

Abstract
The biosynthesis of ribosomally synthesized and posttranslationally modified peptide (RiPP) natural products typically involves a precursor peptide that contains a leader peptide that is important for the modification process, and that is removed in the final step by a protease. Genome mining efforts for new RiPPs are often hampered by the lack of a general method to remove the leader peptides. We describe here the incorporation of hydroxy acids into the precursor peptides in E. coli that results in connection of the leader peptide via an ester linkage that is readily cleaved by simple hydrolysis. We demonstrate the method for two lantibiotics, lacticin 481 and nukacin ISK-1.
Recent genome sequencing efforts have revealed that RiPPs are a much larger class of natural products than previously anticipated.1 The relatively short RiPP biosynthetic pathways lend themselves well to heterologous expression and genome mining,2–6 and the gene-encoded precursor peptides make them attractive for bioengineering efforts.7–15 Based on the currently sequenced bacterial genomes, the lanthionine-containing peptides (lanthipeptides) are the largest class of RiPPs. They are characterized by intramolecular thioether crosslinks called lanthionine (Lan) and methyllanthionine (MeLan). Lanthipetides are biosynthesized from a linear peptide, generically called LanA (e.g. LctA for lacticin 481, NukA for nukacin ISK-1). They consist of a C-terminal core region that is processed into the mature lanthipeptide and an N-terminal leader region important for recognition by the posttranslational modification enzymes (Figure 1A). For class II lanthipeptides, LanM enzymes dehydrate specific Ser and Thr residues to dehydroalanine (Dha) and dehydrobutyrine (Dhb), respectively. The enzyme then promotes Michael-type additions of Cys residues to the dehydro amino acids (e.g. Figure 1A). The complex ring topology provides the lanthipeptides with restricted conformations that bestow antimicrobial properties on a large subset of family members called lantibiotics.16 Several lantibiotics, including derivatives of nukacin ISK-1 and lacticin 481,17,18 have been produced in Escherichia coli by coexpression of the precursor peptides with LanM.19–21 However, the removal of the leader peptide, often presents a major hurdle. In the producing organisms, a protease removes the leader peptide by proteolysis after residue –1, the last amino acid of the leader peptide (Figure 1A). Unfortunately, the protease domain of class II lanthipeptides are membrane associated as part of an ATP-binding cassette transporter (LanT), and the soluble protease domains have low in vitro activity.21,22 The use of commercial proteases (e.g. trypsin, LysC, or GluC) to remove the leader peptide, by mutating the –1 position on LanA to Arg, Lys, or Glu is a common leader peptide removal strategy, but is only possible if the core peptide does not contain the proteolytic cleavage site.19,23–27 Indeed, in previous studies that produced lacticin 481 in E. coli, removal of the LctA leader peptide was achieved with endoproteinase LysC, which cleaved after residue 1, producing analogs lacking the N-terminal lysine residue.28 A longer protease recognition site such as Factor Xa or TEV protease could overcome cleavage in the core peptide, but introduction of longer sites often interferes with posttranslational processing or the proteolysis step is prevented because of the introduction of non-proteinogenic structures at the P1 site (amino acid N-terminal to the cleavage site).24,29,30 A photolabile linker has been employed to produce lacticin 481 that alleviates any sequence dependence, but the linker was introduced by solid phase peptide synthesis and is not amenable to heterologous lanthipeptide biosynthesis.31 In this work we developed a chemical leader peptide removal strategy that can be used with bacterially expressed lanthipeptides, and in principle also other RiPPs.
Figure 1.

A) Biosynthesis of lacticin 481 analogs by incorporating Boc-HO-1, HO-2, or HO-3 at position 1 of the core peptide (X) using the amber stop codon suppression method. WT lacticin 481 contains a lysine at position 1. Also, position –1 (green) was mutated from Ala to Ile as described in the text. B) Structures of non-proteinogenic amino and hydroxy acids introduced into lacticin 481 or nukacin ISK-1 at residue 1 (X).
Previous studies have demonstrated that the ribosomal peptide synthesis machinery of E. coli can incorporate hydroxy acids.32,33 The resulting ester bonds can then be site-specifically cleaved by alkaline hydrolysis.34,35 Therefore, we investigated whether a hydroxy acid could be incorporated in the first position of a lanthipeptide without interfering with the posttranslational modifications. We demonstrate herein that this methodology is successful for the in vivo production and subsequent leader peptide removal of analogues of lacticin 481 and nukacin ISK-1.
