

Abstract
Importance
Understanding molecular mechanisms of response and resistance to anticancer therapies requires prospective patient follow-up and clinical and functional validation of both common and low-frequency mutations. We describe a whole-exome sequencing (WES) precision medicine trial focused on patients with advanced cancer.
Objective
To understand how WES data affect therapeutic decision making in patients with advanced cancer and to identify novel biomarkers of response.
Design, Setting, and Patients
Patients with metastatic and treatment-resistant cancer were prospectively enrolled at a single academic center for paired metastatic tumor and normal tissue WES during a 19-month period (February 2013 through September 2014). A comprehensive computational pipeline was used to detect point mutations, indels, and copy number alterations. Mutations were categorized as category 1, 2, or 3 on the basis of actionability; clinical reports were generated and discussed in precision tumor board. Patients were observed for 7 to 25 months for correlation of molecular information with clinical response.
Main Outcomes and Measures
Feasibility, use of WES for decision making, and identification of novel biomarkers.
Results
A total of 154 tumor-normal pairs from 97 patients with a range of metastatic cancers were sequenced, with a mean coverage of 95× and 16 somatic alterations detected per patient. In total, 16 mutations were category 1 (targeted therapy available), 98 were category 2 (biologically relevant), and 1474 were category 3 (unknown significance). Overall, WES provided informative results in 91 cases (94%), including alterations for which there is an approved drug, there are therapies in clinical or preclinical development, or they are considered drivers and potentially actionable (category 1-2); however, treatment was guided in only 5 patients (5%) on the basis of these recommendations because of access to clinical trials and/or off-label use of drugs. Among unexpected findings, a patient with prostate cancer with exceptional response to treatment was identified who harbored a somatic hemizygous deletion of the DNA repair gene FANCA and putative partial loss of function of the second allele through germline missense variant. Follow-up experiments established that loss of FANCA function was associated with platinum hypersensitivity both in vitro and in patient-derived xenografts, thus providing biologic rationale and functional evidence for his extreme clinical response.
Conclusions and Relevance
The majority of advanced, treatment-resistant tumors across tumor types harbor biologically informative alterations. The establishment of a clinical trial for WES of metastatic tumors with prospective follow-up of patients can help identify candidate predictive biomarkers of response.
High-throughput next-generation sequencing has provided enormous insight into the genomic landscape of several tumor types, illuminating molecularly defined tumor subtypes, identifying new druggable targets, and providing insights into the heterogeneity of many tumors.1 Metastatic tumors often undergo genomic evolution during progression and resistance, and therefore genomic drivers may not always be evident in the primary tumor. Furthermore, no specific guidelines exist to help clinicians interpret and contextualize individual patients' genomic information when making therapeutic decisions. Herein, we describe an evidence-based precision medicine trial for patients with metastatic or treatment-resistant disease using a whole-exome sequencing (WES) clinical test called EXaCT-1, developed and validated by our group. Unique aspects include analysis of more than 21 000 genes of the cancer exome rather than a targeted hot-spot gene approach, complete disclosure of results through a WES clinical report, incorporation of metastatic and serial biopsies, use of fresh/frozen and formalin-fixed tissue, and development of patient-derived organoids and xenografts for co-clinical trials. Integral to the study are a comprehensive computational pipeline capable of categorizing mutations and generating a report for discussion in a multidisciplinary precision medicine tumor board and clinical follow-up to determine the clinical impact of mutations on subsequent response to therapies and patient outcomes. The overarching goals of our trial are to understand how WES affects therapeutic decision making in the context of advanced cancer care and to identify novel bio-markers of response.
