

Abstract
RNA-sequencing of a case of acute myeloid leukemia with the bone marrow karyotype 46,XY,t(2;14)(q22;q32)[5]/47,XY,idem,+?4,del(6)(q13q21)[cp6]/46,XY[4] showed that the t(2;14) generated a ZEB2-BCL11B chimera in which exon 2 of ZEB2 (nucleotide 595 in the sequence with accession number NM_014795.3) was fused to exon 2 of BCL11B (nucleotide 554 in the sequence with accession number NM_022898.2). RT-PCR together with Sanger sequencing verified the presence of the above-mentioned fusion transcript. All functional domains of BCL11B are retained in the chimeric protein. Abnormal expression of BCL11B coding regions subjected to control by the ZEB2 promoter seems to be the leukemogenic mechanism behind the translocation.
Introduction
Acute myeloid leukemia (AML, also known as acute myelogenous leukemia or acute nonlymphocytic leukemia) is a malignancy of the myeloid line of blood cells, characterized by the rapid growth of abnormal immature white blood cells (blasts) that accumulate in the bone marrow and interfere with the production of normal blood cells. According to World Health Organization (WHO) criteria, a diagnosis of AML is established by demonstrating presence of 20% or more blasts in the blood and/or bone marrow [1]. AML is more common in adults, in whom the incidence rises steeply after the age of 55–60 years. The median age is 65 years and men are more often affected than women [2].
Chromosome banding analysis of bone marrow cells has revealed acquired clonal chromosomal rearrangements in most AML patients. The types of aberrations and the frequency with which they are found are influenced by factors such as age, previous treatment or other exposure to genotoxics, gender, ethnic or geographic origin, and constitutional genetics [3]. Structural chromosomal aberrations in AML such as translocations, inversions, and deletions often result in the generation of a fusion gene, i.e., a hybrid gene formed from two previously separate genes, which affects cellular pathways of myeloid maturation and proliferation [3]. In the WHO classification of AML (and myeloid neoplasms in general), the chromosomal rearrangements/fusion genes play an important role in the grouping of AMLs into diagnostic-prognostic-therapeutic entities; for example, rearrangements such as t(8;21)(q22;q22) (fusion gene RUNX1-RUNX1T1), inv(16)(p13.1q22) (fusion gene CBFB-MYH11), and t(15;17)(q22;q12) (fusion gene PML-RARA) define specific AML subsets and their finding is sufficient for an AML diagnosis regardless of the blast percentage in the blood or bone marrow [1]. The three above-mentioned chromosomal aberrations/fusion genes are also associated with favorable prognosis [3] whereas others, e.g., KAT6A-CREBBP, the result of a t(8;16)(p11;p13) chromosomal translocation, are associated with poor prognosis [4]. Furthermore, fusion genes can be the targets of specific molecular therapy, the paradigmatic example being the use of all-trans retinoic acid (ATRA)/arsenic trioxide (ATO) in the treatment of acute promyelocytic leukemia (APL) carrying the PML—RARA fusion which is key to APL leukemogenesis generated by t(15;17) [5]. Hence, the identification and characterization of novel leukemia-specific fusion genes and the study of their effects on cellular processes have clinical significance.
The “traditional” methodology to detect fusion genes in cancer begins with cytogenetic analysis to find the chromosomal rearrangement, followed by utilization of fluorescence in situ hybridization (FISH) techniques to find a probe which spans the chromosomal breakpoint. Eventually, molecular cloning is performed to localize the breakpoint more precisely and identify the genes fused by the chromosomal rearrangement. Although laborious, the above-mentioned sequential approach is quite robust and reliable and a number of fusion genes have been cloned by such means [3]. The introduction of next generation sequencing has opened up new possibilities to detect fusion genes in cancer [6]. The search for fusion genes presently in the main proceeds along two main axes, either using brute molecular genetic force (i.e., high throughput sequencing) in the common cancers where cytogenetic guidance is sparse at best, or by combination of cytogenetics and sequencing so that the karyotypic information can be used to select among the numerous candidate fusion genes offered by the latter method to find only those few that map to chromosomal breakpoints [7]. The actual presence of the fusion genes thus detected is then confirmed by other molecular genetic (PCR and Sanger sequencing analyses) and molecular cytogenetic methods (FISH).
We and others have used combinations of cytogenetics and RNA-sequencing to detect the fusion genes associated with acquired cancer-specific chromosomal rearrangements [7]. These fusion genes were considered to be primary tumorigenic events because the chromosomal rearrangements generating them were often seen as the sole aberration by cytogenetic analysis.
Recently, a recurrent t(2;14)(q22;q32) chromosomal translocation was reported in immature early T-cell precursor leukemia (ETP-ALL) [8]. Using FISH, the translocation was shown to affect the BCL11B and ZEB2 loci on chromosome bands 14q32 and 2q22, respectively, resulting in overexpression of ZEB2. However, neither BCL11B-ZEB2 nor ZEB2-BCL11B (nor their respective fusion transcripts) was demonstrated in that study.