We chose lacticin 481 because of the difficulty of removing its leader peptide without removing the N-terminal Lys. Recently, the pyrrolysyl-tRNA synthetase (PylT) pair,36 which naturally incorporates pyrrolysine (Pyl) in response to the amber stop codon (UAG), was shown to incorporate the hydroxy acid of Bocε-L-lysine (Boc-1, Figure 1B).37 To ensure that the lacticin synthetase LctM would fully modify the LctA precursor peptide analog, we first attempted incorporation of Boc-1 into LctA using PylRS. Duet plasmids were designed to contain pylRS, pylT, his6-lctA(A-1I/K1X), and lctM for coexpression system in E. coli (Supporting Information). The codon for the first amino acid of the LctA core region was replaced with the amber stop codon tag such that non-canonical amino or hydroxy acids (X, Figure 1) could be incorporated. In addition, the last residue of the leader peptide was mutated from Ala to Ile (A–1I) because previous studies have shown that cleavage of ester bonds in a peptide backbone is slow when the ester is preceded by a large hydrophobic residue.35
N-terminally His6-tagged LctA(A–1I/K1X), LctM, PylRS, and PylT was expressed in the presence of Boc-1 in the media. After purification of the peptide by immobilized metal affinity chromatography (IMAC), analysis by matrix-assisted laser-desorption time-of-flight (MALDI-TOF) mass spectrometry (MS) demonstrated that the peptide was fully modified LctA (mLctA) with Boc-1 incorporated (Boc-1-mLctA; Figure S1). When the coexpression was performed in the absence of Boc-1, the full-length LctA analogue was not detected. Incorporation of hydroxy acid Boc-HO-1 (Figure 1B) was attempted next. His6-LctA(A–1I/K1X) was coexpressed with LctM, PylRS, and PylT in the presence of Boc-HO-1, purified by IMAC and reversed phase-high performance liquid chromatography (RP-HPLC), and analyzed by MALDI-TOF MS. Indeed, the hydroxy acid was incorporated into LctA, which was fully modified by LctM (Boc-HO-1-mLctA; Figure 2A).
Figure 2.
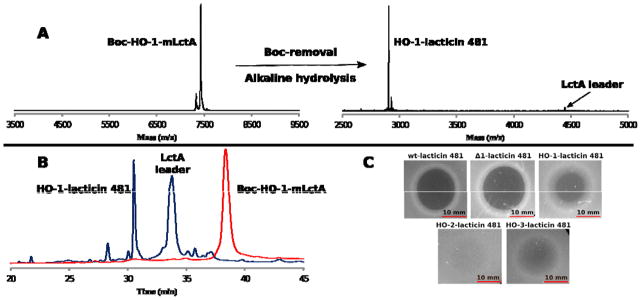
A) MALDI-TOF MS spectra depicting fully modified LctA containing Boc-HO-1 at position 1 (Boc-HO-1-mLctA), and the Boc-removal from and alkaline hydrolysis of Boc-HO-1-mLctA yielding HO-1-lacticin 481 and LctA leader peptide. The leader peptide ionizes poorly in the mass spectrum, but is seen clearly by HPLC. Boc-HO-1-mLctA ([M+H]+; calc’d = 7432 Da; observed = 7428 Da). HO-1-lacticin 481 ([M+H]+; calc’d = 2901.3 Da; observed = 2901.3 Da; monoisotopic mass spectrum). B) Analytical RP-HPLC trace of HO-1-mLctA before (red) and after (blue) Boc removal and alkaline hydrolysis. C) Antimicrobial assays of lacticin 481 and analogues against L. lactis HP. A total of 10 μL of 12.5 μM compound solution was added to each spot. Plates were incubated at 30 °C for 18 h.
To extend the ester backbone insertion to amino acids other than Lys, we next investigated a mutant form of PylRS (mut-PylRS) that can load analogues of the amino acids Phe and Tyr onto including 3-bromo-L-phenylalanine (H-Phe(3-Br)-OH; 2) and O-propargyl-L-tyrosine (H-Tyr(propargyl)-OH; 3).38–40 Indeed, compounds 2 and 3 were successfully incorporated in position 1 of LctA and the resulting mutants were fully modified by LctM in E. coli to produce 2-mLctA and 3-mLctA (Figure S1). Unlike the observations with Boc-1, a very small peak corresponding to L-Phe incorporation at the amber stop codon was also observed by MALDI-TOF MS (Figure S1), consistent with weak recognition of L-Phe by mut-PylRS.38 However, incorporation of L-Phe was very minor compared to incorporation of 2 or 3, suggesting the non-proteinogenic amino acid outcompetes L-Phe in vivo. Introduction of the corresponding hydroxy acids was then investigated by coexpression of His6-LctA(A–1I/K1X) with LctM, mut-PylRS, and PylT in media containing HO-Phe(3-Br)-OH (HO-2) or HO-Tyr(propargyl)-OH (HO-3; Figure 1B). IMAC and RP-HPLC purification followed by MALDI-TOF MS analysis demonstrated that the major products were fully modified LctA analogues containing HO-2 and HO-3 (HO-2-mLctA and HO-3-mLctA; Figures S2 and S3).
