

Abstract
Hypoxia inducible factors (HIF) are the master transcriptional regulators of angiogenesis and energy metabolism in mammals. Histone deacetylase inhibitors (HDAIs) are among the promising anti-cancer compounds currently in clinical trials. In addition to inducing hyperacetylation of histones, HDAIs have been found to repress HIF function, which has been construed as an important pharmacological mechanism underlying the HDAI-mediated repression of tumor growth and angiogenesis. While HDAIs are potent inhibitors of HIF function and thus may be useful in the prevention and treatment of cancers, a major dilemma is that they may induce hyperacetylation of nonspecific targets thus causing side effects. A better understanding is now required of the molecular and biochemical mechanisms underlying the anti-HIF effects of these compounds. Here we summarize the recent advances towards a better understanding of these molecular and biochemical mechanisms.
Keywords: HIF-1, HIF-2, acetylation, deacetylase, HDAC, HDAI, Hsp90, ARD1, VHL, Ubiquitination, p300/CBP, tubulin
HYPOXIA INDUCIBLE FACTORS AND THE CONVENTIONAL REGULATORY PATHWAYS
Hypoxia-inducible factors 1 and 2 (HIF-1 and HIF-2) are the most important regulators of angiogenesis and energy metabolism in tumors, thus having become two of the major targets for cancer therapy.1–5 As heterodimeric transcription factors, function of HIF-1 and HIF-2 is determined by their alpha subunits, HIF-1α and HIF-2α respectively. Collectively, HIF-α activity is controlled by two well known mechanisms6–8 (Fig. 1). The first mechanistic regulation is that HIF-α is rapidly degraded under normoxic conditions through a hydroxylation-ubiquitination-proteasomal system (HUPS). In the presence of an adequate supply of oxygen, newly translated HIF-α is hydroxylated at its oxygen-dependent degradation domain (ODD) by a family of proline hydroxylases (PHD).6–8 Hydroxylated HIF-α is recognized by VHL as a substrate for ubiquitination,9–12 and is degraded by the ubiquitination proteasome system (UPS). Secondly, HIF-α activity is controlled by its transactivation potential (TAP) which depends on formation of a complex including coactivator p300/CBP and other factors. Interaction between the C-terminal activating domain of HIF-α (HIF-αCAD, aa786-826 in HIF-1α, the major transactivation activity) and the CH1 domains of p300/CBP involves a hydrophobic interface and a charge-mediated interaction. Factor inhibiting HIF-1 (FIH), an oxygen-dependent asparagine hydroxylase,13 modifies HIF-1α, hence affecting the hydrophobic interface and disrupting its interaction with p300/CBP.14,15 Lack of oxygen (hypoxia, anoxia), a common pathophysiologic condition frequently associated with neoplastic, cardiovascular, hematologic and respiratory disorders, represses the activity of the hydroxylases and activates HIF function.6 Oxygen and these oxygen-dependent, hydroxylation-triggered events form the conventional regulatory pathways of HIF function (Fig. 1). In tumors, a combination of multiple factors, including hypoxia, growth factors, mitogenic signaling (MAPK, PI3K/Akt), activation of oncogenes and loss of tumor suppressors (VHL, p53, and PTEN), activates HIF-α by acting on various points of the conventional pathways.8,16–20 Thus, it is generally difficulty to repress HIF function in tumors by restoring the conventional regulatory pathways.
Figure 1.
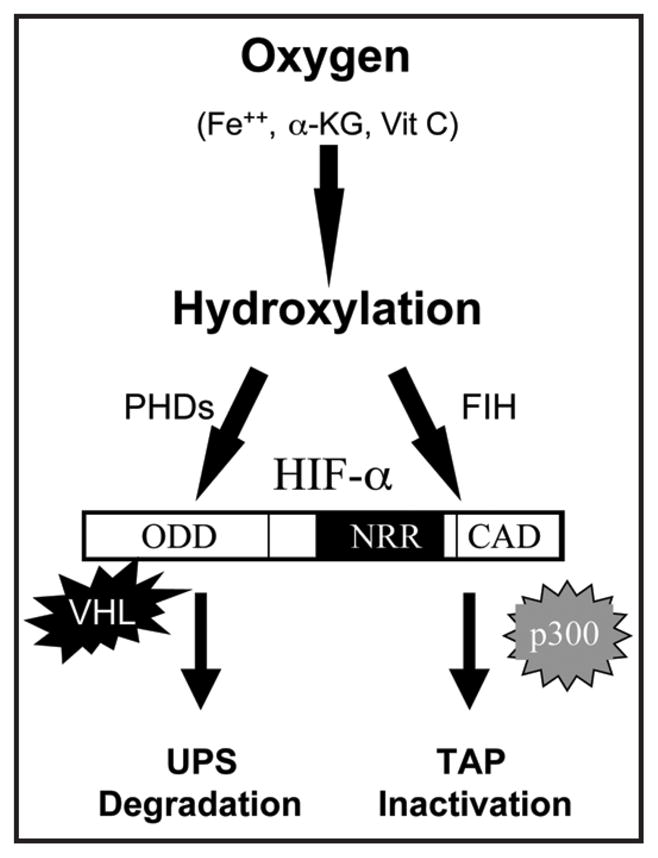
Oxygen-dependent conventional regulatory pathways of HIF-α. In the presence of sufficient amount of oxygen and other substrates or cofactors, PHDs hydroxylate HIF-α at two prolyl sites of ODD (Pro402 and Pro564 in HIF-1α). VHL-containing E3 ubiquitin ligase complex recognizes the hydroxylated prolyl sites and leads to HIF-α degradation through the UPS system. Similarly, FIH catalyzes the hydroxylation of an Asn residue (Asn803 in HIF-1α) in the CAD, which blocks the interaction between HIF-αCAD and p300/CBP.