We here report an AML case carrying the same t(2;14)(q22;q32) chromosomal translocation in bone marrow cells as that reported in ETP-ALL. Using RNA-sequencing, the translocation was shown to result in generation of a ZEB2-BCL11B fusion gene.
Materials and Methods
Ethical approval
The study was approved by the Regional Committee for Medical and Health Research Ethics, South-East Norway (REK Sør) http://helseforskning.etikkom.no). Written informed consent was obtained from the patient prior to his death. The consent sought from the participant included consent for the publication of his clinical details. The ethics committee’s approval included a review of the consent procedure. All patient information has been anonymized and de-identified.
Patient
A 28-year-old man was transferred to our institution with a diagnosis of AML (Table 1 and Fig 1A). He presented with signs of infection (fever and cough) and a remarkable swelling on the neck with slight rubor and tenderness. Computer tomography (CT) demonstrated bilateral massive lymphadenopathy with necrosis. His AML turned out to be chemoresistant as he failed three induction courses (Table 1). Further leukemia-directed treatment was therefore discontinued, and the patient succumbed within short of infections.
Table 1. Hematological data and treatment on the AML patient with the t(2;14) chromosomal aberration.
| Blood analysis | Smear | Molecular genetic analysis in BM | Immunophenotype in BM | Treatment | ||||
|---|---|---|---|---|---|---|---|---|
| Hb | 8.0 g/dL (13.4–17.0) | BM | 90% blasts with a sparse cytoplasmic brim and no granulation | Positive | FLT3-ITD | Positive | Strongly CD34+ and CD117+ (88%) | 1. Idarubicin 12mg/m2 day 1–3, cytarabin 200 mg/m2 day 1–7 |
| Plc | 291 x 109/L (145–390) | PB | 85% blasts | Negative | FLT3-TKD, EVI-1, CEBPalfa, NPM1, CBFB-MYH11, RUNX1-RUNX1T1 | Coexpression | HLA-DR, CD38, CD13, CD71, CD7, Tdt | 2. Cytarabin 1g/m2 x 2 day 1–6, amsakrin 120 mg/m2 day 4–6 |
| WBC | 29.7 x 109/L (3.5–10) | 3. Amsakrin 150 mg/m2 day 1–5, etoposide 110 mg/m2 day 1–5, cytarabin 200 mg/m2 day 1–5 | ||||||
| LD | 677 U/L (105–205) | |||||||
Hb, hemoglobin; Plc, platelet count; WBC, white bloood cells; LD, lactate dehydrogenase; BM, bone marrow; PB, peripheral blood.
Fig 1. Hematological and cytogenetic data on the AML patient with t(2;14).
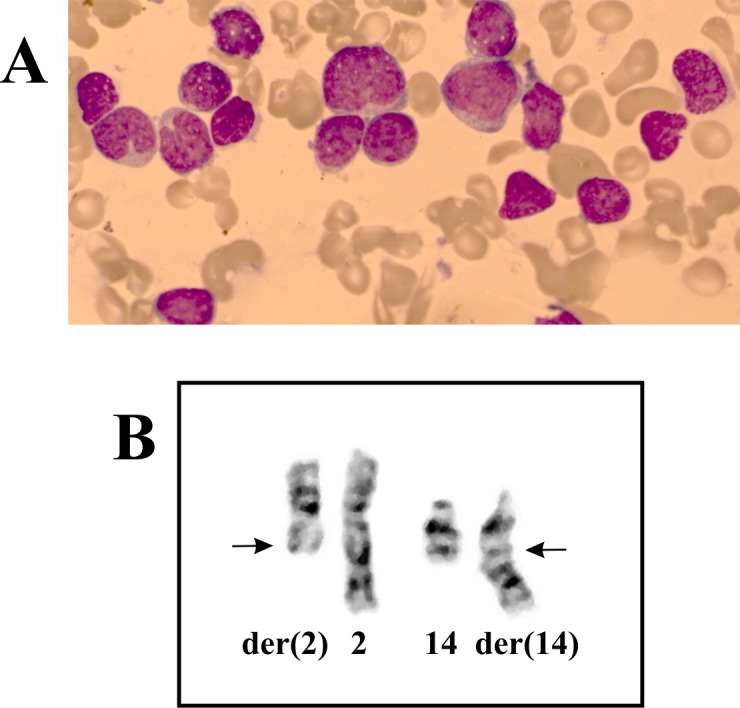
A) Blasts without granulation and with relatively little cytoplasm in the patient´s bone marrow. B) Partial karyotype showing the der(2)t(2;14)(q22;q32) and der(14)t(2;14)(q22;q32) together with the corresponding normal chromosome homologs; breakpoint positions are indicated by arrows.
