

Abstract
Multiple pathogen-associated molecular pattern–induced TLR pathway cross-talk provokes proinflammatory cytokine synergy in macrophages, which is important for pathogen resistance and immune homeostasis. However, the detailed mechanisms are unclear. In this article, we demonstrate viral RNA analog–induced transcription synergy of Il6 and Il12b via IFN regulatory factor (IRF)1 (TLR3–TIR domain–containing adaptor inducing IFN-β [TRIF] responsive), C/EBPβ (TLR7-MyD88 responsive), and JunB (all responsive). Coactivation of the TLR3 and TLR7 pathways synchronizes the interaction of IRF1, JunB, and C/EBPβ with the Il6 and Il12b promoters, facilitating maximal gene expression. MyD88 pathway activation suppresses TRIF-induced IRF1 in a delayed manner, controlling the magnitude and timing of cytokine expression. Our findings provide novel mechanisms of cooperation of different TLR pathways to achieve optimal immune responses, with the potential for immunomodulatory strategies.
Introduction
Tissue-resident macrophages are deployed in frontline immunity, providing surveillance of invading pathogens. Macrophages use TLRs as central sensors of pathogens (1) to recognize pathogen-associated molecular patterns (PAMPs) unique to the microbes (e.g., viral ssRNAs and dsRNAs). Activated resident macrophages produce cytokines to recruit and communicate with other immune cells. Effective and coordinated cytokine production contributes to elimination of the infectious agents, builds up memory, and repairs tissues without compromising homeostasis. Any imbalance or imprecision in cytokine production could reduce resistance to pathogen infection or cause a fatal cytokine storm and chronic inflammatory diseases (2–4).
The production of cytokines induced by PAMP-activated TLRs occurs primarily through the recruitment of two adaptors: MyD88 and TIR domain–containing adaptor inducing IFN-β (TRIF) (5). MyD88, used by all TLRs with the exception of TLR3, activates NF-κB, which is a core transcription factor (TF) for proinflammatory cytokines. In contrast, TRIF, the adaptor for TLR3 and TLR4 (1), activates IFN regulatory factor (IRF)3, a master transcription controller of antiviral responses. In addition, both MyD88 and TRIF activate the MAPK pathway (6). Invading pathogens are likely to interact with multiple TLRs, and the magnitude and timing of cytokine production, which determine the quality of the immune response, are dependent on the coordinated sum of the signals induced by these different TLRs. Costimulation with MyD88- and TRIF-activating TLR ligands was observed to synergize cytokine production in innate immune cells (7–9), indicating cross-talk between the MyD88 and TRIF pathways. By costimulation of mouse macrophages with R848 (R; an ssRNA analog recognized by TLR7) and polyinosinic-polycytidylic acid (I; a dsRNA analog recognized by TLR3), we demonstrate that TLR3-TRIF and TLR7-MyD88 pathways collaborate to induce synergistic and timely expression of proinflammatory cytokines through the coordinated actions of the TFs JunB, C/EBPβ, and IRF1.
Materials and Methods
All experiments were carried out in compliance with Institutional Animal Care and Use guidelines (IACUC Protocol Ref. 049/11).
Reagents and assays
QuantiGene Plex 2.0 (Affymetrix) was used to profile cytokine expression in murine bone marrow–derived macrophages (BMDMs). TaqMan Gene Expression Assays (Life Technologies) Mm00446190_m1, Mm00434174_m1, and Mm00446968_m1 were used to quantify the expression of Il6 and Il12b relative to Hprt. In knockdown experiments, IL-6 and IL-12p40 were measured using ELISA kits (BD OptEIA). Scramble control or ON-TARGETplus SMARTpool siRNA (Dharmacon) against mouse Junb, Irf1, and/or Cebpb (50 nM each) was transfected into J774 macrophages using X-tremeGENE HP DNA Transfection Reagent (Roche). Abs against JunB (sc73), RelB (sc226), IRF1 (sc640), IRF8 (sc6058), and GAPDH (sc32233) were from Santa Cruz Biotechnology; Abs against RelA (ab7970), TBP (ab818), and IRF3 (ab25950) were from Abcam; Ab against C/EBP β (MA1-827) was from Thermo; and Abs against NF-κB p50 (14-6732-63) and cREL (14-6111-82) were from eBioscience.
