

Abstract
Purpose
To study retinal function and morphology in ABCA4 carriers to investigate if ABCA4 carriership is associated with any functional or morphological changes and, if so, to explore whether certain mutations may be associated with particularly severe alterations.
Methods
Eighteen subjects were recruited by means of being the parents of 10 teenagers/young adults with genetically confirmed ABCA4-associated retinal degenerations. The teenagers/young adults are well-known patients and have been followed in our clinic for many years. The eighteen subjects underwent careful ophthalmological examinations, including fundus photography and autofluorescence imaging, Goldmann perimetry, optical coherence tomography (OCT), full-field electroretinography (ffERG), multifocal electroretinography (mERG), and ABCA4 gene sequencing. The ffERG and mERG results were compared with those of healthy controls.
Results
All subjects carried at least one ABCA4 mutation. Two subjects were compound heterozygous and therefore were excluded from the group-wise statistical analysis. Thirteen different ABCA4 mutations were found. C.2894 A>G (5/18) and c.768 G>T (4/18) were most common. Fourteen of 16 ABCA4 carriers demonstrated significantly altered mERG parameters (reduced amplitudes and/or delayed implicit times (ITs)) compared to normal values. In addition, the two subjects with compound heterozygous ABCA4 mutations had altered mERG parameters. A statistical comparison to the control group showed significantly reduced amplitudes and delayed ITs; p≤0.003 for all mERG parameters. FfERG parameters were altered in two ABCA4 carriers and one of the subjects with compound heterozygous ABCA4 mutations (reduced amplitude and delayed IT for the 30 Hz flicker ERG). No significant alterations were found for the whole group of ABCA4 carriers compared to the ffERG control group. Fundus photographs showed subtle to extensive pigmentary changes in several ABCA4 carriers.
Conclusions
In this study, ABCA4 carriers demonstrated reduced macular function measured by mERG along with none to subtle and even extensive morphological retinal changes. The c.768 G>T, c.5461–10T>C, and c.319 C>T mutations were associated with the most deviant ERGs, including both significant reduction of mERG amplitudes and prolongation of mERG ITs, as well as with reduced amplitude or delayed IT for the 30 Hz flicker ffERG in a few cases. They may therefore be considered serious mutations. The c.5917delG and c.4469 G>A mutations were associated with milder or no macular alteration. Long-term follow-up of these and other ABCA4 carriers may be of importance to elucidate the role of ABCA4 mutations in age-related macular degeneration. Moreover, improved knowledge of separate ABCA4 mutations may help us to better understand their role in ABCA4-associated retinal degenerations.
Introduction
ABCA4-associated retinal degenerations are inherited as autosomal recessive traits. The most common form of the disorder is Stargardt macular dystrophy (STGD), which is typically a juvenile-onset macular degeneration presenting with central vision loss in late childhood or the early teenage years [1-3]. Usually, STGD leads to symmetric bilateral atrophy of the retinal pigment epithelium (RPE) in the fovea and posterior pole, along with orange-yellow flecks in the same areas [4]. ABCA4 mutations also can lead to cone-rod dystrophy (CRD) [5-7], a more severe form of retinal degeneration causing reduced central vision and progressive reduction of general cone function with severe central and paracentral scotomas and reduced color vision. Eventually, rod function is also reduced, resulting in visual field constriction and night blindness [8]. In CRD, more unspecific pigmentary changes are frequently seen in the macula and mid-periphery. More seldom, ABCA4 mutations can cause autosomal recessive retinitis pigmentosa (arRP) [6,9,10], starting with reduced night vision combined with visual field constriction and in time also causing reduced central vision and defective color vison. Fundus changes can be typical of RP, with a pale optic nerve head, attenuated retinal vessels, and peripheral bone spicule pigmentations [11] but in some cases may be more unspecific with spread atrophies in the macula and posterior pole. It has also been discussed that some ABCA4 mutations could be important in age-related macular degeneration (AMD) [2,4]. The carrier frequency of possible disease-generating ABCA4 alleles has been found to be quite high at around 1:20 in different populations [12,13], and over 700 ABCA4 variants have been identified to date.
The ABCA4 gene encodes the ABCA4 protein, which is an ATP-binding cassette (ABC) transporter protein located in the rims of the photoreceptor discs [14-16]. The ABC transporter proteins comprise a superfamily of proteins with the important function of transferring various biologic compounds, such as amino acids, polypeptides, vitamins, and lipids, across cell membranes [17]. The main tasks for the ABCA4 protein in photoreceptors are to remove N-retinylidene-phosphatidylethanolamine (NRPE) and phosphatidylethanolamine (PE) from the lumen of the outer segment disc membranes to the cytoplasmic leaflet during photo transduction [18,19]. In this way, the ABCA4 protein facilitates the elimination of potentially toxic retinoid compounds from photoreceptors. ABCA4-associated retinal degenerations are consequently proposed to be caused by a buildup of a lipofuscin fluorophore, N-retinylidene-N-retinylethanolamine (A2E), which is produced when the ABCA4 rim protein is dysfunctional or missing and therefore cannot clear the photoreceptor outer segment of NRPE. Eventually, A2E accumulates in RPE cells, leading to RPE cell death and probably the secondary loss of photoreceptors [19,20]. Lately, it has also been proposed that direct photoreceptor cell death may precede alterations in the RPE [21,22].
Electrophysiological (ERG) evaluation is important in both diagnosing and following up ABCA4-associated retinal disorders. The different entities can be separated by ERG results [23]. In STGD, typically, multifocal ERG (mERG) is reduced while full-field ERG (ffERG) is normal or very mildly affected [23,24]. In CRD, mERG is always significantly reduced along with decreased cone responses in the ffERG. Later on, rod responses may also be affected [8,23]. ArRP is associated with an initial reduction of ffERG rod responses, followed by reduced cone responses [4,23]. Thus, ABCA4 mutations can lead to very diverse phenotypes, making it difficult to predict the course of the disease or visual outcome when a doctor first sees a patient. Some studies propose models that correlate severity of disease to estimated residual function of ABCA4 based on the suggested effect of individual mutations on gene transcription and protein translation [13,25,26]. Other studies instead suggest that the rate of ABCA4 activity correlates with the timing of disease onset rather than to a specific form of retinal degeneration (STGD, CRD or arRP) [27]. In this survey, we wanted to study separate ABCA4 mutations more closely. Therefore, we investigated ABCA4 carriers to explore if they have any functional retinal changes and if any specific mutations are associated with particularly serious deficits indicating severe mutations.
