:122–129. doi: 10.1016/j.imlet.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR. 2007. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol 81:1401–1411. doi: 10.1128/JVI.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng R, Cui Y, Hai Y, Liu Y. 2012. Pattern recognition receptors for respiratory syncytial virus infection and design of vaccines. Virus Res 167:138–145. doi: 10.1016/j.virusres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 17.Lay MK, Gonzalez PA, Leon MA, Cespedes PF, Bueno SM, Riedel CA, Kalergis AM. 2013. Advances in understanding respiratory syncytial virus infection in airway epithelial cells and consequential effects on the immune response. Microbes Infect 15:230–242. doi: 10.1016/j.micinf.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Ravi LI, Li L, Sutejo R, Chen H, Wong PS, Tan BH, Sugrue RJ. 2013. A systems-based approach to analyse the host response in murine lung macrophages challenged with respiratory syncytial virus. BMC Genomics 14:190. doi: 10.1186/1471-2164-14-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman SJ, Laham FR, Polack FP. 2004. Mechanisms of illness during respiratory syncytial virus infection: the lungs, the virus and the immune response. Microbes Infect 6:767–772. doi: 10.1016/j.micinf.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Ramet M, Korppi M, Hallman M. 2011. Pattern recognition receptors and genetic risk for RSV infection: value for clinical decision-making? Pediatric Pulmonol 46:101–110. doi: 10.1002/ppul.21348. [DOI] [PubMed] [Google Scholar]
- 21.Garofalo R, Mei F, Espejo R, Ye G, Haeberle H, Baron S, Ogra PL, Reyes VE. 1996. Respiratory syncytial virus infection of human respiratory epithelial cells up-regulates class I MHC expression through the induction of IFN-beta and IL-1 alpha. J Immunol 157:2506–2513. [PubMed] [Google Scholar]
- 22.Tregoning JS, Yamaguchi Y, Wang B, Mihm D, Harker JA, Bushell ES, Zheng M, Liao G, Peltz G, Openshaw PJ. 2010. Genetic susceptibility to the delayed sequelae of neonatal respiratory syncytial virus infection is MHC dependent. J Immunol 185:5384–5391. doi: 10.4049/jimmunol.1001594. [DOI] [PubMed] [Google Scholar]
- 23.Jamaluddin M, Wang S, Garofalo RP, Elliott T, Casola A, Baron S, Brasier AR. 2001. IFN-beta mediates coordinate expression of antigen-processing genes in RSV-infected pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol 280:L248–L257. [DOI] [PubMed] [Google Scholar]
- 24.Hansen TH, Bouvier M. 2009. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol 9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 25.Antoniou AN, Powis SJ. 2008. Pathogen evasion strategies for the major histocompatibility complex class I assembly pathway. Immunology 124:1–12. doi: 10.1111/j.1365-2567.2008.02804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yewdell JW, Hill AB. 2002. Viral interference with antigen presentation. Nat Immunol 3:1019–1025. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi KS, van den Elsen PJ. 2012. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol 12:813–820. doi: 10.1038/nri3339. [DOI] [PubMed] [Google Scholar]
- 28.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. 2010. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A 107:13794–13799. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Elsen PJ. 2011. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front Immunol 2:48. doi: 10.3389/fimmu.2011.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reith W, Satola S, Sanchez CH, Amaldi I, Lisowska-Grospierre B, Griscelli C, Hadam MR, Mach B. 1988. Congenital immunodeficiency with a regulatory defect in MHC class II gene expression lacks a specific HLA-DR promoter binding protein, RF-X. Cell 53:897–906. [DOI] [PubMed] [Google Scholar]
- 31.Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, Reith W. 1995. A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev 9:1021–1032. [DOI] [PubMed] [Google Scholar]
- 32.Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, Sanchez JC, Hochstrasser DF, Mach B, Reith W. 1998. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet 20:273–277. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- 33.Nagarajan UM, Louis-Plence P, DeSandro A, Nilsen R, Bushey A, Boss JM. 1999. RFX-B is the gene responsible for the most common cause of the bare lymphocyte syndrome, an MHC class II immunodeficiency. Immunity 10:153–162. [DOI] [PubMed] [Google Scholar]