Macrophages and stimulations
BMDMs were isolated as described (10). Isolated BMDMs or mouse macrophage cells (J774) were cultured and stimulated with 10 μg/ml I alone, 25 ng/ml R alone, or both (IR simultaneous costimulation), as described previously (9).
Chromatin-binding protein extraction and isobaric tag for relative and absolute quantitation analysis
Chromatin-binding proteins were purified and analyzed by isobaric tag for relative and absolute quantitation (iTRAQ) (11).
Nuclear extract, promoter cloning and 3′-end biotin labeling, and pull-down assay
Details of Il6 and Il12b promoter cloning and 3′-end biotin labeling are described in the legend for Supplemental Fig. 2A. Nuclear extracts (NEs) were prepared from J774 cells that were treated with I, R, or IR for 5 h, as described previously (12) with modifications (pelleting nuclei by a 14,000 × g pulse spin for 15 s). A total of 100 fmol biotin-labeled promoters was incubated with 25 μg NE in 100 μl binding mixture consisting of 10 mM Tris-HCl (pH 7.5), 50 mM KCl, 1 mM DTT, 12.5% glycerol, 0.05% Nonidet P-40, and 5 μg poly(deoxyinosinic-deoxycytidylic) acid (Sigma) at 25°C for 25–30 min. Biotin-labeled promoters were pulled down from the binding mixture with streptavidin-conjugated magnetic Dynabeads M-280 (Life Technologies) at 4°C for 3 h with agitation.
Results
Costimulation of TLR7 and TLR3 induces transcriptional synergy of Il6 and Il12b
To investigate whether cytokine synergy is a prevalent event in the activated macrophages (9), we compared the fold synergy of macrophage-specific major chemokines and cytokines in mouse BMDMs. Fold synergy was evaluated as described in the legend for Fig. 1. Proinflammatory cytokines (Il6 and Il12b) and anti-inflammatory Il10 elicited the highest synergy (>5-fold) (Fig. 1). This study focused on proinflammatory cytokines. The synergistic expression of Il6 and Il12b is not attributable to chromatin remodeling (Supplemental Fig. 1A) or to enhanced mRNA stability (Supplemental Fig. 1B), indicating elevated transcriptional activity under costimulation.
FIGURE 1.
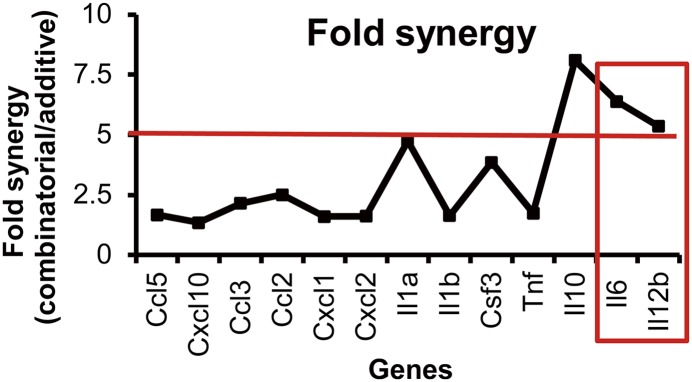
Transcriptional synergy of Il6 and Il12b under TLR7-TLR3 costimulation. BMDMs were treated with I and/or R for 8 h, followed by QuantiGene analysis. Fold synergy was calculated as: mRNA induced by IR/(mRNA induced by I + mRNA induced by R). Proinflammatory cytokine genes with fold synergy > 5 (red line) (e.g., Il6 and Il12b [red box]) were studied further.