Methods
Subjects and control groups
In this study, 18 tentative ABCA4 carriers (nine men and nine women aged 40–61, median age 51, mean age 52) were included, and two control groups were used (29 controls for mERG, aged 38–69, median and mean age 51, and 20 controls for ffERG, aged 42–67 years, median age 47 and mean age 51). The ABCA4 carriers were recruited from our clinic by means of being the parents of ten teenagers/young adults with ABCA4-associated retinal degenerations. The teenagers/young adults have been patients in our clinic for many years and have been followed closely. All the teenagers/young adults have two confirmed mutations in the ABCA4 gene, and mutations in other genes associated with retinal degenerations have been ruled out. Data concerning genotypes and phenotypes for the teenagers/young adults are presented in Table 1. Two parents of the teenagers/young adults (the mother of patient C and the father of patient D) were also already diagnosed with typical STGD and therefore were excluded from the study. Consequently, information about them is not presented. Subjects 1 and 2 only had the right eye examined. The rest of the subjects (3–18) had both eyes tested. The study was conducted in accordance with the Tenets of the Declaration of Helsinki and was approved by the Ethical Committee for Medical Research at Lund University. All subjects gave their written informed consent to participate in the study.
Table 1. Demographics and results of examinations for the teenagers/young adults.
| Patient/ Gender | The child of subject | Age | Age at onset | Mutations | BVA (RE, LE) | Goldmann Visual field | ffERG | mERG Ampl | mERG IT | Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|
| A/F |
1 & 2 |
23 |
7 |
c.768 G>T
c.768 G>T |
0.04, 0.05 |
Large central and nasal paracentral scotomas for V4e. Constricted II4e isopters. |
Flicker Ampl ↓ Flicker IT ↑ |
↓ |
↑ |
CRD |
| B/M |
3 |
13 |
10 |
c.768 G>T c.768 G>T |
0.1, 0.1 |
Large central scotomas for V4e RE, and I4e LE. |
N |
↓ |
N |
STGD |
| C/M |
4 & 5 |
17 |
7 |
c.5461–10T>C c.319 C>T |
0.02, 0.05 |
Large central scotomas for V4e. |
Flicker Ampl ↓ Flicker IT ↑ |
↓ |
↑ |
CRD |
| D/F |
6 |
22 |
20 |
c5917 delG c5882 G>A |
0.16, 0.13 |
Small central scotomas for 04e. |
N |
↓ |
N |
STGD |
| E/F |
7 & 8 |
20 |
11 |
c.2894 A>G c.768 G>T |
0.06, 0.06 |
Large nasal paracentral scotomas for II4e. |
Flicker Ampl ↓ Flicker IT ↑ |
not tested |
not tested |
CRD |
| F/F |
9 &10 |
16 |
6 |
c.1622 T>C c.1804 C>T |
0.06, 0.06 |
Large central and nasal paracentral scotomas for I4e. |
N |
↓ |
↑ |
STGD |
| G/F |
11 & 12 |
20 |
12 |
c.4469 G>A c.6079 C>T |
0.13, 0.16 |
Central scotomas for 03e. |
N |
↓ |
N |
STGD |
| H/M |
13 & 14 |
23 |
10 |
c.2894 A>G c.2894 A>G |
0.1, 0.1 |
Large central scotomas for 03e. |
N |
↓ |
N |
STGD |
| I/M |
15 & 16 |
13 |
6 |
c.2894 A>G
c.5714+5G>A |
0.25, 0.25 |
Small central scotoma for 03e. |
N |
↓ |
N |
STGD |
| J/F | 17 & 18 | 22 | 13 | c.2894 A>G c.4667+2T>C | 0.04, 0.08 | RE scotoma within 10° for V4e. LE large scotoma for I4e and small central scotoma within 5° for V4e. | Flicker IT ↑ | ↓ | N | STGD |
F=female, M=male, BVA=best corrected Snellen visual acuity, RE=right eye, LE=left eye, Ampl=amplitude, IT=implicit time, ↓ reduced, ↑ prolonged, n=normal, CRD=cone rod dystrophy, STGD=Stargardts disease
Ophthalmological examination
Best monocular visual acuity (BVA) was tested separately in both eyes using the Snellen chart at five meters. Visual fields were mapped with Goldmann perimetry using standardized objects V4e, I4e, and 04e. For some subjects, II4e, 03e, 02e, and 01e were also applied. Fundus photographs, including color photographs, red-free photographs, and autofluorescence (AF) images, were captured with a Topcon TRC 50DX fundus camera (Topcon, Inc., Oakland, NJ). For AF images, Spaide filters with an excitation wavelength of 530–580 nm and a barrier filter of 600–720 nm were used, and the field of view was 50×50°. All subjects also had a thorough slit- lamp and fundus examination.
Genetic Analysis
A sequence analysis of the entire coding region of the ABCA4 gene was performed at Asper Biotech (Asper Ophthalmics, Tartu, Estonia). The same analysis was performed for both the teenagers/young adults with ABCA4-associated retinal degenerations (Table 1) and their parents (the tentative ABCA4 carriers; Table 2). No other genetic analyses were performed.
Table 2. Demographics and examination results for the subjects.
| Subject/Gender | Age | Mutation | BCVA (RE, LE) | Goldmann visual field | ffERG | mERG Ampl | mERG IT | Fundus photos | AF |
|---|---|---|---|---|---|---|---|---|---|
| 1/F |
60 |
c.768 G>T |
0.8, 0.4 |
RE small scotoma 03e inf temp between 10 and 20° a, LE normal |
flicker Ampl RE ↓ |
RE ↓
LE not tested |
RE ↑
LE not tested |
N |
not tested |
| 2/M |
61 |
c.768 G>T |
1.0, 1.0 |
N |
N |
RE ↓ |
RE ↑ |
N |
not tested |
| 3/M |
45 |
c.768 G>T |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
N |
N |
| 4/F |
59 |
c.5461–10T>C |
1.0, 0.9 |
RE+LE 30e, 20e, 10e constricted mostly 30e sup and more in LE |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
N |
N |
| 5/M |
61 |
c.319 C>T |
1.0, 1.0 |
N |
flicker IT RE ↑
flicker IT LE ↑ |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
LE; pigmentary changes in the macula |
LE; hyper-fluorescence in the macula |
| 6/F |
41 |
c.5917delG |
0.7, 1.0 |
N |
N |
N |
N |
N |
not tested |
| 7/F |
57 |
c.2894 A>G |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
OA; pigmentary changes in the macula and along the vascular arcades |
LE; hyper- and hypo fluorescence around the vascular arcades |
| 8/M |
61 |
c.768 G>T |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
OA; pigmentary changes in the macula and deep yellow spots in the mid-periphery |
OA; multiple foci of high and low AF signals |
| 9/F |
47 |
c.1622 T>C/
c.3113 C>T |
1.0, 0.9 |
N |
flicker IT LE ↑ |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
N |
N |
| 10/M |
51 |
c.1804 C>T |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
OA; pigmentary changes in the macula |
N |
| 11/F |
49 |
c.4469 G>A |
1.0, 1.0 |
N |
N |
N |
N |
N |
N |
| 12/M |
51 |
c.6079 C>T |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
OA; pigmentary changes in the macula |
N |
| 13/F |
47 |
c.2894 A>G |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
N |
N |
| 14/M |
53 |
c.2894 A>G |
0.8, 1.0 |
N |
N |
N |
RE ↑ LE ↑ |
RE; pigmentary changes in the macula |
RE; high AF signals in the macula and the temp midperiphery |
| 15/F |
41 |
c.2894 A>G |
1.0, 1.0 |
N |
N |
RE ↓ LE ↓ |
RE ↑ LE ↑ |
LE; pigmentary changes sup to the macula |
LE; hyper-fluorescence sup to the macula |
| 16/M |
57 |
c.5714+5G>A |
1.0, 1.0 |
N |
N |
N |
RE ↑ LE ↑ |
OA; very subtle pigmentary changes in the macula |
N |
| 17/F |
47 |
c.2894 A>G |
0.9, 0.9 |
N |
N |
N |
RE ↑ LE ↑ |
N |
N |
| 18/M | 48 | c.4297 G>A/ c.4667+2T>C | 1.0, 1.0 | N | N | RE ↓ LE ↓ | RE ↑ LE ↑ | RE; subtle pigmentary changes in the macula | RE; hyper-fluorescence in the macula |
F=female, M=male, BVA=best corrected Snellen visual acuity, RE=right eye, LE=left eye, Ampl=amplitude, IT=implicit time, AF=autofluorescence, inf=inferior, temp=temporal, ↓ reduced, ↑prolonged, n=normal, sup=superior, OA=both eyes a The patient has myelinated nerve fibers corresponding to the scotoma.
