

Abstract
Focal adhesion kinase (FAK) is a protein tyrosine kinase that is ubiquitously expressed, recruited to focal adhesions, and engages in a variety of cellular signaling pathways. Diverse cellular responses, such as cell migration, proliferation, and survival, are regulated by FAK. Prior to activation, FAK adopts an autoinhibited conformation in which the FERM domain binds the kinase domain, blocking access to the activation loop and substrate binding site. Activation of FAK occurs through conformational change, and acidic phospholipids such as phosphatidylinositol 4,5-bisphosphate (PIP2) are known to facilitate this process. PIP2 binding alters the autoinhibited conformation of the FERM and kinase domains and subsequently exposes the activation loop to phosphorylation. However, the detailed molecular mechanism of PIP2 binding and its role in FAK activation remain unclear. In this study, we conducted coarse-grained molecular dynamics simulations to investigate the binding of FAK to PIP2. Our simulations identified novel areas of basic residues in the kinase domain of FAK that potentially undergo transient binding to PIP2 through electrostatic attractions. Our investigation provides a molecular picture of PIP2-initiated FAK activation and introduces promising new pathways for future studies of FAK regulation.
Introduction
Focal adhesions (FA) are integrin-mediated protein complexes that are found peripheral to the cell membrane at sites of cell attachment to the extracellular matrix (ECM) [1,2]. Focal adhesions establish a direct mechanical link between the actin cytoskeleton and the ECM and also act as cellular signaling integrators, sensing biochemical and mechanical stimuli from the extracellular environment. Diverse and critical biological events, including cell migration, proliferation, differentiation, and survival, are mediated by proteins localized to focal adhesions [3].
Focal adhesion kinase (FAK) is an essential component in the macromolecular assembly of focal adhesions, as well as a crucial kinase that participates in a variety of cellular signaling pathways [4]. FAK is involved in several developmental processes including angiogenesis [5] and axonal outgrowth [6]. In addition, dysregulation of FAK activity is linked to numerous pathological events such as tumorigenesis and metastasis [7]. Understanding the structural underpinnings of FAK activation is essential to development of therapeutic treatments for these diseases. FAK is composed of 4 major domains: an N-terminal band 4.1, Ezrin, Radixin, Moesin (FERM) domain, a central tyrosine kinase domain followed by an unstructured proline-rich region, and a C-terminal focal adhesion targeting (FAT) domain. The FERM and kinase domains are separated by a linker containing the autophosphorylation site, Y397 (Fig 1). The FERM domain plays an essential autoinhibitory role in the regulation of the catalytic activity of FAK [8], in which the F2 subdomain contacts the C-lobe of the kinase domain, blocking access to the catalytic cleft and the activation loop (A-loop) [9]. As a result, the kinase domain remains locked in an inactive, unphosphorylated conformation. In addition, the linker between the FERM and kinase domains forms a beta strand that is part of a beta sheet within the F1 lobe, preventing autophosphorylation at Y397. It has been proposed [9,10] that a conformational change in the autoinhibited form of the FERM-kinase domain is the key initial step in FAK activation, exposing Y397 within the linker to phosphorylation [11] and subsequent recruitment of Src. Once Src binds to the linker, it phosphorylates two tyrosines within the exposed A-loop (Y576 and Y577) and renders FAK fully active [12].
Fig 1. Domain structure of FAK and illustration of the simulation system.
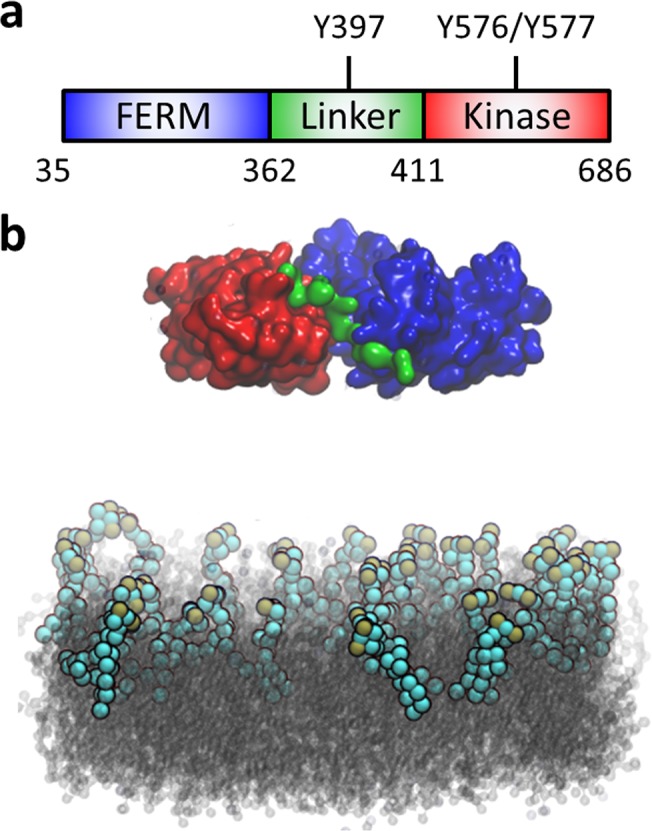
(a) The two N-terminal domains of FAK form the autoinhibited conformation and are connected via a linker. FERM domain: blue; kinase domain: red; linker: green. (b) FAK lies above a DOPC bilayer (gray) with 10% PIP2 (spheres, cyan and ochre) in the upper leaflet. Ions and water molecules are omitted for clarity. All molecular graphics are rendered in VMD [13].