HDAIs REPRESS ANGIOGENESIS THROUGH HIF
Accumulating evidence supports that HDAIs repress the function of HIF in tumor cells through yet unclear pathways.21–24 HDAIs are in clinical trials for cancer therapy and demonstrate anti-cancer and anti-angiogenic features.25–27 The direct targets of HDAIs, histone deacetylases (HDACs), include a large family of enzymes that remove the acetyl groups from N-ε-lysines of histones, transcription factors, coactivators and other proteins.28–30 Acetylation status of these proteins affects their function and is, in most cases, reversibly regulated by a dynamic balance between histone acetyl transferases (HATs) and HDACs. Exposure to HDAIs represses HDACs, modulates the balance and induces hyperacetylation of these proteins. Generally, HDACs are considered to be repressive factors for gene expression as they are present in transcriptionaly repressive complexes such as SMRT, Sin3 and NCoR.28 On the other hand, HDAIs and HATs are believed to promote transcription by enhancing acetylation of histones, transcription factors and coactivators. Based on genetic homology and phylogenetic analysis, mammalian HDACs are classified into three classes.29,30 Those HDAIs showing anti-HIF activity in clinical trials generally block Class I and II HDACs, while most are not specific for a particular HDAC.31
Trichostatin A (TSA) is among the early HDAIs reported to repress angiogenesis in vitro and in vivo.21 Other HDAIs,25,31 including FK228 (depsipeptide, FR901228),23 butyrate,24 and LAQ82432 were found to repress angiogenesis and expression of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF). While HIF-1 is well documented as the mediator of these observed effects, the explanation of such an effect varies, perhaps because of the pleiotropic nature of HDAIs. Here we discuss two mechanistic explanations that are better supported by available data: (1) HDAI-mediated destabilization of HIF-1α; and, (2) HDAI-mediated repression of the TAP of HIF-αCAD (Fig. 1). We discuss data consistent with or contrary to these two views in more detail. Interested readers are referred to other proposed mechanisms including repressing DNA binding ability23 and inhibiting nuclear translocation of HIF-1α24,33 for a complete view.
HDAIs PROMOTE VHL-INDEPENDENT PROTEASOMAL DEGRADATION OF HIF-1α
An early report describing the anti-angiogenic effect of TSA suggested that TSA destabilized HIF-1α.21 Indeed, when degradation was completely blocked by proteasome inhibitors, a comparison of the rate of de novo HIF-1α accumulation in the presence or absence of TSA reveals that TSA has no effect on HIF-1α translation.34 The generally agreed fact that HDAIs destabilize HIF-1α indicates that a deacetylation event is required to stabilize HIF-1α. Perhaps influenced by the documented role of VHL and p53 in the degradation of HIF-1α and the effect of HDAIs in stimulating transcription, the HDAI-mediated destabilization of HIF-1α was originally explained as a result of enhanced expression of VHL and p53.21 Later the same group reported that direct acetylation of Lys532 of HIF-1α by ARD122 promoted its interaction with, and ubiquitination by VHL,22 proposing an acetylation-ubiquitination proteasomal system (AUPS) (Fig. 2). Since ARD1 was reported to be regulated by oxygen supply,22 if proven, ARD1 would represent another VHL-dependent oxygen sensor, mimicking PHDs in the HUPS pathway. While HDAI-mediated destabilization of HIF-1α and repression of angiogenesis are consistently observed by several independent laboratories, it seems difficult to confirm the HDAI-enhanced expression of VHL and p53 under hypoxia conditions as originally reported.34 More importantly, TSA and FK228 were found to induce HIF-1α degradation in VHL-null RCC4 cells,35 suggesting a VHL-independent mechanism. Using 8-hour exposure with 500 nM of TSA, we also observed that TSA decreased the hypoxia-induced HIF-1α levels in most tumor cell lines tested, including Caki, Hep3B, DU145, PC3, U87, BT20, MCF7, and particularly, VHL−/− cells such as RCC4 and C2.34 Similarly, TSA repressed HIF-1α levels in HCT116 cells (p53+/+) and an isogenic p53−/− HCT116-derived cells.34,36 These observations suggest that HDAI-mediated destabilization of HIF-1α is through a mechanism existing in various tumors and is independent of VHL and p53 function.