Control samples
Two controls were used: A previously reported AML with karyotype at diagnosis 46,XY,add(1)(p13),t(8;21)(p11;q22),der(16)t(1;16)(p13;p13)[9]/46,XY[1] which was shown to carry a cryptic KAT6A-CREBBP fusion gene [9], and human bone marrow total RNA (Clontech Laboratories, Inc., Mountainview, CA, USA). According to the company’s information, it is a mixture of normal bone marrow pooled from 6 male/female Caucasians, ages: 38–59.
G-banding and karyotyping
Bone marrow cells were cytogenetically investigated by standard methods. Chromosome preparations were made from metaphase cells of a 24-hours culture, G-banded using Leishman stain, and karyotyped according to ISCN 2009 guidelines [10].
RNA-sequencing
Total RNA was extracted from the patient’s bone marrow at the time of diagnosis using miRNeasy Mini Kit according to the manufacturer’s instructions (Qiagen Nordic, Oslo, Norway). Subsequently, total RNA was purified using QIAcube (Qiagen). The RNA quality was evaluated using the Experion Automated Electrophoresis System (Bio-Rad Laboratories, Oslo, Norway). The RNA Quality Indicator (RQI) was 7.8. Three μg of total RNA were sent for high-throughput paired-end RNA-sequencing at the Norwegian Sequencing Centre, Ullevål Hospital (http://www.sequencing.uio.no/). The RNA was sequenced using an Illumina HiSeq 2000 instrument and the Illumina software pipeline was used to process image data into raw sequencing data. The TruSeq Stranded mRNA sample preparation protocol was used (http://support.illumina.com/downloads/truseq_stranded_mrna_sample_preparation_guide_15031047.ilmn) giving reads of a length of 100 base pairs. A total of 70 million reads were obtained. The quality of the raw sequence data was assessed using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Two softwares were used for the discovery of fusion transcripts: FusionCatcher (version 0.99.3a beta-April 15, 2014) with the associated ENSEMBL, UCSC, and RefSeq databases automatically downloaded by FusionCatcher (https://code.google.com/p/fusioncatcher/) [11] and ChimeraScan (https://code.google.com/p/chimerascan/) [12]. To verify further the fusion genes which were found by FusionCatcher and ChimeraScan, the “grep” command (http://en.wikipedia.org/wiki/Grep) was used to search the fastq files of the sequence data (http://en.wikipedia.org/wiki/FASTQ_format). Our “specific expression” was a sequence of 20 nucleotides at the fusion point, 10 bases upstream (5´-end gene), and 10 bases downstream from the junction (3´-end gene). The expressions were: "AGGAAAAACGCAGAGGCTGA" (type 1), "AGGAAAAACGAGGCTGACCA" (type 2), "GACTTCGCAGAGGCTGACCA" (type 3), and "AGGAAAAACGAGCACAAAAG" (type 4) for the four alternative fusion transcripts of ZEB2-BCL11B which were found with FusionCatcher (see S1 Table). The sequences obtained by “grep” were blasted against the human genomic plus transcript database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) as well as the reference sequences NM_014795.3 (ZEB2) and NM_022898.2 (BCL11B).
RT-PCR analysis
The primers used for PCR amplification and Sanger sequencing are listed in Table 2.
Table 2. Primers used for PCR amplification and Sanger sequencing analyses.
| Name | Sequence (5´->3´) | Direction | Position / Exon | Reference Sequence | Gene |
|---|---|---|---|---|---|
| ZEB2-383F1 | CACACTTCGCGGCTTCTTCATGCTT | Forward | 383–407 / 1 | NM_014795.3 | ZEB2 |
| ZEB2-485F1 | CGAGTCCATGCGAACTGCCATCTG | Forward | 485–508 / 2 | NM_014795.3 | ZEB2 |
| ZEB2-672R1 | TGGTCCAGAGGGTTGGCAATACCG | Reverse | 695–672 / 3 | NM_014795.3 | ZEB2 |
| ZEB2-985R1 | GCTGACTGCATGACCATCGCGTTC | Reverse | 985–1008 / 5 | NM_014795.3 | ZEB2 |
| BCL11B-464F1 | CTCCGGGCGATGCCAGAATAGATG | Forward | 464–487 / 1 | NM_022898.2 | BCL11B |
| BCL11B-654R1 | CACTGGCCACAGGTGAGCAGGTCA | Reverse | 677–654 / 2 | NM_022898.2 | BCL11B |
| BCL11B-969R1 | GCAGGAACCACGCGCTGTTGAAG | Reverse | 991–969 / 3 | NM_022898.2 | BCL11B |
| ABL1-91F1 | CAGCGGCCAGTAGCATCTGACTTTG | Forward | 280–304 / 2 | NM_005157.5 | ABL1 |
| ABL1-404R1 | CTCAGCAGATACTCAGCGGCATTGC | Reverse | 617–593 / 3 | NM_005157.5 | ABL1 |
