

ABSTRACT
Nasopharyngeal carcinoma (NPC) is closely associated with latent Epstein-Barr virus (EBV) infection. Although EBV infection of preneoplastic epithelial cells is not immortalizing, EBV can modulate oncogenic and cell death mechanisms. The viral latent membrane proteins 1 (LMP1) and LMP2A are consistently expressed in NPC and can cooperate in bitransgenic mice expressed from the keratin-14 promoter to enhance carcinoma development in an epithelial chemical carcinogenesis model. In this study, LMP1 and LMP2A were coexpressed in the EBV-negative NPC cell line HK1 and examined for combined effects in response to genotoxic treatments. In response to DNA damage activation, LMP1 and LMP2A coexpression reduced γH2AX (S139) phosphorylation and caspase cleavage induced by a lower dose (5 μM) of the topoisomerase II inhibitor etoposide. Regulation of γH2AX occurred before the onset of caspase activation without modulation of other DNA damage signaling mediators, including ATM, Chk1, or Chk2, and additionally was suppressed by inducers of DNA single-strand breaks (SSBs) and replication stress. Despite reduced DNA damage repair signaling, LMP1-2A coexpressing cells recovered from cytotoxic doses of etoposide; however, LMP1 expression was sufficient for this effect. LMP1 and LMP2A coexpression did not enhance cell growth, with a moderate increase of cell motility to fibronectin. This study supports that LMP1 and LMP2A jointly regulate DNA repair signaling and cell death activation with no further enhancement in the growth properties of neoplastic cells.
IMPORTANCE NPC is characterized by clonal EBV infection and accounts for >78,000 annual cancer cases with increased incidence in regions where EBV is endemic, such as southeast Asia. The latent proteins LMP1 and LMP2A coexpressed in NPC can individually enhance growth or survival properties in epithelial cells, but their combined effects and potential regulation of DNA repair and checkpoint mechanisms are relatively undetermined. In this study, LMP1-2A coexpression suppressed activation of the DNA damage response (DDR) protein γH2AX induced by selective genotoxins that promote DNA replication stress or SSBs. Expression of LMP1 was sufficient to recover cells, resulting in outgrowth of LMP1 and LMP1-2A-coexpressing cells and indicating distinct LMP1-dependent effects in the restoration of replicative potential. These findings demonstrate novel properties for LMP1 and LMP2A in the cooperative modulation of DDR and apoptotic signaling pathways, further implicating both proteins in the progression of NPC and epithelial malignancies.
INTRODUCTION
Epstein-Barr virus (EBV) is a human gammaherpesvirus that establishes lifelong latency in memory B cells, with sporadic reactivation and transmission from oral epithelia (1). More than 90% of the adult population is latently infected, and a subset can develop EBV-associated malignancies, including nasopharyngeal carcinoma (NPC), gastric cancer, Burkitt lymphoma, Hodgkin lymphoma, and lymphomas in the immunocompromised, including AIDS-associated lymphoma and posttransplant lymphoproliferative disease (2, 3). Epithelial cell infection in vitro frequently results in productive replication, and latently infected oral epithelial cells are rare in persistently infected healthy individuals (4, 5). However, epithelial tumors such as NPC consistently express a type II latency program, which includes latent membrane protein 1 (LMP1), LMP2A, and LMP2B (1, 5). Additionally, monoclonal EBV episomes are detected in NPC, suggesting that NPC tumors are the clonal outgrowth of an initially infected cell likely predisposed to oncogenic transformation from additional genetic and environmental cofactors, such as the loss of p16 and exposure to dietary nitrosamines (2, 3). In contrast to the immortalizing properties of EBV to primary B cells, the contribution of EBV infection to epithelial cell oncogenesis is less understood, as infection alone is insufficient to immortalize or induce oncogenic potential in preneoplastic cell lines from the nasopharynx (5, 6).
LMP1 and LMP2A transcripts are consistently expressed in NPC tumors with more variable detection of LMP1 protein by immunohistochemistry, suggesting that LMP1 protein levels are regulated and may be required to balance the cytotoxic effects of high-level LMP1 expression (2, 7, 8). LMP1 and LMP2A are transmembrane proteins that signal constitutively from lipid rafts in a ligand-independent manner and may contribute to NPC pathogenesis by modulating signaling pathways involved in cell growth, motility, survival, and differentiation (9). Through interactions of the C-terminal activation regions (CTAR1 and CTAR2) with cellular signaling molecules, including NF-κB, phosphoinositol 3-kinase (PI3K)/Akt, STAT, Jun N-terminal protein kinase (JNK), extracellular signal-regulated kinase (ERK), and mitogen-activated protein kinase (MAPK), LMP1 promotes cell growth, motility, and epithelial-mesenchymal transition (EMT) (2, 9–12). Expression of LMP1 can transform Rat-1 fibroblasts to form foci in soft agar and tumors in nude mice, as well as induce anchorage-independent growth in human epithelial cells (2, 9, 13). However, the oncogenic potential of LMP2A is less defined and may be cell type dependent (9). In epithelial cells, LMP2A promotes cell motility, resistance to cell death, and, in specific cell types, cell growth through activation of various signaling pathways, including PI3K/Akt and ERK/MAPK, via N-terminal immunoreceptor tyrosine activation (ITAM), PY, and YEEA motifs (9, 14–17).
Activation of oncogenes in cancer cells can induce aberrant proliferation that causes replicative stress. In normal cells, replicative stress activates the DNA damage response (DDR) pathway and initiates repair mechanisms; however, excessive DNA damage can lead to checkpoint activation, cell cycle arrest, and apoptosis (18). Additionally, exogenous genotoxins employed in cancer therapies, including ionizing radiation and chemotherapeutics, produce DNA strand breaks (DSBs) (18). Most preneoplastic lesions maintain intact DDR and apoptotic response mechanisms; however, these pathways may be modulated in later stages of cancer development (18). NPC cells express functional p53 that can be functionally antagonized by LMP1 and specific microRNAs, but potential mutations in DDR genes have not been thoroughly characterized (19–24). EBV, as well as other oncogenic viruses, can modulate members of the DDR pathway, specifically the histone variant H2AX, to promote oncogenesis. Phosphorylation of H2AX can be critical for the maintenance of latent gammaherpesvirus infections (25, 26). In contrast, EBV-mediated B cell immortalization requires the attenuation of DDR by EBNA3C, which may be mediated by downregulation of H2AX expression (27, 28).
The in vivo properties of LMP1 and LMP2A coexpression have been investigated in transgenic mice (29, 30). Spontaneous carcinomas are not induced by transgene expression, but the development of papillomas and carcinomas can be induced by the chemical initiators and promoters dimethylbenz(a)anthracene–12-O-tetradecanoylphorbol 13-acetate (DMBA–TPA). Dysplastic papillomas were increased by LMP1; however, invasive carcinomas were increased in LMP1-2A bitransgenic mice, suggesting that LMP1 and LMP2A cooperate specifically in the latter stages of carcinoma development (29). However, the mechanism by which LMP1 and LMP2A combined expression enhances tumorigenesis is not understood. In the present study, the oncogenic potential of LMP1 and LMP2A were investigated by expression in the EBV-negative NPC cell line HK1 and examined for potentially combined effects in response to cell death activation, cell growth, and motility. HK1 cells are one of the few authenticated NPC cell lines that are sensitive to serum deprivation and chemotherapeutics but can resist these cell death treatments by infection with EBV (6, 23, 31). Additionally, HK1 cells are free of HeLa contamination, which confounds the interpretation of many NPC-derived cell lines (32, 33). To investigate cell death responses activated by DNA damage, HK1 cell lines were treated with several DNA-damaging reagents and replication stress inducers, including the topoisomerase II inhibitor etoposide, ionizing radiation, methyl methanesulfonate (MMS), and aphidicolin. LMP1-2A coexpression reduced caspase activation and γH2AX phosphorylation induced by low-dose etoposide, MMS, or aphidicolin treatments but not by ionizing radiation. Other DDR signaling proteins were not modulated by LMP1-2A coexpression. Additionally, expression of LMP1 (in LMP1 or LMP1-2A coexpressing cells) was sufficient to recover cytostatic cells from etoposide treatment. These findings indicate that LMP1 and LMP2A jointly modulate activation of DNA damage repair and apoptotic mediators, suggesting that these two viral proteins collaborate in the progression and aberrant survival of cancer cells in NPC.