Statistical analysis
The Mann–Whitney U-test was used to compare ffERG and mERG results in ABCA4 carriers, with ffERG and mERG measurements for the control group members. All statistical analyses were performed using SPSS 20.0. Statistical significance was defined as p<0.05. The limits of normality for ERG parameters were defined by the means ± two standard deviations (2SD). The lower limits of normality were used for amplitudes and the upper limits of normality for implicit times (ITs). This method could be used because ffERG parameters have only a slightly skewed distribution.
Full-field electroretinography
FfERGs were recorded using an Espion E2 analysis system (Diagnosys, Lowell, MA, USA) according to the standardized protocol for clinical electroretinography recommended by the International Society for Clinical Electrophysiology of Vision (ISCEV) [28]. Measurements were recorded with a Burian–Allen bipolar ERG contact lens electrode after 40 min of dark adaptation and with maximally dilated pupils (1% cyclopentolate and 10% phenylephrine hydrochloride). To ensure reproducibility, the recordings were repeated for all stimulus intensities until two successive identical curves were obtained.
Multifocal electroretinography
MERGs were recorded with a Visual Evoked Response Imaging System (VERIS Science 6; EDI, San Mateo, CA, USA) using settings that adhere to the ISCEV guidelines [28]. The stimulus matrix consisted of 103 hexagonal elements, scaled with eccentricity to elicit approximately equal amplitude responses at all locations. Each hexagon independently alternated between black and white according to a pseudorandom binary m-sequence at 75 Hz. The pupils were maximally dilated with 1% cyclopentolate and 10% phenylephrine. Retinal activity was registered using a Burian–Allen bipolar ERG contact lens electrode that was placed on the anesthetized (oxybuprocaine) cornea. Fixation was monitored with an IR eye camera. The first order component of the mERG was analyzed regarding amplitudes (A) and ITs of P1 (first positive peak) within the five concentric rings (A 1–5 and IT 1–5) around the fovea. Ring 1, the innermost ring, represents the summed responses from the central hexagon and the first ring (A1 and IT1).
Optical coherence tomography
Optical coherence tomography (OCT) images were obtained with a Topcon 3D OCT-1000 (Topcon, Inc., Paramus, NJ). This is a Fourier domain-based OCT that captures 3D scans with scan size 6×6 mm and a scan density of 512×128. The internal fixation target (pre-set for macular imaging) was used. Macular morphology was studied on B-scan images, and macular thickness was further analyzed using the standard retinal thickness map of the OCT software (version 3.51 or 8.11) consisting of three circles with diameters of 1 mm, 3 mm, and 6 mm, respectively. The two outer rings are further divided into four segments, corresponding to a superior, a nasal, an inferior, and a temporal segment. For each segment of the retinal map, the mean thickness of the retina from the RPE to the inner limiting membrane (ILM) can be measured. The mean thickness of each segment is also automatically compared to an age-matched normative database and results outside normal limits are indicated. Retinal thickness was considered abnormally attenuated if values below the fifth percentile of the age-related normal material were obtained. Likewise, it was considered abnormally thickened if values over the 95th percentile were found.
Results
Genetic analysis
Genetic analysis was performed for all subjects (Table 2). Of the 18 subjects, 16 (89%) carried one ABCA4 mutation and were defined as ABCA4 carriers. Two subjects (9 and 18) were found to be compound heterozygous with ABCA4 mutations on both alleles. They were consequently not considered as ABCA4 carriers and their data were therefore excluded from further analysis, although some of their test results are presented in Table 2. The most common ABCA4 mutation was c.2894 A>G, which was found in five of the 18 (28%) subjects. Four (22%) of the subjects carried the c.768 G>T mutation. In all, 13 different ABCA4 mutations were identified (Table 1). All mutations except c.2894 A>G and c.768 G>T were found in one subject only.
Ophthalmological examination
Data concerning demographics and visual parameters for the ABCA4 carriers are presented in Table 2. Eleven (69%) of the 16 ABCA4 carriers had BVA 1.0 in both eyes, while five (31%) demonstrated reduced visual acuity (VA). Only one of the ABCA4 carriers (1) complained about reduced vision. She had had cataract surgery in her right eye and still had a moderate cortical cataract in her left eye. Fundus examinations demonstrated subtle to extensive retinal changes (Table 2, Figure 1, Figure 2, Figure 3). There was no significant statistical difference in age between the ABCA4 carriers with and without fundus changes (Mann–Whitney U test; p=0.54). In addition to the pigmentary changes, subject 8 had numerous orange-yellow retinal flecks around the macula and in the posterior pole. Myelinated nerve fibers were found along the superior vascular arcade in subject 1, and she also had a corresponding small scotoma for object 03e inferiorly and temporally between the 10° to 20° isopters. AF images were obtained for 13 of the 16 ABCA4 carriers and in some cases showed hyper- and/or hypofluorescence.
Figure 1.