Proteomic analysis reveals potential synergy factors
Next, we searched for newly synthesized/activated TFs that may play a role in the observed synergy. We compared the profiles of chromatin-binding proteins from macrophages treated with I and/or R or NT for 1 or 5 h, based on the premise that transcriptional synergy might be facilitated either by the collaboration of TFs induced by R or I single stimulation or by TFs that are activated under IR costimulation. J774 cells, which exhibit a synergy consistent with that of primary BMDMs (Supplemental Fig. 1C), were used for this study. iTRAQ (13) analysis of chromatin-binding proteins from two biological replicates identified proteomes of ∼1100 proteins with false-discovery rates < 1%. As anticipated, the majority were nucleic acid–binding proteins and TFs (Supplemental Fig. 1D). The enriched functional clusters within the proteins perturbed were sorted (Supplemental Fig. 1E, 1F). Interestingly, the gene ontology functional annotation cluster of the “regulation of transcription” was enriched in the proteins upregulated by 5 h of costimulation but not by other treatments. We list the IR-5–upregulated proteins from the “regulation of transcription” cluster and their iTRAQ ratios under different treatments in Table I, with the potential synergy factors noted. C/EBPβ and NF-κB–p105 were more responsive to R stimulation, whereas IFN-activated protein 204 (Ifi204) responded to I only. JunB and ATF3 were upregulated more after costimulation compared with single stimulation, indicating the synergistic actions of two distinct pathways. As a negative regulator of cytokines (14), ATF3 was not studied further. Being relatively uncharacterized, Ifi204 also was excluded. Western blot of NF-κB–p105, JunB, and C/EBPβ (Supplemental Fig. 1G, 1H) validated their changes in expression detected by iTRAQ, and they are shortlisted as potential synergy factors.
Table I. Transcription regulation cluster enriched in proteins upregulated after 5 h of costimulation.
| Gene Symbol (Accession Number) | I1/NT1 | R1/NT1 | IR1/NT1 | I5/NT5 | R5/NT5 | IR5/NT5 |
|---|---|---|---|---|---|---|
| Atf3 (Q60765)a | 1.23 | 1.02 | 0.90 | 0.97 | 1.16 | 2.42 |
| Baz1a (O88379) | 1.06 | 1.15 | 1.09 | 1.23 | 1.29 | 1.55 |
| Cebpb (P28033)a | 1.05 | 1.10 | 1.16 | 1.58 | 3.65 | 2.73 |
| Ctbp1 (O88712) | 0.87 | 1.00 | 0.95 | 1.61 | 1.49 | 1.58 |
| Ctbp2 (P56546) | 0.87 | 1.01 | 0.91 | 1.34 | 1.33 | 1.57 |
| Ddx5 (Q8BTS0) | 1.37 | 1.36 | 1.21 | 4.27 | 4.47 | 2.83 |
| Etv3 (Q8R4Z4) | 0.87 | 1.17 | 1.10 | 1.42 | 1.12 | 1.69 |
| Foxp1 (P58462) | 1.03 | 1.00 | 1.24 | 0.91 | 1.35 | 1.52 |
| Gatad2a (Q8CHY6) | 0.68 | 0.93 | 0.94 | 1.72 | 1.71 | 1.70 |
| Ifi204 (P15092)a | 1.06 | 1.32 | 1.17 | 3.61 | 0.99 | 3.08 |
| Irf5 (P56477) | 1.08 | 1.20 | 1.24 | 1.66 | 1.54 | 1.53 |
| Junb (P09450)a | 1.54 | 2.81 | 2.91 | 3.48 | 6.49 | 11.27 |
| Nfkb1(P25799)a | 1.92 | 3.45 | 3.93 | 1.35 | 2.70 | 3.58 |
| Parp14 (Q2EMV9) | 1.35 | 1.36 | 1.16 | 2.70 | 1.28 | 1.80 |
| Smad4 (P97471) | 0.65 | 1.07 | 1.06 | 1.56 | 1.35 | 1.53 |
| Zeb2 (Q9R0G7) | 1.03 | 1.12 | 1.15 | 1.28 | 1.60 | 1.61 |
Data are fold change in proteins perturbed by I, R, or IR treatment compared with untreated control (NT). All of the genes can be accessed in the Uniprot database by accession number.
Potential synergy factor.
JunB, C/EBPβ, and IRF1 are the core interaction partners of Il6 and Il12b promoters
By promoter affinity pull down of NEs, we examined the direct or indirect involvement of TFs in the regulation of Il6 and Il12b transcription. Candidates shortlisted by iTRAQ and other low-abundance cytokine regulatory factors, which might have been missed in iTRAQ analysis, were analyzed. In total, we examined JunB, C/EBPβ, NF-κB family members (RelA, RelB, c-Rel, and NF-κB–p50), and IRF1, IRF3, and IRF8, which are known to be downstream of the TRIF signaling pathway and are involved in the production of type I IFN. We observed a consistent association of JunB, IRF1, and C/EBPβ with the Il6 and Il12b promoters (Fig. 2) compared with controls (Supplemental Fig. 2A). The intensities of the detection signal appeared roughly proportional to their respective levels in the NE, suggesting that these TFs associate with the respective promoter independently of each other. Notably, the dominant upper band of JunB was dose dependently sensitive to phosphatase treatment (Supplemental Fig. 2B), indicating that JunB was extensively phosphorylated upon induction. Surprisingly, Rel proteins were not significantly associated with either Il12b or Il6 promoter, although NF-κB is functionally important for proinflammatory responses, and there is a characterized κB-binding element in the Il12b (15) and Il6 (16) promoters.