MATERIALS AND METHODS
Cell lines and reagents.
The NPC cell line HK1 (gift from George Tsao, Hong Kong University) and Akata and LCL5000 B cells (gift from Kenzo Takada, Hokkaido University, and Nancy Raab-Traub, University of North Carolina) were maintained in RPMI 1640 media as previously described (31). HK1 stable cell lines were generated by transduction with pBabe retroviruses expressing hemagglutinin (HA)-tagged LMP1 or LMP2A and selected with 500 μg/ml neomycin for LMP1 or 1 μg/ml puromycin for LMP2A (12). Pooled stable HK1 cell lines were maintained in neomycin and puromycin selection media and used for further analysis. EBV-infected HK1 (gift from George Tsao, Hong Kong University) and 293 (gift from Wolfgang Hammerschmidt, Helmholtz Center) cells were maintained in neomycin (800 μg/ml) and hygromycin (100 μg/ml) selection media, respectively (34, 35). Infected cell lines were treated with 10 μM MG132 for 8 h or 2 mM methyl-beta cyclodextrin overnight for LMP1 and LMP2A expression analyses. Expression of LMP1 and LMP2A was knocked down using the pSilencer 5.1-U6 retro (Ambion) plasmid expressing either short hairpin RNA (shRNA) targeted at the LMP1 sequence (5′ AACTGGTGGACTCTATTGGTT 3′) or LMP2A sequence (5′ AACTCCCAATATCCATCTGCT 3′). A universal scrambled shRNA sequence (Ambion) was used as a negative control. To investigate the DNA damage response, HK1 stable cell lines were treated with ionizing radiation (2 Gy and 5 Gy and recovered for 1 h), methyl methanesulfonate (10 mg/ml for 30 min; Sigma), or aphidicolin (1 μM for 1 to 4 h; Sigma). Etoposide treatment of HK1 stable cell lines and HEK293 cells induced for LMP1 and LMP2A expression by doxycycline is described below.
Etoposide treatment, analysis, and recovery.
HK1 stable cell lines were seeded at 0.8 × 104 to 2 × 104 cells/cm2. When optimum cell density was reached (40 to 50% confluence), cells were treated with dimethyl sulfoxide (DMSO) or etoposide (Sigma) for up to 24 h and analyzed by immunoblotting and immunofluorescence. In recovery assays, cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) after etoposide treatment and recovered in standard culture media. From the time of treatment to 1 to 2 weeks postrecovery, cells were analyzed for dehydrogenase activity by the resazurin assay to determine metabolically viable cells according to the manufacturer's protocol (Cell Signaling Technology) or analyzed for long-term recovery at 1 to 2 weeks by crystal violet staining (0.1% crystal violet in 10% ethanol). For each resazurin assay, measurements were calculated for a minimum of triplicate wells. For cytotoxicity assays, HK1 cell lines were treated with 5 μM etoposide or DMSO for 24 h and analyzed immediately or washed in DPBS and replaced with fresh media for analysis at days 1 and 2 after recovery from etoposide treatment. Cells and debris were pelleted by centrifugation, and 100 μl of supernatant was removed for analysis using a cytotoxicity (lactate dehydrogenase [LDH] release) assay kit by following the manufacturer's specifications (Roche). For cell cycle arrest and reentry experiments, HK1 stable cell lines were seeded at low density (1.7 × 104 cells/cm2) and incubated in starvation (0.1% serum) media for 36 h before etoposide treatment under complete (10% serum) growth conditions to release cells from arrest. For transient induction experiments in 293 cells, stable cell lines expressing the tetracycline transactivator plasmid (pLVX-Tet3G; Clontech) or coexpressing a tetracycline-regulated LMP1 expression plasmid (pTRETight-BI; Clontech) were transfected with a tetracycline-regulated LMP2A expression plasmid or vector control (pTRETightBI-RY-O; 31463; Addgene). Transfected cells were transiently induced with 200 ng/ml doxycycline for 48 h, and lysates were analyzed after etoposide treatment at the first detection of DNA damage signaling (5 μM, 2 h).
Immunoblot analysis.
Whole-cell lysates were made and immunoblot analysis was performed as previously described (36). Briefly, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1 mM EDTA) supplemented with 1 mM phenylmethylsulfonyl, 2 mM activated sodium orthovanadate, and a 1:100 dilution of protease and phosphatase inhibitor cocktails (Sigma). Lysates were prepared with 2.5% β-mercaptoethanol and boiled before loading or by warming to 70°C for 15 min to prevent protein aggregation for LMP2A immunoblots. Equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblotting was performed according to the manufacturer's instructions with the following antibodies: HSC70, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and PCNA (Santa Cruz Biotechnology); cleaved caspase 3, cleaved caspase 8, cleaved poly(ADP-ribose) polymerase (PARP), PARP, cyclin D1, cyclin B1, p21, pATM (S1981), pATR (S428), pChk1 (S345), and pChk2 (Thr68) (Cell Signaling Technology); γH2AX (Ser139) (Millipore and Cell Signaling Technology); H2AX (Bethyl); LMP2A (14B7 clone; Thermo Scientific); and LMP1 (S12 clone hybridoma tissue culture supernatant, 1:10) (36). Secondary antibodies were conjugated to horseradish peroxidase (Jackson ImmunoResearch) and developed with a WesternBright ECL detection kit (Bioexpress) or with secondary antibodies conjugated to IRDyes 680CW and 800CW (LI-COR) and scanned by near-infrared imaging (LI-COR Odyssey). Immunoblots were quantified by densitometry on Image Studio software (LI-COR) for near-infrared blots or on Image J software for ECL-developed blots and compared by unpaired two-tailed t test.
Immunofluorescence.
For LMP1 and LMP2A immunofluorescence staining, HK1 stable cell lines were seeded on glass chamber slides and fixed in 4% paraformaldehyde. After permeabilization with 0.1% Triton X-100, slides were blocked with 5% donkey serum followed by incubation with primary antibodies, anti-LMP1 (1:100 dilution of CS1-4 clone; Santa Cruz Biotechnology) and anti-LMP2A (1 μg/ml of 14B7 clone; Thermo Scientific), at 4°C overnight. Multiple labeling secondary antibodies with minimal cross-reactivity to other species (Cy3 anti-mouse IgG and Alexa Fluor 488 anti-rat IgG; Jackson ImmunoResearch) were incubated for 1 h at room temperature, and nuclei were counterstained with 2.5 μM Draq5 (Thermo Scientific). Staining was imaged with a Leica TCS SP2 upright confocal microscope and software (Center for Biologic Imaging, University of Pittsburgh). For analysis of DNA damage foci by γH2AX and 53BP1 staining, HK1 stable cell lines were seeded on glass-bottom Mattek dishes to reach 40 to 50% confluence and treated with DMSO or 5 μM etoposide for up to 12 h. For γH2AX and 53BP1 immunostaining, cells were fixed in 3.7% formaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 1% bovine serum albumin at 30°C for 30 min. Blocked samples were incubated with primary antibodies to γH2AX (Ser139) (Millipore) and 53BP1 (Novus Biologicals) overnight at 4°C, followed by incubation with the secondary antibodies Alexa Fluor 488 anti-mouse IgG and Alexa Fluor 594 anti-rabbit IgG. Samples were mounted in PermaFluor (Immunon) and imaged with an Olympus FV2000 confocal microscope.
Cell growth and transwell migration assays.
HK1 stable cell lines were seeded in full (10%) or reduced (0.1%) serum conditions at 2 × 104 cells/cm2 for cell-counting assays or 3 × 104 to 6 × 104 cells/cm2 for the resazurin assay (Cell Signaling Technologies) to determine metabolically viable cells according to the manufacturer's protocol. For each cell counting or resazurin assay, measurements were calculated for triplicate wells. Cell motility was determined using transwell migration to fibronectin as previously described (12).