Methods
Patients with advanced treatment-resistant cancer were prospectively enrolled from February 2013 through September 2014 for WES of tumor and normal tissue samples under a protocol approved by the institutional review board of Weill Cornell Medical College. Written informed consent was obtained, including discussion of risks associated with germline sequencing with an optional opportunity for rapid autopsy at the time of death for research to assess tumor extent, heterogeneity, and molecular mechanisms of resistance.2,3 Germline DNA was obtained using peripheral blood mononuclear cells, buccal swab sampling, or adjacent benign tissues. Fresh tissue samples were collected and processed using internal standard operating procedures (e Appendix in the Supplement) and used for WES, as well as patient-derived organoids and xenografts.4,5 We developed and validated a novel clinical-grade WES-based test for this trial, in agreement with Clinical Laboratory Improvement Amendments/Clinical Laboratory Evaluation Program (CLIA/CLEP) requirements and College of American Pathologists guidelines. During the validation phase, we observed sensitivity and specificity of 100% in detecting specific clinically relevant copy number changes (HER2), point mutations (BRAF, KRAS, EGFR, JAK2), and indels (EGFR), with no significant differences between formalin-fixed paraffin-embedded (FFPE) and fresh-frozen specimens independent of DNA quantity (H.R. et al, unpublished data, 2015). At least 200 ng of DNA was required to proceed with WES. DNA quality was confirmed for all samples by means of real-time PCR. Both tumor and normal DNA were sequenced by means of targeted capture of 21 522 genes using the HaloPlex System (Agilent), followed by next-generation sequencing (2 × 100 bp with 4-plexing) using Illumina HiSeq 2500. Short reads were aligned to GRC37/hg19 reference using Burrows-Wheeler Aligner (BWA) and processed accordingly by IPM-Exomepipeline, version 0.9. For details regarding quality control, data processing, and detection of point mutations and copy number alterations, see eMethods in the Supplement. Each tumor/normal pair was interrogated using an analytical algorithm for prioritization of 94 alterations in 49 actionable genes, and these were designated as category 1 alterations (eTable 1 in the Supplement). Category 2 alterations consisted of 508 known cancer-associated genes according to the Sanger center Cancer Gene Census. All other somatic alterations of unknown clinical or biologic significance are annotated as category 3. Patient results were discussed in tumor board, and clinical practice changes based on WES results were documented. Patients were observed prospectively for response to therapy with duration of follow-up of 7 to 25 months and an optional rebiopsy at progression. A program schematic is shown in Figure 1.
Figure 1. Overall Schematic of the Precision Medicine Trial.
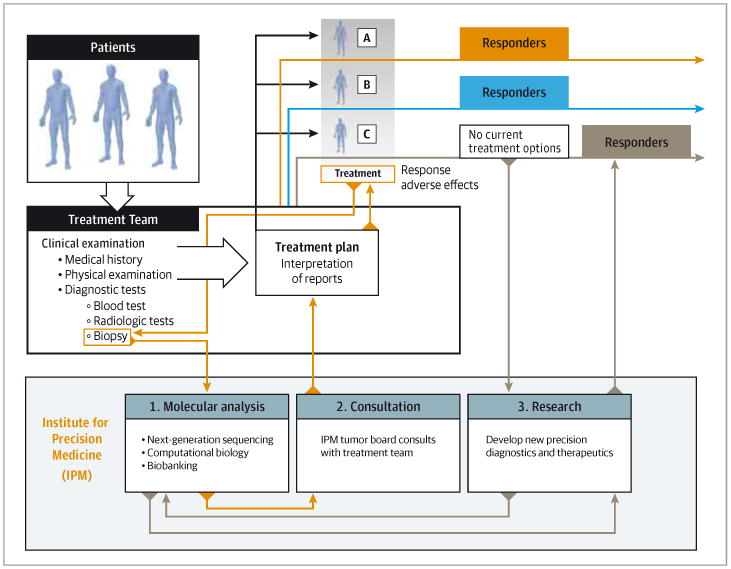
The process starts with clinical examination and consent, followed by metastatic tumor biopsy for whole-exome sequencing and biobanking. Results are discussed in tumor board, returned to the patient and referring physicians to guide treatment, and also used to fuel translational research and the development of new diagnostics and therapeutics.
All samples were analyzed using the same, locked-down computational pipeline and procedures (IPM-Exome-pipeline, version 0.9) (eFigure 1 and eMethods in the Supplement) in agreement with CLIA/CLEP requirements and College of American Pathologists guidelines. Accordingly, software versions and databases remained unchanged during the entire course of the analysis. Likewise, analysis parameters were optimized during the CLIA/CLEP validation phase and remained unchanged during the course of the analysis. We use software version control (gitlab), and as a result the computational pipeline and run parameters used for this study can be recovered in the future if we make parameter and/or version changes and revalidate the analytical pipeline. Raw and aligned short read data, run parameters, and output files are securely stored for an indefinite period and can be easily retrieved on the basis of unique identifiers.
