

Abstract
Hypoxia inducible factor-1α (HIF-1α) is a central component of the cellular responses to hypoxia. Hypoxic conditions result in stabilization of HIF-1α and formation of the transcriptionally active HIF-1 complex. It was suggested that mammalian ARD1 acetylates HIF-1α and thereby enhances HIF-1α ubiquitination and degradation. Furthermore, ARD1 was proposed to be downregulated in hypoxia thus facilitating the stabilization of HIF-1α. Here we demonstrate that the level of human ARD1 (hARD1) protein is not decreased in hypoxia. Moreover, hARD1 does not acetylate and destabilize HIF-1α. However, we find that hARD1 specifically binds HIF-1α, suggesting a putative, still unclear, connection between these proteins.
Keywords: Protein acetylation, Acetyltransferase, Hypoxia inducible factor-1α, Hypoxia, Human ARD1, Degradation
1. Introduction
Survival of mammalian cells is dependent on the adequate supply of oxygen and nutrients which are provided by a well organized vascular network. Precise homeostatic control mechanisms continuously adjust the blood flow to maintain adequate oxygen tension, pH and appropriate concentrations of glucose and other nutrients [1]. This balance is lost in solid tumours, where the process of tumour expansion exceeds the development of blood vessels resulting in a hypoxic microenvironment [2,3]. Successful tumour progression depends on biochemical adaptation and genetic alterations that will promote tumour survival despite this unfavourable microenvironment [4]. Central to the adaptation of cells to hypoxia is the activation of the transcriptional complex HIF-1 (hypoxia-inducible complex-1) [5]. HIF-1 controls the transcriptional response of a plethora of genes involved in angiogenesis and glucose metabolism that tend to ameliorate the damaging effects of hypoxia and promote tumour survival [6,7]. The HIF complex is composed of an oxygen-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit [8]. In normoxic conditions the HIF-1α protein is degraded by the ubiquitinproteasome system in a process that involves its interaction with VHL (Von Hippel Lindau), a tumour suppressor protein that acts as a ubiquitin-ligase (E3) [9,10]. HIF-1α/VHL interactions depend on the oxygen-mediated hydroxylation of prolyl-residues in the oxygen degradation domain of HIF-1α (HIF-ODD). In the presence of oxygen, HIF-1α is hydroxylated at prolyl-residues P402 and P564 by specific oxygen-and iron-dependent hydroxylases (PHDs), thus promoting HIF-1α ubiquination and degradation [11]. During hypoxia, HIF-1α is not degraded and interacts with HIF-1β to form a transcriptionally active DNA-binding complex. Although hypoxia is the main regulator of HIF-1α, an array of factors including cytokines, growth factors and oncogenic mutations have also been shown to induce HIF-1α and control HIF-1 complex formation. These factors appear to have an important role in the non-hypoxic expression of HIF-1α in tumours and inflammatory responses [12,13]. The mechanisms of HIF-1α induction in these conditions is not clear and may involve other post-translational modifications of the HIF-1α protein like phosphorylation or acetylation.
One of the proposed non-hypoxic regulators of HIF-1α is the acetyltransferase hARD1 [14]. hARD1 was shown to acetylate the ε-amino group of lysine residue K532 within the ODD domain of HIF-1α, enhancing the binding of pVHL and the subsequent proteasomal degradation of HIF-1α. During hypoxia, hARD1 mRNA was found to decrease and the interaction between HIF-1α and hARD1 was diminished. Thus, the conclusion was made that in hypoxia the hARD1 mediated negative regulation was reduced and stabilization of HIF-1α was allowed. hARD1 has been described to be a subunit of a protein N-α-acetyltransferase complex [15]. The two proteins constituting the enzyme complex, hARD1 and NATH, were found to interact with ribosomes, probably facilitating the acetylation of the α-amino groups of newly synthesized proteins. However, a fraction of hARD1 was also found to be localized in the nucleus where partial colocalization with HIF-1α was observed [15].
