

Abstract
The therapeutic landscape for advanced melanoma has expanded in recent years. This expansion has largely been driven by investigational work in melanoma tumor biology and immunology that has been successfully translated to the clinical setting. Molecular evidence generated through benchside experimentation identified BRAF and MEK as key molecular targets in melanoma. This commentary will highlight this work and provide a rationale for the continued importance of translational work in the field of targeted melanoma therapies.
Introduction
Recent progress in melanoma drug development highlights the critical impact that translational research plays in advancing patient care. Prior to 2011, dacarbazine, interleukin (IL)-2, and interferon (IFN)α-2b were the only Food and Drug Administration (FDA) approved treatment options for metastatic melanoma. These early therapies resulted in poor and inconsistent overall response rates (~10–15% (Eggermont and Kirkwood, 2004)). A renaissance in melanoma therapeutics occurred with the recognition that molecular aberrations in the mitogen-activated protein kinase (MAPK) pathway (Figure 1) were present in a majority fraction of melanomas. (Davies et al., 2002) These investigations resulted in the vigorous pursuit of small molecule kinase inhibitors and the eventual FDA approval of 3 novel MAPK pathway inhibitors for the treatment of advanced melanoma (Figure 1). Although key insights into immune checkpoint blockade have generated a similar number of recent immunotherapeutic breakthroughs (Brahmer et al., 2012; Hamid et al., 2013; Hodi et al., 2010), this commentary will focus on the development of molecular targeted therapies.
Figure 1.
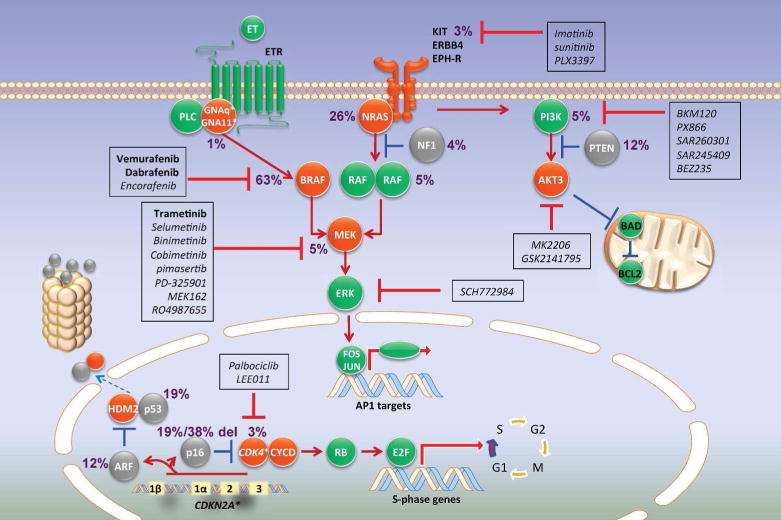
Molecular targeting of the mitogen-activated protein kinase (MAPK), PI3K, and CDK4 pathways and the associated mutation rates of potential molecular targets in advanced melanoma (Hodis et al., 2012). Activation of a receptor tyrosine kinase such as c-KIT results in the propagation of signal via the MAPK pathway leading to activation of RAS, RAF, MEK and ERK. This ultimately results in the gene expression and promotes cellular proliferation and survival. Mutated BRAF bypasses this ordered pathway and stimulates constitutive signaling, making it a prime target for vemurafenib and dabrafenib. Trametinib targets the downstream effector molecule MEK. Dysregulation of the PI3K pathway promotes melanoma progression. Small molecule inhibitors of the PI3K pathway are being clinically tested in combination with MAPK pathway inhibition. Similarly, CDK4 is an attractive candidate molecule to target in melanoma and is the focus of multiple clinical trials. Red=activated; gray=inactivated; green=normal function. Drugs (shown in boxes) that have been approved (bold), or in trial (non-bold, italics), are indicated.
Melanoma arises from the activation, or inactivation, of genes that regulate critical cellular functions including proliferation, cell cycle regulation, survival, angiogenesis, and cell migration. Efforts to systematically codify these changes have uncovered molecularly discrete subsets of melanoma (Curtin et al., 2005). For instance, melanomas arising in the context of non-chronic sun-damaged (CSD) skin are associated with BRAF and NRAS mutations while lesions that arise from mucosal and acral surfaces are linked to KIT alterations (Curtin et al., 2005). Moreover, some melanomas that lack NRAS or BRAF mutations harbor deleterious lesions in NF1 or amplifications of CCND1 and CDK4 (Lin et al., 2008), which are downstream mediators of cell cycle progression in the MAPK pathway (Figure 1). These data suggests that inasmuch as melanomas ought to be classified histologically, molecular subtyping may provide a more pragmatic approach as specific therapies targeting receptor tyrosine kinases, downstream kinases, and other signaling molecules become available. Table 1 highlights key clinical trials that have expanded the treatment landscape of melanoma with targeted therapies.