To test the hydrolytic cleavage of the leader peptides, the Boc group of Boc-HO-1-mLctA was first removed by incubating the modified peptide in 5% TFA at 50 °C. The pH was then increased to 10.5. Analysis by MALDI-TOF MS and RP-HPLC showed consumption of the full-length LctA analog and production of the leader peptide and the anticipated lacticin analogue with a hydroxyl group at its N-terminus (HO-1-lacticin 481) (Figures 2A,B, S2). The bioactivity of HO-1-lacticin 481 was compared to wild type (wt) lacticin 481, isolated from Lactococcus lactis subsp. lactis 481, and Δ1-lacticin 481 heterologously produced in E. coli and proteolyzed with LysC. Both agar diffusion antimicrobial assays (Figure 2C) and liquid media growth assays (Figure S4) with L. lactis HP cells showed that HO-1-lacticin 481 (IC50 = 200 ± 70 nM) exhibited decreased activity compared to wt lacticin 481 (IC50 = 100 ± 25 nM) and Δ1-lacticin 481 (IC50 = 90 ± 20 nM). Collectively, these data show that the N-terminal Lys is not as important as previously suggested based on studies on a mutant that lacked the N-terminal Lys but also had two other mutations compared to wt lacticin 481.41
Alkaline hydrolysis of HO-2-mLctA and HO-3-mLctA successfully produced lacticin 481 analogues with the desired N-terminal mutations (HO-2-lacticin 481 and HO-3-lacticin 481) (Figure S2). The antimicrobial activities of HO-2- and HO-3-lacticin 481 were compared with that of HO-1-lacticin 481 by agar diffusion assays (Figure 2C) and liquid media growth assays (Figure S4) with L. lactis HP cells. HO-1-lacticin 481 was > 10-fold more active than HO-2-lacticin 481 (IC50 = 3.50 ± 0.15 μM) and more than 3-fold more active than HO-3-lacticin 481 (IC50 = 700 ± 280 nM). We next applied the methodology to nukacin ISK-1 (Figure 3) because like lacticin 481 it has an N-terminal lysine. Moreover, in all previous attempts to biosynthesize nukacin ISK-1 in E. coli the removal of the leader peptide was achieved with endoproteinase LysC, which removed the first three residues of the core peptide yielding Δ1–3-nukacin ISK-1 and decreasing the bioactivity of the compound 32-fold.17,42 A coexpression vector was constructed encoding NukM and His6-NukA. After expression in E. coli, His6-mNukA was purified by IMAC and RP-HPLC. Removal of the leader peptide with LysC resulted in Δ1–3-nukacin ISK-1 as shown by MALDI-TOF MS (Figure S5). Analysis by MALDI-TOF MS of the coexpression of His6-NukA(A–1I/K1X) mutant with NukM, PylRS, and PylT in the presence of Boc-HO-1 resulted in cyclized NukA containing the hydroxy acid (Boc-HO-1-mNukA; Figure S6). The Boc-group of Boc-HO-1-mNukA was removed with TFA followed by alkaline hydrolysis yielding nukacin ISK-1 with an N-terminal hydroxyl group (HO-1-nukacin ISK-1; Figure S6). The nukacin ISK-1 analogues were then tested by agar diffusion antimicrobial assays (Figure 4) and liquid media growth assays (Figure S4) against L. lactis HP and compared to authentic nukacin ISK-1 isolated from the producer strain Staphylococcus warneri JCM 11004. The wt compound (IC50 = 400 ± 80 nM) was more active than HO-1-nukacin ISK-1 (IC50 = 560 ± 100 nM) indicating that the N-terminal amine of nukacin ISK-1 contributes to the bioactivity. Importantly, HO-1-nukacin ISK-1 displayed much stronger bioactivity than Δ1–3-nukacin ISK (IC50 = 5900 ± 2500 nM), which was only weakly bioactive in agreement with a previous study.42 Thus, unlike the observations with lacticin 481, the use of an ester linkage between the leader and core peptides resulted in a considerably more active product than use of endoproteinase LysC, illustrating the utility of this approach for lantibiotics with N-termini important for bioactivity. Another important potential application of this method is in synthetic biology efforts as RiPPs are well suited for generation of non-natural cyclic compounds/scaffolds that can be screened/selected for various purposes.12,13 In this arena, there is no obvious inherent advantage or disadvantage whether the peptide contains an N-terminal amino or hydroxyl group at the start of the selection process. However, the ability to readily remove the leader peptide in a general manner without the need to screen various proteases is highly enabling.