In Saccharomyces cerevisiae, Nat1p forms a heterodimer with Ard1p to generate a functional protein N-acetyl transferase (NatA) [16,17], Ard1p being the catalytic subunit. Proteins with Ser-, Thr-, Gly-, or Ala-N-termini are potential substrates of NatA [18]. NatA was found to directly interact with nascent polypeptides as well as ribosomes through binding of Nat1p to the large ribosomal subunit [19]. From an evolutionary point ARD1 may have evolved from an α-amino acetyltransferase to an ε- and α-amino acetyltransferase, although this is perhaps not a very likely event. Alternatively, this dual function could be present also in S. cerevisiae although no such activity has been described.
Here we provide evidence that contradicts previous reports on the role of ARD1 in the regulation of the HIF-1α. Our results indicate that hypoxia does not regulate the level of endogenous hARD1 protein, and that neither overexpression nor silencing of hARD1 affects the stability of HIF-1α. However, we did find a specific interaction between HIF-1α and hARD1, suggesting that a functional link may exist between these two proteins.
2. Materials and methods
2.1. Cell culture and transfection
The human cell lines HEK293 cells (embryonal kidney, ATCC: CRL-1573), HT1080 (fibrosarcoma), RCC4 (renal carcinoma) and HeLa cells (epithelial cervix adenocarcinoma, ATCC: CCL-2) were cultured at 37 °C, 5% CO2 in DMEM supplemented with 10% FBS and 3% l-glutamine. MCF-7 cells (breast adenocarcinoma, ATCC: HTB-22) were cultured as above, but with RPMI1640. Transfections were performed using Fugene6 (Roche) or Lipofectamine (Invitrogen) according to the instruction manuals. Hypoxia (1% O2) was conducted in an oxygen workstation (InVivoO2, Ruskin Technologies) and hypoxia mimicking agents were added at concentrations of 200 μM for CoCl2 and 150 μM for desferroxamine. Plasmid encoding HA-HIF-1α has been described [20]. Plasmid encoding Xpress-hARD1 was made by subcloning hARD1 cDNA to pcDNA4 MaxHis (Invitrogen). siRNAs were from Dharmacon and transfections were performed using Oligofectamine (Invitrogen) according to the instruction manual. The following siRNA duplex target sequences were used: si-hARD1 (siA1), CCA GAU GAA AUA CUA CUU C and siLamin A/C (siL), GGU GGU GAC GAU CUG GGC U.
Tet-inducible ARD1. A tetracycline inducible ARD1 cell line was established using a modified BD™ Tet-on system from BD-Bioscience. Briefly, HeLa cells (BD cat # 630901) expressing the reverse Tet-repressor (rTetR) were transfected with a pCEP4 expression vector containing the FLAG-hARD1 cDNA under the control of the tetracycline responsive element (TRE) and a minimal CMV promoter, as described [21]. Following selection with G418, the HeLa hARD1 cells were cultured in low Tet-FCS and induced with 1 μg Dox for 48 h before use. Control cells were created by transfection of rTetR-HeLa with a similar pCEP4 Tet-responsive plasmid (TRE-Luc) expressing luciferase.
2.2. Western blotting
Western blotting was performed as previously described [22]. In brief, 12% SDS–PAGE was used for hARD1 and β-tubulin detection, while 7.5% gels were utilized for detection of HIF-1α. Antibody-dilutions: anti-NATH 1:500, anti-hARD1 1:500, anti-V5 (Invitrogen) 1:1000, anti-Xpress (Invitrogen) 1:1000, anti-HIF-1α (Pharmigen) 1:500, anti-β-tubulin (Sigma) 1:1000 (loading control), anti-Acetyl-Lysine (Sigma) 1:500, anti-Acetyl-Lysine (Upstate) 1:1000. Horseradish peroxidase-conjugated anti-mouse and anti-rabbit IgG were from Amersham Lifescience, UK.
2.3. Immunoprecipitation
Immunoprecipitation was performed as described [15]. Approximately 2 × 106 cells were used per experiment. Cells were lysed 48 h post-transfection, and then incubated with 30 μl Protein A/G Agarose beads (Santa Cruz) to preclear the lysate. Then 2 μg antibody was added for 4 h and thereafter 50 μl Protein A/G Agarose for 16 h. After several washing steps in PBS, the beads were added sample buffer and analyzed by 7.5% SDS–PAGE and Western blotting using anti-HA (Sigma) 1:1000.