RESULTS
LMP1 and LMP2A coexpression in NPC cells.
In order to study the combined effects of LMP1 and LMP2A in epithelial cells, LMP1 and LMP2A proteins were stably coexpressed by retroviral transduction in the NPC cell line HK1. HK1 cells are EBV negative; thus, they are naive to the effects of LMP1 and LMP2A expression (31). Stable cell lines individually expressing or coexpressing LMP1 and LMP2A proteins were characterized for expression levels, localization, and coexpression by immunoblot and immunofluorescence analyses. Compared to the individually expressing LMP1 or LMP2A stable cell lines, LMP1 and LMP2A proteins were detected by immunoblotting at comparable levels in the LMP1-2A-coexpressing cell line (Fig. 1A). LMP2A was detected as a single band at the monomeric molecular mass (54 kDa), and LMP1 monomers were detected as multiple bands (∼45 to 66 kDa) (12, 37). Although increased levels of LMP1 were detected consistently in LMP1-2A-coexpressing cells, all of the LMP1 bands were detectable in both LMP1- and LMP1-2A-coexpressing cell lines.
FIG 1.

LMP1 and LMP2A coexpression in the NPC cell line HK1. (A) HK1 cell lines stably transduced by the retroviral pBabe expression vector were analyzed for LMP1 and LMP2A expression by immunoblotting. Arrows indicate the LMP1-specific bands, and the asterisk indicates the nonspecific band detected by S12 antibody. LMP1 and LMP2A levels were normalized to that of the GAPDH loading control by densitometry with Image J software and are represented as relative fold changes in the corresponding bar graph. Results from 3 independent harvests are indicated (n = 3), and error bars denote standard deviations. (B) Immunofluorescent detection of LMP1 and LMP2A in stable HK1 cell lines. LMP1 was detected with the CS1-4 antibody cocktail, and LMP2A was detected with the 14B7 antibody clone. Nuclei were stained with Draq5, and confocal images were analyzed with Image J software. Arrows denote intracellular (1) and cell periphery (2) staining. (C and D) Clonal HK1 cell lines were established and analyzed for LMP1 and LMP2A expression by immunoblotting. The intensity of LMP1 and LMP2A detection was scored (−, +) and is indicated below each immunoblot.
HK1 stable cell lines were analyzed by immunofluorescence to determine coexpression and potential changes in localization of LMP1 and LMP2A. In LMP1-2A-coexpressing cells, both LMP1 and LMP2A staining could be detected in the same cell (Fig. 1B). LMP1 staining localized to intracellular puncta and peripheral regions, indicative of cytoplasmic and plasma membrane localization. LMP2A staining was concentrated at perinuclear regions. Equivalent staining was detected in all HK1 cell lines, indicating that coexpression does not alter the localization of LMP1 and LMP2A proteins. To further characterize LMP1 and LMP2A coexpression, clonal cell lines were established and analyzed by immunoblotting (Fig. 1C and D). The majority of clones expressed detectable levels of LMP1 and LMP2A proteins with consistent coexpression in the LMP1-2A clones (4/4 LMP1 cell lines, 5/5 LMP2A cell lines, and 5/6 LMP1-2A cell lines). LMP1 and LMP2A proteins were detected at similar levels in individually expressing clones, with heterogeneous levels in the LMP1-2A clones. In order to alleviate potential selection for clonal variation, particularly for the heterogeneous expression in LMP1-2A-coexpressing clones, pooled stable cell lines were further characterized in downstream analyses.
To determine the physiological levels of LMP1 and LMP2A expression, HK1 stable cell lines were compared to EBV-infected cells. HK1 and 293 cells were infected with the Akata and B95.8 EBV strains, respectively. The Burkitt lymphoma Akata cell line (infected with Akata strain EBV) also was included as a representative B cell expressing amounts of LMP1 and LMP2A more similar to those of epithelial cells than the high levels expressed from lymphoblastoid cell lines (LCLs) (data not shown). The Akata EBV strain expresses the China1 LMP1 variant containing a 30-bp deletion in the C terminus that generates a protein of slightly lower molecular weight (38). LMP1 protein levels were comparable between the stable cell lines (LMP1 and LMP1-2A) and infected epithelial cells and increased in stable cell lines compared to levels in EBV-infected Akata B cells (Fig. 2A). However, LMP2A expression was undetectable in HK1-EBV or 293-EBV cell lines but was detectable in Akata B cells and LCL (Fig. 2A and B). Although treatment of 293-EBV cells with the proteasome inhibitor MG132 stabilized LMP1 protein levels, LMP2A expression was not enhanced by MG132 in 293- or HK1-infected cells (Fig. 2A and data not shown). LMP2A protein levels, although detectable from NPC biopsy specimens and explanted xenografts, are highly regulated and in low abundance when expressed from infected epithelial cell lines (9, 39, 40). Depletion of cholesterol from the plasma membrane can increase the abundance of LMP2A (41). Therefore, infected cells were treated with methyl-beta-cyclodextrin (MβCD), which resulted in low, detectable levels of LMP2A in 293-EBV cells but not in HK1-EBV cells (Fig. 2B). These data indicate that the HK1 stable cell lines express physiological levels of LMP1, similar to infected epithelial cell lines, with LMP2A levels comparable to those of EBV-infected B cells, although this could not be empirically determined from the variable trace levels of LMP2A detected in infected epithelial cell lines.
FIG 2.
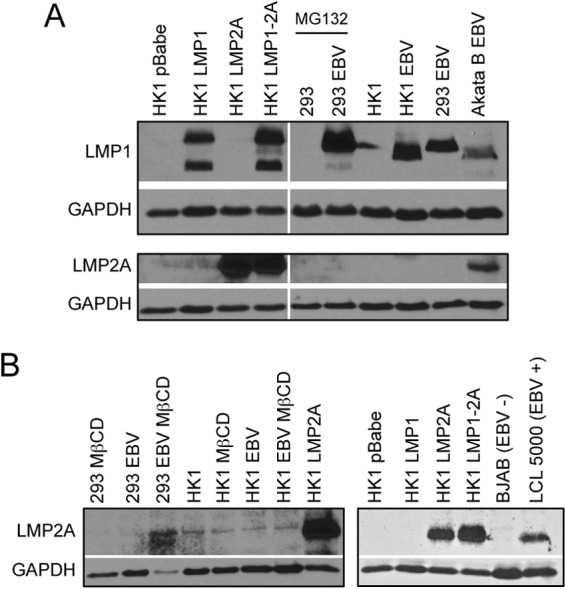
LMP1 and LMP2A expression in EBV-infected epithelial cell lines. (A) LMP1 and LMP2A expression levels were compared by immunoblotting of HK1 stable cell lines and EBV-infected HK1 (Akata strain) and HEK293 (B95.8 strain) cells. (B) EBV-infected HK1 and HEK293 cells were treated with 2 mM methyl-beta cyclodextrin (MβCD) overnight and analyzed for LMP2A expression by immunoblotting. The HK1 LMP2A stable cell line was used as a positive control and compared to levels in EBV-infected LCLs.
Effect on cell growth, motility, and caspase activation by coexpression of LMP1 and LMP2A.
Expression of LMP1 or LMP2A can induce oncogenic properties associated with cell growth, motility, and survival in epithelial cells and rodent fibroblasts (11, 13, 16, 24, 42–44). To investigate if LMP1 and LMP2A coexpression induces differential oncogenic properties in NPC cells, HK1 stable cell lines were analyzed for effects on cell growth and motility. The resazurin assay was used to measure metabolically active dehydrogenase activity, similar to that of the MTT assay, to assess cell viability and growth. In cell counting and metabolic activity assays, comparable rates of cell growth were detected for all HK1 stable cell lines in both full (10%) and reduced (0.1%) serum conditions, indicating that LMP1 and LMP2A do not enhance the growth properties or facilitate serum-independent growth of HK1 cells (Fig. 3A and B). LMP1 and LMP2A can induce transwell migration to extracellular matrices, such as fibronectin (12, 42). In transwell migration assays to fibronectin, the pBabe vector control cells induced high levels of migration (321 cells per field of view), indicating that HK1 cells are highly motile (Fig. 3C). Expression of LMP2A increased migration by >1.5-fold, but coexpression of LMP1 with LMP2A significantly increased migration by >2.5-fold (P = 0.03). However, the induced migration by LMP1-2A coexpression was highly variable, potentially due to the high numbers of trans-migrated cells (877 cells per field of view). These data indicate that LMP1 and LMP2A do not enhance the growth properties of HK1 cells but can further increase the motility to fibronectin.
