

Dear Editor,
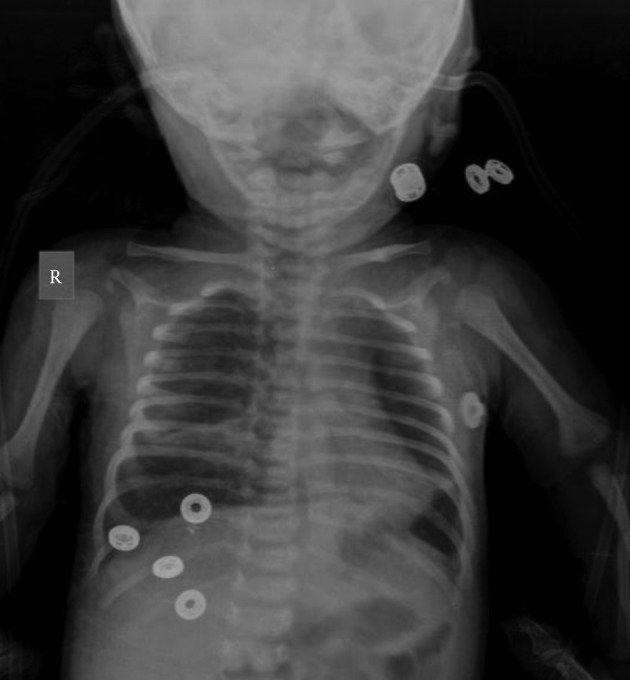
Cystic Fibrosis is an autosomal recessive disorder, which is caused by mutations in CFTR gene on chromosome 7q31.2. The prevalence of CF is approximately 1 in 2500 in Caucasians. In other populations, it has been seen less frequently. Characteristic features include chronic pulmonary disease, pancreatic exocrine insufficiency and secretory systems such as gastro-intestinal and reproductive system function abnormalities. Current guidelines for the diagnosis of CF suggest that a patient must present one or more characteristic phenotypic features or a positive newborn screening test result and a positive sweat test or two CF-causing mutations (1). A screening for CFTR mutations is essential if the sweat test is not conclusive. Here we present a female who was diagnosed with CF at age 1-year with a rare homozygous mutation 3849 + 10 kb C > T and normal sweat chloride concentrations. The proband was a 1-year-old female born to consanguineous parents (Figure 1). She was born at 38 weeks of gestation with a birth weight of 3610 g, birth height of 51 cm and birth head circumference of 37 cm. She was admitted to the hospital at 2 months of age because of respiratory distress syndrome. Her weight was 4700 g (50th percentile), height: 56 cm (75th percentile) and head circumference was 37.5 cm (25th-50th percentile). Intercostal and subcostal retractions were seen and bilateral crackles were heard at the lung in physical examination. Hemoglobin 10.3 g/dL (N: 9-14 g/dL), white blood cells 6670/ µL (N: 6000 - 17500), platelets 577000/µL (N: 150,000 - 450,000), C-reactive protein 21 mg/dL (N: 0-5 mg/dL) and normal blood biochemistry and liver function tests were detected in laboratory investigations. Her chest roentgenogram demonstrated bilateral pulmonary infiltrations and atelectasis (Figure 2). Computerized-tomography (CT) showed fibro-atelectatic bands in right medial lobe and left lower lobe. Echocardiography was normal. Steatorrhoea was detected a few times. Her sweat chloride concentrations were 40 mmol/L (evaluated twice) (N: ≤ 40 mmol/L). CF was suspected due to her clinical features and laboratory findings. Molecular analysis revealed a homozygous mutation 3849 + 10 kb C > T in CFTR. The patient’s mother and father were heterozygous for this mutation (Figure 1) and there were no clinical findings. CF is a complex multi-system disorder. The pathological basis of this disorder is based on mutations in the CFTR gene which encodes the cystic fibrosis transmembrane conductance regulator, a membrane chloride channel located in the apical membrane of secretory epithelia. CF has a higher prevalence in populations of European descent, than in any other population group. The more than 1900 different mutations in the CFTR gene have been reported. The most common mutations in the CFTR gene have been different from one population to the others in the world. The people who are carrying the ∆F508 allel in the Mediterranean population have a relatively low proportion compared with other Caucasoid populations. In the Mediterranean region, more than 200 different CF mutations accounting for about 85% of the CF alleles have been detected indicating the highest heterogeneity and the wide diversity of peoples and cultures. The worldwide frequency of the 3849 + 10 kb C > T mutation which was detected in our case is approximately 0.2%. In Europe, its frequency is 0.15%. The 3849 + 10 kb C > T mutation was first described by Highsmith et al. in 13 patients with chronic pulmonary disease but normal sweat chloride values (2). Feldmann et al. present CF patients who had 3849 + 10 kb C > T mutation. They had normal sweat chloride concentration but had pulmonary symptoms (3). In another sudy, Augarten et al. described patients with the 3849 + 10 kb C > T mutation had been diagnosed at older age while our case was 1 year old. These patients had nomal pancreatic and liver functions (4). Stern et al. reported 8 patients who were compound heterozygous for the 3849 + 10 kb C > T mutation with normal sweat chloride values, but pulmonary disease was seen ranging from mild to severe (5). Gilbert et al. reported 14 patients who were homozygous for the 3849 + 10 kb C > T mutation. The age ranged between 2 - 32 years. Most of them had normal pancreatic functions. Seven patients had normal sweat chloride concentrations (6). In genotype-phenotype correlation study by Dugueperoux and Braekeleer, the 3849 + 10 kb C > T allel had been shown to be associated with mild CF phenotype as in our case (7). Liu et al. reported a 20-year-old female. She had a 3849 + 10 kb C > T homozygous mutation with pulmonary symptoms and normal sweat chloride test (8). Although there are a few studies on CFTR mutation spectrum, 3849 + 10 kb C > T mutation is not reported before in Turkey. Thus our case is the first report of CFTR 3849 + 10 kb C > T homozygous mutation. In Turkey, Halicioglu et al. reported 24 cases with CF. ∆F508 was the most common mutation. Other mutations such as R1066C, 1677 delTA, 2789 + 5G-A (9) were rarely seen. In another study from Turkey, 67 CF patients were screened for CFTR gene mutations. The most common mutation was ∆F508 (28.4%). R347H and N1303K mutations account for 6.7% (10). The C-to-T mutation in intron 19 leads to create an alternative splice acceptor site. Thus it leads to produce an abnormal mRNA but normally spliced mRNA has also occurred (2). Because of this normal transcripts, 3849 + 10 kb C > T mutation is most likely responsible for a milder phenotype.
