

Abstract
The role of brain cholesterol in Alzheimer's disease (AD) is currently a matter of debate. Experimental evidence suggests that reducing circulating and brain cholesterol protects against AD, however recent data indicate that low membrane cholesterol results in neurode-generation and that the cholesterol synthesis catalyst seladin-1 is down-regulated in AD-affected brain regions. We previously reported a significant correlation between resistance to amyloid toxicity and content of membrane cholesterol in differing cultured cell types. Here we provide evidence that Aβ42 pre-fibrillar aggregates accumulate more slowly and in reduced amount at the plasma membrane of human SH-SY5Y neuroblastoma cells overexpressing seladin-1 or treated with PEG-cholesterol than at the membrane of control cells. The accumulation was significantly increased in cholesterol-depleted cells following treatment with the specific seladin-1 inhibitor 5,22E-cholestadien-3-ol or with methyl-β-cyclodextrin. The resistance to amyloid toxicity and the early cytosolic Ca2+ rise following exposure to Aβ42 aggregates were increased and prevented, respectively, by increasing membrane cholesterol whereas the opposite effects were found in cholesterol-depleted cells. These results suggest that seladin-1-dependent cholesterol synthesis reduces membrane-aggregate interaction and cell damage associated to amyloid-induced imbalance of cytosolic Ca2+. Our findings extend recently reported data indicating that seladin-1 overexpression directly enhances the resistance to Aβ toxicity featuring seladin-1/DHCR 24 as a possible new susceptibility gene for sporadic AD.
Keywords: Aβ peptides, Alzheimer's disease, amyloid toxicity, membrane cholesterol, seladin-1
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disease associated with aging; it affects about 5–7% of individuals over 65 years thus representing a major public health problem. An effective pharmacological treatment for AD has not yet been found and progress in deciphering the biological mechanisms of AD pathogenesis can provide significant clues to develop new pharmacological strategies against such severe pathology. Accumulation of amyloid-β peptide (Aβ) aggregates in the central nervous system (CNS) is an inherent feature of AD (reviewed in 1). Aβ40 and Aβ42 peptides arise from intracellular processing of APP carried out by BACE and γ-secretase in the amyloidogenic pathway [1–3].
Cholesterol appears predominantly synthesized de novo in the CNS, where it enhances the production of pre-synaptic components and synaptic vesicles, whereas statins block dendrite outgrowth and axonal branching [4]. The role of cholesterol in AD pathogenesis is still controversial [5]. High cholesterol levels as risk factor for AD were first proposed in the early 1990s and since then considerable biochemical and clinical research has claimed the existence of a link between cholesterol, statin therapy and AD development [6–10]. On the other hand, it is known that hydrophilic statins do not cross the blood brain barrier and that brain cholesterol progressively reduces with increasing age (the main risk factor for sporadic AD), such decrease appearing considerably accelerated in AD people [11]. Presently, the question of whether statins can lower the incident rate of AD remains unresolved [12]. Recent findings suggest that the proteolytic machinery required for Aβ generation is located within lipid rafts or detergent-resistant membranes (DRMs) that are discrete cholesterol-rich microdomains of the cell membrane involved in a wide range of key biological processes [13, 14].
A key role of membrane cholesterol as a modulator of Aβ pep-tide production and clearance [15] as well as aggregation and neu-rotoxicity [16–19] has recently been described. In the hippocampus of healthy humans and transgenic mice expressing moderate levels of human APP only a small pool of endogenous APP and β site APP cleaving Enzyme or β-secretase (BACE) colocalize in the same membrane environment. An increased APP/BACE colocalization, likely caused by lipid raft disorganization, is found in hippocampal membranes from AD patients or in rodent hippocampal neurons with a moderate reduction of membrane cholesterol and is associated with elevated Aβ production. DRMs disruption also results in reduced activity of the Aβ-degrading enzyme plasmin because of decreased plasminogen binding to the plasma membrane [20–22]. Accordingly, rodents treated with lovastatin, the most permeable statin to the blood brain barrier, display increased Aβ production and senile plaque deposition [23]. Altogether, these data support a link between AD pathogenesis and loss of brain cholesterol suggesting that a fine modulation of the levels of this membrane sterol plays a crucial role in neuronal viability.