Results
We report WES data of 154 tumor-normal pairs from 97 patients with advanced cancer. Patient characteristics, including tumor types and sites of metastases, are described in Figure 2. The most common tumor types were prostate cancer and urothelial cancer, which was not reflective of the incidence of cancer types seen at our institution but initially skewed on the basis of differences in referring habits. Twenty-three of 70 approached patients (33%) consented for future rapid autopsy, and 5 rapid autopsies were performed (eTable 2 in the Supplement). Tumor samples were obtained most commonly from new metastatic fresh tissue biopsy, with the sites determined by accessibility and safety. Specific biopsy techniques were developed to ensure adequate tissue from various sites for sequencing, including use of the On Control drill (Vida care, Inc) for sclerotic bone lesions. The most common sites of biopsy were bone (n = 24) or metastatic lymph node (n = 23), with an overall biopsy success rate of greater than 95% with all but 1 yielding sufficient tumor DNA of greater than 200 ng required for WES. In clinical cases when fresh-frozen tumor tissue was not available, FFPE material was used provided that high-quality DNA could be extracted. Estimated tumor purity ranged from 10% to 99%, with greater than 50% in 75% of cases (eFigures 2 and 3 in the Supplement). Mean input DNA was 225 ng, and mean coverage 85× for FFPE and 101× for fresh-frozen tissue (95× for all combined), with comparable sequencing metrics between FFPE and fresh-frozen tissue (eFigure 4 in the Supplement). A custom computational pipeline with alignment, normalization, rigorous quality control, mutation calling, and annotation was implemented for each case-control pair for simultaneous detection of somatic single-nucleotide variation, indels, and copy number alterations (eFigure 1 in the Supplement).
Figure 2. Clinical Demographic Characteristics Including Sequencing From a Wide Range of Metastatic Biopsy Sites and a Predominantly Solid Tumor Population.
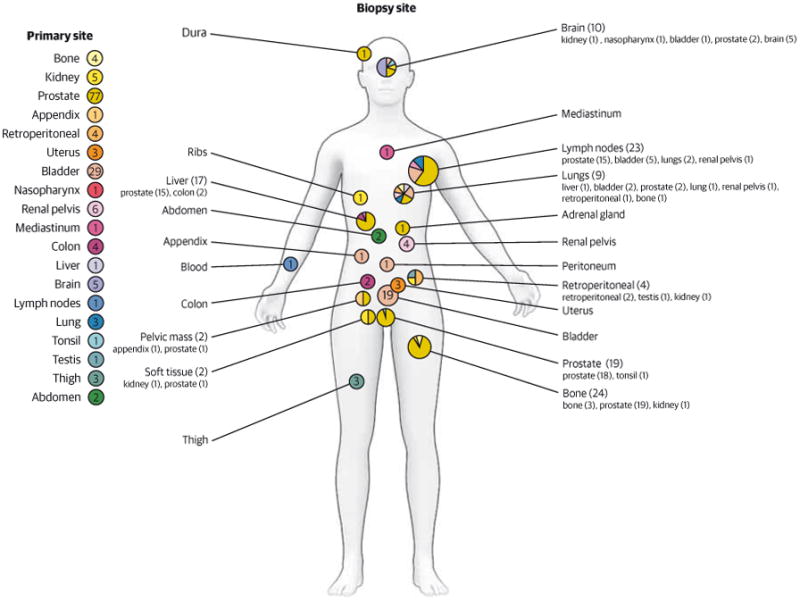
Numbers indicate number of patients with primary tumor types (left) and number of biopsies performed at specific locations (indicated on person). Smaller font indicates primary tumor site.
We interrogated each case for 94 alterations in 49 actionable and clinically relevant genes using an analytical algorithm for prioritization and designated these as category 1 alterations (eTable 1 in the Supplement). In this category are genes with alterations that can be targeted by available drugs. Category 2 alterations consist of 508 known cancer-associated genes that represent targets for therapies in clinical or preclinical development or are considered mutational drivers and potentially actionable. All other somatic alterations of unknown clinical or biologic significance were reported as category 3. A mean of 16 mutations was detected per patient, and a total of 16 category 1, 98 category 2, and 1474 category 3 mutations were observed across tumor types (Figure 3). A summary of the genomic data of the cohort including the most commonly altered genes and number of genes with copy number alterations across patients is presented in eFigures 5 through 8 in the Supplement. When comparing the mutational landscape of 8 patient-matched primary untreated tumors and platinum-resistant metastases for a subset of urothelial carcinomas, we observed a mean of only one-third shared point mutations including cases with targetable alterations present in only the metastases (B.F. et al, unpublished data, 2015). Specific examples of how WES data from serial biopsies were used to assess differences between primaries and metastases and disease evolution are illustrated in eFigures 9 and 10 in the Supplement. A case of rapid autopsy highlights shared mutations between multiple sites of metastasis at time of death from a 55-year-old patient with metastatic prostate cancer (eFigure 11 in the Supplement).
Figure 3. The Mutational Landscape of Precision Medicine Cases.

Figure shows the number of somatic alterations detected per patient in each category by tumor type (51 patients with prostate cancer, 14 with urothelial cancer, and 32 with other cancers).