Table 1.
Table of key clinical trials in targeted melanoma therapy.
| Trial Name | Experimental agent | Control agent | Tumor response (experimental; 95% CI vs control; 95% CI) | PFS (experimental vs control) | OS (experimental vs control) |
|---|---|---|---|---|---|
| BRIM-3 | vemurafenib (N = 337) |
dacarbazine (N = 338) |
48%; 42 – 55 vs 5%; 3 – 9 | 5.3 vs 1.6 months (HR 0.26; 0.20 – 0.33; p < 0.001 | 84% vs 64% (HR 0.37; 0.26 – 0.55; p < 0.001) |
| BREAK-3 | dabrafenib (N = 187) |
dacarbazine (N = 63) |
50%; 42.4 – 57.1 vs 6%; 1.8 – 15.5 | 5.1 vs 2.7 months (HR 0.30; 0.18 – 0.51; p < 0.0001 | Not statistically significant (HR 0.76; 0.48-1.21) |
| METRIC | trametinib (N = 214) |
dacarbazine or paclitaxel (N = 108) |
22%; 17 – 28 vs 8%; 4 – 15 | 4.8 vs 1.5 months (HR 0.45; 0.33 – 0.63; p < 0.001) | 81% vs 67% (HR 0.54; 0.32 – 0.92; p = 0.01) |
| COMBI-d | dabrafenib + trametinib (N = 211) |
dabrafenib (N = 212) |
67%; 60 – 73 vs 51%; 45 – 58 | 9.3 vs 8.8 months (HR 0.75; 0.57 – 0.99; p = 0.03) | 93% vs 85% (HR 0.63, 95% CI 0.42 – 0.94; p = 0.023) |
| coBRIM | vemurafenib + cobimetinib (N = 247) |
vemurafenib (N = 248) |
68%; 61 – 73 vs 45%; 38 – 51 | 9.9 vs 6.2 months (HR 0.51; 0.39 – 0.68; p < 0.001) | 81% vs 73% (HR 0.65; 0.42 – 1.00; p = 0.046) |
| COMBI-v | dabrafenib + trametinib (N = 352) |
vemurafenib (N = 352) |
64%; 59 – 69 vs 51%; 46 – 57 | 11.4 vs 7.3 months (HR 0.56; 0.46 – 0.69; p < 0.001) | 72% vs 65% (HR 0.69; 0.53 – 0.89; p = 0.005) |
BRAF inhibition
The rationale for targeting BRAF is evident since the Val-600-Glu mutation in the BRAF kinase domain is the single most common mutation in cutaneous melanoma (Davies et al., 2002). This substitution constitutively activates BRAF and its attendant downstream MAPK pathway effectors such as MEK (Figure 1). Although 50% of melanomas harbor the BRAF V600E mutation, nearly 80% of benign nevi also contain the identical mutation (Pollock et al., 2003). Thus, it is an early genetic lesion that is neither necessary nor sufficient to fully induce melanoma. Preclinical work demonstrated that BRAF knockdown reduced tumor formation in murine xenograft models, and selective small molecule inhibitors of RAF suppressed BRAF-mutant melanoma cell lines (Hoeflich et al., 2006; Joseph et al., 2010). These translational studies show that BRAF is a well-validated target, and set the stage for the development of BRAF inhibitors for clinical use.
Early studies targeting RAF employed sorafenib, a multi-kinase inhibitor that has activity against BRAF and CRAF, but resulted in little to no clinical activity as monotherapy (Eisen et al., 2006; Ott et al., 2010). Selective BRAF inhibitors (i.e. those agents that specifically target mutant BRAF over wildtype BRAF), however, demonstrated impressive results in melanoma. The small molecule inhibitors vemurafenib and dabrafenib selectively bind the active conformation of BRAF and inhibit signal transduction between BRAF and MEK. A phase III trial, BRIM-3, of vemurafenib versus dacarbazine as first-line therapy for BRAF V600E mutated metastatic melanoma demonstrated improved median progression free survival (PFS; 5.3 vs 1.6 months) and better overall survival (OS; 84% vs 64%) at 6 months in the vemurafenib versus dacarbazine groups, respectively (Chapman et al., 2011). The most commonly detected toxicities of vemurafenib included cutaneous eruptions, arthralgias, photosensitivity reactions, and cutaneous squamous cell carcinomas that were observed in 26% of patients. These results led to the FDA approval of vemurafenib (Zelboraf) in August 2011 for the treatment of unresectable BRAF V600E mutant melanoma.