Seladin-1, a recently characterized gene, is down-regulated in neurons of AD-affected brain areas [24]. Seladin-1 overexpression appears to protect neuroglioma H4 cells against Aβ-mediated toxicity by reducing oxidative stress and caspase 3 activity [24]. Remarkably, the gene product is identical to DHCR24, the enzyme that catalyses the last step of cholesterol biosynthesis by reducing the Δ24 double bond of desmosterol. A mutation of this gene is associated with desmosterolosis, a severe disease which affects, among others, the nervous system [25]. We have recently shown that seladin-1 expression in human mesenchymal stem cells (hMSC) is abundant, but decreases dramatically upon differentiation into a neuronal phenotype (hMSC-n) with concomitant reduction of the total content of cholesterol [26]. This finding provides a new experimental approach to study the role of membrane cholesterol in the early events associated with neurodegeneration. Our recent studies demonstrate an hormonal regulation of seladin-1 expression [27, 28]; oestrogens and selective estrogen receptor modulators (SERMS) up-regulate seladin-1 in a long-term neuroblast cell culture from human foetal olfactory epithelium. This effect results in higher resistance to Aβ-induced toxicity and oxidative stress, featuring seladin-1 as a mediator of the neuro-protective effect of oestrogens [28]. A recent investigation on the involvement of seladin-1 in APP processing in mouse brains and in cultured neuroblastoma cells suggests that seladin-1-dependent cholesterol synthesis is involved in controlling APP location and processing as well as Aβ levels [29].
Whether seladin-1 protects against Aβ by merely controlling APP processing or also by directly enhancing cell resistance to Aβ cytotoxicity remains to be elucidated. It has been proposed that Aβ acts by forming specific channels in the plasma membranes of the exposed cells allowing a toxic flux of Ca2+ ions to penetrate inside them [30]. It has also been shown that enriching with cholesterol the plasma membranes of PC12 and GT1-7 cells modifies membrane fluidity preventing Aβ incorporation into, and perme-abilization of, the cell membrane [16, 17, 31]. We have previously shown that differing cultured cell types exhibit different resistance to amyloid toxicity and that the latter significantly correlates, among others, to the content of membrane cholesterol [32]. Accordingly, in the present study we investigated the relationship between cell resistance to amyloid toxicity, content of membrane cholesterol and seladin-1 levels by modulating the expression or the enzymatic activity of the latter in SH-SY5Y neuroblastoma cells. We provide evidence that in this neurotypic cell model (i) seladin-1 plays a key role as a modulator of the content of membrane cholesterol; (ii) high membrane cholesterol resulting from seladin-1 overexpression or treatment with PEG-cholesterol significantly protects cells against Aβ42 aggregate toxicity; (iii) low membrane cholesterol in cells treated with the seladin-1 inhibitor Δ22 or with β-CD results in increased cell vulnerability to Aβ42 aggregates; (iv) the different viability of cholesterol-enriched or cholesterol-depleted cells matches a reduced or increased interaction of the Aβ42 oligomers with the cell membrane, respectively; (v) the levels of intracellular free Ca2+ results significantly reduced or increased in cholesterol-enriched or cholesterol-depleted cells, respectively. Our results support the idea that seladin-1 protects cells against amyloid toxicity not only by controlling DRMs dynamics, APP processing and Aβ production, as previously shown [29], but also by directly enhancing cell resistance to Aβ toxicity. Both these mechanisms establish a link between seladin-1 down-regulation and AD pathogenesis featuring seladin-1 as a susceptibility gene candidate for sporadic AD and its pharmacological modulation as a possible novel approach in AD therapy.
Materials and methods
Materials and cell culture
All reagents were of analytical grade or the highest purity available. Cholesterol, Bradford reagent and BSTFA, Congo Red, PEG-cholesterol, β-CD and other chemicals were from Sigma, unless otherwise stated. Stigmasterol and Δ22 were from Steraloids Inc. (Newport, RI, USA). Wheat germ agglutinin-conjugated fluorescein, Fluo3-AM, Calcein-AM and pluronic acid F127 were purchased from Molecular Probes (Eugene, OR, USA). Hoechst 33342 was from Immunochemistry Technologies, LLC (Bloomington, MN, USA). Lyophilized Aβ42 and Aβ40 peptides, as trifluoroacetate salts (Bachem, Bübendorf, Switzerland), were stored and reconstituted as previously reported [33]. Lyophilized amylin 1-37 (Sigma, Milan, Italy) was stored and processed as previously reported [34]. Human SH-SY5Y neuroblastoma cells were obtained from A.T.C.C. (Manassas, VA) and were cultured in Dulbecco's modified eagle's medium (DMEM)/F-12 Ham with 25 mM N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid) (HEPES) and NaHCO3 (1:1) supplemented with 10% foetal bovine serum (FBS), 1.0% glutamine and 1.0% antibiotics from Sigma (Milan, Italy) in a 5.0% CO2 humidified atmosphere at 37°C in tissue plastic-ware from PBI international (Milan, Italy).