Figure 2.

Schematic outline of the HUPS and AUPS pathways of HIF-1α degradation. In the AUPS pathway, ARD1 is proposed to acetylate Lys532, and such modification leads to VHL recognition. Both HUPS and AUPS are dependent on functional E1, E2 and VHL for ubiquitination.
HDAIs PROMOTED PROTEASOMAL DEGRADATION OF HIF-1α IS INDEPENDENT OF HYDROXYLATION-UBIQUITINATION
The ubiquitination proteasomal system (UPS) plays essential roles in intracellular degradation of misfolded, toxic or no longer needed cellular proteins. While HDAI-triggered degradation of HIF-1α is clearly independent of VHL function,34,35 and lack of hydroxylation failed to protect HIF-1α from HDAI-mediated degradation, HDAIs failed to decrease HIF-1α levels in the presence of proteasome inhibitors, indicating that proteasomal activity is indispensable for HDAI-mediated HIF-1α degradation. As mentioned above, HIF-1α accumulated by VHL deficiency can be destabilized by HDAIs. It seems unlikely that HDAIs stimulate an alternative E3 activity that compensates for VHL deficiency in these VHL−/ cells, because exposure to HDAIs did not induce ubiquitination of HIF-1α in these cells. More importantly, HDAIs effectively destabilized HIF-1α accumulated by inactivation (39°C) of the ubiquitin-activating enzyme (E1) in Ts20TGR cells34 which contains a temperature-sensitive E1,37 the only ubiquitin-activating enzyme in cells. In addition, HDAI treatment failed to cause ubiquitination of HIF-1α or other proteins in E1-inactivated cells,34 ruling out the possibility that HDAIs somehow restore the ubiquitination process, and demonstrating that E1 is absolutely required for protein ubiquitination in Ts20TGR cells.
ACETYLATION TARGETS INVOLVED IN HDAI-INDUCED DESTABILIZATION OF HIF-1α
Direct acetylation of HIF-1α at Lys532 was proposed in the AUPS model as the underlying reason for HDAI-mediated destabilization of HIF-1α21,22 (Fig. 2). So far the only evidence to support HIF-1α acetylation in vivo is detection of HIF-1α in immunocomplexes precipitated by an anti-acetyl lysine antibody.22 Those results, however, could be interpreted alternatively as an acetylated protein coprecipitating with HIF-1α. It has been demonstrated that at least two HIF-1α interacting proteins, p300 and heat shock protein 90 (Hsp90), are subject to acetylation in vivo.34,38–41 It remains unclear whether full-length HIF-1α is acetylated in vivo, and if yes, its role in HIF-1α stability. In any case, this proposed model can not explain HDAI-mediated, VHL-independent and ubiquitination-independent degradation of HIF-1α.