FIG 3.

Effect of LMP1 and LMP2A coexpression on cell growth, motility, and caspase activation. Cell counting and metabolic activity assay (resazurin) to determine HK1 cell growth under 10% (A) and 0.1% (B) reduced fetal bovine serum (FBS) conditions. Fold change represents the change in cell numbers or relative fluorescence units (RFU) normalized to day 1 and averaged from triplicate wells of 3 independent experiments. (C) Transwell migration assay of HK1 stable cell lines to fibronectin. Fold change represents the number of migrating cells relative (Rel.) to that for pBabe control cells, averaged from duplicate wells of 3 independent experiments. The average number of migrating cells for each cell line was determined by counting the number of DAPI-positive nuclei, averaged from 5 fields of view per transwell, and is displayed above each column. Representative DAPI images taken from a cropped field of view are shown below the graph. Statistical significance was tested using an unpaired 2-tailed t test. NS, not significant. (D) Immunoblot analysis of caspase (caspases 3 and 8) and PARP cleavage in HK1 cells treated with etoposide for 24 h. Shown are representative blots from 3 independent experiments in HK1 stable cell lines.
In order to study the potential enhancement of NPC cell survival by coexpression of LMP1 and LMP2A, HK1 cells were treated with the topoisomerase II inhibitor etoposide and analyzed for etoposide-induced caspase activation by immunoblotting for cleaved caspase 8, caspase 3, and poly(ADP-ribose) polymerase (PARP). Caspase 8 is an effector caspase that mediates the extrinsic apoptotic pathway and also can activate the intrinsic pathway. The downstream executioner caspase 3 can be cleaved by caspase 8, inducing its activation and cleavage of downstream substrates such as PARP (45). Treatment with 5 μM etoposide for 24 h induced cleavage of caspases (caspases 3 and 8) and PARP in pBabe and LMP1- or LMP2A-expressing cell lines (Fig. 3D). In contrast, LMP1-2A coexpression reduced the levels of caspase and PARP cleavage to levels comparable to those of the DMSO control (Fig. 3D). At a higher dose of etoposide (25 μM), caspase and PARP cleavage were restored in LMP1-2A-coexpressing cells, indicating that the decreased activation of caspases was specific to a lower dose of etoposide and that the caspase pathway was intact (Fig. 3D). These data indicate that coexpression of LMP1 and LMP2A specifically reduced the cleavage of caspases following low-dose etoposide treatment.
LMP1 and LMP2A cooperate to suppress γH2AX phosphorylation.
H2AX is a variant of the core histone H2A family and is recruited to sites of DNA damage upon phosphorylation at serine 139 (γH2AX) (46). Immunoblotting and immunofluorescence staining for γH2AX foci identifies sites of DNA damage and is a marker for the induction of DNA damage signaling. To investigate potential effects of LMP1 and LMP2A on DNA damage signaling, HK1 stable cell lines were treated with etoposide to induce genotoxic stress. Etoposide predominantly affects cells in the S/G2 phase of the cell cycle to induce DNA damage, with unrepaired DNA damage leading to caspase activation and cytotoxicity (47). Activation of the DNA damage response, as indicated by increased γH2AX levels in pBabe and LMP1- or LMP2A-expressing cells, was evident with 5 μM etoposide treatment by 12 h (Fig. 4A). With a doubling time of 24 h (Fig. 3A), earlier time points with 5 μM etoposide treatment did not induce a robust increase in γH2AX levels in asynchronous cells (data not shown). However, γH2AX levels were not induced in LMP1-2A-coexpressing cells, and by 24 h of etoposide treatment γH2AX levels still were similar to levels for DMSO treatment (Fig. 4A and B). In immunofluorescence analyses, cells positive for the DDR marker 53BP1 were scored for costained γH2AX foci. In control cells (DMSO treated and pBabe), 53BP1 was stained diffusely in the nucleus with no detectable γH2AX staining. Etoposide treatment induced 53BP1 nuclear foci in pBabe, LMP1, and LMP2A cell lines, with γH2AX foci colocalizing with the majority (75 to 93%) of 53BP1 focus-positive cells (Fig. 4C and D). Strong 53BP1 foci also were detected in LMP1-2A-coexpressing cells; however, weak and diffuse γH2AX foci were detected in 21% of 53BP1 focus-positive cells (Fig. 4C and D). The differential reduction of γH2AX levels in LMP1-2A cells, with intact 53BP1 recruitment, suggests that LMP1 and LMP2A coregulate DNA damage signaling and repair proteins.
FIG 4.

LMP1 and LMP2A expression reduces γH2AX phosphorylation. Immunoblotting of γH2AX levels in HK1 cell lines treated with 5 μM etoposide for 12 (A) or 24 h (B). Changes in γH2AX levels were normalized to those for HSC70 and averaged from 3 independent treatments (n = 3). Quantitation of γH2AX levels was determined by LI-COR infrared imaging. Statistical significance was determined by an unpaired 2-tailed t test (*, P = 0.05; **, P = 0.01; ***, P = 0.001). (C) Immunofluorescence analysis of proteins associated with DNA damage foci in the nucleus of HK1 cell lines treated with 5 μM etoposide for 12 h and sampled at time intervals to identify 53BP1-positive cells. Cells were stained for γH2AX (S139) and 53BP1 and imaged by Olympus FV2000 confocal microscopy. Merged images were generated using Olympus Fluoview 3.0 software. (D) The number of 53BP1-positive nuclei was counted for all time points and analyzed for corresponding γH2AX focus staining, represented as a percentage of nuclei doubly positive for 53BP1 and γH2AX out of the total number of 53BP1-positive nuclei per field of view. Standard errors of the means (SEM) were calculated from 5 representative fields of view per image. A cumulative ratio of total γH2AX-positive nuclei to total number of 53BP1-positive nuclei is indicated above each column.
In order to demonstrate that suppression of γH2AX phosphorylation was dependent on LMP1 and LMP2A coexpression, LMP1 or LMP2A shRNA knockdown was performed in HK1 LMP1-2A stable cells. Immunoblotting indicated successful knockdown of LMP1 (63% reduction) and LMP2A (77% reduction) that was not apparent from the scrambled shRNA transduction (Fig. 5A and B). Knockdown of either LMP1 or LMP2A increased phosphorylation of H2AX compared to that of the scrambled shRNA control following etoposide treatment, further indicating that modulation of γH2AX is dependent on the coexpression of LMP1 and LMP2A (Fig. 5A and B). Activation of p53 may affect DNA damage responses; therefore, to investigate the potential requirement for intact p53 mechanisms, the modulation of γH2AX was examined by transient induction of LMP1 and LMP2A in 293 cells with ablated p53 mechanisms (Fig. 5C). 293 cells were treated with 5 μM etoposide for a shorter period of 2 h, the earliest time point at which DNA damage signaling is activated without detectable caspase activation (data not shown). In contrast to HK1 cells, induced expression of LMP1 or LMP2A in 293 cells upon etoposide treatment increased γH2AX levels above those of pBabe control cells (Fig. 5C). However, LMP1-2A coexpression decreased γH2AX to levels comparable to those of pBabe control cells, supporting that LMP1 and LMP2A also can coregulate γH2AX independent of intact p53 mechanisms (Fig. 5C).
FIG 5.