2.4. In vitro interaction and acetylation assays
Full length or deletion constructs of hARD1 cDNA was cloned into the pET M60 vector (G. Stier, EMBL) using the restriction sites Acc65I and NcoI. Escherichia coli strain BL21 (DE3) pLysS (Invitrogen) was used for the protein expression according to the instruction manual. Purification of recombinant hARD1 protein was performed using several steps of His-affinity columns and gel filtration with or without cleavage from the fusion partner NusA using TEV-protease (Invitrogen). GST-ODD containing the amino acids 393–580 of HIF-1α was constructed by cloning the ODD fragment (393–580) into the pGEX4T-3 vector (Amersham). The GST-ODD K532R mutant was made by sited-directed mutagenesis using a QuickChange kit from Stratagene. GST and GST-fusion-proteins were coupled to Sepharose 4B (Amersham). In the interaction assays (Fig. 3B and C), the components were allowed to interact for 30 min at 30 °C in interaction buffer (2 mM DTT; 10 mM KH2PO4; 10 mM Na2HPO4 · 2 H2O; 200 mM NaCl; 1 tablet Complete EDTA free protease inhibitor (Roche) per 50 ml buffer; pH adjusted to 7.4 before use) before the Sepharose beads were washed three times using the same buffer. NusA-ABD (Actin binding domain) and GST was used as negative controls in the interaction assays. Interaction assays (Fig. 4A) and acetylation assays (Fig. 4B and C) were performed in acetylation buffer (1 mM DTT; 10 mM Na butyrate; 50 mM Tris-HCl; 800 μM EDTA; 10% glycerol; pH adjusted to 8.5 before use) for 1 h at 30 °C. Input was 3 °g GST-ODD and 1 °g hARD1. N-α-acetylation assays were performed as previously described [15]. Briefly, immunoprecipitation was done as above (Section 2.3) without transfection. Pure hARD1 or pellets of Protein A/G-agarose bound NATH-hARD1 complex was added to 10 μl ACTH (0.5 mM, human adrenocorticotropic hormone fragment 1–24, Calbiochem), 4 μl [3-H]acetyl-coenzyme A (1 μCi, 107 GBq/mmol, Amersham Biosciences) and 136 μl of 0.2 M K2HPO4 (pH 8.1). The mixture was incubated at 37 °C and samples were collected after 1 h. After centrifugation the supernatant was added to 150 μl SP Sepharose (50% slurry in 0.5 M Acetic acid, Sigma) and incubated on a rotor for 5 min. The mixture was centrifuged and the pellet was washed three times with 0.5 M acetic acid and finally with methanol. Radioactivity in the ACTH containing pellet was determined by scintillation counting.
Fig. 3.

hARD1 interacts with HIF-1α. (A) MCF-7 cells were cotransfected by plasmids encoding HA-HIF-1α and Xp-hARD1 (or Xp-lacZ as a negative control). Immunoprecipitates of the cell lysates using anti-Xp were analyzed by Western blotting with anti-HA to detect HA-HIF-1α. (B) GST (negative control) or GST-ODD beads were incubated with NusA-hARD1 or NusA-ABD (negative control). After washing steps, the beads were analyzed by SDS–PAGE and Coomassie staining to determine the level of retained NusA-hARD1/ABD. (C) As (B) using pure hARD1 and analysis by Western blotting and anti-hARD1.
Fig. 4.

hARD1 does not acetylate HIF-1α. (A) Interaction assay as Fig. 3C using GST, GST-ODD WT or GST-ODD K532R beads and acetylation buffer with indicated concentrations of Acetyl Coenzyme A (AcCoA). hARD1 retained on beads after washing was analyzed by SDS–PAGE and Western blotting with anti-hARD1. (B) Acetylation assay with GST-ODD and purified hARD1. Detection of acetylated GST-ODD by Western blotting and anti-acetyl lysine and by incorporation of [14-C] acetyl coenzyme A. Coomassie staining verifies equal loading of GST-ODD. Upper panel: Demonstration of hARD1 activity in ACTH (adrenocorticotropin 1–24) N-α-acetylation assay. (C) As (B), but with immunoprecipitated NATH–hARD1 complexes as enzyme. HeLa cellular lysates were immunoprecipitated (IP) with anti-NATH, anti-hARD1 or rabbit immunoglobulins (Ig) as a negative control.