While the acetylated targets underlying HDAI-mediated destabilization of HIF-1α remains a debatable issue, studies with Hsp90 inhibitors revealed a good candidate. Hsp90 inhibitors represent another group of compounds in clinical trials with good anti-cancer efficacy and anti-angiogenic features.42 Hsp90 is a central component of the cellular chaperone machinery which is responsible for the folding, maturation and quality control for its client proteins including various oncogenic signaling proteins.43 Normal function of Hsp90 depends on its intrinsic ATPase activity. Geldanamycin and its derivatives bind to Hsp90 directly, thus blocking its association with ATP. It was observed that when geldanamycin or its derivatives were used to block Hsp90 function, some oncogenic proteins, including viral oncoproteins, activated cellular oncoproteins and mutated tumor suppressors were destabilized, suggesting a general role of Hsp90 in stabilizing its client proteins.40,42,43 Interestingly, HIF-1α was found to bind Hsp90,44,45 and inhibition of Hsp90 function leads to VHL-independent destabilization of HIF-1α.44 When examined in Ts20TGR cells, the destabilization of HIF-1α caused by Hsp90 inhibitors also is independent of E1 function,34 suggesting a common ubiquitination-independent mechanism shared by both HDAIs and Hsp90 inhibitors. In fact, it was reported that Hsp90 is one of the nonhistone targets of HDAC6; and HDAI-induced hyperacetylation of Hsp90 repressess its chaperone function.40,41,46 Therefore, it seems reasonable to speculate that the normal chaperone function of Hsp90 protects its client proteins from degradation. Inactivation of Hsp90 renders its client proteins to be degraded by a ubiquitination-independent proteasomal system (UIPS). Consistent with this speculation, butyrate which inhibits both Class I and Class II HDACs except for HDAC6 shows little effect on HIF-1α stability (Fig. 3A). Moreover, while TSA destabilizes HIF-1α accumulated in VHL−/− cells, in the presence of a translation blocker, TSA did not shorten the half life of HIF-1α accumulated prior to the addition of TSA (Fig. 3B), suggesting that the stability of nascent HIF-1α translated in the presence of HDAIs is affected. A UIPS has been reported to degrade p53, ODC and other proteins through the 20 S proteasome.47–49 Interestingly, it has been reported that HIF-1α directly interacts with PMSA7, a component of the 20 S protea-some.50 Contrary to this speculation, several reports described that HDAIs promote degradation of ubiquitinated client proteins of Hsp90.44 No evidence, however, supports that ubiquitination per se is absolutely required for degradation of these client proteins. It is possible that in the presence of functional Hsp90, its client proteins are adequately folded, thus being protected from degradation unless ubiquitinated. Upon inhibition of Hsp90, its client proteins may not assume the protective folding, so that both UPS and UIPS are engaged in the degradation of those misfolded, wasteful, and sometimes even toxic proteins (Fig. 4).
Figure 3.

Additional evidence to support the HDAC6-Hsp90 model. (A) Hep3B cells were cultured at normal condition (Nmx: normoxia), or exposed to a hydroxylase inhibitor (Dfo, desferioxamine, 100 μM). Whole cell lysates were prepared and the effects of TSA (600 nM), sodium butyrate (NaB, 2.5 mM) and suberoylanilide hydroxamic acid (SAHA, 2.5 μM) on HIF-1α stability were examined by Western blot. (B) VHL−/− RCC4 cells were incubated with DMSO or TSA (600 nM). In the absence of a translation blocker, the HIF-1α levels represent a balance between gradual degradation and de novo synthesis. Addition of TSA broke this balance and caused reduced HIF-1α levels. In the presence of a translation blocker (CHX: Cyclohexamide, 20 μg/ml), however, TSA did not expedite the degradation of HIF-1α synthesized prior to the treatment.
Figure 4.

Schematic outline of the HUPS and the UIPS. Hsp90-containing chaperone function is proposed to be required for the folding and maturation of nascent HIF-α. Upon maturation, correctly folded HIF-α is regulated by the VHL-dependent HUPS pathway. When Hsp90 function is inhibited either by HDAIs or Hsp90 inhibitors, nascent HIF-α cannot be folded correctly, and the misfolded HIF-α may be disposed either by the UIPS pathway or the UPS pathway. The ubiquitination of misfolded HIF-α may be independent of hydroxylation or VHL, the HIF-α specific ubiquitin ligase.
Another acetylation candidate possibly involved in HDAI-induced destabilization of HIF-α is the α subunit of the tubulin heterodimers (α-tubulin), which form microtubules and other cellular structures with a variety of function. α-tubulin is a target of HDAC6 and its acetylation at Lys40 is the marker of stabilized microtubules.51,52 Small molecules disrupting the dynamics of tubulins have been shown to affect the stability of HIF-1α.53,54 However, a defined role of α-tubulin acetylation in either UPS or UIPS-mediated degradation of HIF-1α remains unclear.
It is not clear whether the UIPS pathway is specifically triggered/enhanced by HDAIs, but it seems to be part of a quality control system of protein synthesis which safeguards the cell by responding to stress caused by misfolded proteins.
HISTONE DEACETYLASE INHIBITORS REPRESS THE TRANSACTIVATION POTENTIAL OF HIF-α
Early reports ascribed the repressive effects of HDAIs on HIF transcriptional activity exclusively to destabilizing HIF-α or changing its nuclear localization. A recent intriguing observation is that HDAIs also repress the TAP of the major transactivation domain of HIF-α.38 We recently reported that low doses of HDAIs that were not sufficient to cause HIF-1α degradation were sufficient to repress HIF-1α transactivation potential under both normoxic and hypoxic conditions.38 This effect can be clearly demonstrated by using a recombinant HIF-αCAD construct fused to the DNA binding domain of the yeast GAL4 transcription factor. The protein levels of this fusion protein are not decreased by HDAIs, allowing the examination of its activity by monitoring the expression of a reporter gene.38 All other transactivators tested in the same way, including p300, VP16, MyoD and p53, were enhanced by HDAIs under the same conditions. This is particularly interesting because it shows the uniqueness of HIF-αCAD.