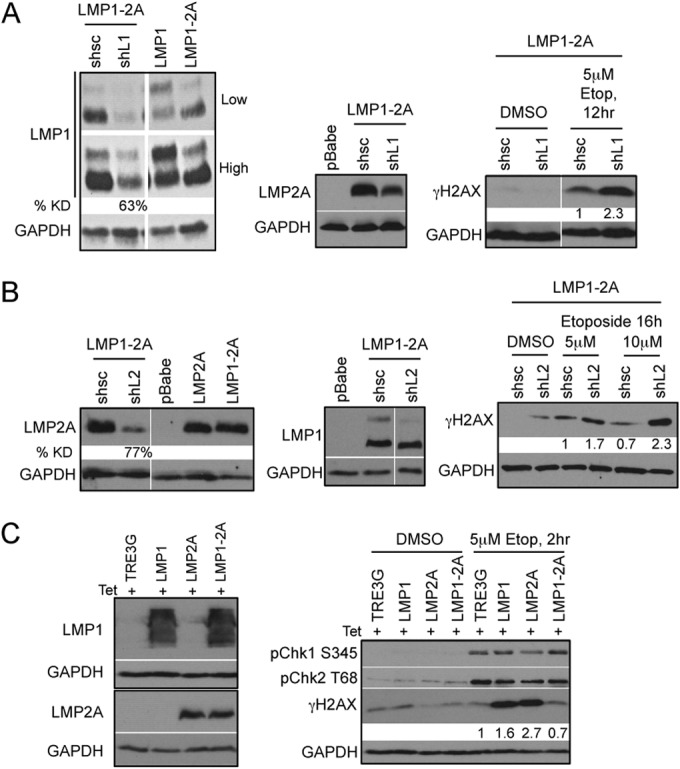
Reduction of γH2AX phosphorylation is dependent on the coexpression of LMP1 and LMP2A. Immunoblot analysis of efficient LMP1 (A) and LMP2A (B) knockdown (KD) 5 days after transduction with control (shsc) and specific (shL1 and shL2) shRNA constructs. LMP1 and LMP2A expression was normalized to that of the GAPDH loading control by densitometry with Image J software and represented as percent knockdown (% KD) compared to that of shsc control cells. HK1 LMP1-2A cells with LMP1 and LMP2A KD treated with 5 μM etoposide (Etop) were analyzed for DDR signaling proteins by immunoblotting. Changes in γH2AX levels were normalized to that of GAPDH by densitometry with Image J software and represented as fold change relative to the level for shsc control cells. (C) Transient expression of LMP1 and LMP2A in HEK293 cells was induced by 200 ng/ml doxycycline treatment for 48 h and analyzed by immunoblotting with LMP1-specific S12 antibody and LMP2A-specific 14B7 antibody. Lysates were analyzed 2 h after treatment with 5 μM etoposide for DNA damage signaling proteins and γH2AX phosphorylation. Changes in γH2AX phosphorylation levels were normalized to that of GAPDH by densitometry with Image J software and represented as fold change relative to the level for TRE3G control cells.
To further distinguish if the LMP1-2A-coregulated effects on DDR occur independently of caspase activation, DDR signaling was analyzed in HK1 cells that were arrested by serum starvation (0.1% for 36 h) and released from arrest in complete growth medium (10% serum) in the presence of etoposide treatment (5 μM for 24 h) (Fig. 6A). These conditions enable the characterization of initial DNA damage effects before the onset of caspase activation. Compared to asynchronous cells treated with 5 μM etoposide for 12 and 24 h, HK1 cells that were released from arrest induced negligible levels of caspase activation when treated with 5 μM etoposide for 24 h, as determined by immunoblotting for cleavage of caspase 8 and PARP (Fig. 6B). Under these conditions, γH2AX also was decreased by LMP1-2A coexpression upon etoposide treatment (Fig. 6C). These data further support that LMP1 and LMP2A coexpression regulates γH2AX levels, and this can occur independently of potential secondary modification of caspase activation (Fig. 3D).
FIG 6.

γH2AX levels are reduced in synchronized LMP1-2A cells in the absence of caspase activation. (A) Graphical timeline for HK1 treatment conditions, including 36 h of serum deprivation and subsequent treatment with 5 μM etoposide in 10% serum media for 24 h. Time points for lysate harvests are indicated. (B) Immunoblot analysis of caspase 8 and PARP cleavage in HK1 cell lines. Results are compared to those from treatment with 5 μM etoposide (12 and 24 h) without prior serum starvation and arrest. As positive controls for caspase and PARP cleavage, pBabe control cells were treated with 25 μM etoposide for 24 h or 0.5 μM staurosporine (Stauro) for 8 h. (C) Immunoblot analysis of DNA damage signaling proteins and H2AX after 24 h of 5 μM etoposide treatment in HK1 cells released from serum deprivation. (D) Analysis of time points for continuous etoposide or DMSO (lanes labeled D) treatment of cells released from serum deprivation up to 24 h. Separate cell lines are grouped by perforated lines.
LMP1 and LMP2A selectively regulate γH2AX phosphorylation.
Due to the undetectable caspase activation in prearrested cells, these conditions were further analyzed for potential modulation of additional DNA damage signaling proteins. Phosphorylation of ATM and ATR, and their downstream targets, including the checkpoint kinases Chk1 and Chk2, was analyzed by immunoblotting upon etoposide treatment. Etoposide induced phosphorylation of ATM (S1981), Chk2 (T68), and Chk1 (S345) but not ATR (S428); however, LMP1-2A coexpression did not reduce phosphorylation of these proteins (Fig. 6C). Transient coexpression of LMP1 and LMP2A in 293 cells also did not reduce Chk1 or Chk2 phosphorylation, supporting that LMP1 and LMP2A specifically modulate γH2AX levels without global effects on canonical DDR signaling mediators (Fig. 5C). Although γH2AX phosphorylation was reduced in LMP1-2A-coexpressing cells, expression of H2AX was equivalent in all cell lines (Fig. 6C). In sum, LMP1 and LMP2A coexpression did not reduce expression of H2AX or phosphorylation of ATM, ATR, Chk2, or Chk1 following etoposide treatment, indicating that modulation of DDR mediators was specific to γH2AX phosphorylation.
HK1 cells released from arrest were further analyzed for earlier time points after etoposide treatment to determine if γH2AX levels are dynamically regulated. To identify susceptible time points, cells were analyzed by immunoblotting for the cell cycle checkpoint proteins p21 and cyclin-D1 and the DNA polδ processivity factor PCNA upon release from arrest and etoposide treatment. The high levels of the cyclin-dependent kinase inhibitor p21 detected at the beginning of the experiment (0 to 3 h) indicate effective cell cycle arrest (Fig. 6D). Increased cyclin D1 beginning at 3 h further indicates exit from G1 arrest (Fig. 6D). At subsequent time points, accumulation of PCNA and cyclin B1 demonstrate enrichment of cells in S phase and mitosis, with the highest levels detected at 24 h (18). Levels of γH2AX were not strongly induced in any of the HK1 cell lines until 24 h. Although pChk2 was activated by 3 h and continued to be at levels higher than basal levels until the end of treatment, γH2AX levels varied over the 24-h period, suggesting that γH2AX levels are dynamically and perhaps cell cycle regulated despite persistent DNA damage signaling activation (Fig. 6D).
LMP1 and LMP2A effects upon DNA replication stress.