Figure 1. The Pedigree of the Patient.
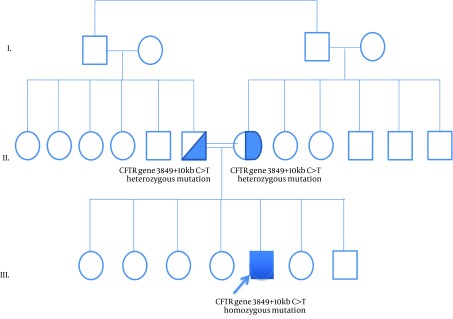
Figure 2. The Patient’s Chest Roentgenogram Demonstrated Bilateral Pulmonary Infiltrations and Atelectasis.
References
- 1.Egan M. Cystic fibrosis. In: Kliegman RM, Stanton B. F., Geme JS, Schor NF, Behrman RE, editors. Nelson Textbook of Pediatrics. USA: Judith Fletcher; 2011. pp. 1481–97. [DOI] [Google Scholar]
- 2.A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. Highsmith WE, Burch LH, Zhou Z, Olsen JC, Boat TE, Spock A, et al. N Engl J Med. 1994;331(15):974. doi: 10.1056/NEJM199410133311503. [DOI] [PubMed] [Google Scholar]
- 3.CFTR genotypes in patients with normal or borderline sweat chloride levels. Feldmann D, Couderc R, Audrezet MP, Ferec C, Bienvenu T, Desgeorges M, et al. Hum Mutat. 2003;22(4):340. doi: 10.1002/humu.9183. [DOI] [PubMed] [Google Scholar]
- 4.Mild cystic fibrosis and normal or borderline sweat test in patients with the 3849 + 10 kb C → T mutation. Augarten A, Yahav Y, Szeinberg A, Noiman S, Gazit E, Kerem BS, et al. Lancet. 1993;342(8862):25. doi: 10.1016/0140-6736(93)91885-p. [DOI] [PubMed] [Google Scholar]
- 5.3849+10kbC>T mutation and disease severity in cystic fibrosis. Stern RC, Doershuk CF, Drumm ML. Lancet J. 1995;346(8970):274. doi: 10.1016/s0140-6736(95)92165-6. [DOI] [PubMed] [Google Scholar]
- 6.Clinical spectrum in homozygotes and compound heterozygotes inheriting cystic fibrosis mutation 3849+10kbC>T: significance for geneticists. Gilbert F, Li Z, Arzimanoglou II, Bialer M, Denning C, Gorvoy J. Am J Med Genet. 1995;58(4):356. doi: 10.1002/ajmg.1320580411. [DOI] [PubMed] [Google Scholar]
- 7.The CFTR 3849+10kbC->T and 2789+5G->A alleles are associated with a mild CF phenotype. Dugueperoux I, De Braekeleer M. Eur Respir J. 2005;25(3):468. doi: 10.1183/09031936.05.10100004. [DOI] [PubMed] [Google Scholar]
- 8.Cystic fibrosis: experience in one institution. Liu LC, Shyur SD, Chu SH, Huang LH, Kao YH, Lei WT. J Microbiol Immunol Infect. 2014;47(4):358. doi: 10.1016/j.jmii.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Diverse genotypical features and impacts on clinical course and severity of cystic fibrosis: early childhood experience. Halicioglu O, Akman SA, Sutcuoglu S, Coker I. Minerva Pediatr. 2011;63(3):169. [PubMed] [Google Scholar]
- 10.Study of 12 mutations in Turkish cystic fibrosis patients. Yilmaz E, Erdem H, Ozguc M, Coskun T, Ozcelik U, Gocmen A, et al. Hum Hered. 1995;45(3):175. doi: 10.1159/000154281. [DOI] [PubMed] [Google Scholar]