HDAIs repress HIF-1α-p300CH1 function independently of hydroxylation, because mutation of Asn803 of HIF-1α to Ala does not abolish HDAI-mediated repression.38 In addition, FIH levels were not affected by HDAIs. Furthermore, a minimal CAD domain (HIF-1α786-826) lacks the normoxic repressive region; and, thus being constitutively active15 can be repressed by HDAIs, so the HDAI-mediated repression is distinct from the oxygen-mediated repression. Similar to the destabilization, the HDAI-mediated repression of HIF-α potential is independent of VHL or p53.38 Because the highly conserved, constitutively active HIF-1αCAD (aa786-826) is repressed by HDAIs but contains no lysyl residue, it is clear that the HDAIs repress HIF-αCAD by somehow targeting the HIF-αCAD/p300 complex, not HIF-αCAD itself.
Finally, HIF-1α possesses a p300/CBP CH1-independent trans-activating mechanism which is sensitive to HDAIs.55,56 Because HIF-αCAD has been demonstrated to be absolutely dependent on p300/CBP CH1,55 the p300/CBP CH1-independent mechanism might implicate the N-terminal transactivation domain (NAD) of HIF-α. Indeed, we found that HDAIs repress HIF-1αNAD in a similar dose-dependent manner. So it is likely that HDAIs repress both NAD and CAD of HIF-α.
POSSIBLE MECHANISMS INVOLVED IN HDAI-MEDIATED REPRESSION OF HIF-αCAD
The fact that HDAIs may alter the acetylation status of both histones and nonhistone proteins complicates further dissection of underlying mechanism. Because HDAIs may repress HIF-1αCAD without changing of nuclear localization of HIF-1α, a hyper-acetylation event in nuclei might be involved. As discussed above, it is clear that no direct acetylation of HIF-αCAD can explain the HDAI-mediated repression of its TAP. Based on analysis of published and unpublished data, several possibilities, but neither exclusive nor exhaustive are proposed.38 First of all, HDAIs can enhance the acetylation status of p300 in vivo, and p300CH1 can be acetylated by p300HAT in vitro, suggesting a direct role of p300CH1 acetylation in HIF-1α/p300 function. One possibility is that such acetylation directly affects the dynamics of HIF-1α/p300 complex formation. Supporting this model, three highly conserved Lysyl residues of CH1 have been proposed to be essential for the HIF-αCAD/p300 interaction.57,58 Alternatively, acetylation of p300 (CH1 and other regions) or p300-interacting factors may enhance their interactions, thus competitively sequestrating p300 to non-HIF proteins. It is also possible that HDAIs promote an inhibitory factor, either by directly hyperacetylating this factor or by increasing its expression via histone acetylation, to target the HIF-αCAD/p300 complex (Fig. 5). The observation that proteasomal inhibitors increase HIF-1α levels but repress its TAP59,60 is consistent with this model, in which a HIF-1α inhibitor is constitutively synthesized and needs to be actively disposed by proteasomes.
Figure 5.
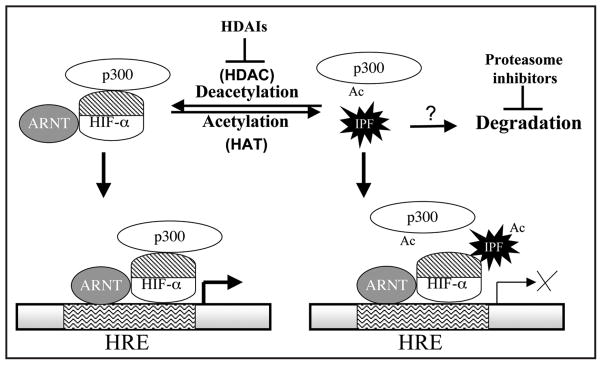
Possible molecular basis underlying the HDAI-mediated repression of HIF-αCAD TAP. A deacetylation event is proposed to be essential for the function of HIF-αCAD/p300 complex. The addition of HDAIs blocks the deacetylation event and causes the hyperacetylation of an inhibitory protein factor (IPF) or p300. The eventual consequence would be a change in the formation of HIF-αCAD/p300 complex. Proteasome inhibitors may enhance the levels of IPF, thus repressing HIF function in a similar model. ARNT: aryl hydrocarbon receptor nuclear translocator, the dimerization partner of HIF-α, also known as HIF-1β.