Etoposide predominantly induces DNA replication stress and double-strand breaks (DSBs), but at low doses it also can induce single-strand breaks (SSBs) by incomplete inhibition of topoisomerase II homodimers (48). In order to investigate DNA damage mechanisms that may contribute to LMP1-2A effects on γH2AX, HK1 stable cell lines were treated with ionizing radiation and SSB or DNA replication stress inducers (MMS and aphidicolin) to probe for responses to DSBs and SSBs, respectively. Upon ionizing radiation at doses that do not activate caspases (2 Gy and 5 Gy), phosphorylation of ATM, Chk2, and Chk1 was detected for all cell lines with comparable levels induced in LMP1-2A cells (Fig. 7A). Although concurrent induction of γH2AX over basal levels did not occur at 2 Gy, LMP1-2A coexpression reduced the basal level of γH2AX (Fig. 7A). At 5 Gy, γH2AX levels were moderately induced to equivalent levels in all cell lines (Fig. 7A). Etoposide inhibits the unwinding of replicating DNA; hence, to investigate if regulation of γH2AX by LMP1 and LMP2A is mediated in response to SSBs or replication fork stress, HK1 cells were treated with MMS and aphidicolin. MMS treatment induced phosphorylation of ATM, Chk1, Chk2, and γH2AX, indicating the successful activation of DDR signaling (Fig. 7B). Although ATR is activated in response to canonical SSB repair, basal levels of ATR were high in the highly replicative HK1 cells and were not induced by MMS treatment (Fig. 7B). Coexpression of LMP1-2A suppressed γH2AX induction upon MMS treatments, with no changes in other DNA damage mediators, further supporting that modulation of DDR signaling by LMP1-2A is specific to H2AX phosphorylation (Fig. 7B). Similar effects on γH2AX were observed with the DNA polymerase α/δ inhibitor aphidicolin in the presence of LMP1-2A coexpression, and this occurred in the absence of significant ATM, ATR, or Chk2 activation but with enhanced Chk1 phosphorylation, suggesting that modulation of γH2AX occurs independently of these checkpoints (Fig. 7C). In these experiments, basal levels of γH2AX also were consistently lower in LMP1-2A-coexpressing cells, suggesting that modulation of H2AX phosphorylation is more relevant to endogenous DNA stress encountered during normal replication and cell growth (Fig. 6D and 7A to C). These data support that LMP1 and LMP2A cooperate to suppress γH2AX phosphorylation in response to DNA replication stress inducers but not in response to DSBs induced by ionizing radiation.
FIG 7.

LMP1 and LMP2A coregulate γH2AX phosphorylation in response to DNA replication stress. (A) Immunoblot analyses of DNA damage signaling proteins in HK1 cell lines treated with ionizing radiation doses of 2 Gy and 5 Gy. Lysates were harvested after 1 h of incubation. To induce DNA replication stress, HK1 cells were treated with either 10 mg/ml MMS for 30 min (B) or 1 μM aphidicolin (Aph) for 4 h (C). As positive controls for DNA damage signaling responses, pBabe control cells were treated with 25 μM etoposide for 24 h or 10 mg/ml MMS for 30 min.
LMP1-expressing cells recover from etoposide-induced cytotoxicity.
To further examine the effect of LMP1-2A coexpression on the cytotoxic effects of etoposide, HK1 cells were analyzed for cell viability by resazurin and for long-term recovery following 24 h of etoposide treatment. The pan-kinase inhibitor staurosporine was included as a positive control for the loss of cell viability. The cytotoxic effect of etoposide at 5 μM and 25 μM doses initially was determined in pBabe cells. A decrease in cell viability was not immediately apparent until 1 to 2 days after the removal of etoposide at both 5 μM and 25 μM doses (Fig. 8A and B). The cytotoxic effects then were titrated with increasing doses of etoposide (1 μM to 50 μM) and analyzed for recovery by crystal violet staining of recovered cells by day 12 postremoval. At 5 μM, the cytotoxic effects of etoposide were apparent in pBabe and LMP2A cells (Fig. 8C). However, cells expressing LMP1 (LMP1-expressing or LMP1-2A-coexpressing cells) recovered from cytotoxic doses of etoposide with isolated colonies appearing in up to 25 μM treatment (Fig. 8C). To determine if the increased recovery mediated by LMP1 is due to a change in cell viability, HK1 cell lines were analyzed by resazurin immediately following treatment with 5 μM etoposide and monitored for a recovery period of 16 days (Fig. 8A and D). The initial cytotoxic effects of etoposide were similar for all cell lines during the 24-h treatment, with continued loss of viability for 10 days after etoposide removal. Despite the reduction of caspase activation in LMP1-2A cells immediately after etoposide treatment (Fig. 3D), the decreased resazurin activity beginning at 2 to 4 days postrecovery correlated with enhanced cytotoxicity, as determined by LDH release, with comparable cell death (50 to 70%) for all cell lines (Fig. 8E). This suggests that the molecular changes induced by LMP1-2A are separate from the phenotypic effects induced by etoposide cytotoxicity. At 10 to 16 days of recovery, restoration of cell growth was measurable in cell lines expressing LMP1 (LMP1 and LMP1-2A), correlating with the isolated colonies detected by crystal violet staining at day 12. These data reveal a distinct phenotype mediated by LMP1 expression alone in the recovery phase, such that despite activation of caspases, DNA damage signaling, and equivalent cytotoxicity induced by etoposide, LMP1 can restore the replicative potential of cytostatic cells.
FIG 8.

LMP1-expressing cells recover from etoposide-induced cytotoxicity. (A) Graphical timeline for 24-h etoposide (5 μM) treatment of HK1 cell lines in 10% serum media and subsequent recovery. Treatment and recovery time points are indicated. (B) Etoposide-induced cytotoxicity was assessed by the resazurin metabolic activity assay and analyzed after 24 h of etoposide treatment (5 μM and 25 μM), followed by up to 2 days of recovery in etoposide-free medium. The relative fluorescence units (RFU) and standard deviations were determined from triplicate wells of 3 independent experiments. Staurosporine treatment was used as a positive control for cytotoxicity at the 24-h time point. (C) The cytotoxic effects of etoposide on HK1 stable cell lines were analyzed by crystal violet staining of cells recovered under normal growth conditions for 12 days following treatment with increasing doses of etoposide for 24 h. Shown is a representative plate from 3 biological replicas. (D) HK1 cell lines treated with 5 μM etoposide for 24 h were analyzed for recovery by measuring viability and proliferating cells as determined by the resazurin assay. Each data point represents the means from 3 biological replicas. (E) After 5 μM etoposide treatment for 24 h, HK1 cell lines were analyzed for cytotoxicity by measuring the release of lactate dehydrogenase (LDH) from dying cells. Each data point represents the means from 3 biological replicas.
DISCUSSION
LMP1 and LMP2A transcripts are consistently expressed in NPC tumors, and although the signaling and biological properties of LMP1 and LMP2A have been investigated individually, their combined effects in epithelial cells have not been thoroughly examined (2, 7, 9). In this study, several considerations for utilizing HK1 include its origin from an EBV-negative NPC and p53 status, such that the cell line would be naive to the effects of EBV and mimic intact p53 mechanisms in NPC tumors (19, 20, 31). Although heterozygous for a mutation in p53 (codon 130), HK1 cells still can activate p53-dependent targets such as PUMA and still are responsive to p53 stabilization and growth arrest induced by Nutlin-3 treatment (20, 23, 31–33 and data not shown). In stably transduced HK1 cells, LMP1 and LMP2A coexpression did not affect cell growth, with a subtle increase in motility to fibronectin (Fig. 3). Intriguingly, only low doses of etoposide resulted in reduced caspase cleavage in the LMP1-2A-coexpressing cells (Fig. 3D), but this did not result in a difference in cytotoxicity (Fig. 8D and E). Additionally, coexpression of LMP1 and LMP2A selectively reduced γH2AX without affecting H2AX expression, 53BP1 focus formation, or phosphorylation of other DDR pathway mediators, suggesting the selective modulation of H2AX phosphorylation. In response to DSBs and SSBs induced by exogenous agents, such as irradiation (DSBs), or endogenous agents, such as replication fork stress (SSBs), H2AX is phosphorylated on S139 (γH2AX) by the phospholinositide-3-kinase-related protein kinases (PIKKs) ATM and ATR (ATM and Rad3 related), respectively (46, 49). Drugs that induced replication fork stress or SSBs selectively affected γH2AX levels in LMP1-2A-coexpressing cells, suggesting that LMP1 and LMP2A reduce H2AX phosphorylation in response to DNA replication stress but not in response to canonical ATM-dependent DSB repair pathways. Increased phosphorylation and activation of ATR was not detected in any of the replication fork stress-inducing treatments (Fig. 6C and 7A to C); however, γH2AX phosphorylation also can be mediated by the related PIKK family protein member DNA-dependent protein kinase (DNA-PK) (50). In addition to the H2AX phosphorylation on S139, which promotes the recruitment of DNA repair proteins to sites of DNA damage, a second phosphorylation on H2AX (Y142) is dephosphorylated by the phosphatase EYA to promote DNA repair (46, 51). The data in this study indicate that suppression of γH2AX by LMP1 and LMP2A is not mediated by modulation of ATM and ATR kinase activation and is regulated by other kinases, such as DNA-PK, or dephosphorylation mechanisms.