Another phase III trial, BREAK-3, compared dabrafenib to dacarbazine in the treatment of patients with unresectable, metastatic, BRAF V600E mutation positive melanoma. BREAK-3 demonstrated similarly impressive results as BRIM-3. Patients in the dabrafenib arm had improved median PFS when compared to those in the dacarbazine arm, 5.1 versus 2.7 months, respectively, with a hazard ratio (HR) for progression of 0.30 (95% CI 0.18 – 0.51; p<0.0001) (Hauschild et al., 2012). However, one important distinction between the 2 trials is that the primary endpoint for BREAK-3 was PFS, whereas the co-primary endpoint for BRIM-3 was PFS and OS. Dabrafenib also demonstrated remarkable efficacy in the treatment of intracranial metastases (Long et al., 2012). Though vemurafenib and dabrafenib appear to have similar efficacy with respect to overall response rates, patients in the vemurafenib trials had higher rates of cutaneous squamous cell carcinomas, 18 – 25%, when compared to those in the dabrafenib trials, 6 – 11% (Chapman et al., 2011; Hauschild et al., 2012). BREAK-3 led to the FDA approval of dabrafenib (Tafinlar) in May of 2013 for the treatment of unresectable melanoma harboring BRAF V600E.
MEK inhibition
Solit et al. reported early pre-clinical results that melanoma sensitivity to MEK inhibition was also correlated with the presence of the BRAF V600E mutation (Solit et al., 2006). Thus, pharmacologic attenuation of MEK signaling represents another possible approach for BRAF-mutated tumors. Exome sequencing of metastatic melanoma specimens identified somatic mutations in MEK1 and MEK2 as potential clinically significant aberrations, characterizing MEK1 and MEK2 mutations in 8% of melanomas (Nikolaev et al., 2012). Moreover, pharmacological MEK blockade completely abrogated tumor growth in BRAF mutant xenografts (Solit et al., 2006). These data provided the rationale for a phase III trial, METRIC, which compared trametinib, a small molecule selective MEK1/2 inhibitor, to chemotherapy (dacarbazine or paclitaxel) in the treatment of patients with BRAF V600E/K mutant positive metastatic melanoma. Compared with patients receiving chemotherapy, patients treated with trametinib demonstrated significant improvement in median PFS (1.5 versus 4.8 months; HR 0.45; 95% CI 0.33 – 0.63; p<0.001) and 6-month OS (67% versus 81%; HR 0.54; 95% CI 0.32 – 0.92; p=0.01), despite being permitted to crossover to trametinib. Though cutaneous eruptions were observed as an adverse effect in 87% of patients, trametinib treatment was minimally associated with the development of cutaneous squamous cell carcinomas. Other toxic effects such as diarrhea and peripheral edema occurred in 35% and 27% of patients, respectively (Flaherty et al., 2012b). Trametinib (Mekinist) gained FDA approval in May 2013 for the first-line treatment of patients with unresectable, BRAF V600E/K mutant positive melanoma.
Combination BRAF and MEK inhibition
Despite the impressive levels of tumor shrinkage observed in BRAF mutant melanoma patients treated with small molecule BRAF inhibitors, responses are typically short-lived with a PFS of approximately 7 months (Chapman et al., 2011; Hauschild et al., 2012). Importantly, molecular studies characterized a number of potential mechanisms of resistance and demonstrated that combined BRAF and MEK inhibition effectively abrogated resistance mediated by MEK1 mutations, BRAF truncation, and acquired NRAS mutations (Paraiso et al., 2010; Poulikakos et al., 2011; Wagle et al., 2011). In order to “over-suppress” MAPK signaling, a phase I/II trial demonstrated that combination dabrafenib and trametinib at full monotherapy doses improved response rate (76% vs 54%, p=0.03) and PFS (10.5 versus 5.6 months; HR 0.39; 95% CI 0.25 – 0.52; p<0.001) when compared to dabrafenib alone, respectively (Flaherty et al., 2012a). Based on these results, both dabrafenib and trametinib received accelerated FDA approval in January of 2014 for use in the treatment of patients with metastatic melanoma with a BRAF V600E/K mutation. Three recent phase III trials corroborated these early results. The combination of vemurafenib and the MEK inhibitor cobimetinib improved PFS (9.9 versus 6.2 months; HR 0.51; 95% CI 0.39 – 0.68; p<0.001) and OR (68% versus 45%; p<0.001) when compared to vemurafenib alone (Larkin et al., 2014). In another expanded study, the combination of dabrafenib and trametinib improved PFS (9.3 versus 8.8 months, HR 0.75; 95% CI 0.57 – 0.99; p=0.03) and OR (67% versus 51%, p = 0.002) when compared to dabrafenib alone (Long et al., 2014). The much anticipated trial of the combination dabrafenib and trametinib demonstrated improved PFS (11.4 vs 7.3 months, HR 0.56, 0.46 – 0.69; p < 0.001) and OS (72% vs 65%, HR 0.69; 0.53 – 0.89, p = 0.005) when compared to vemurafenib as monotherapy (Robert et al., 2014). Consistent with earlier studies, combination treatment led to lower rates of cutaneous squamous cell carcinoma formation compared to BRAF monotherapy. Thus, at this juncture, dual BRAF and MEK inhibition is quickly emerging as a standard-of-care for BRAF V600E mutated melanomas.