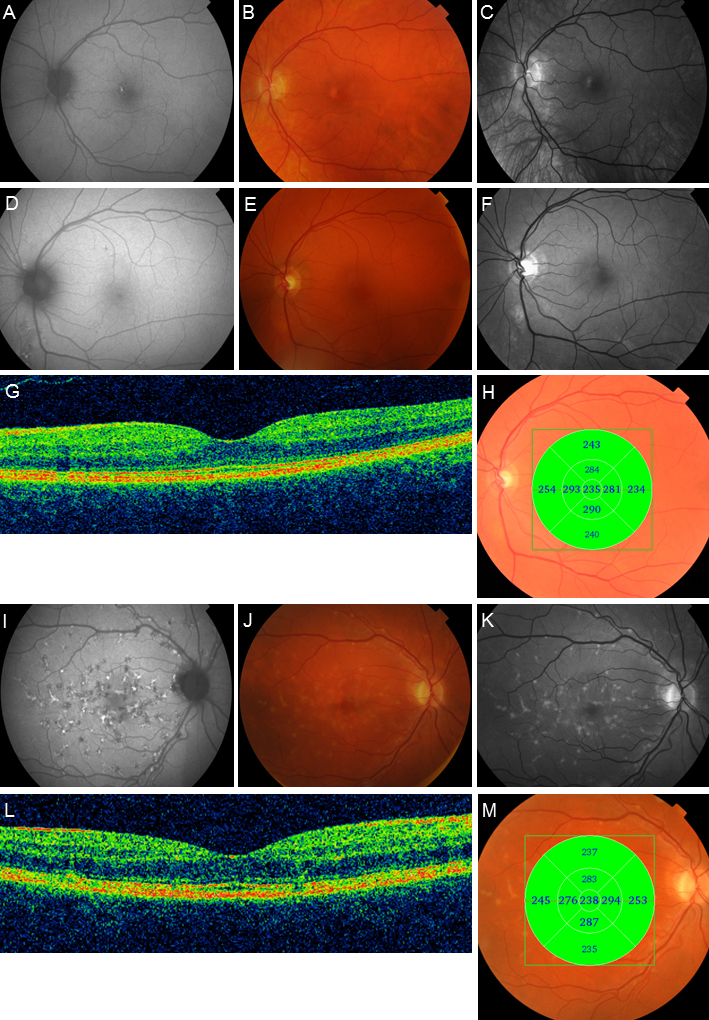
Autofluorescence (AF) images, color photographs, red-free photographs, and OCT examinations of subjects 5, 7, and 8. In subject 5, (A–C), the AF (A) image shows a hyperfluorescent spot in the left macula corresponding with pigmentary changes on fundus photos (B and C). In subject 7 (D–H), the AF image (D) shows discrete hyper- and hypofluorescence around the vascular arcades. E and F show subtle pigmentary changes in the macula and along the vascular arcades, seen most clearly in the red-free photo (F). The OCT B-scan (G) shows subtle outer retinal disruption, but the retinal thickness (H) is within normal limits. The AF image (I) from subject 8 demonstrates multiple foci with mixed hyper- and hypofluorescence. Fundus photographs (J and K) also reveal widespread orange-yellow flecks in the posterior pole, as well as macular pigmentary changes. The OCT B-scan (L) shows outer retinal disruption, although the retinal thickness (M) is within normal limits.
Figure 2.
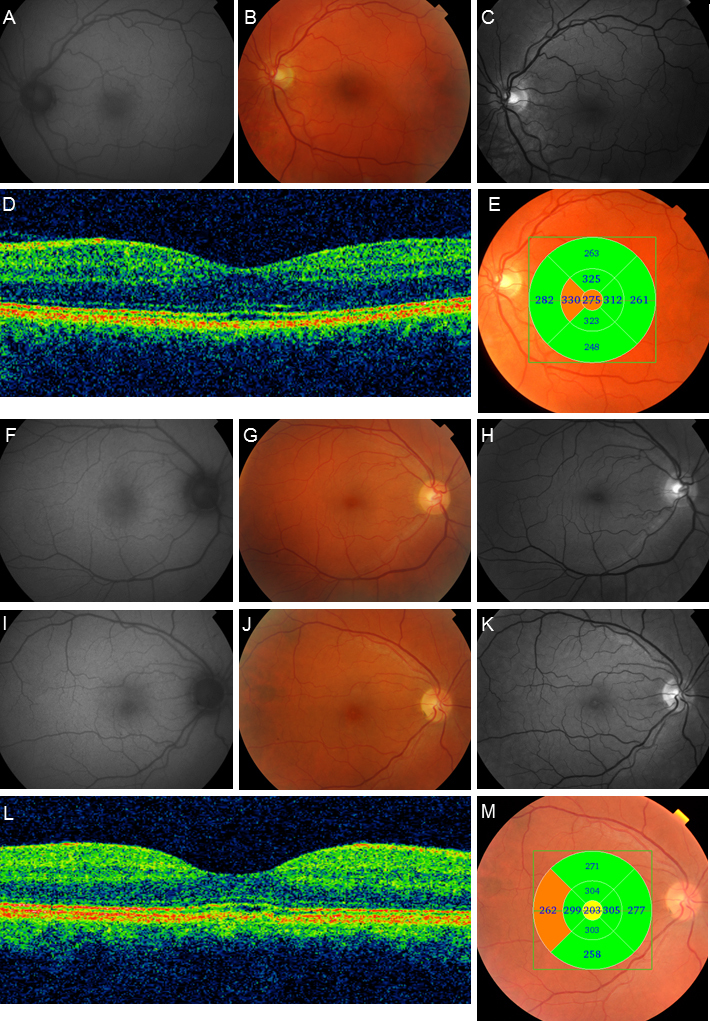
AF images, color photographs, red-free photographs, and OCT examinations from subjects 10, 11, and 12. In subject 10 (A–E), the AF image (A) is normal. Fundus photographs (B and C) show subtle pigmentary changes in the macula. The OCT B-scan (D) reveals subtle central outer retinal disruption with some central thickening (E). Subject 11 has normal FA image (F) and fundus photographs (G and H). In subject 12 (I–M), the AF image (I) is normal while the fundus photographs (J and K) show pigmentary changes in the macula, and the OCT B-scan reveals some degree of central outer retinal disruption (L) and central retinal attenuation (M).
Figure 3.
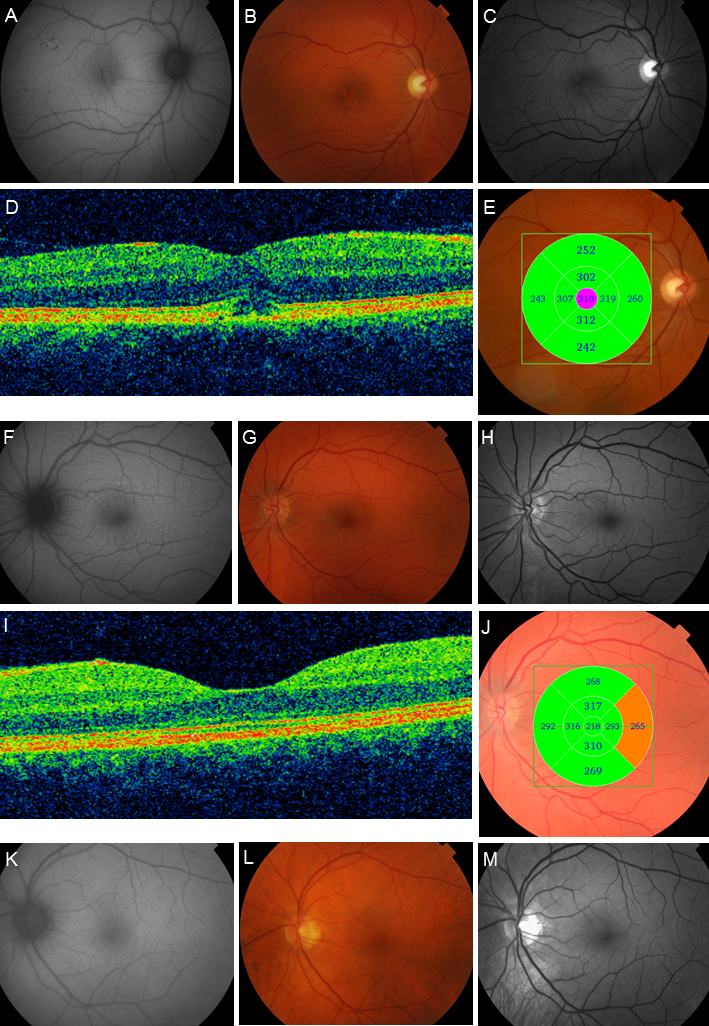
AF images, color photographs, red-free photographs, and OCT examinations from subjects 14, 15, and 16. In subject 14 (A–E), the AF image (A) demonstrates foci with mixed high and low AF signals in the macula and in the more peripheral parts of the posterior pole inside the vascular arcades. The fundus photographs (B and C) show pigmentary changes in the macula. The OCT B-scan (D) shows outer retinal disruption and some degree of edema, and there is thickening of the retina in the most central ring of the macular map (E). In subject 15, the AF image (F) shows some discrete hyperfluorescent spots superior to the macula, with corresponding subtle pigmentary changes on fundus photographs (G and H). The OCT B-scan is normal (I). On the retinal map (J), the outermost temporal segment is slightly thickened. Subject 16 (K–M) has no changes on the AF image (K) or fundus photographs (L and M).