Overall, WES provided clinically or biologically informative results in 91 patients (94%); this includes alterations in which there is an approved drug available or therapies in clinical or preclinical development or alterations considered mutational drivers and potentially actionable (category 1 or 2 alterations). Our tumor board was able to make treatment recommendations in the majority of these cases; however, treatment was most often not guided by these recommendations (only in 5 patients [5%]) because of patient access to clinical trials and/or off-label use of drugs. After discussion in tumor board, a clinical report in PDF format was uploaded into the patient's electronic medical record and discussed with the referring physician (s) and patient. A representative precision medicine EXaCT-1 sequencing report is shown in the eAppendix in the Supplement. This report details the sequencing of a metastatic inguinal lymph node biopsy from a patient with recurrent, platinum-refractory metastatic urothelial carcinoma to liver and lungs, for which there are no Food and Drug Administration–approved therapies available. In categories 1 and 2, ERBB2 (HER2) amplification, FGFR1 amplification, KITpE76K mutation, NKX2-1, TP53 mutation, TSC1 mutation, and ATM mutation were reported, as well as a substantial number of category 3 alterations. Overexpression of HER2 was confirmed as 3+ by immunohistochemical analysis (image shown on the report in the eAppendix in the Supplement). On the basis of these findings, the decision was made to start the patient on HER2-based therapy with trastuzumab and paclitaxel. The patient showed significant clinical improvement with combination therapy with complete response seen on imaging including resolution of extensive pulmonary and liver metastases (eFigure 12 in the Supplement) and has since been maintained in complete response with trastuzumab monotherapy at more than 9 months follow-up.
By focusing on patients with advanced refractory disease, we intended to develop a trial that could help identify novel targets, as well as mutations associated with drug resistance, and, by following patients prospectively, also identify and learn from patients with preferential response to either standard-of-care drugs or those in development. An example of this last scenario is illustrated by the case of PM12, a patient in his 60s with an aggressive variant of prostate cancer. He initially received a diagnosis of localized, high-grade (Gleason score 9) prostate cancer with focal neuroendocrine differentiation and underwent radical prostatectomy. Approximately 6 months later, he developed back pain and was found to have a lumbar spinal metastasis, local recurrence, and extensive liver and lung metastases; serum prostate-specific antigen level was undetectable. Biopsy revealed high-grade prostate cancer with predominantly neuroendocrine carcinoma features that was androgen receptor negative (Figure 4A). Small-cell neuroendocrine prostate cancer is a relatively uncommon, aggressive prostate cancer phenotype with limited available treatment options and poor overall survival.6 He was treated with cisplatin-docetaxel chemotherapy with palliative intent. Scans after cycle 3 showed tumor shrinkage, and quite surprisingly, scans after cycle 5 revealed a near complete response including resolution of his liver and lung metastases. His condition was clinically stable, and follow-up after this consisted of surveillance only. Approximately 1 year later, he experienced relapse with an isolated parenchymal brain metastasis, which was completely resected. He remained without evidence of systemic recurrence at more than 2 years follow-up. His dramatic and durable remission after systemic chemotherapy was exceptional.
Figure 4. Whole-Exome Sequencing of 1 Patient's Primary and Metastatic Tumors.
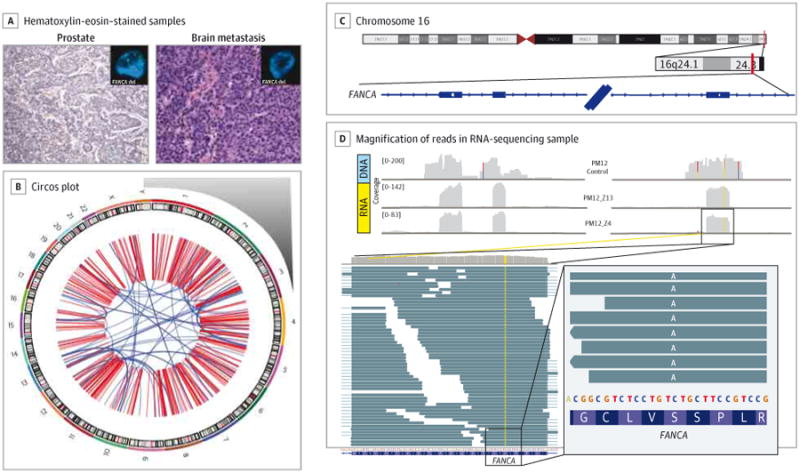
A, Both primary tumor (prostate, histologic subtype Gleason 9 adenocarcinoma with focal neuroendocrine differentiation) and metastatic tumor (brain, histologic subtype neuroendocrine prostate cancer) of patient PM12 were sequenced by means of whole-exome sequencing (WES). A substantial number of somatic mutations and copy number alterations were observed by both WES and whole-genome sequencing. Images are original magnification ×200. B, The whole-genome sequencing Circos plot of PM12's metastatic tumor illustrated a highly altered genome with complex structural variations and rearrangements. Chromosome number is indicated outside the circle. One important finding from WES was hemizygous deletion of the DNA repair gene FANCA in both primary tumor and metastasis, which was confirmed by fluorescence in situ hybridization (tumor panel insets; green probe = control centromeric; red probe = FANCA). C and D, Loss of heterozygosity in FANCA. In the tumor cells, in the heterozygous germline single-nucleotide polymorphism on exon 33 of FANCA (rs17233497), only the missense variant is expressed. C, Coverage of the region for germline (DNA) and 2 tumor samples (RNA). D, Magnification of the reads in 1 RNA-sequencing sample. Reads are reported as on the forward strand; hence, the A variant is indicated, corresponding to a T in the mRNA molecule because FANCA is transcribed from the reverse strand.