THE DEACETYLASES AND ACETYLASES INVOLVED IN HIF-α FUNCTION
In the AUPS model (Fig. 2), HIF-1α directly undergoes acetylation and deacetylation, and such modification involves HDAC1 (HIF-α deacetylase) and ARD1 (acetylase), respectively.21,22 Supporting that model, it was reported that hypoxia stimulates expression of HDAC1, which in turn, stabilizes HIF-1α. A recent report described that hypoxia induced expression of MTA1 (metastasis associated protein 1), which interacts with and causes deacetylation of HIF-1α by increasing the levels of HDAC1.61 We found that HDAC1 is not stimulated by hypoxia, and disrupting HDAC1 is not sufficient to destabilize HIF-1α under conditions where the HUP pathway is repressed (38, and unpublished observation). Data from Kim’s group indicate that ARD1, the mammalian orthologue of a yeast N-α-acetylase,62 catalyzes N-ε-acetylation of HIF-1α at Lys532, promoting HIF-1α for recognition and ubiquitination by VHL.22 The same report described that TSA (about 1000 nM) destabilized HIF-1α in HT1080 cells by inducing hyperacetylation of HIF-1α. However, because mutation of Lys532 to Arg failed to abolish the HDAI-induced HIF-1α degradation, we believe that Lys532 acetylation, if it does happen in vivo, is unlikely involved in HDAI-triggered destabilization of HIF-1α.34 Moreover, two independent groups found that ARD1 could not acetylate Lys532 in HIF-1α in vitro,63,64 which is inconsistent with the role of ARD1 as a bona fide N-ε-acetylase of HIF-1α. Functionally, ARD1 expression status has been shown to have no effect on the expression of either HIF-1α or HIF-regulated genes.65,66 The recent identification of ARD1 variants in mice67 may not provide an explanation, because the siRNA used65,66 should disrupt all ARD1 variants. Whether ARD1 cotranslationally modifies the N-termini of nascent HIF-1α (N-α-acetylation) remains to be investigated. On the other hand, considering the HDAC6-Hsp90 model,34 the acetylase involved should be one that acetylates Hsp90. Identification of the acetylation site of Hsp90 and the responsible acetylase would eventually confirm the mechanistic involvement of Hsp90 in HDAI-induced degradation of HIF-1α and other client proteins.
The acetylase and deactylase involved in the HDAI-mediated repression of TAP of HIF-αCAD remain unclear. HDAC6 seems not to be the major player in the HDAI-mediated repression of HIF-1αCAD,38 because butyrate does not efficiently inhibit HDAC6 is equally effective in repressing HIF-1αCAD. However, HDAC1 may play a role in HIF-1α TAP, because cotransfection of HIF-1αCAD with HDAC1 increased TAP. Supporting this model, HDAC1 has been reported to interact with p300.68 The report that HDAC7 interacts with a repressive domain of HIF-1α and regulates the subcellular translocation of HIF-1α upon hypoxic stimulation33 raises the possibility that HDAC7 may regulate its TAP. However, the facts that HDAIs repress HIF-1αCAD independently of direct HDAC7 association, and under certain conditions HDAIs can repress HIF-1 function without changing its protein levels or nuclear localization38 support the idea that other HDACs are involved in this regulation. Knowledge of the acetylated protein involved will facilitate identification of the acetylase and deacetylase responsible for regulation of HIF-αCAD activity.
ACETYLATION IN OXYGEN SENSING
Albeit the destabilizing and repressing effects of HDAIs on HIF-1α, a role for acetylation in oxygen sensing is unlikely. The AUPS model indicates that hypoxia stimulates expression of HDAC1 and ARD1, implying a role for acetylation in oxygen sensing.21,22,61 We and others observed that hypoxia or hypoxic mimics do not notably affect levels of ARD1 or HDAC1.38,63,66 Similarly, so far there is no evidence to support that HDAC6 activity is regulated by oxygen.34 Since HIF-1αCAD (aa786-826) can interact with p300/CBP efficiently and is constitutively active under normoxic conditions,15 it is reasonable to believe that HIF-αCAD-stimulating deacetylase maintains a considerable level of activity even under normoxic conditions. Therefore, while deacetylase activity is absolutely required for the activity of HIF-αCAD, it is unlikely that it is an oxygen sensor. It is more likely that the acetylation-mediated modulation of HIF function represents a novel signaling pathway. Nevertheless, the levels of deacetylase activity in cells may affect the magnitude of the HIF response upon HIF-stimulating signaling.