Full-field electroretinography
FfERGs were recorded for all subjects. Of the 16 ABCA4 carriers, 14 (88%) had normal results (Table 2). The ITs of the 30 Hz flicker were delayed in subject 5, and the amplitude of the 30 Hz flicker was reduced in subject 1 (Table 2). The two subjects (9 and 18) with compound heterozygous mutations were excluded from the group-wise statistical analysis. A comparative analysis (Mann–Whitney U test) demonstrated no significant changes in ffERG parameters for the ABCA4 carriers compared to the control group (p>0.05 for all ffERG parameters). Subject 9, who was compound heterozygous, also had delayed ITs for the 30 Hz flicker.
Multifocal electroretinography
MERGs were recorded for all subjects. Examples of mERG registrations are shown in Figure 4. The two subjects with compound heterozygous mutations were excluded from the group-wise statistical analysis. The Mann–Whitney U test revealed significant reduction of the amplitudes for all rings (A1–A5) of the mERG when comparing the measurements in ABCA4 carriers to the control group (right eye: A1, p<0.0001; A2, p=0.003; A3, p=0.002; A4, p=0.001; A5, p<0.0001; left eye: A1, p=0.001; A2, p=0.001; A3, p=0.002; A4, p<0.0001; A5, p<0.0001; Figure 5). A corresponding statistical comparison of ITs showed significant delay in study subjects compared to the control group (right eye: IT1, p=0.002; IT2–5, p<0.0001; left eye: IT1–5, p<0.0001; Figure 6). Only two (13%) of 16 subjects (6 and 11) had both normal amplitudes and ITs compared to controls. Eleven of the 16 subjects (69%) had significantly reduced mERG amplitudes, and 14 (89%) had significantly delayed ITs (Table 2). The two subjects that were compound heterozygous (9 and 18) both had reduced mERG amplitudes and delayed ITs.
Figure 4.
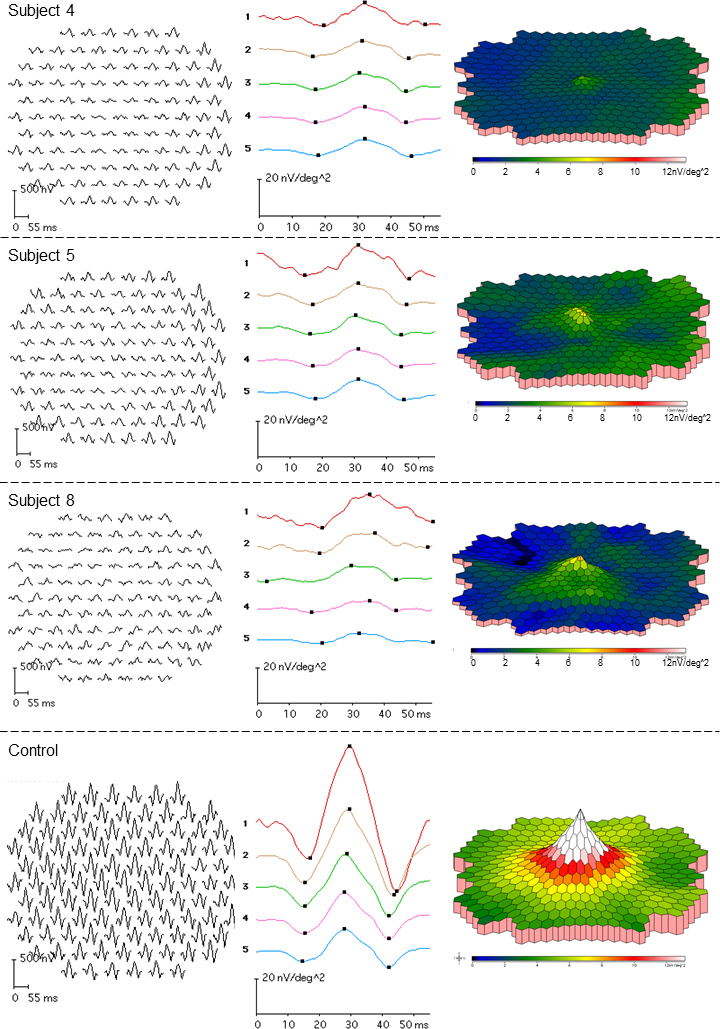
MERGs from three of the ABCA4 carriers (subject 4, top row; subject 5, second row; and subject 8, third row). The bottom row illustrates a normal mERG in one of the controls. Subject 4 has reduced mERG amplitudes and delayed ITs, although the fundus photos do not show any changes. Subject 5 also has reduced mERG amplitudes and delayed ITs. In this subject, the mERG alterations are accompanied by pigmentary changes in the macula (Figure 1B, C). Subject 8 with quite widely spread retinal flecks (Figure 1J, K) demonstrates delayed mERG ITs but normal mERG amplitudes.
Figure 5.

MERG P1 amplitudes (nV/deg2) in rings 1–5 for ABCA4 carriers and controls. Boxes show the median and interquartile ranges. Bars illustrate range. Circles indicate outliers.
Figure 6.

MERG P1 implicit times (ms) in rings 1–5 for ABCA4 carriers and controls. Each box shows the median and interquartile ranges. Bars illustrate range. Circles indicate outliers.