Seladin-1 up regulation by transient transfection and selective inhibition by Δ22
Cells were transiently transfected with a construct, kindly provided by Dr. I. Greeve [24], containing the seladin-1 open reading frame cloned in pEGFP-N1 vector (Clontech) using Lipofectamine 2000 (Invitrogen Carlsbad, CA, USA) on cells plated at 90% confluence in DMEM/F12 supplemented with 10% FBS without antibiotics. 48 hrs after transfection the cells were used for the Aβ42 toxicity experiments. Selective inhibition of seladin-1/DHCR24 by Δ22 was carried out by cell exposure to 10 μM Δ22 in ethanol (final ethanol concentration 0.2%) for 48 hrs replacing the medium after 24 hrs.
Membrane cholesterol enrichment with PEG-cholesterol or depletion with methyl-β-cyclodextrin
The increase of membrane cholesterol content was achieved by supplementing cell culture media with 0.1 mM PEG-cholesterol for 1 hr at 37°C, before cell exposure to the aggregates, as previously described [34]. Membrane cholesterol depletion was performed by incubating the cells with 1.0 mM β-CD for 30 min. at 37°C in the presence of 1.0% FBS.
Cholesterol and desmosterol measurements
The amount of cholesterol in the cells was determined by gas chromatography-mass spectrometry (GC-MS) as previously described with minor modifications [26]. Briefly, cells were maintained in medium without FBS for at least 6 hrs, then harvested in Phosphate Buffered Saline (PBS), pelletted and frozen in liquid nitrogen for storage at −80°C. Cell pellets were homogenized in PBS containing 9.0% sucrose with three short pulses of a polytron homogenizer (Ultraturrax T8, IKA LABORTECHNIK, Germany) in ice. The samples were centrifuged at 700x g for 10 min. at 4°C and the supernatant was considered as total extract. A further centrifugation of the supernatant was performed at 110,000x g for 1 hr at 4°C to pellet the membrane fraction. Aliquots corresponding to 5.0|μg of protein, as determined by the method of Bradford [35], were used for cholesterol determination in total extract or membrane fraction. After the addition of stigmasterol (1000 ng) as an internal standard, sterols were extracted, derivatized and automatically injected in a Hewlett-Packard GC-MS system. A six-point calibration curve in the 50-2000 ng cholesterol range was used for cholesterol quantification. The peak area ratios (PAR) were calculated using the signals at 458 m/z and 484 m/z for cholesterol and stigmasterol, respectively. Desmosterol was quantified by using the same procedure of cholesterol on sample aliquots containing 70 μg of protein. The PAR were calculated using the signal at 343 m/z.
Aggregate binding to the cell membrane and plasmin activity
The interaction of Aβ42 aggregates with plasma membranes was monitored in neuroblastoma cells seeded on glass coverslips by confocal scanning microscopy as previously described [33]. Briefly, after treatment with 1.0 μM Aβ42 pre-fibrillar aggregates for 10, 20, 30 and 60 min., the cells were counterstained with fluorescein-conjugated wheat germ agglutinin (50 μg/ml) for 10 min. to detect plasma membrane profiles and fixed in 2.0% buffered paraformaldehyde for 10 min. at room temperature. After plasma membrane permeabilization with a 3.0% glycerol solution, the coverslips were incubated for 60 min. with 1:1000 diluted mouse monoclonal anti-Aβ antibodies 6E10 (Signet, DBA, Italy) and then with 1:1000 diluted Texas Red-conjugated anti-mouse secondary antibodies (Vector Laboratories, DBA, Italy) for 90 min. Cell fluorescence was analysed by a confocal Bio-Rad MCR 1024 ES scanning microscope (Hercules, CA, USA) equipped with a Nikon Plan Apo 60× oil immersion objective and a krypton/argon laser source (15 mW) using two emission lines at 568 nm and 488 nm for Texas Red and fluorescein excitation, respectively. Fluorescence was achieved by Java image processing program (ImageJ) and expressed as fractional change above the resting baseline, ΔF/F, where F is the average baseline fluorescence before the application of peptide and ΔF represents the fluorescence changes over the baseline. The quantitation of aggregate adsorption to the cell surface was performed in neuroblastoma cells using the specific Congo Red dye as previously described [32]. Briefly, the same number of cells, representative of differing cholesterol enrichment or depletion conditions, was treated for 0, 5, 10, 20, 30, 60 min. with 1.0 μM Aβ42 aggregates in a 96-well plate and then washed twice with PBS. The residual aggregate-cell complexes were stained with 100 μL of 1.0 μM Congo Red in PBS for 20 min. at 37°C and measured photometrically at 550 nm with an ELISA plate reader according to Datki [36]. Congo Red values are reported as percent increases with respect to untreated cells (assumed as 100%). Endogenous plasmin enzymatic activity was assayed using the chromogenic substrate S-2251 (Chromogenix-Instrumentation Laboratory, Milan, Italy), according to Crameri with minor modification [29]. Briefly, cell pellets were homogenized in Hank's balanced saline solution (HBSS) (Sigma) with three short pulses of the ultraturrax T8 in ice, then 200 μg of the total cell lysate was placed in a 96-well plate in the presence of 2.0 mM chromogenic peptide. Absorbance was measured at 37°C and 405 nm every 5 min. for 60 min. on a Victor3 multi-label reader (Perkin Elmer, Milan, Italy).