Looking forward
Indeed, molecular targeted therapy has proven successful in the treatment of melanoma. Future directions include optimizing the currently available drugs for maximal clinical benefit and also the identification of novel therapeutic targets in melanoma. The above discussion highlights the significant clinical benefit of selective BRAF inhibitors and the eventual relapse that occurs in patients treated with this modality. Combination therapy targeting BRAF and MEK is one potential avenue to abrogate this resistance that has been explored clinically. Work in human melanoma xeno-graft models suggests another approach to delay BRAF inhibitor resistance. These studies demonstrate that vemurafenib-resistant melanoma cells maintain dependency on BRAF V600E signaling via BRAF V600E overexpression. Intriguingly, the vemurafenib-resistant tumors in this model demonstrate dependency on vemurafenib for continued proliferation, such that cessation of the drug leads to tumor regression (Das Thakur et al., 2013). This suggests that a pulsed dosing strategy may forestall eventual vemurafenib-resistant tumors. As novel targeted therapies for melanoma emerge, one key clinical challenge will be developing optimal therapeutic regimens or combinations of therapies that will provide the most durable clinical response.
Inasmuch as the ideal treatment regimen of the existing melanoma drugs are being investigated, therapies targeting novel melanoma targets are in development. Given the genetic diversity of melanoma cells, there exist other attractive therapeutic targets including PI3K, CDK4, ERK, NF1, PPP6C, BCL-2, HSP90, mTOR, PDGF, Notch, MITF, and RAC1. Pre-clinical studies have begun to lay a foundation for further efforts to confirm clinical relevance.
PTEN/PI3K
Molecular aberrations in the PI3K pathway play an important role in the pathogenesis of melanoma (Figure 1). The tumor suppressor PTEN negatively regulates PI3K signaling and has emerged as the dominant genetic target in the PI3K pathway for melanoma therapies. PTEN mutations were found in 40% of melanoma cell lines and 10% of primary melanomas (Guldberg et al., 1997; Tsao et al., 1998b). Moreover, PTEN forced expression of PTEN in PTEN-deficient melanoma tumor cells abrogate activation of the downstream PI3K effector molecule AKT and cell growth (Robertson, 2005). Mouse models have suggested that PTEN dysregulation acts in concert with BRAF V600E to promote melanoma tumorigenesis and that targeting both MEK and the PI3K pathway in these mice inhibits tumor growth (Dankort et al., 2009). These data strongly implicate PTEN and the PI3K pathway in the development of melanoma. Clinical trials targeting the PI3K pathway in combination with inhibiting the MAPK pathway in melanoma are currently underway (Table 2).
Table 2.
Table of combined melanoma targeted therapies in progress.