For RT-PCR, one μg of total RNA was reverse-transcribed in a 20 μL reaction volume using iScript Advanced cDNA Synthesis Kit for RT-qPCR according to the manufacturer’s instructions (Bio-Rad Laboratories, Oslo, Norway). The 25 μL PCR volume contained 12.5 μL Premix Ex Taq DNA Polymerase Hot Start Version (Takara Bio Europe/SAS, Saint-Germain-en-Laye, France), 1 μL of cDNA, and 0.4 μM of each of the forward and reverse primer. The primer sets ZEB2-383F1/BCL11B-654R1 and ZEB2-485F1/BCL11B-654R1 were used to detect possible ZEB2-BCL11B fusion transcripts. The primer sets BCL11B-464F1/ZEB2-985R1 and BCL11B-464F1/ZEB2-672R1 were used to detect possible BCL11B-ZEB2 fusion transcripts. To detect the expression of the normal ZEB2 gene, the primer set ZEB2-383F1/ZEB2-985R1 was used. To detect expression of the normal BCL11B gene, the primer set BCL11B-464F1/BCL11B-969R1 was used. The quality of the cDNA synthesis was examined by amplification of a cDNA fragment of the ABL1 gene using the primers ABL1-91F1 and ABL1-404R1. The PCR amplifications were run on a C-1000 Thermal cycler (Bio-Rad Laboratories) with an initial denaturation at 94°C for 30 sec, followed by 35 cycles of 7 sec at 98°C, 2 min at 68°C, and a final extension for 5 min at 68°C. Three μL of the PCR products were stained with GelRed (Biotium, Hayward, CA, USA), analysed by electrophoresis through 1.0% agarose gel, and photographed. The remaining 22 μL PCR products were purified using the MinElute PCR purification kit (Qiagen Nordic, Oslo, Norway) and sequenced at GATC Biotech (Germany, http://www.gatc-biotech.com/en/home.html). The BLAST software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for computer analysis of sequence data.
Results
G-banding
G-banding analysis of bone marrow cells at diagnosis showed two cytogenetically related subclones. In the first subclone, five cells carried the chromosomal translocation t(2;14)(q22;q32) as the sole cytogenetic abnormality (Fig 1B). In the second subclone, six cells carried an extra chromosome, presumably #4, and an interstitial deletion in chromosome 6 together with the above-mentioned translocation. Thus, G-banding analysis of bone marrow cells at diagnosis indicated that t(2;14)(q22;q32) was the primary cytogenetic abnormality. The resulting karyotype was 46,XY,t(2;14)(q22;q32)[5]/47,XY,idem,+?4,del(6)(q13q21)[cp6]/46,XY[4].
RNA-sequencing
Using FusionCatcher on the raw sequencing data obtained from the Genomics Core Facility, 103 potential fusion transcripts were found (S1 Table), among them the ZEB2-BCL11B. Using ChimeraScan with the same fastq file, 263 potential fusion transcripts were found (S2 Table), among them the ZEB2-BCL11B. Neither FusionCatcher nor ChimeraScan found the reciprocal BCL11B-ZEB2 (S1 and S2 Tables).
Further analysis of the fusion transcripts identified with the two softwares showed that FusionCatcher identified four types (types 1–4) of ZEB2-BCL11B (S1 Table). Type 1 corresponds to the fusion of exon 2 of ZEB2 with exon 2 of BCL11B. Type 2 corresponds to the same fusion but without the first three, CAG, nucleotides of exon 2 of BCL11B. Type 3 corresponds to the fusion of exon 1 of ZEB2 with exon 2 of BCL11B without the first three bases, CAG, of exon 2 of BCL11B. Type 4 corresponds to the fusion of exon 2 of ZEB2 with nt 719 within exon 2 of BCL11B. Only the type 1 ZEB2-BCL11B fusion transcript was found by ChimeraScan (S2 Table).
In order to verify the fusion obtained with the FusionCatcher and ChimeraScan softwares, we used the “grep” command utility to search for expressions composed of 10 nt of ZEB2 and 10 nt of BCL11B upstream and downstream of the fusion point (Table 3). Using the expression "AGGAAAAACGCAGAGGCTGA", which is composed of 10 nt, "AGGAAAAACG", from ZEB2 and 10 nt, "CAGAGGCTGA", from BCL11B, 18 sequences were retrieved. With the expression "AGGAAAAACGAGGCTGACCA", which is composed of 10 nt, "AGGAAAAACG", from ZEB2 and 10 nt, "AGGCTGACCA", from BCL11B, 4 sequences were retrieved. The expression "GACTTCGCAGAGGCTGACCA", which is composed of 10 nt, "GACTTCGCAG", from ZEB2 and 10 nt, "AGGCTGACCA", from BCL11B, retrieved 2 sequences. Finally, "AGGAAAAACGAGCACAAAAG", which is composed of the 10 nt "AGGAAAAACG" from ZEB2 and the 10 nt "AGCACAAAAG" from BCL11B, retrieved 1 sequence (Table 3).