Both his primary prostate tumor and metastatic tumor recurrence demonstrated hypermutated genotype, with a more than 5-fold greater total number of point mutations compared with other metastatic prostate cancers and more than 3-fold greater number of indels in both his primary tumor and metastasis. Whole-genome sequencing also revealed a significantly greater number of copy number alterations and complex genomic rearrangements compared with the genome sequences of other metastatic prostate tumors (Figure 4A and eTable 2 in the Supplement). These findings were suggestive of genomic instability and pointed to a possible DNA repair defect. Notably, there was a hemizygous deletion detected in both primary and metastatic tumors involving a region on chromo some 16 with the DNA repair gene FANCA. FANCA deletion demonstrated significant clonality in his tumor using the CLONET (CLONality Estimate in Tumors) algorithm7 (eFigure 13 in the Supplement), suggesting that it was an early event. In addition, a germline variant of reported potential clinical relevance for Fanconi anemia (S1088F) was detected within the second allele on chromosome 16 at position 89815152 (Figure 4B). This S1088F mutation has been described in Fanconi anemia as potentially deleterious in function without associated loss of expression8,9 and is included in Fanconi screening panels. Consistent with this, FANCA was still expressed in this patient's tumor, but only the mutated allele was expressed (Figure 4B and eFigure 14 in the Supplement) and the wild-type allele was absent. Therefore, a possible complete loss of function of FANCA was suspected within this tumor on the basis of the patient's germline S1088F mutation and a somatic hemizygous copy number loss of FANCA.
FANCA encodes for a Fanconi anemia family protein involved in removal of the DNA interstrand cross links, which covalently link the 2 strands of DNA, preventing transcription and replication.10 Germline biallelic deletion or mutation in FANCA results in Fanconi anemia, characterized by a developmental phenotype, predisposition for malignant neoplasm, and hypersensitivity to DNA cross linking chemotherapy such as cisplatin. FANCA-deficient cells demonstrate chromosome instability and altered mutability, thus pointing to a potential initiator of the unstable genomic profile seen in patient PM12's primary and metastatic tumors. The “second hit” somatic alteration in FANCA seen in PM12 had not been reported as commonly altered in prostate cancer. Therefore, to assess the overall frequency of somatic deletion of FANCA in prostate cancer, we developed a dual-color, locus-specific fluorescence in situ hybridization (FISH) assay (eFigure 15 in the Supplement). FANCA deletion was not detected in benign prostate tissue samples (0 of 69 cases) and was present in 16% of localized prostate adenocarcinomas (11 of 69 cases) and 14% of advanced prostate cancers (4 of 29 cases).