FERM-interacting molecules, including growth factor receptors, p53, Ezrin, and acidic phospholipids, are known to play key roles in FAK regulation [14]. In particular, phosphatidylinositol 4,5-bisphosphate (PIP2) directly binds FAK in the FERM domain, inducing a conformational change and subsequent activation [10,15]. While PIP2 binding is likely driven by electrostatic forces [16], the molecular details of FAK binding to PIP2, the stoichiometry of binding, and the binding kinetics remain unknown. Determination of these binding characteristics is essential in order to understand PIP2-mediated activation of FAK.
We performed a series of modeling studies to investigate the molecular details of interactions between FAK and PIP2. Coarse-grained molecular dynamics (CGMD) [17,18] allows modeling of length and timescales that are significantly larger than all-atom molecular dynamics and are ideal for phenomena such as PIP2 activation of FAK. CGMD simulations were carried out to examine the binding of FAK to bilayers containing PIP2 lipids. Binding to the KAKTLRK basic patch within the FERM domain was observed, in agreement with experimental studies [10,15]; surprisingly, we also identified PIP2 binding in the kinase domain. These results provide novel insight into the mechanisms controlling FAK activation, and could potentially identify new strategies to manipulate FAK regulation and treat disease.
Computational Methods
FAK simulations were performed using the crystal structure of the autoinhibited conformation [9] (PDB 2J0J). Missing residues in the A-loop of the kinase domain (residues 574–583) were modeled as a random coil and further minimized using the molecular simulation package CHARMM c37b1 [19]. Missing residues from the linker region (residues 363–393) were not modeled due to its length and structural flexibility. The all-atom model was converted to a coarse-grained model with the MARTINI v2.2 force field [20], and backbone Cα atoms were constrained by an elastic network [21] with a cutoff of 0.9 nm and a force constant of 500 kJ-mol-1. Simulation of FAK in water was initially carried out with the FERM and kinase domains allowed to move independently of one another. Deformation of the global structure occurred (results not shown), limiting all subsequent simulations with the FERM and kinase domains constrained as a single unit in the autoinihibited form. Another 1-μs-long simulation of FAK in water with the added constraint was carried out to verify that the system would remain stable.
To model the PIP2-containing lipid bilayer, we used an equilibrated DOPC bilayer of 15.2 nm × 15.2 nm in the x-y dimension and randomly replaced 10% of the DOPC molecules on the upper leaflet of the bilayer with PIP2, as per experimental studies [15], resulting in a lipid bilayer system composed of 629 DOPC molecules and 33 PIP2 molecules. Finally, FAK was placed at distances of 4.5 nm (simulation I), 2.5 nm (simulation II), and 1.5 nm (simulation III), respectively, from the surface of the lipid bilayer, and 170 sodium ions and ≈27,000 polarized water molecules were added to the simulation box to make the final dimension of each system 15.2 nm × 15.2 nm × 19.1 nm with ≈92,000 particles (Fig 1).
All CGMD simulations were run in GROMACS v4.6 [22]. Simulations were performed in the isothermal-isobaric ensemble (NPT) with nonbond cutoff at 1.2 nm. Temperature was coupled at 320 K using a Berendsen thermostat with a coupling constant of 1 ps. The pressure was coupled to a Berendsen barostat at 1 bar with a relaxation time of 1 ps. Each simulation was run for 1 μs with an integration time step of 10 fs and system coordinates were collected every 10 ps. Given the relatively fast system equilibration and initial ligand-binding timescales, as well as the simulation length, the full trajectory is used for analysis unless stated otherwise. (Discussion of simulation convergence can be found in S5 Table.) Analysis was carried out with GROMACS analysis tools and custom-made scripts.