Table 3. Sequences retrieved with the «grep» command using the expressions "AGGAAAAACGCAGAGGCTGA", "AGGAAAAACGAGGCTGACCA", "GACTTCGCAGAGGCTGACCA", and "AGGAAAAACGAGCACAAAAG" which correspond to type 1, type 2, type 3 and type 4 of ZEB2-BCL11B fusion transcript, respectively.
The sequences of BCL11B are in bold.
| Type (expression) | Sequences |
|---|---|
| Type 1 (AGGAAAAACG CAGAGGCTGA) | ATCCGCTCTTATCAATGAAGCAGCCGATCATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCAT |
| CAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCC | |
| CCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGGGCTGATGGTG | |
| ATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGGGCTGATG | |
| CAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGGGCTGATGGTGG | |
| AGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGAGCACAAAAGGAAGCAGTGTGGCGGCAGCTTGGGTGCCTGCTA | |
| GGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGT | |
| GATCCGCTCTTATCAATGAAGCAGCCGATCATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCA | |
| CGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGA | |
| ATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGA | |
| GGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTC | |
|
GGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGG
CAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGG | |
| TATCAATGAAGCAGCCGATCATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCC | |
| AAGCAGCCGATCATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCT | |
| ATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGA | |
| ATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGA | |
| CAAGCCAATCCCAGGAGGAAAAACGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGG | |
| Type 2 (AGGAAAAACG AGGCTGACCA) | CAAGCCAATCCCAGGAGGAAAAACGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGGGCT |
| TCTGATCCGCTCTTATCAATGAAGCAGCCGATCATGGCGGATGGACCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGAGGCTGACCA | |
| CAAGCCAATCCCAGGAGGAAAAACGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGGGCT | |
| AAACAAGCCAATCCCAGGAGGAAAAACGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGG | |
| Type 3 (GACTTCGCAG AGGCTGACCA) | CTACAAACAAGACTTCGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGATAGAGGAGCCAAGTGGCCTGGGGCTGATGG |
| GCTTTTTCTTCTCACCATTTCTGGCCAAAACTACAAACAAGACTTCGCAGAGGCTGACCATGTGGAGGCCGCCATCCTCGAAGAAGACGAGGGTCTGGAGA | |
| Type 4 (AGGAAAAACG AGCACAAAAG) | ATGAAGCAGCCGATCATGGCGGATGGCCCCCGGTGCAAGAGGCGCAAACAAGCCAATCCCAGGAGGAAAAACGAGCACAAAAGGAAGCAGTGTGGCGGCAG |
Because the ZEB2 and BCL11B genes map to chromosome bands 2q22 and 14q32, respectively, and the chromosomal translocation t(2;14)(q22;q32) was the primary cytogenetic aberration, we decided to investigate further the patient’s bone marrow for the presence of the ZEB2-BCL11B fusion transcript using molecular techniques. No other fusions were examined.
Molecular Genetic Confirmation of the Fusions
PCR with the primers ABL1-91F1 and ABL1-404R1 amplified a 338 bp ABL1 fragment from the cDNAs of the AML-control (patient with KAT6A-CREBBP fusion) and the normal bone marrow (Fig 2). From the cDNA of the patient with t(2;14), two fragments were amplified: 338 bp and 901 bp in size, respectively. The 338 bp fragment corresponds to the processed mRNA (exon 2/exon 3 of ABL1) while the 901 bp fragment corresponds to the presence of an unspliced intron 2 (exon 2-intron 2- exon 3) of ABL1. The amplified cDNA fragments of ABL1 indicated that the synthesized cDNAs were of good quality.
Fig 2. RT-PCR amplification.

Gel electrophoresis for the ZEB2-BCL11B, BCL11B-ZEB2, ZEB2, BCL11B, and ABL1 cDNA transcripts of AML patient with t(2;14), control AML patient with KAT6A-CREBBP fusion, and normal bone marrow.
PCR with the ZEB2-383F1 and BCL11B-654R1 primer combination amplified two cDNA fragments of size 337 bp and 192 bp only from the cDNA of the patient with t(2;14) (Fig 2). Sanger sequencing of the long, 337 bp, fragment showed that it was a type 1 ZEB2-BCL11B chimeric cDNA fragment with the fusion point identical to that found with FusionCatcher, i.e., exon 2 of ZEB2 (nucleotide 595 in the sequence with accession number NM_014795.3) was fused to exon 2 of BCL11B (nucleotide 554 in the sequence with accession number NM_022898.2) (Fig 3A). Sanger sequencing of the short, 192 bp, fragment showed that it was a type 3 ZEB2-BCL11B chimeric cDNA fragment in which the first three bases, CAG, of exon 2 of BCL11B were also present, i.e., exon 1 of ZEB2 (nucleotide 453 in the sequence with accession number NM_014795.3) was fused to exon 2 of BCL11B (nucleotide 554 in the sequence with accession number NM_022898.2) (Fig 3B).