On the basis of this newly observed overall frequency of FANCA alteration in prostate cancer and the extreme and exceptional response of patient PM12's disease to chemotherapy, we sought to evaluate the impact of FANCA loss on platinum sensitivity in prostate cancer using both in vitro models and a patient-derived xenograft derived from patient PM12's initial relapse biopsy (before chemotherapy). Although the patient also received a taxane, we chose to focus on platinum sensitivity given the sensitivity of Fanconi germline mutation carriers to DNA cross linking agents. Genome editing of FANCA using CRISPR resulted in cisplatin hypersensitivity (half maximal inhibitory concentration [IC50], 0.82 μM for 22Rv1-FANCA-KO1; IC50, 2.58 μM for 22Rv1-control cells) (Figure 5A), which was confirmed in an independent cell population, 22Rv1-FANCA-KO2 (eFigure 16 in the Supplement). Similarly, small interfering RNA– mediated depletion of FANCA in both LNCaP and VCaP prostate cancer cells resulted in a greater than 2-fold increase in cisplatin sensitivity compared with nontargeted scrambled small interfering RNA (LNCaP: IC50, 16.67 vs 24.92 μM, respectively; VCaP: IC50, 2.18 vs 12.56 μM, respectively), and comparable to control isogeneic fibroblast cells with and without FANCA expression (IC50, 2.6 vs 6.0 μM, respectively) (eFigure 16 in the Supplement). Moreover, FANCA loss led to a significant decrease in activity of the FANC core complex as measured by FANCD2 or FANCI foci formation in the presence or absence of a DNA-damaging agent (Figure 5B and eTables 3 and 4 in the Supplement). To determine the functional impact of the S1088F mutation, we generated isogenic cell populations that overexpress the wild type or S1088F mutant (wild-type FANCA or FANCA S1088F) in FANCA-negative RA3087 fibroblasts. Expression of FANCA S1088F resulted in an increased sensitivity to cisplatin (IC50, 0.8 μM) compared with cells overexpressing wild-type FANCA (IC50, 1.8 μM) (eFigure 16 in the Supplement). The patient-derived xenograft of our patient PM12 (annotated LTL545) appeared morphologically indistinguishable from his meta static tumor and also harbored the FANCA deletion by means of FISH (eFigure 17 in the Supplement). We compared the cisplatin response of his patient-derived xenograft to that of a control patient-derived xenograft derived from another patient with advanced neuroendocrine prostate cancer of similar morphology but lacking the FANCA deletion (annotated LTL352). Time course experiments showed that the LTL545 (from our patient's tumor) was significantly more sensitive to cisplatin (overall tumor growth inhibition, 47.6% in geometric mean of tumor size, mixed-effect analysis, df = 18, P < .001) compared with control LTL352 (overall tumor growth inhibition, 20.4% in geometric mean of tumor size, mixed-effect analysis, df = 11, P = .06) (Figure 5B and Figure e18 in the Supplement) and as-measured final tumor weight (LTL545, Wilcoxon rank-sum test P < .001; LTL352, Wilcoxon rank-sum test P = .73) (eFigure 18 and eTable 5 in the Supplement), and consistent with our patient's clinical response.
Figure 5. Cisplatin Sensitivity In Vitro and In Vivo.
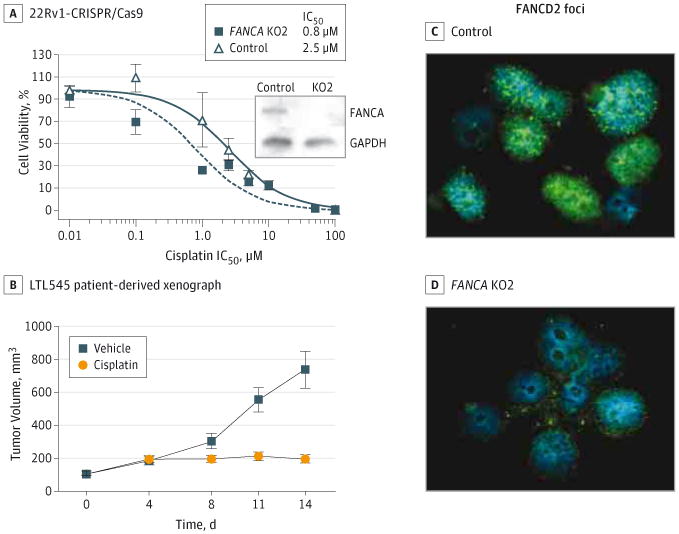
A, Cisplatin sensitivity in 22Rv1 cells following genome editing of FANCA (KO1) or a control sequence by CRISPR. Inset, Western blot of FANCA and GAPDH expression in indicated cell lines. IC50 indicates half maximal inhibitory concentration. B, Mean tumor size of the LTL545 xenograft before, during, and after treatment with vehicle (blue lines) or cisplatin (4 mg/kg, day 1 and day 8, intraperitoneal injection). C and D, FANCD2 foci formation following genome editing of FANCA (KO2) or a control sequence by CRISPR.
We interrogated publicly available sequencing data of cell lines and clinical samples across common tumor types to assess the frequency of FANCA alterations within the cancer population. We also queried germline data within the 1000 Genome Project and the Single-Nucleotide Polymorphism Database and found the S1088F FANCA germline variant to be present in 2.3% to 3.0% of the general population. In cancers, we found that FANCA expression correlated negatively with cisplatin sensitivity in cell lines (eFigure 19 in the Supplement), and somatic alterations involving FANCA were most frequent in ovarian cancers (eFigure 20 in the Supplement), a well-known platinum chemotherapy–sensitive tumor in which platinum-based combination therapy is standard front-line therapy. Surprisingly, the second most frequent tumor type to harbor FANCA deletion was prostate cancer, a tumor not known to be particularly platinum sensitive.11,12 Our findings suggest that perhaps a subset of prostate cancers may be particularly vulnerable to cytotoxic chemotherapy and provide a potential biologic rationale to help explain the exceptional response of our patient to platinum-based chemotherapy, although we cannot definitively rule out the additional contribution of other somatic alterations also present in his tumor that may be cooperating with FANCA. These data warrant further investigation of FANCA as a potential candidate predictive biomarker in cancer.