3. Results
3.1. Hypoxia does not decrease the level of endogenous hARD1 protein
According to an early work from Jeong et al., hypoxic conditions decreased the level of hARD1 mRNA. Although a recent study partially confirmed this finding, downregulation of hARD1 mRNA was not consistent in all cell lines [23]. To further test the role of hypoxia on ARD1, we used a recently developed antibody against hARD1 and evaluated the effect of hypoxia and hypoxia-mimicks (cobalt and desferroxamine) in several cell lines. As shown in Fig. 1, the endogenous hARD1 protein did not decrease significantly in different cell lines tested: HT1080 and RCC4 cells treated with hypoxia (1% O2) for 18 h (Fig. 1A) or HeLa cells and HEK293 cells treated with CoCl2 for 6 h (Fig. 1B). Although the stimuli stabilized HIF-1α protein, no decrease in the level of hARD1 protein was observed. MCF-7 cells treated with CoCl2 at various times between 0 and 18 h showed a peak of HIF-1α protein level at 6 h after treatment, while the level of hARD1 was quite stable throughout the treatment (Fig. 1C). Hypoxia treatment of HEK293 cells and HeLa cells, and desferroxamine treatment of HT1080 cells showed similar results (data not shown). The results using different human cell lines and different hypoxia-mimics strongly suggest that the level of endogenous hARD1 protein is not regulated by hypoxia.
Fig. 1.
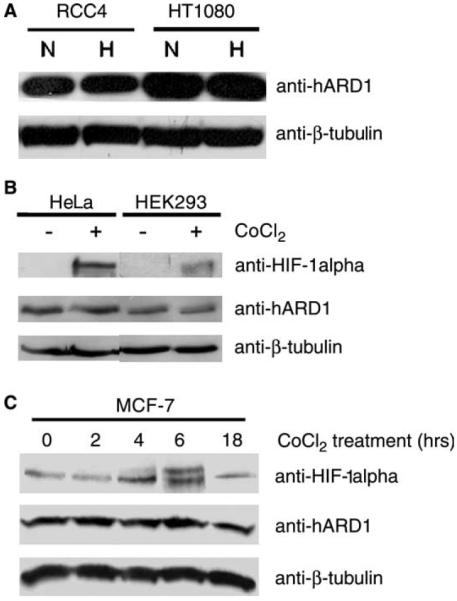
The level of hARD1 protein is stable during hypoxia. (A) RCC4 and HT1080 cells were exposed to either normoxia (N) or hypoxia (H) for 18 h and the cell lysates were analyzed by Western blotting with anti-hARD1. (B) As (A) using HeLa and HEK293 cells and CoCl2 treatment for 6 h. (C) As (A) adding CoCl2 for the indicated times to MCF-7 cells.
3.2. Stability of HIF-1α is not regulated by hARD1
Jeong et al. had found that hARD1 negatively regulated the protein stability of HIF-1α and that induced levels of hARD1 inversely correlated with the level of HIF-1α [14]. We performed overexpression and knock-down of hARD1 and monitored the levels of endogenous HIF-1α protein. Using Tet-inducible HeLa cells, we induced overexpression of FLAG-hARD1 and tested for its effect on HIF-1α under hypoxic and normoxic conditions. Western blot analysis of cell lysates following induction of ARD1 by Tet-treatment revealed no significant changes in HIF-1α levels compared with their control cell lines (Fig. 2A). Similarly, transient overexpression of hARD1 in HEK293 cells had no significant effect on HIF-1α levels (not shown).
Fig. 2.