Fig 3. Sanger sequence of the amplified products from the AML patient with the chromosomal aberration t(2;14).

A) Partial sequence chromatogram of the cDNA fragment showing that exon 2 of ZEB2 is fused to exon 2 of BCL11B (type 1 fusion transcript). B) Partial sequence chromatogram of the cDNA fragment showing that exon 1 of ZEB2 is fused to exon 2 of BCL11B (type 3 fusion transcript). C) Sequence of the amplified cDNA fragment using the primers ZEB2-485F1 and BCL11B-654R1 (shown in box). The fusion point “gc” is double underlined. The open reading frame is also shown.
PCR with the ZEB2-485F1 and BCL11B-654R1 primer combination amplified a 235 bp cDNA fragment only from the cDNA of the patient with t(2;14) (Fig 2). Sanger sequencing showed that it was a type 1 ZEB2-BCL11B chimeric cDNA fragment (fusion of exon 2 of ZEB2 with exon 2 of BCL11B) (Fig 3A and 3C). No cDNA amplified product was obtained when BCL11B forward and ZEB2 reverse primers were used, suggesting that BCL11B-ZEB2 was absent or not expressed (Fig 2).
Normal ZEB2 cDNA fragments were amplified from the cDNAs of the patient with t(2;14), the AML-control, and the normal bone marrow suggesting that ZEB2 is expressed in the three samples (Fig 2). BCL11B cDNA fragments were amplified from the cDNAs of the AML-control and the normal bone marrow, whereas no BCL11B cDNA fragments were amplified from the patient´s cDNA suggesting that BCL11B was not expressed or had very low expression in the bone marrow of the patient with t(2;14)-ZEB2-BCL11B fusion (Fig 2).
Discussion
We present a case of AML which had FLT3-internal tandem duplication mutation (FLT3-ITD) and cytogenetic aberrations at diagnosis. The primary cytogenetic abnormality was a t(2;14)(q22;q32) chromosomal translocation which rearranged and fused the ZEB2 gene in 2q22 with the BCL11B gene in 14q32. The translocation resulted in the expression of chimeric ZEB2-BCL11B transcripts whereas the reciprocal BCL11B-ZEB2 fusion was either absent or not expressed.
FLT3-ITD mutation is an important prognostic factor but the prognostic impact must be interpreted against the genetic context [13]. In patients with normal karyotype, FLT3-ITD is associated with poor prognosis [14–17]. In acute promyelocytic leukemia with t(15;17), the prognostic impact of FLT3-ITD mutation is minimal [18]. In other contexts, both the mutant to wild type allelic ratio and the position of the ITD within the FLT3 gene may carry prognostic impact [19, 20]. The present case of AML turned out to be chemoresistant as it failed three induction courses. The FLT3-ITD mutations might have played a role in this but so may the presence at diagnosis of the chromosomal aberration t(2;14)(q22;q32) and its molecular consequence, the generation of a ZEB2-BCL11B. One cannot at present say which is more prognostically important.
The same chromosomal translocation as the one we found, t(2;14)(q22;q32), was recently reported in ETP-ALL [8]. In that study, neither BCL11B-ZEB2 nor ZEB2-BCL11B (nor their fusion transcripts) was detected although the translocation was shown to result in overexpression of ZEB2 [8].
The ZEB2 gene (in 2q22) codes for a protein which is a member of the Zfh1 family of 2-handed zinc finger/homeodomain proteins. ZEB2 protein is located in the nucleus and functions as a DNA-binding transcriptional repressor that interacts with activated SMADs, the transducers of TGF-beta signaling, and interacts with the nucleosome remodeling and histone deacetylation (NURD) complex (http://omim.org/entry/605802). ZEB2 and its paralogue ZEB1 play a pivotal role in vertebrate embryogenesis [21]. Recent evidence shows that both proteins also drive the process of epithelial-mesenchymal transition during cancer progression [22]. The two proteins are involved in the control of several cancer cell capabilities, including proliferation, senescence, apoptosis, angiogenesis, resistance to chemotherapy and radiotherapy, and tumor invasiveness and metastasis [22].