Results
A. FAK binds to PIP2-containing bilayers
As a system validation, we first simulated FAK in polarized water. The protein was stable with an average RMSD of ≈0.3 nm. Our next set of simulations allowed FAK to freely diffuse above a lipid bilayer. Radial distribution functions (RDF) of PIP2 and ions to PIP2 with and without FAK were calculated (Fig 2), and there is no notable difference in distribution in the presence or absence of FAK. This result indicates that the presence of FAK does not globally perturb the distribution of PIP2 within the lipid bilayer, ruling out the possibility that FAK actively recruits large clusters of PIP2 lipids. However, it is possible for PIP2 to form clusters in the presence of divalent cations such as calcium, meaning that FAK could potentially interact with multiple PIP2 molecules [23].
Fig 2. FAK does not actively recruit PIP2.
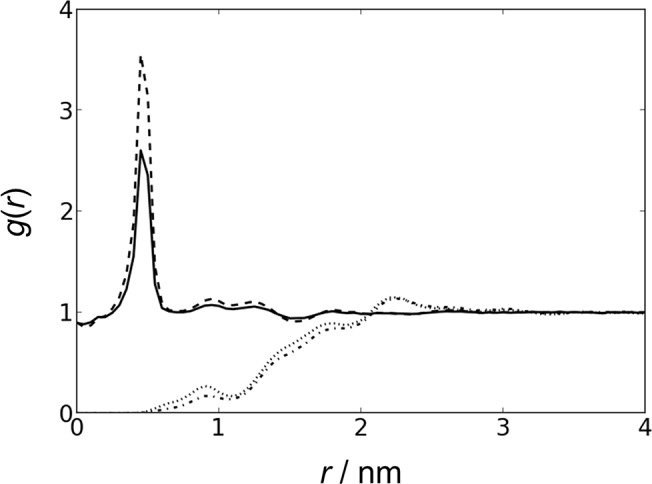
RDF of ions as a function of distance to PIP2 in the presence (solid line) and absence (dashed line) of FAK. RDF of PIP2 as a function of distance to PIP2 in the presence (dash-dotted line) and absence (dotted line) of FAK. All distances are measured with respect to the 1’-phosphate of PIP2. The final 900 ns of each simulation was used for analysis. Simulations II and III had very similar results and are therefore not shown.
For all simulations, FAK was placed far enough from the lipid bilayer to ensure that binding to PIP2 would occur through random diffusion, independent of initial orientation. Due to the use of periodic boundary conditions, FAK could freely diffuse and interact with each leaflet of the lipid bilayer (one leaflet containing PIP2 and DOPC, the other leaflet containing only DOPC). This design was intentional, since PIP2 is asymmetrically distributed in vivo [24–26]. Our simulations showed that FAK preferentially interacted with the PIP2-containing leaflet (Fig 3).
Fig 3. FAK preferentially interacts with the PIP2-containing bilayer leaflet.
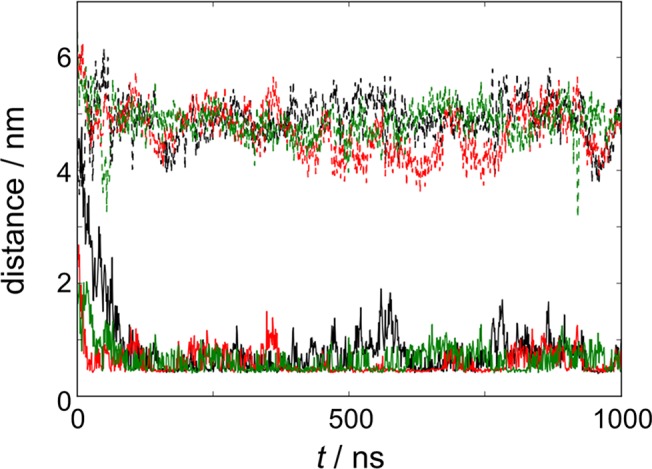
Minimal distance between FAK and DOPC leaflet with (solid line) and without PIP2 (dashed line) in simulation I (black), II (red), and III (green)
B. Multiple association and dissociation events occur between FAK and PIP2
We next examined the propensity for FAK to interact with PIP2 lipids (Table 1). FAK was considered to bind with PIP2 if the minimal distance between any coarse-grained bead of FAK and any phosphate bead of PIP2 was less than 0.52 nm. (Results are not sensitive to the cutoff—see S1–S4 Tables.) The average distance between PIP2 and FAK was 0.48 nm ±0.02 nm. FAK was bound to PIP2 for at least 2/3 of the total simulation time (Table 2), with initial binding events occurring at 80, 18, and 30 ns, respectively (Fig 3). Binding was dynamic, as multiple events of PIP2 association and dissociation were observed throughout the course of the simulations. In addition, when FAK was bound to PIP2, it can interact with multiple PIP2 lipids (usually between 1–3 molecules), as determined by the percentage of time when FAK binds n P number of PIP2 lipids (n P = 1–6) (Table 2).