Mice deficient for H2AX or 53BP1 are defective in cell cycle checkpoints and are predisposed to cancer (52). Therefore, the cooperative modulation of H2AX phosphorylation by LMP1 and LMP2A may contribute to increased genome instability and cancer predisposition. ATM transcription is reduced and protein levels are undetectable in the majority of microdissected NPC tumor cells, and this also can occur in NPC cells infected by EBV in vitro (21). The lack of changes to ATM phosphorylation in HK1 stable cell lines expressing LMP1 and LMP2A suggest an alternative mechanism. It also would be of interest to investigate LMP1-2A codependent effects on DNA damage signaling in the absence of an intact ATM-dependent pathway. Individual expression of LMP1 and other latent EBV proteins can increase the number of spontaneous and mutagen-induced chromosomal aberrations, further supporting EBV latent proteins in promoting genome instability (22).
Activation of the DDR pathway is critical for lytic replication of several DNA tumor viruses. In herpesvirus infection, including EBV, activation of the DDR and ATM signaling is critical for efficient lytic replication (53). Additionally, the lytic products of murid herpesvirus 68 (MHV68) (Orf36 kinase) and its EBV homolog (BGLF4) induce γH2AX, and this modification is critical for efficient MHV68 replication and also may be required for EBV (54). The phosphorylation of H2AX also contributes to LANA-mediated episomal maintenance and may be important in the establishment of Kaposi's sarcoma-associated herpesvirus (KSHV) latency (25). Although activation of DDR signaling is required for replication of many DNA viruses, DDR signaling can result in deleterious effects, including triggering of apoptosis and cell cycle arrest. Therefore, many DNA viruses have evolved mechanisms to antagonize the DDR pathway in order to prevent activation of cell death pathways (53). Establishment of EBV latency in B cells and subsequent outgrowth of LCLs requires the attenuation of DDR signaling by EBNA3C (27, 28). However, NPC tumors do not express EBNA3C or other C promoter (Cp) transcripts characteristic of type III latency in LCLs. Therefore, the coregulation of γH2AX by LMP1 and LMP2A may be more relevant to type II latency in the absence of EBNA3C expression. The cellular and viral determinants for establishment of latency in epithelial cells are not well defined, and it remains to be determined if it similarly requires the attenuation of DDR signaling (5). Interestingly, the basal levels of γH2AX also were reduced by coexpression of LMP1 and LMP2A, and this may explain the differential response to low, but not high, doses of etoposide (Fig. 6C and 7A to C). This indicates relevance to physiological levels of endogenous stress induced by host replication or maintenance of EBV genomes rather than exogenous stress induced by therapeutic agents. It would be important to establish the effects of LMP1 and LMP2A in the context of viral infection, and additional work is necessary to establish EBV-infected epithelial cell lines that coexpress LMP1 and LMP2A proteins to mirror their codetection in NPC biopsy specimens.
This study addresses three aspects of responses to genotoxic stress: molecular changes in DDR mediators and phenotypic cell death responses, including cytotoxicity and recovery. The cytotoxic response to etoposide was not affected by expression of LMP1 or LMP2A, suggesting that additional mechanisms are involved. However, a distinct property dependent on LMP1 resulted in enhanced recovery of cells from etoposide despite activation of apoptotic mediators in the cell population (Fig. 3D and 8C and D). Only a subset of cells expressing LMP1 survive etoposide treatment and regain replicative potential, suggesting that LMP1 recovers cytostatic cells. This LMP1-dependent recovery is independent of genotoxins and was recapitulated with other cell cycle arrest treatments (data not shown). This underscores that the molecular effects described in this study likely result in phenotypic changes to endogenous stress. It would be of interest to characterize these distinguishing properties and potential mechanisms of LMP1 recovered cells. Although LMP1 is required for the immortalization and hyperproliferation of primary B cells, the viral products that contribute to neoplastic transformation of epithelial cells are not well established (55, 56). The present data support that EBV LMP1 and LMP2A proteins jointly contribute to oncogenic mechanisms by modulating DNA repair, and LMP1 can independently contribute to the aberrant recovery of unchecked cytostatic cells.
ACKNOWLEDGMENTS
We thank George Tsao (University of Hong Kong) for providing the HK1 cells.
This research was supported by the Hillman Foundation, a Cancer Center Support Grant, and UPCI shared resources that are supported in part by NIH award P30CA047904.
REFERENCES
- 1.Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Raab-Traub N. 2002. Epstein-Barr virus in the pathogenesis of NPC. Semin Cancer Biol 12:431–441. doi: 10.1016/S1044579X0200086X. [DOI] [PubMed] [Google Scholar]
- 3.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 4.Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. 2014. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci U S A 111:16544–16549. doi: 10.1073/pnas.1400818111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsao SW, Tsang CM, Pang PS, Zhang G, Chen H, Lo KW. 2012. The biology of EBV infection in human epithelial cells. Semin Cancer Biol 22:137–143. doi: 10.1016/j.semcancer.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Tsang CM, Zhang G, Seto E, Takada K, Deng W, Yip YL, Man C, Hau PM, Chen H, Cao Y, Lo KW, Middeldorp JM, Cheung AL, Tsao SW. 2010. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: regulation of infection and phenotypic characterization. Int J Cancer 127:1570–1583. doi: 10.1002/ijc.25173. [DOI] [PubMed] [Google Scholar]
- 7.Brooks L, Yao QY, Rickinson AB, Young LS. 1992. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol 66:2689–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pratt ZL, Zhang J, Sugden B. 2012. The latent membrane protein 1 (LMP1) oncogene of Epstein-Barr virus can simultaneously induce and inhibit apoptosis in B cells. J Virol 86:4380–4393. doi: 10.1128/JVI.06966-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson CW, Port RJ, Young LS. 2012. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin Cancer Biol 22:144–153. doi: 10.1016/j.semcancer.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, Kikutani H. 1999. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286:300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 11.Mainou BA, Everly DN Jr, Raab-Traub N. 2007. Unique signaling properties of CTAR1 in LMP1-mediated transformation. J Virol 81:9680–9692. doi: 10.1128/JVI.01001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shair KH, Schnegg CI, Raab-Traub N. 2008. EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer Res 68:6997–7005. doi: 10.1158/0008-5472.CAN-08-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mainou BA, Everly DN Jr, Raab-Traub N. 2005. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene 24:6917–6924. doi: 10.1038/sj.onc.1208846. [DOI] [PubMed] [Google Scholar]
- 14.Scholle F, Bendt KM, Raab-Traub N. 2000. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol 74:10681–10689. doi: 10.1128/JVI.74.22.10681-10689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison JA, Klingelhutz AJ, Raab-Traub N. 2003. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J Virol 77:12276–12284. doi: 10.1128/JVI.77.22.12276-12284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fotheringham JA, Raab-Traub N. 2013. Epstein-Barr virus latent membrane protein 2 effects on epithelial acinus development reveal distinct requirements for the PY and YEEA motifs. J Virol 87:13803–13815. doi: 10.1128/JVI.02203-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fruehling S, Swart R, Dolwick KM, Kremmer E, Longnecker R. 1998. Tyrosine 112 of latent membrane protein 2A is essential for protein tyrosine kinase loading and regulation of Epstein-Barr virus latency. J Virol 72:7796–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kastan MB, Bartek J. 2004. Cell-cycle checkpoints and cancer. Nature 432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 19.Effert P, McCoy R, Abdel-Hamid M, Flynn K, Zhang Q, Busson P, Tursz T, Liu E, Raab-Traub N. 1992. Alterations of the p53 gene in nasopharyngeal carcinoma. J Virol 66:3768–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spruck CH III, Tsai YC, Huang DP, Yang AS, Rideout WM III, Gonzalez-Zulueta M, Choi P, Lo KW, Yu MC, Jones PA. 1992. Absence of p53 gene mutations in primary nasopharyngeal carcinomas. Cancer Res 52:4787–4790. [PubMed] [Google Scholar]