Optical coherence tomography
OCT scanning of the macular region was performed for all subjects. In two subjects, only the right eye was tested (1 and 2). Outer retinal disruption was found in several ABCA4 carriers. Examples of OCT images are found in Figures 1G,H,L,M, Figures 2 D,E,L,M, and Figures 3D, E,I,J. Measurements of retinal thickness were performed for 34 eyes. Fifteen had retinal thickness within normal limits compared to the normative database of the Topcon 3D OCT-1000. Thirteen showed thickening of one or more segments, three showed attenuation of one or more segments, and three had both attenuated and thickened segments.
Genetics, full-field electroretinography, multifocal electroretinography, optical coherence tomography, and ophthalmological examination
The three subjects (1, 5, and 9) with altered ffERG parameters all carried different ABCA4 mutations. One (9) was compound heterozygous for c.1622 T>C and c.3113 C>T, and in addition to delayed ffERG IT, she had severely reduced mERG amplitudes, prolonged mERG ITs, and bilateral outer retinal disruption on OCT images. The other two (1 and 5) carried the c.768 G>T and c.319 C>T mutations, respectively. Subject 1 with c.768 G>T also had severely reduced mERG amplitudes, prolonged mERG ITs, and outer retinal loss on an OCT B-scan. Subject 5 with the c.319 C>T mutation had altered mERG, AF changes, and pigmentary changes in the fundus (Figure 1A-C), and the OCT B-scan detected subtle outer retinal disruption in her left eye. Subject 4, who carried the mutation c.5461–10T>C, had normal ffERG but marked mERG changes, including reduced amplitudes and delayed ITs for all rings of the mERG and constricted 03e, 02e, and 01e isopters on Goldmann perimetry. Her OCT images also showed outer retinal disruption in both eyes. Two of the ABCA4 carriers (6 and 11) had normal mERG and ffERG results. Subject 6 with the c.5917delG mutation also had normal visual fields and fundus images but reduced BVA in her right eye, for which the OCT B-scan showed subtle outer retinal disruption. Subject 11, who had a c.4469 G>A mutation, presented with normal BVA, visual fields, AF, and fundus findings, (Figure 2F-H) including OCT images. Subject 10 (Figure 2A-E) with c.1804 C>T and subject 12 (Figure 2I-M) with c.6079 C>T both had normal ffERG and BVA but reduced mERG amplitudes, delayed mERG ITs, subtle pigmentary macular changes on fundus photos, and discrete outer retinal disruption on OCT B-scans. In three subjects (2, 3, and 13), the mERG showed reduced amplitudes and delayed ITs, and OCT images identified outer retinal changes; the other tests were normal. Two of these subjects carried c.768 G>T (2 and 3), and one had the c.2894 A>G mutation (13). Subjects 16 (Figure 3K-M) and 17 that carried the c.5714+5G>A and c.2894 A>G mutations, respectively, only showed delayed mERG ITs; otherwise, all their examinations were normal.
Discussion
ABCA4-associated retinal degenerations are recessively inherited disorders that lead to a wide spectrum of visual handicaps, from juvenile macular degeneration that affects central vision to CRD and recessive forms of RP with more widespread reduction of retinal function. The combination of ABCA4 mutations seems to influence the course of the disease [13,25,26] but in what way is still not fully understood. The conventional view on recessive traits is that carriers are free of symptoms. Still, in this study we wanted to investigate ABCA4 carriers to evaluate their visual function and, in the case of reduction, see if any particular mutations may be linked to particularly severe or mild changes. All but one subject (1) regarded themselves as free of visual complaints. Despite this, mERG recordings revealed clearly reduced macular function in the majority of the subjects and for the group of ABCA4 carriers as a whole compared to controls, with strong statistical significance for both P1 amplitudes and P1 ITs. Genetic testing revealed ABCA4 mutations in all the subjects. Two were even found to be compound heterozygous, which explains their reduced retinal function; therefore, they were not included in further group analysis. Still, another 14 ABCA4 carriers showed abnormal mERG results along with other altered parameters, such as AF, fundus appearance, and OCT, suggesting that carriership of ABCA4 mutations is associated with a risk of retinal dysfunction. This is in agreement with findings by Maia-Lopes et al. [29] and has to our knowledge been shown only once before. The most frequent mutations were c.2894 A>G and c.768 G>T, which have been found at a high frequency in other studies as well [9,30,31]. For example, c.2894 A>G was the most common mutation in a study done in Denmark [30]. In our study, the c.768 G>T mutation seemed to be associated with serious macular dysfunction, including both widespread reduced mERG amplitudes and delayed ITs. In one subject (1), even ffERG 30 Hz flicker amplitudes were reduced. This is in agreement with previous results [9,31,32] that found c.768 G>T to be a serious mutation. The c.768 G>T mutation was also associated with outer retinal disruption in all carriers. The c.2894 A>G mutation seemed to be associated with slightly milder but still severe macular dysfunction with delayed mERG ITs in all five subjects, but two had normal mERG amplitudes. AF was abnormal in three of the c.2894 A>G carriers, and OCT showed outer retinal changes in three c.2894 A>G subjects. Previous studies [30,32,33] have also suggested that the c.2894 A>G mutation is quite serious, leading to reduced mERGs and ffERGs.
The remaining ABCA4 mutations each occurred in only one subject and were therefore difficult to evaluate more closely. Nevertheless, some of them seemed to stand out in certain ways. For example, subject 5, who carried the quite uncommon mutation c.319 C>T, demonstrated severe changes with delayed ffERG 30 Hz flicker ITs, abnormal mERG parameters, an aberrant AF pattern, and degenerative macular changes. Based on this, c.319 C>T may be regarded as a likely serious mutation. Subject 4 showed constricted visual fields, reduced mERG amplitudes, delayed mERG ITs, and slightly reduced BVA. She carried the c.5461–10T>C mutation that has been found to be rather common in many populations [34-36] and that has also been associated with severe disease and early presentation [35,36], facts that support the seriousness of this mutation.
Subjects 6 and 11 had normal electrophysiological results, indicating that they may carry milder mutations. The c.5917delG mutation was found in subject 6. This mutation has previously been suggested to be quite severe in the homozygous state [32,33,37] but has also been associated with late onset STGD in compound heterozygous patients [32,37,38]. Thus, the severity is still difficult to define, but in the present study it seemed to be associated with normal retinal function in the carrier state. Subject 11 carried the c.4469 G>A mutation, a mutation that has been described in patients with STGD [34,39], but thorough phenotypic descriptions are few. Testa et al. [33] have described one compound heterozygous patient who had normal ffERG but abnormal mERG, and Fujunami et al. [34] have reported another compound heterozygous subject with abnormal pattern ERG and fundus changes who had a late onset foveal-sparing phenotype without any subjective visual complaints. The latter case supports the observation of this study that c.4469 G>A might be fairly mild.
Previous studies of patients with ABCA4-associated retinal degenerations have shown disruption of outer retinal layers and atrophy in the RPE and neurosensory retina on OCT scans [33,40-42]. Similar but less severe occurrences of outer retinal disruption were also found in subjects in the present study. Moreover, ABCA4-associated retinal degenerations have been associated with foveal attenuation and reduced macular volume [25,32,33,43]. In our study, OCT measurements of retinal thickness instead showed a mix of macular attenuation and thickening, and no correlations were found between macular thickness and mERG results. Perhaps this is because early retinal changes are very subtle and sometimes include some degree of edema before degeneration is manifest. Similar phenomena are seen in other retinal degenerations, such as RP [44-47].