Table 1. Comparison of PIP2 and PC lipids involved in FAK binding.
| Simulation | % time of binding | N L a (x i > 10%) b | N L (x i > 5%) | |||
|---|---|---|---|---|---|---|
| PIP2 | PC | PIP2 | PC | PIP2 | PC | |
| I | 68.1 | 31.8 | 9 | 0 | 12 | 7 |
| II | 88.2 | 58.4 | 8 | 1 | 13 | 17 |
| III | 84.6 | 39.1 | 7 | 0 | 12 | 1 |
a N L is defined as the number of lipids that make contact over 10% or 5% of simulation time.
b The contact percentage of PIP2/PC lipids is calculated as the number of times a specific PIP2/PC lipid made contact with FAK normalized by the total number of times FAK established contact with PIP2/PC lipids.
Table 2. Quantification of FAK binding to PIP2.
| Simulation | % time of binding a | n P PIP2 bound to FAK, % time of binding b | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4–6 | ||
| I | 68.1 | 40.5 | 35.5 | 17.6 | 6.4 |
| II | 88.2 | 28.7 | 41.0 | 24.1 | 6.3 |
| III | 84.6 | 38.9 | 37.3 | 17.9 | 5.9 |
a Percentage of time FAK is bound to PIP2 during simulation.
b Percentage of time FAK is bound to n P number of PIP2.
C. Observed PIP2 binding sites on FAK
To identify PIP2 binding sites on FAK, the percentage of time x i each residue interacts with PIP2 is monitored using x i = n i/N, where n i is the number of times residue i makes contact with PIP2 and N is the total number of times FAK makes contact with PIP2 during the simulation (Table 3). All identified interacting residues are basic (lysine or arginine) and thus capable of establishing electrostatic attractions with the highly negatively-charged PIP2 head groups. Among these residues, we identified known PIP2-binding residues K216, K218, R221, and K222 (part of the KAKTLRK ridge) [15,27]. These residues lie on the surface of the F2 subdomain of the FERM domain and are critical for FAK activation. Surprisingly, we also observed additional basic residues not previously known to interact with PIP2, as will be detailed below.
Table 3. Percentage of time (x i) individual residues contact PIP2.
| Simulation | K191 a | K216 | K218 | R221 | K222 | R229 | R508 | R514 | K515 | K578 | K621 | K627 | R640 | K657 | R665 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | 7.8 | 8.6 | 18.2 | 1.3 | 20.6 | 17.4 | 7.3 | 4.8 | 19.0 | 27.7 | 22.3 | 35.6 | 18.9 | 5.4 | 10.4 |
| II | 4.1 | 5.9 | 12.2 | 1.9 | 7.3 | 4.8 | 27.8 | 11.2 | 13.1 | 37.8 | 53.1 | 39.4 | 16.5 | 6.9 | 11.0 |
| III | 11.0 | 7.3 | 23.4 | 10.5 | 21.7 | 16.6 | 0.0 | 3.5 | 22.6 | 35.4 | 15.7 | 48.0 | 19.5 | 6.4 | 10.5 |
a Residues with x i > 5% in at least two simulations or x i > 10% in any simulation.
To further characterize the binding sites, PIP2-interacting residues were grouped based upon their location on the protein surface, designated Group I and Group II (Table 4and Fig 4). Group I residues are located at the interface between the F2 subdomain of the FERM domain along the functionally important KAKTLRK ridge [15,27] and directly adjacent in the C-lobe of the kinase domain. These two clusters in Group I (the KAKTLRK ridge and the C-lobe of the kinase domain) are proximal to each other and may form part of a larger phospholipid binding site. Group II residues are completely new and represent a potential binding site suggested by our simulations, located near the interface between the N- and C-lobes of the kinase domain. Both Group I and Group II binding sites are tightly correlated to the two basic patches on the electrostatic potential surface of the protein (Fig 4).
Table 4. Grouping of PIP2 interaction sites.
| Group I | F2 a | K191 | K216 | K218 | R221 | K222 | R229 |
| C-lobe | R640 | K657 | R665 | ||||
| Group II | N-lobe & C-lobe | R508 | R514 | K515 | K578 | K621 | K627 |
a F2 is part of the FERM domain, N- and C-lobes are part of the kinase domain.