- 21.Bose S, Yap LF, Fung M, Starzcynski J, Saleh A, Morgan S, Dawson C, Chukwuma MB, Maina E, Buettner M, Wei W, Arrand J, Lim PV, Young LS, Teo SH, Stankovic T, Woodman CB, Murray PG. 2009. The ATM tumour suppressor gene is down-regulated in EBV-associated nasopharyngeal carcinoma. J Pathol 217:345–352. doi: 10.1002/path.2487. [DOI] [PubMed] [Google Scholar]
- 22.Gruhne B, Sompallae R, Masucci MG. 2009. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 28:3997–4008. doi: 10.1038/onc.2009.258. [DOI] [PubMed] [Google Scholar]
- 23.Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, Kwong DL, Tsao SW, Jin DY. 2008. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med 205:2551–2560. doi: 10.1084/jem.20072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fries KL, Miller WE, Raab-Traub N. 1996. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J Virol 70:8653–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jha HC, Upadhyay SK, Prasad AJM, Lu M, Cai Q, Saha A, Robertson ES. 2013. H2AX phosphorylation is important for LANA-mediated Kaposi's sarcoma-associated herpesvirus episome persistence. J Virol 87:5255–5269. doi: 10.1128/JVI.03575-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarakanova VL, Stanitsa E, Leonardo SM, Bigley TM, Gauld SB. 2010. Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo. Virology 405:50–61. doi: 10.1016/j.virol.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 27.Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, Allday MJ, Patel A, Dave SS, Kim W, Hu K, Guo J, Tainter D, Rusyn E, Luftig MA. 2010. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8:510–522. doi: 10.1016/j.chom.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jha HC, Aj MP, Saha A, Banerjee S, Lu J, Robertson ES. 2014. Epstein-Barr virus essential antigen EBNA3C attenuates H2AX expression. J Virol 88:3776–3788. doi: 10.1128/JVI.03568-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shair KH, Bendt KM, Edwards RH, Nielsen JN, Moore DT, Raab-Traub N. 2012. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) and LMP2A function cooperatively to promote carcinoma development in a mouse carcinogenesis model. J Virol 86:5352–5365. doi: 10.1128/JVI.07035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longan L, Longnecker R. 2000. Epstein-Barr virus latent membrane protein 2A has no growth-altering effects when expressed in differentiating epithelia. J Gen Virol 81:2245–2252. doi: 10.1099/vir.0.17101-0. [DOI] [PubMed] [Google Scholar]
- 31.Huang DP, Ho JH, Poon YF, Chew EC, Saw D, Lui M, Li CL, Mak LS, Lai SH, Lau WH. 1980. Establishment of a cell line (NPC/HK1) from a differentiated squamous carcinoma of the nasopharynx. Int J Cancer 26:127–132. doi: 10.1002/ijc.2910260202. [DOI] [PubMed] [Google Scholar]
- 32.Chan SY, Choy KW, Tsao SW, Tao Q, Tang T, Chung GT, Lo KW. 2008. Authentication of nasopharyngeal carcinoma tumor lines. Int J Cancer 122:2169–2171. doi: 10.1002/ijc.23374. [DOI] [PubMed] [Google Scholar]
- 33.Strong MJ, Baddoo M, Nanbo A, Xu M, Puetter A, Lin Z. 2014. Comprehensive high-throughput RNA sequencing analysis reveals contamination of multiple nasopharyngeal carcinoma cell lines with HeLa cell genomes. J Virol 88:10696–10704. doi: 10.1128/JVI.01457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo AK, Lo KW, Tsao SW, Wong HL, Hui JW, To KF, Hayward DS, Chui YL, Lau YL, Takada K, Huang DP. 2006. Epstein-Barr virus infection alters cellular signal cascades in human nasopharyngeal epithelial cells. Neoplasia 8:173–180. doi: 10.1593/neo.05625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feederle R, Bartlett EJ, Delecluse HJ. 2010. Epstein-Barr virus genetics: talking about the BAC generation. Herpesviridae 1:6. doi: 10.1186/2042-4280-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shair KH, Bendt KM, Edwards RH, Bedford EC, Nielsen JN, Raab-Traub N. 2007. EBV latent membrane protein 1 activates Akt, NFkappaB, and Stat3 in B cell lymphomas. PLoS Pathog 3:e166. doi: 10.1371/journal.ppat.0030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mainou BA, Raab-Traub N. 2006. LMP1 strain variants: biological and molecular properties. J Virol 80:6458–6468. doi: 10.1128/JVI.00135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sung NS, Edwards RH, Seillier-Moiseiwitsch F, Perkins AG, Zeng Y, Raab-Traub N. 1998. Epstein-Barr virus strain variation in nasopharyngeal carcinoma from the endemic and non-endemic regions of China. Int J Cancer 76:207–215. doi:. [DOI] [PubMed] [Google Scholar]
- 39.Shair KH, Schnegg CI, Raab-Traub N. 2009. Epstein-Barr virus latent membrane protein-1 effects on junctional plakoglobin and induction of a cadherin switch. Cancer Res 69:5734–5742. doi: 10.1158/0008-5472.CAN-09-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moody CA, Scott RS, Su T, Sixbey JW. 2003. Length of Epstein-Barr virus termini as a determinant of epithelial cell clonal emergence. J Virol 77:8555–8561. doi: 10.1128/JVI.77.15.8555-8561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikeda M, Longnecker R. 2007. Cholesterol is critical for Epstein-Barr virus latent membrane protein 2A trafficking and protein stability. Virology 360:461–468. doi: 10.1016/j.virol.2006.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fotheringham JA, Coalson NE, Raab-Traub N. 2012. Epstein-Barr virus latent membrane protein-2A induces ITAM/Syk- and Akt-dependent epithelial migration through alphav-integrin membrane translocation. J Virol 86:10308–10320. doi: 10.1128/JVI.00853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dawson CW, Laverick L, Morris MA, Tramoutanis G, Young LS. 2008. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J Virol 82:3654–3664. doi: 10.1128/JVI.01888-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukuda M, Longnecker R. 2004. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J Virol 78:1697–1705. doi: 10.1128/JVI.78.4.1697-1705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degterev A, Yuan J. 2008. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 46.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10:886–895. doi: 10.1016/S0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 47.Montecucco A, Biamonti G. 2007. Cellular response to etoposide treatment. Cancer Lett 252:9–18. doi: 10.1016/j.canlet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Muslimovic A, Nystrom S, Gao Y, Hammarsten O. 2009. Numerical analysis of etoposide induced DNA breaks. PLoS One 4:e5859. doi: 10.1371/journal.pone.0005859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norbury CJ, Zhivotovsky B. 2004. DNA damage-induced apoptosis. Oncogene 23:2797–2808. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- 50.An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B, Wang Y, Liu XD, Wu DC, Zhou PK. 2010. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol 11:18. doi: 10.1186/1471-2199-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. 2009. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. 2003. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 114:371–383. doi: 10.1016/S0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luftig MA. 2014. Viruses and the DNA damage response: activation and antagonism. Annu Rev Virol 1:605–625. doi: 10.1146/annurev-virology-031413-085548. [DOI] [PubMed] [Google Scholar]
- 54.Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HWT. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaye KM, Izumi KM, Kieff E. 1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U S A 90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsao SW, Yip YL, Tsang CM, Pang PS, Lau VM, Zhang G, Lo KW. 2014. Etiological factors of nasopharyngeal carcinoma. Oral Oncol 50:330–338. doi: 10.1016/j.oraloncology.2014.02.006. [DOI] [PubMed] [Google Scholar]