Fundus photographs showed pigmentary changes in several ABCA4 carriers. Subject 8 even had deep yellow spots in the posterior pole and mid-periphery, pigmentary changes in the macula, and corresponding AF hyper - and hypofluorescence. This brings up the suspicion that he may have another as yet undetected ABCA4 mutation on his other allele and actually has late onset STGD [32,37,38]. However, because we found only one mutation he is considered a carrier in this study.
The findings of reduced retinal function and fundus changes in ABCA4 carriers again bring up the question of a possible role for ABCA4 mutations in AMD, and our results may support this hypothesis. Early studies on AMD propose the role of genetic factors in the cause of the disease, as the prevalence of AMD has been shown to be significantly higher in relatives of patients having the disease [48,49]. Allikmets et al. [50] have suggested that the risk of AMD is elevated threefold in c.6529 G>A carriers and fivefold in subjects carrying c.5882 G>A, as those mutations are correspondingly overrepresented in AMD patients compared to control subjects. Fritsche et al. [51] have reported that one subtype of AMD with typical fundus changes characterized by a fine granular pattern and peripheral punctate spots is significantly associated with heterozygous ABCA4 mutations. Moreover, Maia-Lopes et al. [29] have also demonstrated reduced retinal function in ABCA4 carriers, suggesting a predisposition for AMD in this group. Yet, other studies screening AMD populations for ABCA4 mutations have failed to show any increased frequency of ABCA4 mutations [39,52]. The discrepancy between results may be due to differences in the groups of AMD patients that have been included; perhaps ABCA4 mutations only occur in certain sub-groups of AMD [51]. To further elucidate the role of the ABCA4 gene in AMD, we plan to follow our subjects over time, repeatedly monitor the course of functional and morphological changes, and repeat the genetic testing as new ABCA4 mutations are continuously identified. Advanced knowledge of separate ABCA 4 mutations and their roles in various retinal degenerations is important to improve our counselling of patients and their relatives. One drawback of this study is the small sample size, although this shortcoming is partly compensated for by the evident statistical results concerning retinal function clearly reached with non-parametric tests.
To conclude, this study demonstrated reduced retinal function along with none to subtle or more widespread morphological retinal changes in ABCA4 carriers, again indicating a possible role for the ABCA4 gene in AMD. However, further follow-up of these and other ABCA4 carriers is needed. It also identified some ABCA4 mutations (c.768 G>T, c.5461–10T>C, and c.319 C>T) that seem to be associated with more severe retinal changes, making them likely severe mutations in ABCA4-associated retinal degenerations. Moreover, some mutations were instead correlated to milder or no retinal changes in the heterozygous state (c.5917delG and c.4469 G>A).
Acknowledgments
I thank Prof. Sten Andréasson for fruitful collaboration and Ing-Marie Holst and Boel Nilsson for skillful technical assistance. This study was supported by grants from Skåne County Council Research and Development Foundation, Stiftelsen för synskadade i fd Malmöhuslän, Stiftelsen Synfrämjandets Forskningsfond/Ögonfonden, the Cronqvists foundation, ARMEC Lindebergs stiftelse (2015-3), the Swedish Society of Medicine and Skane University Hospital foundations and donations. Disclosure: The author has full control of all primary data and agrees to allow the journal to review the data on request. The author has no conflict of interest to disclose.
References
- 1.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 2.Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, Lupski JR, Leppert M, Dean M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64:422–34. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. J Med Genet. 2003;40:641–50. doi: 10.1136/jmg.40.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allikmets R. Simple and complex ABCR: genetic predisposition to retinal disease. Am J Hum Genet. 2000;67:793–9. doi: 10.1086/303100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF, Travis GH. Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp Eye Res. 2001;73:877–86. doi: 10.1006/exer.2001.1093. [DOI] [PubMed] [Google Scholar]
- 6.Cremers FP, Van De Pol DJ, Van Driel M, Den Hollander AI, Van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 7.Fishman GA, Stone EM, Eliason DA, Taylor CM, Lindeman M, Derlacki DJ. ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch Ophthalmol. 2003;121:851–5. doi: 10.1001/archopht.121.6.851. [DOI] [PubMed] [Google Scholar]
- 8.Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, Rohrschneider K. Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci. 2002;43:1980–5. [PubMed] [Google Scholar]
- 9.Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 10.Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, Del Rio T, Dean M, Vilageliu L, Gonzalez-Duarte R, Balcells S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–2. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 11.Maugeri A, Van Driel MA, Van De Pol DJ, Klevering BJ, Van Haren FJ, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Dahl N, Brunner HG, Deutman AF, Hoyng CB, Cremers FP. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet. 1999;64:1024–35. doi: 10.1086/302323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC, Testa F, Maugeri A, Hoyng CB, Gouras P, Simonelli F, Lewis RA, Lupski JR, Cremers FP, Allikmets R. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 13.Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet. 2001;108:346–55. doi: 10.1007/s004390100493. [DOI] [PubMed] [Google Scholar]
- 14.Azarian SM, , Travis GH . The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR). FEBS Lett. 1997;409:247–52. doi: 10.1016/s0014-5793(97)00517-6. [DOI] [PubMed] [Google Scholar]
- 15.Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000;25:257–8. doi: 10.1038/77004. [DOI] [PubMed] [Google Scholar]
- 16.Sun H, Nathans J. Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet. 1997;17:15–6. doi: 10.1038/ng0997-15. [DOI] [PubMed] [Google Scholar]
- 17.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–8. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012;3:925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 20.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA. 2000;97:7154–9. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomes NL, Greenstein VC, Carlson JN, Tsang SH, Smith RT, Carr RE, Hood DC, Chang S. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2009;50:3953–9. doi: 10.1167/iovs.08-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullins RF, Kuehn MH, Radu RA, Enriquez GS, East JS, Schindler EI, Travis GH, Stone EM. Autosomal recessive retinitis pigmentosa due to ABCA4 mutations: clinical, pathologic, and molecular characterization. Invest Ophthalmol Vis Sci. 2012;53:1883–94. doi: 10.1167/iovs.12-9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119:359–69. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- 24.Cella W, Greenstein VC, Zernant-Rajang J, Smith TR, Barile G, Allikmets R, Tsang SHG. 1961E mutant allele in the Stargardt disease gene ABCA4 causes bull's eye maculopathy. Exp Eye Res. 2009;89:16–24. doi: 10.1016/j.exer.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burke TR, Tsang SH. Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet. 2011;32:165–74. doi: 10.3109/13816810.2011.565397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Driel MA, Maugeri A, Klevering BJ, Hoyng CB, Cremers FP. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet. 1998;19:117–22. doi: 10.1076/opge.19.3.117.2187. [DOI] [PubMed] [Google Scholar]