Figure 3.

Structures of the lanthipeptides lacticin 481 and nu-kacin ISK-1. Residues in nukacin ISK-1 that differ from lacticin 481 are highlighted in red.
Figure 4.

Zones of growth inhibition by nukacin ISK-1 and analogues against L. lactis HP. A total of 10 μL of solution at the concentrations listed was applied to each spot.
In conclusion, we produced analogs of lacticin 481 and nukacin ISK-1 by hydrolytic removal of the leader peptide. Unlike methods using commercial proteases, the ester bond hydrolysis reaction is not sequence dependent and is completely site-selective. Furthermore, the method can be readily multiplexed. This general methodology may therefore find use for lanthipeptides and other classes of RiPPs, and many applications can be envisioned in synthetic biology when an N-terminal amino group is not important.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM58822 to W.A.V. and CA161158 to W.R.L.).
Footnotes
Experimental procedures, supporting figures, and full author list for reference 1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
Contributor Information
Wenshe R. Liu, Email: wliu@chem.tamu.edu.
Wilfred A. van der Donk, Email: vddonk@illinois.edu.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Nat Prod Rep. 2013;30:108. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velásquez JE, van der Donk WA. Curr Opin Chem Biol. 2011;15:11. doi: 10.1016/j.cbpa.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohimani H, Kersten RD, Liu WT, Wang M, Purvine SO, Wu S, Brewer HM, Pasa-Tolic L, Bandeira N, Moore BS, Pevzner PA, Dorrestein PC. ACS Chem Biol. 2014;9:1545. doi: 10.1021/cb500199h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maksimov MO, Link AJ. J Ind Microbiol Biotechnol. 2014;41:333. doi: 10.1007/s10295-013-1357-4. [DOI] [PubMed] [Google Scholar]
- 5.Letzel AC, Pidot SJ, Hertweck C. BMC Genomics. 2014;15:983. doi: 10.1186/1471-2164-15-983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Doroghazi JR, Zhao X, Walker MC, van der Donk WA. Appl Environ Microb. 2015 doi: 10.1128/AEM.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levengood MR, van der Donk WA. Bioorg Med Chem Lett. 2008;18:3025. doi: 10.1016/j.bmcl.2008.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortés J, Appleyard AN, Dawson MJ. Methods Enzymol. 2009;458:559. doi: 10.1016/S0076-6879(09)04822-8. [DOI] [PubMed] [Google Scholar]
- 9.Rink R, Arkema-Meter A, Baudoin I, Post E, Kuipers A, Nelemans SA, Akanbi MH, Moll GN. J Pharmacol Toxicol Methods. 2010;61:210. doi: 10.1016/j.vascn.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Knappe TA, Manzenrieder F, Mas-Moruno C, Linne U, Sasse F, Kessler H, Xie X, Marahiel MA. Angew Chem Int Ed. 2011;50:8714. doi: 10.1002/anie.201102190. [DOI] [PubMed] [Google Scholar]
- 11.Tianero MD, Donia MS, Young TS, Schultz PG, Schmidt EW. J Am Chem Soc. 2012;134:418. doi: 10.1021/ja208278k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houssen WE, Bent AF, McEwan AR, Pieiller N, Tabudravu J, Koehnke J, Mann G, Adaba RI, Thomas L, Hawas UW, Liu H, Schwarz-Linek U, Smith MC, Naismith JH, Jaspars M. Angew Chem Int Ed. 2014;53:14171. doi: 10.1002/anie.201408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Heel AJ, Mu D, Montalban-Lopez M, Hendriks D, Kuipers OP. ACS Synth Biol. 2013;2:397. doi: 10.1021/sb3001084. [DOI] [PubMed] [Google Scholar]
- 14.Deane CD, Mitchell DA. J Ind Microbiol Biotechnol. 2014;41:315. doi: 10.1007/s10295-013-1361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molloy EM, Ross RP, Hill C. Biochem Soc Trans. 2012;40:1492. doi: 10.1042/BST20120193. [DOI] [PubMed] [Google Scholar]
- 16.Bierbaum G, Sahl HG. Curr Pharm Biotechnol. 2009;10:2. doi: 10.2174/138920109787048616. [DOI] [PubMed] [Google Scholar]