FIGURE 2.

C/EBPβ, IRF1, and JunB bind to the Il6 and Il12b promoter. Cells were stimulated with I, R, or IR for 5 h, followed by nuclear extraction. Control cells were not treated (NT). Biotin-labeled Il6 and Il12b promoters were incubated separately with NEs to pull down TFs. Pull-down samples were subjected to Western blot analysis. The data shown are representative of three independent experiments.
Costimulation synchronizes the activation of C/EBPβ, IRF1, and JunB
IRF1 and C/EBPβ were strongly induced by I and R, respectively (see NEs, Fig. 2), and no additive effect was observed using IR. This suggests that IRF1 and C/EBPβ are regulated separately under the TRIF and MyD88 signaling pathways, respectively. In contrast, JunB expression, which was induced weakly by I but strongly by R, was substantially higher under costimulatory conditions.
Next, we examined the dynamic profiles of IRF1, C/EBPβ, and JunB under different treatments (Fig. 3). C/EBPβ responded primarily to R stimulation. IRF1 was upregulated and sustained by I, but was transiently (1 h) upregulated by R, likely due to the rapid primary activation of NF-κB, which is known to be required for IRF1 expression (17). However, R-induced MyD88 pathway activation apparently repressed the I-induced expression of IRF1 in a delayed manner. IRF1 returned to a low basal level at 12 h post-costimulation, correlating with subdued cytokine gene transcriptions. In contrast, JunB was upregulated rapidly within 1 h by all stimulations. Notably, over 4–8 h, R induced a stronger JunB response than did I. By 12 h, I and R induced comparable levels of JunB, indicating equally limited potentials in single stimulations. Nevertheless, in costimulated macrophages, JunB expression was sustained and JunB protein was accumulated over 8–24 h. Taken together, the coincidence of IRF1, C/EBPβ, and Junb expression in the 4–12-h post–combinatorial stimulation and the obvious correlation with the mRNA peaks of Il6 and Il12b (Supplemental Fig. 1C) strongly indicate their cooperation in synergistic transcription.
FIGURE 3.

Costimulation synchronizes C/EBPβ, IRF1, and JunB. J774 cells were stimulated with I, R, or IR for 5 or 30 min or for 1, 4, 8, 12, or 24 h or were not treated (NT). Whole-cell lysates were collected to check the dynamic profiles of C/EBPβ, JunB, and IRF1 by Western blot immunodetection using the respective Abs. The detected protein bands were densitometrically quantified relative to GAPDH. The data are representative of three independent experiments.
TRIF and MyD88 signaling pathways collaborate to synergize IL-6 and IL-12p40 expression through synchronized IRF1 and C/EBPβ activities
The functional cooperation between TRIF-responsive IRF1 and MyD88-responsive C/EBPβ in macrophages was examined using single and double knockdown with gene-specific siRNAs for Irf1 and Cebpb (Supplemental Fig. 2C). Irf1 and/or Cebpβ knockdown significantly attenuated Il6 and Il12b, at the mRNA and protein levels, in R- and IR-stimulated cells (Fig. 4). In I-stimulated cells, Il6 and Il12b mRNA levels remained at baseline. In IR-stimulated cells, the effect of single knockdown was only incremental, whereas double knockdown reduced Il12b and Il6 further (IL-12p40: ∼50%; IL-6: ∼75%). This indicates that costimulation of TLR3 and TLR7 significantly induced the expression of Il6 and Il12b and that the copresence of a certain level of IRF1 and C/EBPβ is necessary and sufficient for the optimal transcription of Il6 and Il12b.
FIGURE 4.

IRF1 and C/EBPβ collaborate to synergize IL-6 and IL-12p40 production. J774 cells were transfected with sequence-specific siRNA targeting Irf1 or Cebpb, singly or doubly, or were transfected with a nontargeting scramble siRNA control for 24 h. After transfection, cells were not treated (NT) or were stimulated with I, R, or IR for 8 h to test for cytokine mRNA (A) or for 12 h to measure cytokine protein (B). Data are mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, two-tailed Student t test.