| Clinical trial number | Description |
|---|---|
| NCT01512251 | BKM120 (PI3K inhibitor) + vemurafenib in BRAF V600E/K advanced melanoma |
| NCT01616199 | PX866 (PI3K inhibitor) + vemurafenib in advanced melanoma |
| NCT01363232 | Safety and pharmacodynamics of BKM120 (PI3K inhibitor) + MEK162 (MEK1/2 inhibitor) in advanced solid tumors |
| NCT01673737 | Phase I/Ib trial of SAR260301 (PI3K inhibitor) +/− vemurafenib in advanced cancers |
| NCT01820364 | LGX818 (RAF inhibitor) + MEK162, BKM120, LEE011, BGJ398, or INC280 in advanced BRAF melanoma |
| NCT02065063 | Safety, anti-cancer activity, and pharmacodynamics of trametinib + palbociclib (CDK4/6 inibitor) in solid tumors |
| NCT01777776 | Safety and efficacy of LEE011 (CDK4/6 inhibitor) + LGX818 (RAF inhibitor) in BRAF melanoma |
| NCT01826448 | Phase1b trial of PLX3397 (Kit inhibitor) + vemurafenib in BRAF melanoma |
| NCT01928940 | Japanese Phase I/II trial of GSK2118436 (dabrafenib) + GSK1120212 (trametinib) in BRAF solid tumors and cutaneous melanoma |
| NCT01433991 | E7050 (cMET + VEGF inhibitor) + E7080 (VEGF inhibitor) in glioblastoma or advanced melanoma |
| NCT01909453 | LGX818 (RAF inhibitor) +/− MEK162 (MEK1/2 inhibitor) vs vemurafenib in BRAF melanoma |
| NCT01701037 | Dabrafenib +/− trametinib before surgery in advanced melanoma that can be removed surgically |
| NCT01562899 | MEK162 (MEK1/2 inhibitor) + AMG479 (IGFR-1 mAb) in solid tumors |
| NCT01519427 | Selumetinib (MEK inhibitor) + MK2206 (AKT inhibitor) in advanced melanoma that failed vemurafenib or dabrafenib |
| NCT01271803 | Vemurafenib + GDC0973 (MEK inhibitor) in BRAF advanced melanoma |
| NCT01380818 | MEK inhibitor + PI3K/mTOR inhibitor in advanced solid tumors |
| NCT01781572 | Phase Ib/II trial of LEE011 (CDK4/6 inhibitor) + MEK162 (MEK1/2 inhibitor) in NRAS melanoma |
CDK4/6
Inappropriate CDK4/6 activity can result from rare activating mutations of CDK4 (Tsao et al., 1998a; Zuo et al., 1996), or more commonly, loss of p16 (Figure 1). Knock-in mice expressing CDK4 mutations display enhanced melanoma in response to carcinogen exposure (Sotillo et al., 2001). Thus, small molecule inhibitors targeting CDK4/6 may also be an effective strategy to target the cell cycle, and several agents are currently in clinical trials (Table 2).
The development of novel targeted therapeutic interventions in the treatment of advanced melanoma demonstrates the importance of translational studies. Characterization of the molecular aberrations present in different subsets of melanoma cells has driven the development of therapies targeted specifically at mutations proven to contribute to melanomagenesis. Pre-clinical and clinical efforts have led to the recent approval of a number of therapeutic agents that have improved the PFS and OS of advanced melanoma patients. Despite these successes, there remains much work to be done to characterize and exploit other molecular targets, to overcome resistance and adverse effects associated with the recently approved agents, and to evaluate how best to combine these new interventions to maximal benefit. The “bench to bedside” model provides an effective and efficient means to achieve these goals.
Acknowledgments
Mentorship during the writing of this work was supported in part by the National Institutes of Health (K24 CA149202 to H.T.).
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. The New England journal of medicine. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Eggermont AM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? European journal of cancer. 2004;40:1825–36. doi: 10.1016/j.ejca.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Eisen T, Ahmad T, Flaherty KT, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. British journal of cancer. 2006;95:581–6. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine. 2012a;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. The New England journal of medicine. 2012b;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- Guldberg P, thor Straten P, Birck A, et al. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer research. 1997;57:3660–3. [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Gray DC, Eby MT, et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer research. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- Joseph EW, Pratilas CA, Poulikakos PI, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14903–8. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. The New England journal of medicine. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Lin WM, Baker AC, Beroukhim R, et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer research. 2008;68:664–73. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. The New England journal of medicine. 2014;371:1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. The Lancet Oncology. 2012;13:1087–95. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- Nikolaev SI, Rimoldi D, Iseli C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nature genetics. 2012;44:133–9. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- Ott PA, Hamilton A, Min C, et al. A phase II trial of sorafenib in metastatic melanoma with tissue correlates. PloS one. 2010;5:e15588. doi: 10.1371/journal.pone.0015588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. British journal of cancer. 2010;102:1724–30. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nature genetics. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. The New England journal of medicine. 2014 doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer metastasis reviews. 2005;24:273–85. doi: 10.1007/s10555-005-1577-9. [DOI] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Garcia JF, Ortega S, et al. Invasive melanoma in Cdk4-targeted mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13312–7. doi: 10.1073/pnas.241338598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Benoit E, Sober AJ, et al. Novel mutations in the p16/CDKN2A binding region of the cyclin-dependent kinase-4 gene. Cancer research. 1998a;58:109–13. [PubMed] [Google Scholar]
- Tsao H, Zhang X, Benoit E, et al. Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene. 1998b;16:3397–402. doi: 10.1038/sj.onc.1201881. [DOI] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Weger J, Yang Q, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nature genetics. 1996;12:97–9. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]