Fig 4. Group I and II interaction sites correspond to large basic patches in FAK.

a1, b1, and c1: Mapping of Group I (green) and Group II (yellow) residues onto the crystal structure of FAK (F1 (violet), F2 (blue), and F3 (cyan) subdomains of the FERM domain, N-lobe (red), A-loop (iceblue), and C-lobe (magenta) of the kinase domain, and linker (gray)). a2, b2, and c2: Electrostatic potential surface of FAK kinase calculated using APBS [28]. The view of the electrostatic potential surface in a2, b2, and c2 corresponds to the orientation shown in a1, b1, and c1, respectively.
D. FAK adopts multiple binding poses with PIP2
Group I and Group II interactions with PIP2 account for the majority of FAK-PIP2 contacts and are independent of one another (Table 5). However, preferential orientations of FAK with respect to the lipid bilayer do occur. Interactions involving only Group I required both the KAKTLRK ridge and C-lobe of the kinase domain to be effaced with the lipid bilayer (Fig 5A). When both Group I and Group II sites interacted with PIP2, a tilted orientation with the Group II site on the C-lobe facing the bilayer surface occurred (Fig 5B). Finally, when only Group II preferentially interacted with PIP2, it required an orientation in which the C-lobe was heavily tilted towards and almost parallel to the bilayer (Fig 5C).
Table 5. Percentage of time Group I and Group II sites contacted PIP2 during FAK-PIP2 interactions.
| Simulation | Only Group I | Only Group II | I and II | Neither I or II |
|---|---|---|---|---|
| I | 31.4 | 42.6 | 23.2 | 2.8 |
| II | 22.7 | 60.3 | 15.6 | 1.4 |
| III | 25.3 | 40.7 | 32.6 | 1.4 |
Fig 5. FAK adopts a set of preferential orientations toward PIP2-containing lipid bilayers.
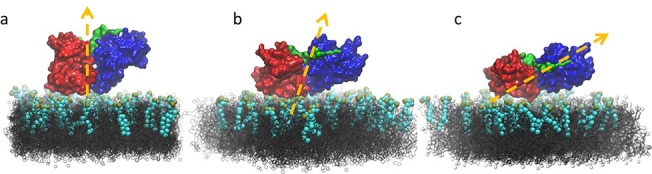
Arrows represent the direction from the center of mass of the F2 subdomain of the FERM domain and C-lobe of the kinase domain to the COM of the F1 subdomain of the FERM domain and N-lobe of the kinase domain. Color scheme is the same as Fig 1.
Discussion
The computational studies carried out have led us to more carefully consider the mechanism of PIP2 binding to FAK and the potential effects on FAK activation. When PIP2-FAK interactions were first discovered, it was shown that the highly basic KAKTLRK ridge (part of the F2 lobe in the FERM domain) was necessary for PIP2-FAK binding in vitro and FAK activation in vivo [15,27]. More recently, vesicle pull-down and surface plasmon resonance studies identified that the combined kinase and FERM domains of FAK in the autoinhibited conformation bind less effectively to PIP2-containing bilayers than the FERM domain alone, indicating that the autoinhibitory conformation of FAK is predisposed to decreasing the favorability of PIP2 binding [10]. The motivation for our CGMD studies was to gain molecular insight into these interactions, with the expectation that the interactions would remain localized to the FERM domain. Our simulation faithfully reproduced the known PIP2 binding sites located on the basic patch of the FERM domain (Group I). However, observing interactions of Group II residues within the kinase domain with PIP2 was completely unexpected. Although the previously-mentioned studies indicate the majority of PIP2 binding to FAK occurs within the vicinity of the KAKTLRK ridge, residual binding observed in the KAKTLRK mutant [10] may be due to additional interaction sites as proposed here. Our results indicate that the kinase domain (Group II) could represent a viable site for secondary binding of PIP2.