TRIF and MyD88 signaling pathways collaborate to synergize cytokine expression through enhancing JunB production and synchronizing IRF1-JunB activation
To functionally characterize the JunB-mediated regulation of Il6 and Il12b expression and its potential cooperation with IRF1, we knocked down Junb alone or together with Irf1 (Supplemental Fig. 2D). A significant reduction in R- or IR-induced Il6 and Il12b was observed in Junb-knockdown cells (Fig. 5) at both the mRNA and protein levels. With I stimulation, both Il6 and Il12b mRNAs remained extremely low, similar to nontreated cells (Fig. 4); thus, the impact of JunB knockdown on I-induced cytokine expression was not assessed further. The drastic difference in the induction of cytokines between single stimulation (I or R) and costimulation (IR) compared with the smaller difference caused by Junb knockdown clearly indicates the involvement of additional critical cofactors induced by the TRIF pathway, which is necessary for the synergistic transactivation of Il6 and Il12b. This is supported by the double knockdown of Junb and Irf1, which yielded a more pronounced attenuation of Il12b and Il6 (IL-12p40: ∼75%, IL-6: ∼80%) at both the mRNA and protein levels (Fig. 5) compared with single knockdown. Thus, costimulation-mediated enhancement of JunB itself promoted cytokine expression and, at the same time, Junb collaborates with TRIF-responsive IRF1 to augment cytokine production.
FIGURE 5.

IRF1 and JunB collaborate to synergize IL-6 and IL-12p40 production. J774 cells were transfected with sequence-specific siRNA targeting Irf1 or Junb singly or doubly or were transfected with a nontargeting scramble siRNA control for 24 h. Cells were then stimulated with R or IR for 8 h to test for cytokine mRNA (A) or for 12 h to measure cytokine protein (B). Data are mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, two-tailed Student t test.
Taken together, R strongly upregulates and sustains MyD88-responsive JunB and C/EBPβ but only transiently activates IRF1, resulting in mild transcription activation of Il6 and Il12b. In contrast, I induces a high level of TRIF-responsive IRF1 but only minimal levels of JunB and C/EBPβ. On its own, IRF1 appeared insufficient to drive the basal transcription. However, costimulation synchronized the activation of IRF1, JunB, and C/EBPβ for up to 8–12 h, whereby these TFs probably worked in concert to achieve optimal cytokine expression.
Discussion
Thus far, the precise molecular mechanisms governing the expression of individual cytokine genes are not well elucidated, although NF-κB, AP1, and/or IRF activities are known to be involved. Also, it remains unknown how cytokine synergy is induced by TRIF- and MyD88-coupled TLR signaling pathways. Our findings demonstrated that the TRIF and MyD88 signaling pathways synergize directly at the level of the transcription of Il6 and Il12b, although the activation of NF-κB members and chromatin remodeling appeared redundantly or additively induced by PAMP stimulation. Of particular significance is our identification of the critical active core transcription complex (IRF1, C/EBPβ, and JunB) of Il6 and Il12b promoters. IRF1 and C/EBPβ are responsive to TLR3/TRIF and TLR7/MyD88 activation, respectively, whereas JunB is responsive to both TLR3/TRIF and TLR7/MyD88 activation, but it is preferentially more responsive to and sustained by TLR7-MyD88 signaling and enhanced by costimulation. Small interfering RNA (siRNA) knockdown further demonstrated that all of these TFs are crucial for optimal transcription of Il6 and Il12b. Thus, we propose that the TLR3/TRIF and TLR7/MyD88 signaling pathways synergize the transcription of Il6 and Il12b through contributing distinct, newly synthesized TFs as the core components of optimal transcriptional complexes (Supplemental Fig. 2E). However, we cannot preclude other yet-to-be-defined “transcriptional synergy factors” (X) that may be the products of cooperation of distinct pathways in costimulation (18). Thus, both mechanisms (TLR3/TRIF and TLR7/MyD88 each contribute newly synthesized TFs to cooperate in cytokine regulation or TLR3/TRIF and TLR7/MyD88 cooperate to produce a new TF that is not present with single stimulation but has a regulatory role in cytokine production) could participate in a network to regulate Il6 and Il12b expression in vivo.