- 27.Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, Lupski JR. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 2005;14:2769–78. doi: 10.1093/hmg/ddi310. [DOI] [PubMed] [Google Scholar]
- 28.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 29.Maia-Lopes S, Silva ED, Silva MF, Reis A, Faria P, Castelo-Branco M. Evidence of widespread retinal dysfunction in patients with stargardt disease and morphologically unaffected carrier relatives. Invest Ophthalmol Vis Sci. 2008;49:1191–9. doi: 10.1167/iovs.07-1051. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg T, Klie F, Garred P, Schwartz M. N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol Vis. 2007;13:1962–9. [PubMed] [Google Scholar]
- 31.Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, Den Hollander AI, Hoyng CB. Clinical and genetic characteristics of late-onset Stargardt's disease. Ophthalmology. 2012;119:1199–210. doi: 10.1016/j.ophtha.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Kjellström U. Association between genotype and phenotype in families with mutations in the ABCA4 gene. Mol Vis. 2014;20:89–104. [PMC free article] [PubMed] [Google Scholar]
- 33.Testa F, Rossi S, Sodi A, Passerini I, Di Iorio V, Della Corte M, Banfi S, Surace EM, Menchini U, Auricchio A, Simonelli F. Correlation between photoreceptor layer integrity and visual function in patients with Stargardt disease: implications for gene therapy. Invest Ophthalmol Vis Sci. 2012;53:4409–15. doi: 10.1167/iovs.11-8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujinami K, Sergouniotis PI, Davidson AE, Wright G, Chana RK, Tsunoda K, Tsubota K, Egan CA, Robson AG, Moore AT, Holder GE, Michaelides M, Webster AR. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am J Ophthalmol 2013; 156:487–501 e1. [DOI] [PubMed]
- 35.Jonsson F, Burstedt MS, Sandgren O, Norberg A, Golovleva I. Novel mutations in CRB1 and ABCA4 genes cause Leber congenital amaurosis and Stargardt disease in a Swedish family. Eur J Hum Genet. 2013;21:1266–71. doi: 10.1038/ejhg.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miraldi Utz V, Coussa RG, Marino MJ, Chappelow AV, Pauer GJ, Hagstrom SA, Traboulsi EI. Predictors of visual acuity and genotype-phenotype correlates in a cohort of patients with Stargardt disease. Br J Ophthalmol. 2014;98:513–8. doi: 10.1136/bjophthalmol-2013-304270. [DOI] [PubMed] [Google Scholar]
- 37.Rivera A, White K, Stohr H, Steiner K, Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP, Apfelstedt-Sylla E, Weber BH. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet. 2000;67:800–13. doi: 10.1086/303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerth C, Andrassi-Darida M, Bock M, Preising MN, Weber BH, Lorenz B. Phenotypes of 16 Stargardt macular dystrophy/fundus flavimaculatus patients with known ABCA4 mutations and evaluation of genotype-phenotype correlation. Graefes Arch Clin Exp Ophthalmol. 2002;240:628–38. doi: 10.1007/s00417-002-0502-y. [DOI] [PubMed] [Google Scholar]
- 39.Webster AR, Heon E, Lotery AJ, Vandenburgh K, Casavant TL, Oh KT, Beck G, Fishman GA, Lam BL, Levin A, Heckenlively JR, Jacobson SG, Weleber RG, Sheffield VC, Stone EM. An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2001;42:1179–89. [PubMed] [Google Scholar]
- 40.Ducroq D, Shalev S, Habib A, Munnich A, Kaplan J, Rozet JM. Three different ABCA4 mutations in the same large family with several consanguineous loops affected with autosomal recessive cone-rod dystrophy. Eur J Hum Genet. 2006;14:1269–73. doi: 10.1038/sj.ejhg.5201691. [DOI] [PubMed] [Google Scholar]
- 41.Fujinami K, Sergouniotis PI, Davidson AE, Mackay DS, Tsunoda K, Tsubota K, Robson AG, Holder GE, Moore AT, Michaelides M, Webster AR. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology. 2013;120:2324–31. doi: 10.1016/j.ophtha.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 42.Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets R, Michaelides M, Moore AT. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015;122:326–34. doi: 10.1016/j.ophtha.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hargitai J, Zernant J, Somfai GM, Vamos R, Farkas A, Salacz G, Allikmets R. Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2005;46:4402–8. doi: 10.1167/iovs.05-0504. [DOI] [PubMed] [Google Scholar]
- 44.Hwang YH, Kim SW, Kim YY, Na JH, Kim HK, Sohn YH. Optic nerve head, retinal nerve fiber layer, and macular thickness measurements in young patients with retinitis pigmentosa. Curr Eye Res. 2012;37:914–20. doi: 10.3109/02713683.2012.688163. [DOI] [PubMed] [Google Scholar]
- 45.Kim YJ, Joe SG, Lee DH, Lee JY, Kim JG, Yoon YH. Correlations between spectral-domain OCT measurements and visual acuity in cystoid macular edema associated with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54:1303–9. doi: 10.1167/iovs.12-10149. [DOI] [PubMed] [Google Scholar]
- 46.Lupo S, Grenga PL, Vingolo EM. Fourier-domain optical coherence tomography and microperimetry findings in retinitis pigmentosa. Am J Ophthalmol. 2011;151:106–11. doi: 10.1016/j.ajo.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 47.Testa F, Rossi S, Colucci R, Gallo B, Di Iorio V, Della Corte M, Azzolini C, Melillo P, Simonelli F. Macular abnormalities in Italian patients with retinitis pigmentosa. Br J Ophthalmol. 2014;98:946–50. doi: 10.1136/bjophthalmol-2013-304082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hyman LG, Lilienfeld AM, Ferris FL, 3rd, Fine SL. Senile macular degeneration: a case-control study. Am J Epidemiol. 1983;118:213–27. doi: 10.1093/oxfordjournals.aje.a113629. [DOI] [PubMed] [Google Scholar]
- 49.Seddon JM, Ajani UA, Mitchell BD. Familial aggregation of age-related maculopathy. Am J Ophthalmol. 1997;123:199–206. doi: 10.1016/s0002-9394(14)71036-0. [DOI] [PubMed] [Google Scholar]
- 50.Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–91. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fritsche LG, Fleckenstein M, Fiebig BS, Schmitz-Valckenberg S, Bindewald-Wittich A, Keilhauer CN, Renner AB, Mackensen F, Mossner A, Pauleikhoff D, Adrion C, Mansmann U, Scholl HP, Holz FG, Weber BH. A subgroup of age-related macular degeneration is associated with mono-allelic sequence variants in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2012;53:2112–8. doi: 10.1167/iovs.11-8785. [DOI] [PubMed] [Google Scholar]
- 52.De La Paz MA, Guy VK, Abou-Donia S, Heinis R, Bracken B, Vance JM, Gilbert JR, Gass JD, Haines JL, Pericak-Vance MA. Analysis of the Stargardt disease gene (ABCR) in age-related macular degeneration. Ophthalmology. 1999;106:1531–6. doi: 10.1016/S0161-6420(99)90449-9. [DOI] [PubMed] [Google Scholar]