Our simulations indicate that FAK-PIP2 interaction is mainly nonspecific due to the electrostatic nature of binding, and agrees well with the lack of influence on FAK activation in the presence of neutrally-charged phospholipids such as DOPC [10,15]. A large number of basic residues on the FAK surface established contact with PIP2. The residues engaging frequent contact in each simulation remain largely consistent (S5 Table), indicating a converged identification of PIP2-binding residues. Due to the constraints of our protein model, there are potential PIP2-interacting residues that may not have been identified by our CGMD simulation. For example, R184 and K190 in the FERM domain (the KAKTLRK ridge), which were shown to be key residues in the allosteric relationship between ATP and PIP2 binding [29], and R634 on the kinase domain bridge the Group I and Group II sites but are inaccessible to PIP2 in the autoinhibited conformation. However, multiple association and dissociation events were observed, which is supported by the fact that the probability distribution of the principal axis of FAK with respect to the membrane normal showed that no prolonged binding pose existed (S1 Fig). These transient association dynamics mainly stem from the fact that the friction originating from fine-grained degrees of freedom present in an all-atom model is missing in the coarse-grained model used in this study [18,30,31]. Instead of association with a defined binding pocket, PIP2 molecules maintain “stable” contact with residues located within Group I and Group II, interacting with the basic surfaces and sampling multiple binding complexes. This observed binding phenomena suggests mutations that could be tested experimentally to further characterize PIP2 binding to FAK in the Group II basic patch, in particular R508, R514, K515, K621, and K627.
Our study has provided novel insight into potential PIP2 interactions that have been shown to promote the conformational change responsible for FAK activation. The autoinhibitory interaction between the F2 subdomain of the FERM domain and the C-lobe of the kinase domain is formed by a network of charge complementarity, including interactions mediated by the KAKTLRK ridge and R634, a possible secondary PIP2-binding target that becomes exposed during separation of the two domains. PIP2 binding could potentially weaken the interactions that stabilize the KAKTLRK ridge and R634 in the inactive conformation, leading to conformational change and phosphorylation of Y397. This secondary PIP2 binding site may also stabilize FAK in its active conformation. The discovery of a second PIP2 binding site on the FAK kinase domain (i.e., Group II) suggests additional mechanisms contributing to conformational change that could operate independently or in concert with the FERM domain. While the initial interaction might resemble the pose illustrated in Fig 5B, twisting the domains to simultaneously maximize KAKTLRK ridge and Group II interactions with PIP2 could promote conformational change that affects the linker, exposing Y397 for phosphorylation. Further modeling and experimental studies are currently being conducted to fully elucidate the mechanism(s) of FAK activation in response to PIP2 binding.
Supporting Information
As the contact cutoff increases, the percentage of time when individual residues make contacts increase accordingly.
(DOCX)
(DOCX)
(DOCX)
The residues identified to bind PIP2 remain unchanged, meaning our results are insensitive to the cutoff distance.
(DOCX)
Identified residues in simulation are well-converged. Ignoring the equilibration period in original simulations or starting an independent simulation from a PIP2-bound conformation has minimal effects on the amino acid residues from FAK that interact with PIP2.
(DOCX)
Probability (p) of the first principal axis of FAK (V z) with respect to the bilayer normal (z). The broad distribution indicates there is no preferential binding pose. black: simulation I; red: simulation II; green: simulation III.
(TIFF)
Interaction energies between FAK and PIP2 in simulation I. red: van der Waals energy; black: electrostatic energy. Several PIP2-FAK dissociation and association events occur, as indicated by the significant changes in the interaction energy.
(TIFF)
A sequence similarity search was carried out on the human version of focal adhesion kinase 1 (Uniprot Q05397) using the BLAST server [32]. Clustal Omega [33] was used to perform a multiple sequence alignment (MSA) on 25 different species from the animal kingdom (ranging from mammals to sea urchins), with all alignment variables set to their default values. The alignment was visually inspected and manually adjusted in Seaview [34], with the final alignment input into Scorecons [35]. Scorecons was used to quantify residue conservation with respect to the human version of FAK used in the sequence alignment. Each residue in Group II was highly conserved, with the majority being completely conserved. What is interesting to note is that K578 and K621 are both located in flexible loop regions (on the A-loop and near the C-lobe, respectively). When K578 is mutated along with K581 to glutamic acid, FAK’s enzymatic activity is greatly enhanced (‘SuperFAK’ [36]), underscoring the key role K578 plays in activation of FAK, since it is next to the tyrosine residues that are phosphorylated by Src kinase (Y576 and Y577). In addition, the A-loop of FAK kinase was shown to be intimately involved in the allosteric pathway that exists between ATP and PIP2 binding recently revealed by all-atom molecular dynamics simulations [37]. The role of K621 in FAK function, if any, is unclear. This residue was in contact with PIP2 molecules for the longest duration in our simulations, and this behavior could be linked to the flexible nature of the long loop that connects two of the α-helices in the C-lobe of the kinase domain. Each of the Group II residues (except for K578, which is a well-characterized mutation) appear to be promising candidates for further experimental investigations into the effect they have on binding to PIP2. MSA was drawn using the ESPript 3.0 server [38] using default values for residue similarity scores (0.7). Similar residues are shown in bold type with white background, completely conserved residues are shown in white font with black background. First row: annotated secondary structure based on PDB 2J0J from avian Gallus gallus. All other rows: primary amino acid sequences of known FAK1 genes, listed by Uniprot identification numbers. Green background and black triangles: Group II residues. Scorecons results for Group II residues are listed below the MSA.