The BCL11B gene and its paralogue BCL11A code for krueppel-like C2H2-type zinc finger proteins of the BCL11 family of transcription factors [23]. Although the specific function of BCL11B has not been determined, the encoded protein is known to be a bi-functional transcriptional regulator that acts as a repressor and transactivator [24]. BCL11B protein is essential for T-cell development and for maintenance of T-cell identity; in fact, T-cells acquire NK cell properties upon BCL11B deletion [23]. BCL11B gene alterations are related to malignant T-cell transformation in hematological malignancies [24]. The fusion genes BCL11B-TLX3, BCL11B-NKX2-5, BCL11B-TRD, and IKZF2-BCL11B have all been reported in T-cell acute lymphoblastic leukemia [24]. An RN28S1-BCL11B fusion transcript was also identified in a case of mixed-lineage (T/myeloid) acute leukemia with t(6;14)(q25;q32) [25]. The RN28S1 gene is not translated to protein and BCL11B and RN28S1 were fused in opposite transcription directions. Thus, disruption of the normal functions of BCL11B seemed to contribute to leukemogenesis in this case [25]. BCL11B was also involved in a case of AML with a similar chromosomal aberration, a t(6;14)(q25~q26;q32), but there was not enough material to identify the partner genomic locus [26]. Very recently, BCL11B was shown to be involved in 14q32 translocations with different chromosomal partners in AML [27].
In the ZEB2-BCL11B fusion transcripts 1 and 2, the sequence of ZEB2 involved codes only for the first 24 amino acids (MKQPIMADGPRCKRRKQANPRRKN) of the ZEB2 protein (starting ATG codon in exon 2, on position 523–525 of the ZEB2 sequence with accession number NM_014795.3) (Fig 3C). This amino acid sequence does not contain any functional domain except nuclear localization signals (KRRK, PRRK, and PRCKRRK), but replaces the first 19 amino acids of BCL11B (MSRRKQGNPQHLSQRELIT) in the ZEB2-BCL11B chimeric protein. The BCL11B in the ZEB2-BCL11B chimeric protein retains all functional domains, i.e., the 6 krueppel-like Zn-finger domains and the proline-rich region [24]. In the ZEB2-BCL11B fusion transcript 3, the untranslated exon 1 of ZEB2 is fused to exon 2 of BCL11B.
Taking all the data into consideration, i.e., presence of ZEB2-BCL11B fusion transcript and absence of BCL11B-ZEB2 fusion transcript and wild type BCL11B, we believe that placing all functional domains of BCL11B under the control of the ZEB2 promoter represents the leukemogenic mechanism for the ZEB2-BCL11B chimera. Consequently, the principal result should be abnormal expression of BCL11B. Integrative genomic analyses of ZEB2 showed that the gene’s proximal promoter region contains a conserved Ets-Smad-binding CGGAGAC motif as well as bHLH-, POU/OCT-, and HIF1-binding sites [28]. Nevertheless, one cannot rule out the possible importance of the 24 amino acids (MKQPIMADGPRCKRRKQANPRRKN) from ZEB2 in the chimeric protein.
According to the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman, last updated on May 7, 2015), a t(2;14)(q21;q32) chromosome translocation was reported in three cases of AML and a t(2;14)(q23;q32) in one case. No information is at hand about the molecular consequences of the translocation in the four cases, but we surmise that it could be generation of a ZEB2-BCL11B fusion gene. The difference between two neighboring bands (2q21 and 2q22 or 2q22 and 2q23) would be close to or beyond the resolution level of G-banding. Worthy of mention is furthermore that all five patients (including the present case) so far reported with the t(2;14) aberration were male; this may or may not be coincidental.
In conclusion, we show here that the chromosomal translocation t(2;14)(q22;q32) in AML results in ZEB2-BCL11B fusion transcripts the leukemogenic mechanism of which seems to be that the coding regions of the BCL11B gene come under the control of the ZEB2 promoter.
Supporting Information
Fusion transcripts detected using FusionCatcher.
(XLSX)
Fusion transcripts detected using ChimeraScan.
(XLSX)
Data Availability
All data underlying the findings in the study are freely available in the paper and in the Supporting Information.