In conclusion, HDAIs inhibit HIF activity by destabilizing HIF-α and repressing its TAP. The HDAI-induced repressive pathways are distinct from the oxygen-dependent conventional regulatory pathways. The novel pathways may be explored as targets for modulation of HIF function in vivo.
Acknowledgments
We thank Drs. J. Caro and S Surrey for helpful discussion and critical reviewing of the manuscript before submission, and Ms. Y Jiang and Z Lin for technical assistance. Research in Dr. Sang’s laboratory is supported by grants from NIH (CA098809) and the W.W. Smith Charitable Trust (C#0505).
References
- 1.Welsh SJ, Powis G. Hypoxia inducible factor as a cancer drug target. Curr Cancer Drug Targets. 2003;3:391–405. doi: 10.2174/1568009033481732. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 3.Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nat Rev Drug Discov. 2003;2:803–11. doi: 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- 4.Powis G, Kirkpatrick L. Hypoxia inducible factor-1α as a cancer drug target. Mol Cancer Ther. 2004;3:647–54. [PubMed] [Google Scholar]
- 5.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 6.Lando D, Gorman JJ, Whitelaw ML, Peet DJ. Oxygen-dependent regulation of hypoxia-inducible factors by prolyl and asparaginyl hydroxylation. FEBS J. 2003;270:781–90. doi: 10.1046/j.1432-1033.2003.03445.x. [DOI] [PubMed] [Google Scholar]
- 7.Semenza GL. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiol. 2004;19:176–82. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 8.Kaelin WG., Jr The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338:627–38. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- 9.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 10.Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA. 2001;98:9630–5. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 12.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim AV, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 13.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 15.Sang N, Fang J, Srinivas V, Leshchinsky I, Caro J. Carboxyl terminal transactivation activity of HIF-1α is governed by VHL-independent, hydroxylation regulated association with p300/CBP. Mol Cell Biol. 2002;9:2984–92. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 17.Blagosklonny MV, An WG, Romanova LY, Trepel J, Fojo T, Neckers L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J Biol Chem. 1998;273:11995–8. doi: 10.1074/jbc.273.20.11995. [DOI] [PubMed] [Google Scholar]
- 18.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 19.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK Signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–9. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–6. [PMC free article] [PubMed] [Google Scholar]
- 21.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nature Med. 2001;7:437–43. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 22.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell. 2002;111:709–20. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 23.Lee YM, Kim SH, Kim HS, Son MJ, Nakajima H, Kwon HJ, Kim KW. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1α activity. Biochem Biophys Res Commun. 2003;300:241–6. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 24.Zgouras D, Wächtershäuser A, Frings D, Stein J. Butyrate impairs intestinal tumor cell-induced angiogenesis by inhibiting HIF-1α nuclear translocation. Biochem Biophys Res Commun. 2003;300:832–8. doi: 10.1016/s0006-291x(02)02916-9. [DOI] [PubMed] [Google Scholar]
- 25.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477–83. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Ann Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 27.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell. 2003;4:13–8. doi: 10.1016/s1535-6108(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 28.Ahringer J. NuRD and SIN3-histone deacetylase complexes in development. TIG. 2000;16:351–5. doi: 10.1016/s0168-9525(00)02066-7. [DOI] [PubMed] [Google Scholar]
- 29.Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143–53. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- 30.Yang XJ, Gregoire S. Class II histone deacetylases: From sequence to function, regulation and clinical implication. Mol Cell Biol. 2005;25:2873–84. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minucci S, Pelicci PG. histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 32.Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, Atadia P, Pili R. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64:6626–34. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 33.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004;279:41966–74. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 34.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demidenko ZN, Rapisarda A, Garayoa M, Giannakakou P, Melillo G, Blagosklonny MV. Accumulation of hypoxia-inducible factor-1alpha is limited by transcription-dependent depletion. Oncogene. 2005;24:4829–38. doi: 10.1038/sj.onc.1208636. [DOI] [PubMed] [Google Scholar]
- 36.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW. arrest after DNA damage. Science Vogelstein B. Requirement for p53 and p21 to sustain G2. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 37.Chowdary DR, Dermody JJ, Jha KK, Ozer HL. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fath DM, Kong X, Liang D, Lin Z, Chou A, Jiang Y, Fang J, Caro J, Sang N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J Biol Chem. 2006;281:13612–9. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovacs JJ, Murphy PJM, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 40.Blagosklonny MV, Trostel S, Kayastha G, Demidenko ZN, Vassilev LT, Romanova LY, Bates S, Fojo T. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res. 2005;65:7386–92. doi: 10.1158/0008-5472.CAN-04-3433. [DOI] [PubMed] [Google Scholar]