Although IRF1 is known to be strongly induced in macrophages by IFN-γ, the detailed functional role and the mechanisms of IRF1 in regulating immune responses are poorly understood (19). IRF1 plays a major role in the transcriptional regulation of Il12a but not Il12b (20), although an IRF1-binding element was characterized in the Il12b promoter (21). The current investigation revealed that IRF1 is the principal TF induced by the TLR3-TRIF signaling pathway, and it binds both Il6 and Il12b promoters. Because IRF1 was not reported to bind the mouse Il6 promoter before, we used EMSA and identified a 142-bp region (−344 to –203) in the mouse Il6 promoter that binds IRF1 (Supplemental Fig. 2F, 2G). Binding of JunB and C/EBPβ to the mouse Il6 and Il12b promoters was reported previously (22–24). An earlier investigation using an Il12b promoter reporter assay demonstrated cooperation between the AP1 binding site and the adjacent C/EBP binding site (24). However, we found that IRF1 is required to achieve optimal transcription of both Il6 and Il12b. Surprisingly, we did not detect a significant association between the well-recognized master regulator NF-κB with the Il6 or Il12b promoter. Plausibly, as a primary-response regulator, NF-κB members function at the outset of the assembly of the preinitiation transcriptional complex for Il6 and Il12b (14). The promoter binding of NF-κB could be transient and highly dynamic and, thus, not captured by pull down.
In summary, we show for the first time, to our knowledge, that the proinflammatory cytokine genes Il6 and Il12b share a similar core configuration of a transcriptional complex (including IRF1, C/EBPβ, and JunB), which facilitates an optimal and timely expression of IL-6 and IL-12p40 in macrophages to respond precisely to viral signals of infection. The delayed inhibition of IRF1 expression by TLR7-MyD88 signaling correlates with the progressive waning of Il6 and Il12b transcription. Thus, the cross-talk between TLR3-TRIF and TLR-MyD88 signaling pathways probably controls the timing and magnitude of IL-6 and IL-12p40 production through downregulating IRF1, a critical event that warrants future investigation. A comparison of the Il6 and Il12b transcriptional proteomes would provide information on additional regulatory factors, specific to Il6 or Il12b or both, which are anticipated to exist in vivo. These factors could fine-tune the regulation, leading to a specific outcome for each cytokine expression. Such insights about how cytokine synergy is orchestrated will help us to develop novel strategies to resolve immune overactivation and regain homeostasis. Furthermore, knowledge about cytokine synergy could be used in the design of improved vaccines and immunotherapeutics (25).
Supplementary Material
This work was supported by grants from the A*STAR Biomedical Research Council (BMRC 10/1/21/19/658) and the National Medical Research Council (NMRC/CBRG/0055/2014).
The online version of this article contains supplemental material.
- BMDM
- bone marrow–derived macrophage
- I
- polyinosinic-polycytidylic acid
- IRF
- IFN regulatory factor
- iTRAQ
- isobaric tag for relative and absolute quantitation
- NE
- nuclear extract
- PAMP
- pathogen-associated molecular pattern
- R
- R848
- siRNA
- small interfering RNA
- TF
- transcription factor
- TRIF
- TIR domain–containing adaptor inducing IFN-β.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Ishii K. J., Koyama S., Nakagawa A., Coban C., Akira S. 2008. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3: 352–363. [DOI] [PubMed] [Google Scholar]
- 2.Berg D. J., Davidson N., Kühn R., Müller W., Menon S., Holland G., Thompson-Snipes L., Leach M. W., Rennick D. 1996. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Invest. 98: 1010–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parrillo J. E. 1993. Pathogenetic mechanisms of septic shock. N. Engl. J. Med. 328: 1471–1477. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R. 2008. Origin and physiological roles of inflammation. Nature 454: 428–435. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T., Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11: 373–384. [DOI] [PubMed] [Google Scholar]
- 6.Honda K., Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6: 644–658. [DOI] [PubMed] [Google Scholar]