Discussion
Precision medicine is an approach intended to match the best drug with the right patient on the basis of the specific molecular alterations in an individual patient's tumor. This requires systematic integration of clinical and molecular information. There are a number of potential benefits of precision medicine,1 and several centers and companies now offer molecular cancer tests using tumor or blood, FFPE or frozen tissues. Our approach of using WES for a population with advanced, treatment-resistant disease rather than a focused gene assay is supported by the realization that the pace of genomic discoveries and development of novel targeted therapies is rapid. What may not be considered actionable today, and therefore missed on a highly targeted panel, could become actionable tomorrow. Furthermore, there is potential to identify novel resistance-associated mutations and/or pathways using this approach.
Overall, we detected biologically informative genomic alterations in most patients, and treatment recommendations were offered on the basis of these findings in more than 90% of cases; however, treatment was changed in only 5% of cases because of patient access to therapeutic trials. With a projected increased clinical use of genomic testing in cancer care, these findings highlight the growing clinical demand for n of 1 or biomarker-driven trials that are accessible to patients with tumors harboring low-frequency mutations. It is also important to observe patients prospectively to help elucidate how mutations of low frequency or of unknown significance may contribute toward driving tumor progression or response to therapy if evaluated in the appropriate clinical context. The case of patient PM12 is an example of how an alteration in the DNA repairgene FANCA might have gone unnoticed or had different clinical meaning if not explored in the context of a real patient. Although a double hit with both somatic and germ line alterations involving FANCA may be uncommon across the cancer population, understanding how such infrequent mutations might correlate with extreme response will be essential in adopting precision medicine into routine cancer care and can provide new opportunities for target biomarker development. Extensive work is required to credential novel mutations as potentially clinically significant, including rapid translation of clinical and molecular findings into the laboratory for in vitro and in vivo validation. Using matched tissue for patient-derived organoids and xenografts will become an important and growing resource for the development of co-clinical trials and larger-scale validation studies, as well as elucidating the biologic impact of novel resistance mutations discovered through sequential patient biopsies. Prioritization of candidates will greatly facilitate the design of genomically driven clinical trials and inform which of the less common of category 2 or 3 mutations to eventually move into category I.
We opted to sequence both tumor and normal tissue to en-sure that data generated from both somatic and germline DNA could potentially be used for future patient queries. An example illustrating the utility of sequencing both tumor and normal tissue is in the FANCA case presented, in which germline information was critical in elucidating biologic mechanisms to help understand clinical response to cisplatin therapy. One potential limitation to a WES approach is the inability to detect gene fusions; to overcome this, we perform RNA sequencing in select clinical cases (such as sarcomas and leukemias) in addition to EXaCT-1 for fusion identification. Therefore, refinement of the assay and/or future automated integration with other rapid tests including potentially RNA-sequencing and epi genomic assays may allow for a more customized approach to cancer care.
Conclusions
This precision medicine trial provides a rigorous proof of principle for developing a precision medicine approach to clinical cancer care with the capability to identify novel biomarkers of response and therapeutic targets. With rapidly advancing technologies and decreasing costs, the use of genomic information to select the most effective treatment options for patients and minimize adverse effects will soon become routinely feasible. This effort, while exciting, will require integrative studies dedicated toward correlation of clinical and molecular information and clinical follow-up of individual patients.
Supplementary Material
At a Glance.
We describe a whole-exome sequencing precision medicine trial that captures a diverse range of patients with advanced, treatment-resistant cancer with prospective clinical follow-up.
More than 90% of patients harbored actionable or biologically informative alterations, although treatment was guided by this information in only 5% of cases.
A novel alteration involving FANCA was found in a patient with prostate cancer who had an exceptional response to cisplatin therapy. Preclinical studies including patient-derived xenograft models support potential biologic relevance and predictive value.
This study highlights both the opportunities and challenges involved with bringing whole-exome sequencing into precision medicine clinical cancer care.
Acknowledgments
Funding/Support: This work was supported by Damon Runyon Cancer Research Foundation– Gordon Family Clinical Investigator Award CI-67-13 (H.B.), Department of Defense PC121341 (H.B.), Ann and William Bresnan Foundation (H.B., D.M.N.), Starr Cancer Consortium I7-A771 (H.B., M.A.R.), R01 CA116337 (H.B., F.D., M.A.R), Prostate Cancer Foundation (H.B., M.A.R.), Early Detection Research Network US NCI CA111275 (J.M.M., M.A.R.), Weill Cornell Medical College Clinical and Translational Science Center KL2TR000458 (B.F.), Canadian Institutes of Health Research (Y.W.), Weill Cornell Medical College Clinical and Translational Science Center UL1 RR024996 (F.C.), National Science Foundation CAREER (O.E.), Leukemia and Lymphoma Society Specialized Center of Research (O.E.), Hirschl Trust (O.E.), and Starr Cancer Consortium I6-A618 (O.E.).