- 41.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 42.Isaacs JS. Heat-shock protein 90 inhibitors in antineoplastic therapy: Is it all wrapped up? Expert Opin Investig Drugs. 2005;14:569–89. doi: 10.1517/13543784.14.6.569. [DOI] [PubMed] [Google Scholar]
- 43.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–51. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 44.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem. 2002;277:29936–44. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 45.Katschinski DM, Le L, Schindler SG, Thomas T, Voss AK, Wenger RH. Interaction of the PAS B domain with HSP90 accelerates hypoxia-inducible factor-1alpha stabilization. Cell Physiol Biochem. 2004;14:351–60. doi: 10.1159/000080345. [DOI] [PubMed] [Google Scholar]
- 46.Yu X, Guo ZS, Marcu MG, Nechers L, Nguyen DM, Chen GA, Schrump DS. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in Lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504–13. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 47.Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor-suppressors p53 and p73. Genes Dev. 2005;19:316–21. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asher G, Bercovich Z, Tsvetkov P, Shaul Y, Kahana C. 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol Cell. 2005;17:645–55. doi: 10.1016/j.molcel.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 50.Cho S, Choi YJ, Kim JM, Jeong ST, Kim JH, Kim SH, Ryu SE. Binding and regulation of HIF-1alpha by a subunit of the proteasome complex, PSMA7. FEBS Lett. 2001;498:62–6. doi: 10.1016/s0014-5793(01)02499-1. [DOI] [PubMed] [Google Scholar]
- 51.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Mattias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–79. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mabjeesh N, Escuin D, Lavallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–72. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 54.Escuin D, Kline ER, Giannakakou P. Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1alpha accumulation and activity by disrupting microtubule function. Cancer Res. 2005;65:9021–8. doi: 10.1158/0008-5472.CAN-04-4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005;24:3846–58. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kasper LH, Brindle PK. Mammalian gene expression program resiliency: The roles of multiple coactivator mechanisms in hypoxia-responsive transcription. Cell Cycle. 2006;5:142–6. doi: 10.4161/cc.5.2.2353. [DOI] [PubMed] [Google Scholar]
- 57.Dames SA, Martinez-Yamout M, DeGuzman RN, Dyson HJ, Wright PE. Structural basis for HIF-1/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA. 2002;99:5271–6. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freedman SJ, Sun ZYJ, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1. Proc Natl Acad Sci USA. 2002;99:5367–72. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salceda S, Caro J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. J Biol Chem. 1997;272:22642–7. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 60.Kaluz S, Kaluzova M, Stanbridge EJ. Proteasomal inhibition attenuates Transcriptional activity of hypoxia-inducible factor 1 (HIF-1) via specific effect on the HIF-1alpha C-terminal activation domain. Mol Cell Biol. 2006;26:5895–907. doi: 10.1128/MCB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. 2006;25:1231–41. doi: 10.1038/sj.emboj.7601025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park EC, Szostak JW. ARD1 and NAT1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 1992;11:2087–93. doi: 10.1002/j.1460-2075.1992.tb05267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arnesen T, Kong X, Evjenth R, Gromyko D, Vargaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. Interaction between HIF1α (ODD) and hARD1 does not induce acetylation and destabilization of HIF1α. FEBS Lett. 2005;579:6428–32. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murray-Rust TA, Oldham NJ, Hewitson KS, Schofield CJ. Purified recombinant hARD1 does not catalyse acetylation of Lys532 of HIF-1alpha fragments in vitro. FEBS Lett. 2006;580:1911–8. doi: 10.1016/j.febslet.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 65.Fisher TS, Des Etages S, Hayes L, Crimin K, Li B. Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J Biol Chem. 2005;280:17749–57. doi: 10.1074/jbc.M412055200. [DOI] [PubMed] [Google Scholar]
- 66.Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF-1alpha) and is not induced by hypoxia or HIF. J Biol Chem. 2005;280:31132–40. doi: 10.1074/jbc.M504482200. [DOI] [PubMed] [Google Scholar]
- 67.Kim SH, Park JA, Kim JH, Lee JW, Seo JH, Jung BK, Chun KH, Jeong JW, Bae MK, Kim KW. Characterization of ARD1 variants in mammalian cells. Biochem Biophys Res Commun. 2006;340:422–7. doi: 10.1016/j.bbrc.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 68.Simone C, Stiegler P, Forcales SV, Bagella L, De Luca A, Sartorelli V, Giordano A, Puri PL. Deacetylase recruitment by the C/H3 domain of the acetyltransferase p300. Oncogene. 2004;23:2177–87. doi: 10.1038/sj.onc.1207327. [DOI] [PubMed] [Google Scholar]