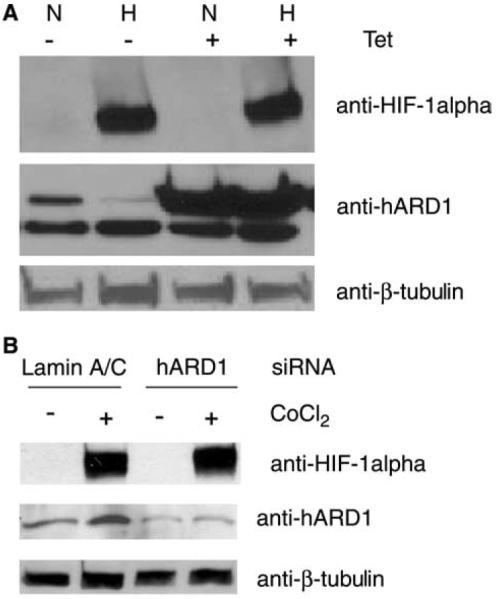
hARD1 does not regulate the stability of HIF-1α. (A) Overexpression of FLAG-hARD1 was induced in a HeLa-Tet inducible cell line under normoxic (N) or hypoxic (H) conditions. Cell lysates were analyzed by Western blotting with anti-HIF-1α. (B) hARD1 was knocked down in HeLa cells by siRNAs in the presence or absence of CoCl2. The level of HIF-1α in the cell lysates was analyzed by Western blotting.
Suppression of hARD1 by siRNA knock-down also failed to affect HIF-1α levels either under hypoxic or in normoxic conditions (Fig. 2B). If hARD1 contributes to the destabilization of HIF-1α in normoxia, then we should have observed a norm-oxic increase in HIF-1α levels when hARD1 is significantly reduced by RNAi. The recent study by Bilton et al. found no regulation of HIF-1α protein as a consequence of up- or downregulation of hARD1 [24]. Also Fisher et al. [23] found no stabilization of HIF-1α by RNAi mediated silencing of hARD1. Taken together, these results indicate that hARD1 is not a regulator of HIF-1α protein stability.
3.3. hARD1 interacts with HIF-1α in vivo and in vitro
Since there seemed to be no regulation of hARD1 level by hypoxia and no regulation of HIF-1α by hARD1, we wanted to investigate whether or not these two proteins interact at all. The in vivo interaction was analyzed by immunoprecipitation experiments using plasmids encoding HA-tagged HIF-1α and Xpress-tagged hARD1. MCF-7 cells cotrans-fected with both plasmids were lysed and subjected to immunoprecipitation using anti-Xpress. Western blot analysis of the immunoprecipitates demonstrated that hARD1 co-immunoprecipitated with HIF-1α (Fig. 3A). To test the direct in vitro interaction between hARD1 and the ODD-domain within HIF-1α, we purified NusA-hARD1 and GST-ODD.
Gluthatione beads containing GST-ODD were incubated with NusA-hARD1 and thereafter washed several times under physiological conditions. The results demonstrated that a portion of the purified NusA-hARD1 bound specifically to GST-ODD as compared to the negative controls (Fig. 3B). Binding studies with a deletion mutant containing amino acids 1–179 of hARD1 also demonstrated specific binding to GST-ODD (not shown). Similarly, parallel assays using pure hARD1 without the NusA fusion partner demonstrated binding of hARD1 to GST-ODD, but not to GST (Fig. 3C). Thus, these two proteins directly interact with each other. The demonstration of a specific in vivo and in vitro interaction between hARD1 and HIF-1α is in accordance with the original findings of Jeong et al. [14].
3.4. HIF-1α is not acetylated by hARD1
Since ODD (HIF-1α) has been proposed to be a substrate of hARD1 acetylation [14], one could expect that the addition of the acetyl donor, acetyl CoA, would allow for the enzymatic reaction to occur and thus release the substrate from its enzyme. The presence or absence of acetyl-CoA in a mixture of hARD1 and GST-ODD in acetylation assay conditions did not have any effect on the binding (Fig. 4A). We also mutated the K532 within ODD reported to be the acetyl acceptor. Interestingly, the GST-ODD K532R interacted equally strong with hARD1 as the GST-ODD WT (Fig. 4A). Furthermore, results of in vitro acetylation assays using purified GSTODD and hARD1 showed no acetylation, as assessed by using anti-acetyl lysine antibodies (Fig. 4B) or by incorporation of [14-C]-acetyl coenzyme A. The proper folding of the purified hARD1 is supported by the fact that the purified protein is active as an N-α-acetyltransferase and that it is able to specifically interact with ODD. We also immunoprecipitated hARD1-NATH complexes from human cell lines and while these complexes were active in N-α-acetylation assays, they were not able to acetylate the ODD (Fig. 4C). Concordingly, the recent study by Bilton et al. also failed in demonstrating any HIF-1α acetylation by hARD1 in vitro. Thus, ODD does not seem to be a substrate of hARD1 or hARD1-NATH mediated acetylation.