- 34.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W. 1997. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. EMBO J 16:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. 2000. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev 14:1156–1166. doi: 10.1101/gad.14.9.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreno CS, Emery P, West JE, Durand B, Reith W, Mach B, Boss JM. 1995. Purified X2 binding protein (X2BP) cooperatively binds the class II MHC X box region in the presence of purified RFX, the X box factor deficient in the bare lymphocyte syndrome. J Immunol 155:4313–4321. [PubMed] [Google Scholar]
- 37.Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman L, Peterlin BM. 2003. MHC class II enhanceosome: how is the class II transactivator recruited to DNA-bound activators? Int Immunol 15:467–475. doi: 10.1093/intimm/dxg048. [DOI] [PubMed] [Google Scholar]
- 38.Biswas A, Meissner TB, Kawai T, Kobayashi KS. 2012. Cutting edge: impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J Immunol 189:516–520. doi: 10.4049/jimmunol.1200064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neerincx A, Jakobshagen K, Utermohlen O, Buning H, Steimle V, Kufer TA. 2014. The N-terminal domain of NLRC5 confers transcriptional activity for MHC class I and II gene expression. J Immunol 193:3090–3100. doi: 10.4049/jimmunol.1401065. [DOI] [PubMed] [Google Scholar]
- 40.Staehli F, Ludigs K, Heinz LX, Seguin-Estevez Q, Ferrero I, Braun M, Schroder K, Rebsamen M, Tardivel A, Mattmann C, MacDonald HR, Romero P, Reith W, Guarda G, Tschopp J. 2012. NLRC5 deficiency selectively impairs MHC class I-dependent lymphocyte killing by cytotoxic T cells. J Immunol 188:3820–3828. doi: 10.4049/jimmunol.1102671. [DOI] [PubMed] [Google Scholar]
- 41.Neerincx A, Rodriguez GM, Steimle V, Kufer TA. 2012. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J Immunol 188:4940–4950. doi: 10.4049/jimmunol.1103136. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Shao F. 2012. NLRC5: a NOD-like receptor protein with many faces in immune regulation. Cell Res 22:1099–1101. doi: 10.1038/cr.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams GS, Malin M, Vremec D, Chang CH, Boyd R, Benoist C, Mathis D. 1998. Mice lacking the transcription factor CIITA—a second look. Int Immunol 10:1957–1967. [DOI] [PubMed] [Google Scholar]
- 44.Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, van den Elsen PJ, Kobayashi KS. 2012. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol 188:4951–4958. doi: 10.4049/jimmunol.1103160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Peters N, Schwarze J. 2006. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol 177:6263–6270. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- 46.Foster S, Bedford KJ, Gould ME, Coward WR, Hewitt CR. 2003. Respiratory syncytial virus infection and virus-induced inflammation are modified by contaminants of indoor air. Immunology 108:109–115. doi: 10.1046/j.1365-2567.2003.01539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang YT, Wertz GW. 1982. The genome of respiratory syncytial virus is a negative-stranded RNA that codes for at least seven mRNA species. J Virol 43:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collins PL, Dickens LE, Buckler-White A, Olmsted RA, Spriggs MK, Camargo E, Coelingh KV. 1986. Nucleotide sequences for the gene junctions of human respiratory syncytial virus reveal distinctive features of intergenic structure and gene order. Proc Natl Acad Sci U S A 83:4594–4598. doi: 10.1073/pnas.83.13.4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindenmann J, Burke DC, Isaacs A. 1957. Studies on the production, mode of action and properties of interferon. Br J Exp Pathol 38:551–562. [PMC free article] [PubMed] [Google Scholar]
- 50.Ranjan P, Singh N, Kumar A, Neerincx A, Kremmer E, Cao W, Davis WG, Katz JM, Gangappa S, Lin R, Kufer TA, Sambhara S. 2015. NLRC5 interacts with RIG-I to induce a robust antiviral response against influenza virus infection. Eur J Immunol 45:758–772. doi: 10.1002/eji.201344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vilcek J. 2003. Novel interferons. Nat Immunol 4:8–9. doi: 10.1038/ni0103-8. [DOI] [PubMed] [Google Scholar]