- 17.Nagao J, Harada Y, Shioya K, Aso Y, Zendo T, Nakayama J, Sonomoto K. Biochem Biophys Res Commun. 2005;336:507. doi: 10.1016/j.bbrc.2005.08.125. [DOI] [PubMed] [Google Scholar]
- 18.Oman TJ, Knerr PJ, Bindman NA, Vélasquez JE, van der Donk WA. J Am Chem Soc. 2012;134:6952. doi: 10.1021/ja3017297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Yang X, Garg N, van der Donk WA. J Am Chem Soc. 2011;133:2338. doi: 10.1021/ja109044r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caetano T, Krawczyk JM, Mosker E, Süssmuth RD, Mendo S. Chem Biol. 2011;18:90. doi: 10.1016/j.chembiol.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Lin Y, Teng K, Huan L, Zhong J. Microbiol Res. 2011;166:146. doi: 10.1016/j.micres.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Furgerson Ihnken LA, Chatterjee C, van der Donk WA. Biochemistry. 2008;47:7352. doi: 10.1021/bi800278n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goto Y, Li B, Claesen J, Shi Y, Bibb MJ, van der Donk WA. PLoS Biol. 2010;8:e1000339. doi: 10.1371/journal.pbio.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plat A, Kluskens LD, Kuipers A, Rink R, Moll GN. Appl Environ Microbiol. 2011;77:604. doi: 10.1128/AEM.01503-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA. Proc Natl Acad Sci USA. 2010;107:10430. doi: 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohans CT, Li JL, Vederas JC. J Am Chem Soc. 2014;136:13150. doi: 10.1021/ja5070813. [DOI] [PubMed] [Google Scholar]
- 27.Ökesli A, Cooper LE, Fogle EJ, van der Donk WA. J Am Chem Soc. 2011;133:13753. doi: 10.1021/ja205783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bindman NA, van der Donk WA. J Am Chem Soc. 2013;135:10362. doi: 10.1021/ja4010706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang W, van der Donk WA. Biochemistry. 2012;51:4271. doi: 10.1021/bi300255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garg N, Tang W, Goto Y, van der Donk WA. Proc Natl Acad Sci U S A. 2012;109:5241. doi: 10.1073/pnas.1116815109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bindman N, Merkx R, Koehler R, Herrman N, van der Donk WA. Chem Commun. 2010;46:8935. doi: 10.1039/c0cc02945a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fahnestock S, Rich A. Science. 1971;173:340. doi: 10.1126/science.173.3994.340. [DOI] [PubMed] [Google Scholar]
- 33.Guo J, Wang J, Anderson JC, Schultz PG. Angew Chem Int Ed Engl. 2008;47:722. doi: 10.1002/anie.200704074. [DOI] [PubMed] [Google Scholar]
- 34.England PM, Lester HA, Dougherty DA. Biochemistry. 1999;38:14409. doi: 10.1021/bi991424c. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe T, Miyata Y, Abe R, Muranaka N, Hohsaka T. Chembiochem. 2008;9:1235. doi: 10.1002/cbic.200700578. [DOI] [PubMed] [Google Scholar]
- 36.Srinivasan G, James CM, Krzycki JA. Science. 2002;296:1459. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi T, Yanagisawa T, Sakamoto K, Yokoyama S. J Mol Biol. 2009;385:1352. doi: 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- 38.Wang YS, Fang X, Wallace AL, Wu B, Liu WR. J Am Chem Soc. 2012;134:2950. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang YS, Fang X, Chen HY, Wu B, Wang ZU, Hilty C, Liu WR. ACS Chem Biol. 2013;8:405. doi: 10.1021/cb300512r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tharp JM, Wang YS, Lee YJ, Yang Y, Liu WR. ACS Chem Biol. 2014;9:884. doi: 10.1021/cb400917a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levengood MR, Kerwood CC, Chatterjee C, van der Donk WA. ChemBioChem. 2009;10:911. doi: 10.1002/cbic.200800752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asaduzzaman SM, Nagao J, Aso Y, Nakayama J, Sonomoto K. Appl Environ Microbiol. 2006;72:6012. doi: 10.1128/AEM.00678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.