4. Discussion
Early studies by Jeong et al. suggested that acetylation of HIF-1α by ARD1 was an important mechanism controlling HIF-1α stability by enhancing its interaction with VHL and promoting its ubiquination and degradation. Furthermore, it was suggested that the levels of ARD1 were negatively regulated by hypoxia conforming to a feedback loop that would allow the hypoxic stabilization of HIF-1α protein. Our results indicate that ARD1 seems not to be regulated by hypoxia at the mRNA or protein level and hARD1 does not appear to have an impact on HIF-1α stability. During preparation of this manuscript similar findings were reported by Bilton et al. [24]. They found that hARD1 remained stable during hypoxic conditions both at the mRNA and at the protein level. Thus, it seems clear that a regulation of hARD1, if any, by hypoxic conditions does not occur through downregulation of hARD1 mRNA or protein. The reported N-ε-acetylation of the HIF-1α-ODD by hARD1 could not be verified by our experiments using in vitro acetylation assays with bacterially and mammalian expressed enzymes. Moreover, a K532R mutation at the putative hARD1 acetylation site did not affect the interaction between the HIF-1α-ODD and hARD1 (Fig. 4A), neither affected the stability of HIF-1α-ODD in vivo (Kong et al., submitted). Thus, hARD1 can in our hands only be regarded as an N-α-acetyltransferase. Our findings are consistent with the recent observations by Fisher et al. and Bilton et al., who reported that suppression and overexpression of ARD1 did not affect basal HIF-1α levels or its response to hypoxia. However, we confirmed that there is a physical interaction between HIF-1α and hARD1, suggesting that they may be functionally connected in a so far uncovered manner. Studies have demonstrated that hARD1 localized in the nucleus as well as the cytoplasm and partially co-localized with HIF-1α in the nucleus [15]. The demonstrated interaction between HIF-1α and hARD1 supports that this may take place. Other studies have shown that little or no hARD1 is present in the nucleus [14,24,25]. The reason for these discrepancies is unknown, but the subcellular localization of hARD1 may be regulated. Future studies are needed to reveal the functional role of the hARD1-HIF-1α interactions. The NATH-hARD1 N-α-acetyltransferase complex represents so far the only known function for hARD1. It will be interesting to determine whether the hARD1-HIF-1α interaction is independent of the NATH-hARD1 complex. As is described herein for ODD, also NATH interacts with amino acids 1–179 within hARD1 [15]. It is therefore possible that hARD1 is not capable of interacting with both NATH and HIF-1α simultaneously. Other proteins, like p53 have also been reported to interact with the ODD domain within HIF-1α [26]. Further interaction studies are required to elucidate whether or not the binding of hARD1 to ODD may affect these interactions.
Acknowledgments
We thank K. Jacobsen and C. Hoff for technical assistance. This work was supported by The Norwegian Cancer Society (Grants to T.A., J.E.V., J.R.L.), The Locus of Experimental Cancer Research (University of Bergen), The Meltzer Foundation (Grant to T.A.) and by the NIH (K01-CA098809) to N.S. and (CA0892212) to J.C.