- 52.Demoor T, Petersen BC, Morris S, Mukherjee S, Ptaschinski C, De Almeida Nagata DE, Kawai T, Ito T, Akira S, Kunkel SL, Schaller MA, Lukacs NW. 2012. IPS-1 signaling has a nonredundant role in mediating antiviral responses and the clearance of respiratory syncytial virus. J Immunol 189:5942–5953. doi: 10.4049/jimmunol.1201763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mejias A, Dimo B, Suarez NM, Garcia C, Suarez-Arrabal MC, Jartti T, Blankenship D, Jordan-Villegas A, Ardura MI, Xu Z, Banchereau J, Chaussabel D, Ramilo O. 2013. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med 10:e1001549. doi: 10.1371/journal.pmed.1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J Gen Virol 43:247–252. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- 55.Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J Virol 2:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosca JD, Pitha PM. 1986. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol Cell Biol 6:2279–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. 2006. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol 80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dave KA, Norris EL, Bukreyev AA, Headlam MJ, Buchholz UJ, Singh T, Collins PL, Gorman JJ. 2014. A comprehensive proteomic view of responses of A549 type II alveolar epithelial cells to human respiratory syncytial virus infection. Mol Cell Proteomics 13:3250–3269. doi: 10.1074/mcp.M114.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spann KM, Tran KC, Chi B, Rabin RL, Collins PL. 2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J Virol 78:4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, Grotzinger J, Fickenscher H, Schreiber S, Rosenstiel P. 2010. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol 184:1990–2000. doi: 10.4049/jimmunol.0900557. [DOI] [PubMed] [Google Scholar]
- 61.Kolli D, Velayutham TS, Casola A. 2013. Host-viral interactions: role of pattern recognition receptors (PRRs) in human pneumovirus infections. Pathogens 2:232–263. doi: 10.3390/pathogens2020232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, Wang R-F. 2010. NLRC5 negatively regulates the NF-κB and type I interferon signaling pathways. Cell 141:483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, Buning H, Schwarzenbacher R, Kufer TA. 2010. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem 285:26223–26232. doi: 10.1074/jbc.M110.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoneyama M, Fujita T. 2007. Function of RIG-I-like receptors in antiviral innate immunity. J Biol Chem 282:15315–15318. doi: 10.1074/jbc.R700007200. [DOI] [PubMed] [Google Scholar]
- 65.Gainey MD, Rivenbark JG, Cho H, Yang L, Yokoyama WM. 2012. Viral MHC class I inhibition evades CD8+ T-cell effector responses in vivo but not CD8+ T-cell priming. Proc Natl Acad Sci U S A 109:E3260–E3267. doi: 10.1073/pnas.1217111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang W, Sung PS, Park SH, Yoon S, Chang DY, Kim S, Han KH, Kim JK, Rehermann B, Chwae YJ, Shin EC. 2014. Hepatitis C virus attenuates interferon-induced major histocompatibility complex class I expression and decreases CD8+ T cell effector functions. Gastroenterology 146:1351-60.e1-4. doi: 10.1053/j.gastro.2014.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lukacs NW, Smit JJ, Mukherjee S, Morris SB, Nunez G, Lindell DM. 2010. Respiratory virus-induced TLR7 activation controls IL-17-associated increased mucus via IL-23 regulation. J Immunol 185:2231–2239. doi: 10.4049/jimmunol.1000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cannon MJ, Openshaw PJ, Askonas BA. 1988. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med 168:1163–1168. doi: 10.1084/jem.168.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Graham BS, Bunton LA, Wright PF, Karzon DT. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin Invest 88:1026–1033. doi: 10.1172/JCI115362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tregoning JS, Yamaguchi Y, Harker J, Wang B, Openshaw PJ. 2008. The role of T cells in the enhancement of respiratory syncytial virus infection severity during adult reinfection of neonatally sensitized mice. J Virol 82:4115–4124. doi: 10.1128/JVI.02313-07. [DOI] [PMC free article] [PubMed] [Google Scholar]