(PDF)
Acknowledgments
JF and BM would like to thank the WVU High Performance Computing shared facility for providing computational resources and J. Hall and M. Schaller for thoughtful discussion and interpretation of our results.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
West Virginia University provided startup funds to conduct this research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Burridge K, Fath K, Kelly T, Nuckolls G, Turner C (1988) Focal adhesions—transmembrane junctions between the extracellular-matrix and the cytoskeleton. Annu Rev Cell Biol 4: 487–525. [DOI] [PubMed] [Google Scholar]
- 2. Burridge K, ChrzanowskaWodnicka M (1996) Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol 12: 463–518. [DOI] [PubMed] [Google Scholar]
- 3. Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6: 56–68. [DOI] [PubMed] [Google Scholar]
- 4. Schlaepfer DD, Hauck CR, Sieg DJ (1999) Signaling through focal adhesion kinase. Prog Biophys Mol Biol 71: 435–478. [DOI] [PubMed] [Google Scholar]
- 5. Vadali K, Cai X, Schaller MD (2007) Focal adhesion kinase: an essential kinase in the regulation of cardiovascular functions. IUBMB Life 59: 709–716. [DOI] [PubMed] [Google Scholar]
- 6. Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M (1999) FAK and PYK2/CAK beta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci 22: 257–263. [DOI] [PubMed] [Google Scholar]
- 7. McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC (2005) The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer 5: 505–515. [DOI] [PubMed] [Google Scholar]
- 8. Cooper LA, Shen TL, Guan JL (2003) Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol 23: 8030–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ (2007) Structural basis for the autoinhibition of focal adhesion kinase. Cell 129: 1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goni GM, Epifano C, Boskovic J, Camacho-Artacho M, Zhou J, Bronowska A, et al. (2014) Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci USA 111: E3177–3186. 10.1073/pnas.1317022111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2 dependent binding of pp60src. Mol Cell Biol 14: 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Calalb MB, Polte TR, Hanks SK (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 15: 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38. [DOI] [PubMed] [Google Scholar]
- 14. Frame MC, Patel H, Serrels B, Lietha D, Eck MJ (2010) The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol 11: 802–814. 10.1038/nrm2996 [DOI] [PubMed] [Google Scholar]
- 15. Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, et al. (2008) Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol 28: 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, et al. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479: 552–555. 10.1038/nature10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH (2007) The MARTINI force field: Coarse grained model for biomolecular simulations. J Phys Chem B 111: 7812–7824. [DOI] [PubMed] [Google Scholar]
- 18. Periole X, Marrink SJ (2013) The Martini coarse-grained force field. Methods Mol Biol 924: 533–565. 10.1007/978-1-62703-017-5_20 [DOI] [PubMed] [Google Scholar]
- 19. Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, et al. (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30: 1545–1614. 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Jong DH, Singh G, Bennett WFD, Arnarez C, Wassenaar TA, Schäfer LV, et al. (2012) Improved parameters for the Martini coarse-grained protein force field. J Chem Theory Comput 9: 687–697. [DOI] [PubMed] [Google Scholar]
- 21. Periole X, Cavalli M, Marrink S-J, Ceruso MA (2009) Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J Chem Theory Comput 5: 2531–2543. [DOI] [PubMed] [Google Scholar]
- 22. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447. [DOI] [PubMed] [Google Scholar]
- 23. Ellenbroek WG, Wang Y-H, Christian DA, Discher DE, Janmey PA, Liu AJ (2011) Divalent Cation-Dependent Formation of Electrostatic PIP2 Clusters in Lipid Monolayers. Biophys J 101: 2178–2184. 10.1016/j.bpj.2011.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bretsche MS (1972) Asymmetrical lipid bilayer structure for biological-membranes. Nature New Biol 236: 11–12. [DOI] [PubMed] [Google Scholar]
- 25. Irino Y, Tokuda E, Hasegawa J, Itoh T, Takenawa T (2012) Quantification and visualization of phosphoinositides by quantum dot-labeled specific binding-domain probes. J Lipid Res 53: 810–819. 10.1194/jlr.D019547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM (2002) Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J 363: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dunty JM, Gabarra-Niecko V, King ML, Ceccarelli DF, Eck MJ, Schaller MD (2004) FERM domain interaction promotes FAK signaling. Mol Cell Biol 24: 5353–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou J, Bronowska A, Le Coq J, Lietha D, Graeter F (2015) Allosteric Regulation of Focal Adhesion Kinase by PIP2 and ATP. Biophys J 108: 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marrink SJ, de Vries AH, Mark AE (2003) Coarse grained model for semiquantitative lipid simulations. J Phys Chem B 108: 750–760. [Google Scholar]