Abbreviations
- ARD
arrest-defective (homologue of yeast Ard1p)
- NAT
N-acetyltransferase
- NATH
NAT human
- hARD1
human ARD1
- HIF-1α
hypoxia inducible factor 1α
References
- [1].Marti HH. Angiogenesis – a self-adapting principle in hypoxia. EXS. 2005:163–180. doi: 10.1007/3-7643-7311-3_12. [DOI] [PubMed] [Google Scholar]
- [2].Acker T, Plate KH. Role of hypoxia in tumor angiogenesis-molecular and cellular angiogenic crosstalk. Cell Tissue Res. 2003;314:145–155. doi: 10.1007/s00441-003-0763-8. [DOI] [PubMed] [Google Scholar]
- [3].Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;(Suppl. 5):10–17. doi: 10.1634/theoncologist.9-90005-10. [DOI] [PubMed] [Google Scholar]
- [4].Bacon AL, Harris AL. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann. Med. 2004;36:530–539. doi: 10.1080/07853890410018231. [DOI] [PubMed] [Google Scholar]
- [5].Metzen E, Ratcliffe PJ. HIF hydroxylation and cellular oxygen sensing. Biol. Chem. 2004;385:223–230. doi: 10.1515/BC.2004.016. [DOI] [PubMed] [Google Scholar]
- [6].Hopfl G, Ogunshola O, Gassmann M. HIFs and tumors – causes and consequences. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;286:R608–R623. doi: 10.1152/ajpregu.00538.2003. [DOI] [PubMed] [Google Scholar]
- [7].Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2005 doi: 10.1016/j.canlet.2005.05.028. In press PMID: 16002209. [DOI] [PubMed] [Google Scholar]
- [8].Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- [9].Poellinger L, Johnson RS. HIF-1 and hypoxic response: the plot thickens. Curr. Opin. Genet. Dev. 2004;14:81–85. doi: 10.1016/j.gde.2003.12.006. [DOI] [PubMed] [Google Scholar]
- [10].Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- [11].Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- [12].Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim. Biophys. Acta. 2005;1755:107–120. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- [13].Brahimi-Horn MC, Pouyssegur J. The hypoxia-inducible factor and tumor progression along the angiogenic pathway. Int. Rev. Cytol. 2005;242:157–213. doi: 10.1016/S0074-7696(04)42004-X. [DOI] [PubMed] [Google Scholar]
- [14].Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- [15].Arnesen T, Anderson D, Baldersheim C, Lanotte M, Varhaug JE, Lillehaug JR. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 2005;386:433–443. doi: 10.1042/BJ20041071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mullen JR, Kayne PS, Moerschell RP, Tsunasawa S, Gribskov M, Colavitoshepanski M, Grunstein M, Sherman F, Sternglanz R. Identification and characterization of genes and mutants for an N-terminal acetyltransferase from yeast. EMBO J. 1989;8:2067–2075. doi: 10.1002/j.1460-2075.1989.tb03615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Park EC, Szostak JW. Ard1 and Nat1 proteins form a complex that has N-terminal acetyltransferase activity. EMBO J. 1992;11:2087–2093. doi: 10.1002/j.1460-2075.1992.tb05267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Polevoda B, Norbeck J, Takakura H, Blomberg A, Sherman F. Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 1999;18:6155–6168. doi: 10.1093/emboj/18.21.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gautschi M, Just S, Mun A, Ross S, Rucknagel P, Dubaquie Y, Ehrenhofer-Murray A, Rospert S. The yeast N(alpha)-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell Biol. 2003;23:7403–7414. doi: 10.1128/MCB.23.20.7403-7414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yu F, White SB, Zhao Q, Lee FS. HIF-1 alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA. 2001;98:14744. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jost M, Kari C, Rodeck U. An episomal vector for stable tetracycline-regulated gene expression. Nucleic Acids Res. 1997;25:3131–3134. doi: 10.1093/nar/25.15.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Arnesen T, Gromyko D, Horvli O, Fluge O, Varhaug JE, Lillehaug JR. Expression of NATH and hARD1 proteins in Thyroid neoplasms. Thyroid. 2005;15:1131–1136. doi: 10.1089/thy.2005.15.1131. [DOI] [PubMed] [Google Scholar]
- [23].Fisher TS, Etages SD, Hayes L, Crimin K, Li B. Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J. Biol. Chem. 2005;280:17749–17757. doi: 10.1074/jbc.M412055200. [DOI] [PubMed] [Google Scholar]
- [24].Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC. ARD1, an acetyltransferase, does not alter stability of hypoxia-inducible factor-1alpha and is not induced by hypoxia or HIF. J. Biol. Chem. 2005;280:31132–31140. doi: 10.1074/jbc.M504482200. [DOI] [PubMed] [Google Scholar]
- [25].Sugiura N, Adams SM, Corriveau RA. An evolutionarily conserved N-terminal acetyltransferase complex associated with neuronal development. J. Biol. Chem. 2003;278:40113–40120. doi: 10.1074/jbc.M301218200. [DOI] [PubMed] [Google Scholar]
- [26].Sanchez-Puig N, Veprintsev DB, Fersht AR. Binding of natively unfolded HIF-1alpha ODD domain to p53. Mol. Cell. 2005;17:11–21. doi: 10.1016/j.molcel.2004.11.019. [DOI] [PubMed] [Google Scholar]