Funding Statement
This work was supported by grants from the Norwegian Cancer Society and the Norwegian Radium Hospital Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4 ed Lyon: IARC; 2008. [Google Scholar]
- 2. Deschler B, Lubbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006;107(9):2099–107. 10.1002/cncr.22233 . [DOI] [PubMed] [Google Scholar]
- 3. Heim S, Mitelman F. Cancer Cytogenetics. Third Edition ed: Wiley-Blackwell; 2009. [Google Scholar]
- 4. Gervais C, Murati A, Helias C, Struski S, Eischen A, Lippert E, et al. Acute myeloid leukaemia with 8p11 (MYST3) rearrangement: an integrated cytologic, cytogenetic and molecular study by the groupe francophone de cytogenetique hematologique. Leukemia. 2008;22(8):1567–75. Epub 2008/06/06. [pii]. . [DOI] [PubMed] [Google Scholar]
- 5. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–21. 10.1056/NEJMoa1300874 . [DOI] [PubMed] [Google Scholar]
- 6. Annala MJ, Parker BC, Zhang W, Nykter M. Fusion genes and their discovery using high throughput sequencing. Cancer Lett. 2013;340(2):192–200. 10.1016/j.canlet.2013.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Panagopoulos I, Thorsen J, Gorunova L, Micci F, Heim S. Sequential combination of karyotyping and RNA-sequencing in the search for cancer-specific fusion genes. Int J Biochem Cell Biol. 2014;53:462–5. 10.1016/j.biocel.2014.05.018 . [DOI] [PubMed] [Google Scholar]
- 8. Goossens S, Radaelli E, Blanchet O, Durinck K, Van der Meulen J, Peirs S, et al. ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat Commun. 2015;6:5794 10.1038/ncomms6794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Panagopoulos I, Torkildsen S, Gorunova L, Tierens A, Tjonnfjord GE, Heim S. Comparison between karyotyping-FISH-reverse transcription PCR and RNA-sequencing-fusion gene identification programs in the detection of KAT6A-CREBBP in acute myeloid leukemia. PLoS One. 2014;9(5):e96570 10.1371/journal.pone.0096570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaffer LG, Slovak ML, Campbell LJ. ISCN 2009: an International System for Human Cytogenetic Nomenclature. Basel: Karger; 2009. [Google Scholar]
- 11. Nicorici D, Satalan H, Edgren H, Kangaspeska S, Murumagi A, Kallioniemi O, et al. FusionCatcher—a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv. 2014. 10.1101/011650 [DOI] [Google Scholar]
- 12. Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27(20):2903–4. 10.1093/bioinformatics/btr467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am Soc Hematol Educ Program. 2013;2013:220–6. 10.1182/asheducation-2013.1.220 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–74. 10.1182/blood-2009-07-235358 . [DOI] [PubMed] [Google Scholar]
- 15. Estey EH. Acute myeloid leukemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(1):89–99. 10.1002/ajh.22246 . [DOI] [PubMed] [Google Scholar]
- 16. Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88(4):318–27. 10.1002/ajh.23404 . [DOI] [PubMed] [Google Scholar]
- 17. Estey EH. Acute myeloid leukemia: 2014 update on risk-stratification and management. Am J Hematol. 2014;89(11):1063–81. 10.1002/ajh.23834 . [DOI] [PubMed] [Google Scholar]
- 18. Barragan E, Montesinos P, Camos M, Gonzalez M, Calasanz MJ, Roman-Gomez J, et al. Prognostic value of FLT3 mutations in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline monochemotherapy. Haematologica. 2011;96(10):1470–7. 10.3324/haematol.2011.044933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114(12):2386–92. 10.1182/blood-2009-03-209999 . [DOI] [PubMed] [Google Scholar]
- 20. Schlenk RF, Kayser S, Bullinger L, Kobbe G, Casper J, Ringhoffer M, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124(23):3441–9. 10.1182/blood-2014-05-578070 . [DOI] [PubMed] [Google Scholar]
- 21. Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66(5):773–87. 10.1007/s00018-008-8465-8 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanchez-Tillo E, Siles L, de Barrios O, Cuatrecasas M, Vaquero EC, Castells A, et al. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res. 2011;1(7):897–912. [PMC free article] [PubMed] [Google Scholar]
- 23. Liu P, Li P, Burke S. Critical roles of Bcl11b in T-cell development and maintenance of T-cell identity. Immunol Rev. 2010;238(1):138–49. 10.1111/j.1600-065X.2010.00953.x . [DOI] [PubMed] [Google Scholar]
- 24. Huang X, Du X, Li Y. The role of BCL11B in hematological malignancy. Exp Hematol Oncol. 2012;1(1):22 10.1186/2162-3619-1-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobayashi S, Taki T, Nagoshi H, Chinen Y, Yokokawa Y, Kanegane H, et al. Identification of novel fusion genes with 28S ribosomal DNA in hematologic malignancies. Int J Oncol. 2014;44(4):1193–8. 10.3892/ijo.2014.2291 . [DOI] [PubMed] [Google Scholar]
- 26. Bezrookove V, van Zelderen-Bhola SL, Brink A, Szuhai K, Raap AK, Barge R, et al. A novel t(6;14)(q25-q27;q32) in acute myelocytic leukemia involves the BCL11B gene. Cancer Genet Cytogenet. 2004;149(1):72–6. . [DOI] [PubMed] [Google Scholar]
- 27. Abbas S, Sanders MA, Zeilemaker A, Geertsma-Kleinekoort WM, Koenders JE, Kavelaars FG, et al. Integrated genome-wide genotyping and gene expression profiling reveals BCL11B as a putative oncogene in acute myeloid leukemia with 14q32 aberrations. Haematologica. 2014;99(5):848–57. 10.3324/haematol.2013.095604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Katoh M, Katoh M. Integrative genomic analyses of ZEB2: Transcriptional regulation of ZEB2 based on SMADs, ETS1, HIF1alpha, POU/OCT, and NF-kappaB. Int J Oncol. 2009;34(6):1737–42. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fusion transcripts detected using FusionCatcher.
(XLSX)
Fusion transcripts detected using ChimeraScan.
(XLSX)
Data Availability Statement
All data underlying the findings in the study are freely available in the paper and in the Supporting Information.