Role of the Funder/Sponsor: The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Drs Beltran and Rubin had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Drs Elemento and Rubin served as co–senior authors, each with equal contribution to the manuscript.
Study concept and design: Beltran, Eng, Sigaras, Greco, Herrscher, MacDonald, Chakravarty, Kim, Campagne, Xiang, Smogorzewska, Demichelis, Elemento, Rubin.
Acquisition, analysis, or interpretation of data: Beltran, Eng, Mosquera, Sigaras, Romanel, Rennert, Kossai, Pauli, Faltas, Fontugne, Park, Banfelder, Prandi, Madhukar, Zhang, Padilla, Greco, McNary, Wilkes, MacDonald, Xue, Vacic, Emde, Oschwald, Tan, Chen, Collins, Gleave, Wang, Chakravarty, Schiffman, Campagne, Robinson, Nanus, Tagawa, Xiang, Smogorzewska, Demichelis, Rickman, Sboner, Elemento, Rubin.
Drafting of the manuscript: Beltran, Eng, Park, Padilla, Greco, Wilkes, Vacic, Emde, Chen, Chakravarty, Xiang, Rickman, Sboner, Elemento, Rubin.
Critical revision of the manuscript for important intellectual content: Beltran, Mosquera, Sigaras, Romanel, Rennert, Kossai, Pauli, Faltas, Fontugne, Banfelder, Prandi, Madhukar, Zhang, McNary, Herrscher, MacDonald, Xue, Emde, Oschwald, Tan, Chen, Collins, Gleave, Wang, Chakravarty, Schiffman, Kim, Campagne, Robinson, Nanus, Tagawa, Smogorzewska, Demichelis, Sboner, Elemento, Rubin.
Statistical analysis: Eng, Sigaras, Romanel, Banfelder, Prandi, Madhukar, Zhang, Greco, Wilkes, Chen, Sboner, Elemento.
Obtained funding: Kim, Rubin.
Administrative, technical, or material support: Beltran, Rennert, Kossai, Pauli, Park, Banfelder, Padilla, Greco, McNary, Herrscher, Wilkes, MacDonald, Xue, Oschwald, Tan, Collins, Kim, Nanus, Tagawa, Xiang, Rickman, Rubin. Study supervision: Beltran, Mosquera, Rennert, Greco, Oschwald, Gleave, Wang, Chakravarty, Kim, Robinson, Tagawa, Smogorzewska, Demichelis, Rickman, Sboner, Elemento, Rubin.
Supplemental content: at jamaoncology.com
Conflict of Interest Disclosures: None reported.
References
- 1.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Rubin MA, Putzi M, Mucci N, et al. Rapid (“warm”) autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res. 2000;6(3):1038–1045. [PubMed] [Google Scholar]
- 3.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64(24):9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 4.Gao D, Vela I, Sboner A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin D, Wyatt AW, Xue H, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74(4):1272–1283. doi: 10.1158/0008-5472.CAN-13-2921-T. [DOI] [PubMed] [Google Scholar]
- 6.Wang HT, Yao YH, Li BG, Tang Y, Chang JW, Zhang J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis—a systematic review and pooled analysis. J Clin Oncol. 2014;32(30):3383–3390. doi: 10.1200/JCO.2013.54.3553. [DOI] [PubMed] [Google Scholar]
- 7.Prandi D, Baca SC, Romanel A, et al. Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol. 2014;15(8):439. doi: 10.1186/s13059-014-0439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levran O, Erlich T, Magdalena N, et al. Sequence variation in the Fanconi anemia gene FAA. Proc Natl Acad Sci U S A. 1997;94(24):13051–13056. doi: 10.1073/pnas.94.24.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wijker M, Morgan NV, Herterich S, et al. Heterogeneous spectrum of mutations in the Fanconi anaemia group A gene. Eur J Hum Genet. 1999;7(1):52–59. doi: 10.1038/sj.ejhg.5200248. [DOI] [PubMed] [Google Scholar]
- 10.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493(7432):356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aparicio AM, Harzstark AL, Corn PG, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res. 2013;19(13):3621–3630. doi: 10.1158/1078-0432.CCR-12-3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sternberg CN, Petrylak DP, Sartor O, et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: the SPARC trial. J Clin Oncol. 2009;27(32):5431–5438. doi: 10.1200/JCO.2008.20.1228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.