- 31. Baron R, Trzesniak D, de Vries AH, Elsener A, Marrink SJ, van Gunsteren WF (2007) Comparison of thermodynamic properties of coarse-grained and atomic-level simulation models. ChemPhysChem 8: 452–461. [DOI] [PubMed] [Google Scholar]
- 32. Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schäffer AA, et al. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS Journal 272: 5101–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sievers F, Higgins DG (2014) Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. In: Russell DJ, editor. Multiple Sequence Alignment Methods. pp. 105–116. [DOI] [PubMed]
- 34. Guoy M, Guindon S, Gascuel O (2010) SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 27:221–224. 10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- 35. Valdar WSJ (2002) Scoring residue conservation. Proteins-Structure Function and Bioinformatics 48: 227–241. [DOI] [PubMed] [Google Scholar]
- 36. Gabarra-Niecko V, Keely PJ, Schaller MD (2002) Characterization of an activated mutant of focal adhesion kinase: 'SuperFAK', Biochem. J, 365:591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou J, Bronowska A, Le Coq J, Lietha D, Graeter F (2015) Allosteric Regulation of Focal Adhesion Kinase by PIP2 and ATP. Biophys J 108: 698–705. 10.1016/j.bpj.2014.11.3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res. 42: W320–W324. 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As the contact cutoff increases, the percentage of time when individual residues make contacts increase accordingly.
(DOCX)
(DOCX)
(DOCX)
The residues identified to bind PIP2 remain unchanged, meaning our results are insensitive to the cutoff distance.
(DOCX)
Identified residues in simulation are well-converged. Ignoring the equilibration period in original simulations or starting an independent simulation from a PIP2-bound conformation has minimal effects on the amino acid residues from FAK that interact with PIP2.
(DOCX)
Probability (p) of the first principal axis of FAK (V z) with respect to the bilayer normal (z). The broad distribution indicates there is no preferential binding pose. black: simulation I; red: simulation II; green: simulation III.
(TIFF)
Interaction energies between FAK and PIP2 in simulation I. red: van der Waals energy; black: electrostatic energy. Several PIP2-FAK dissociation and association events occur, as indicated by the significant changes in the interaction energy.
(TIFF)
A sequence similarity search was carried out on the human version of focal adhesion kinase 1 (Uniprot Q05397) using the BLAST server [32]. Clustal Omega [33] was used to perform a multiple sequence alignment (MSA) on 25 different species from the animal kingdom (ranging from mammals to sea urchins), with all alignment variables set to their default values. The alignment was visually inspected and manually adjusted in Seaview [34], with the final alignment input into Scorecons [35]. Scorecons was used to quantify residue conservation with respect to the human version of FAK used in the sequence alignment. Each residue in Group II was highly conserved, with the majority being completely conserved. What is interesting to note is that K578 and K621 are both located in flexible loop regions (on the A-loop and near the C-lobe, respectively). When K578 is mutated along with K581 to glutamic acid, FAK’s enzymatic activity is greatly enhanced (‘SuperFAK’ [36]), underscoring the key role K578 plays in activation of FAK, since it is next to the tyrosine residues that are phosphorylated by Src kinase (Y576 and Y577). In addition, the A-loop of FAK kinase was shown to be intimately involved in the allosteric pathway that exists between ATP and PIP2 binding recently revealed by all-atom molecular dynamics simulations [37]. The role of K621 in FAK function, if any, is unclear. This residue was in contact with PIP2 molecules for the longest duration in our simulations, and this behavior could be linked to the flexible nature of the long loop that connects two of the α-helices in the C-lobe of the kinase domain. Each of the Group II residues (except for K578, which is a well-characterized mutation) appear to be promising candidates for further experimental investigations into the effect they have on binding to PIP2. MSA was drawn using the ESPript 3.0 server [38] using default values for residue similarity scores (0.7). Similar residues are shown in bold type with white background, completely conserved residues are shown in white font with black background. First row: annotated secondary structure based on PDB 2J0J from avian Gallus gallus. All other rows: primary amino acid sequences of known FAK1 genes, listed by Uniprot identification numbers. Green background and black triangles: Group II residues. Scorecons results for Group II residues are listed below the MSA.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.