- 7.Whitmore M. M., DeVeer M. J., Edling A., Oates R. K., Simons B., Lindner D., Williams B. R. 2004. Synergistic activation of innate immunity by double-stranded RNA and CpG DNA promotes enhanced antitumor activity. Cancer Res. 64: 5850–5860. [DOI] [PubMed] [Google Scholar]
- 8.Napolitani G., Rinaldi A., Bertoni F., Sallusto F., Lanzavecchia A. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6: 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suet Ting Tan R., Lin B., Liu Q., Tucker-Kellogg L., Ho B., Leung B. P., Ling Ding J. 2013. The synergy in cytokine production through MyD88-TRIF pathways is co-ordinated with ERK phosphorylation in macrophages. Immunol. Cell Biol. 91: 377–387. [DOI] [PubMed] [Google Scholar]
- 10.Iglesias M. J., Reilly S. J., Emanuelsson O., Sennblad B., Pirmoradian Najafabadi M., Folkersen L., Mälarstig A., Lagergren J., Eriksson P., Hamsten A., Odeberg J. 2012. Combined chromatin and expression analysis reveals specific regulatory mechanisms within cytokine genes in the macrophage early immune response. PLoS ONE 7: e32306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutta B., Adav S. S., Koh C. G., Lim S. K., Meshorer E., Sze S. K. 2012. Elucidating the temporal dynamics of chromatin-associated protein release upon DNA digestion by quantitative proteomic approach. J. Proteomics 75: 5493–5506. [DOI] [PubMed] [Google Scholar]
- 12.Birbach A., Gold P., Binder B. R., Hofer E., de Martin R., Schmid J. A. 2002. Signaling molecules of the NF-kappa B pathway shuttle constitutively between cytoplasm and nucleus. J. Biol. Chem. 277: 10842–10851. [DOI] [PubMed] [Google Scholar]
- 13.Zieske L. R. 2006. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J. Exp. Bot. 57: 1501–1508. [DOI] [PubMed] [Google Scholar]
- 14.Gilchrist M., Thorsson V., Li B., Rust A. G., Korb M., Roach J. C., Kennedy K., Hai T., Bolouri H., Aderem A. 2006. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441: 173–178. [DOI] [PubMed] [Google Scholar]
- 15.Murphy T. L., Cleveland M. G., Kulesza P., Magram J., Murphy K. M. 1995. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol. Cell. Biol. 15: 5258–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libermann T. A., Baltimore D. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 10: 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su R. C., Sivro A., Kimani J., Jaoko W., Plummer F. A., Ball T. B. 2011. Epigenetic control of IRF1 responses in HIV-exposed seronegative versus HIV-susceptible individuals. Blood 117: 2649–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayama H., Ramirez-Carrozzi V. R., Yamamoto M., Mizutani T., Kuwata H., Iba H., Matsumoto M., Honda K., Smale S. T., Takeda K. 2008. Class-specific regulation of pro-inflammatory genes by MyD88 pathways and IkappaBzeta. J. Biol. Chem. 283: 12468–12477. [DOI] [PubMed] [Google Scholar]
- 19.Ikushima, H., H. Negishi, and T. Taniguchi. 2013. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb. Symp. Quant. Biol. 78: 105–116. [DOI] [PubMed]
- 20.Liu J., Cao S., Herman L. M., Ma X. 2003. Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. J. Exp. Med. 198: 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maruyama S., Sumita K., Shen H., Kanoh M., Xu X., Sato M., Matsumoto M., Shinomiya H., Asano Y. 2003. Identification of IFN regulatory factor-1 binding site in IL-12 p40 gene promoter. J. Immunol. 170: 997–1001. [DOI] [PubMed] [Google Scholar]
- 22.Plevy S. E., Gemberling J. H., Hsu S., Dorner A. J., Smale S. T. 1997. Multiple control elements mediate activation of the murine and human interleukin 12 p40 promoters: evidence of functional synergy between C/EBP and Rel proteins. Mol. Cell. Biol. 17: 4572–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baccam M., Woo S. Y., Vinson C., Bishop G. A. 2003. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-kappa B, AP-1, and C/EBP. J. Immunol. 170: 3099–3108. [DOI] [PubMed] [Google Scholar]
- 24.Zhu C., Gagnidze K., Gemberling J. H., Plevy S. E. 2001. Characterization of an activation protein-1-binding site in the murine interleukin-12 p40 promoter. Demonstration of novel functional elements by a reductionist approach. J. Biol. Chem. 276: 18519–18528. [DOI] [PubMed] [Google Scholar]
- 25.Trinchieri G., Sher A. 2007. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 7: 179–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.