Confocal analysis of cytosolic Ca2+ transients
The modifications of the intracellular free Ca2+ concentration in SH-SY5Y cells were monitored in cells plated on glass coverslips and incubated at room temperature for 10 min. in serum-free DMEM containing 0.1% bovine serum albumin (BSA), 10 μM Fluo3-AM as fluorescent calcium indicator, 0.1% dimethylsulfoxide (DMSO) and Pluronic acid F-127 (0.01% w/v) as previously reported [32]. Briefly, cell fluorescence was continuously monitored at 488-nm excitation in open slide flow-loading chambers mounted on the stage of a confocal Bio-Rad MRC-1024 ES scanning microscope. The time course analysis of Ca2+ waves after cell stimulation with 1.0 μM Aβ42 aggregates was performed using the Time Course Kinetics software (Bio-Rad). A single coverslip with adherent cells was used for each experiment and a variable number of cells ranging from 10 to 22 were analysed. Fluorescence signals are expressed as average of fractional changes in all cells above the resting baseline, ΔF/F, as reported above. Moreover, intracellular free Ca2+ was imaged in cells exposed for 20 min. to Aβ42 aggregates either in Ca2+-containing and in Ca+-free media, the latter including 5mM Mg2+ and 10 mM Ethylene Glycol-bis (β-Aminoethyl ether)N,N,N’,N’-TeTraaceTic Acid (EGTA), according to Demuro [39]. Then the cells were incubated with 10 μM Fluo3-AM for 10 min., fixed with 2.0% paraformaldehyde for 10 min., mounted on glass and analysed on the confocal microscope. In order to assess whether Aβ42 aggregates alter the membrane permeability, Δ22 treated and control cells were loaded with 2.0 μM calcein for 30 min. and then exposed to 1.0 μM Aβ42 aggregates for 20 min.
Amyloid cytotoxicity assay
The effect of aggregate treatments on neuroblastoma cell morphology was investigated by Hoechst 33342 dye staining. Briefly, after the exposure to 1.0 μM Aβ42 aggregates for 24 hrs at 37°C, cells were incubated with 20μg/ml Hoechst for 15 min. at 37°C. Then, cells were fixed as above reported. Blue fluorescence micrographs of cells were obtained under UV illumination in an epifluorescence inverted microscope (Nikon, Diaphot TMD-EF) with an appropriate filter set. Moreover, aggregate cytotoxicity to neuroblastoma cells was assessed in 96-well plates by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously reported [32]. Briefly, after the exposure to 1.0 μM Aβ42, Aβ40 or amylin aggregates for 24 hrs at 37°C, the cell cultures were incubated with 0.5 mg/ml MTT solution at 37°C for 4 hrs and with cell lysis buffer (20% SDS, 50% N,N-dimethylformamide, pH 4.7) overnight. Absorbance values of blue formazan were determined at 590 nm. Cell viability was expressed as percent of MTT reduction in treated cells as compared to cognate untreated cells, where it was assumed as 100%.
Statistical analysis
All data are expressed as mean ± standard deviation (SD). Comparisons between the different groups were performed by anova followed by Bonferroni's t-test. A P-value less than 0.05 was accepted as statistically significant.
Results
Modulation of cholesterol content in cellular compartments of human SH-SY5Y neuroblastoma cells
In the CNS, cell cholesterol is almost entirely provided by de novo synthesis; accordingly, any modulation of the expression or activity of the enzymes involved in cholesterol biosynthesis can rationally produce a variation of its intracellular levels. In our experimental model we modulated the activity of seladin-1, the enzyme that catalyses the last step of cholesterol synthesis, by transiently over-expressing its encoding gene or by the selective inhibition of its enzymatic activity and evaluated the effect on total and membrane cell cholesterol. The transient transfection of seladin-1 in SH-SY5Y cells resulted in a significant increase in cell cholesterol after 48 hrs. The increase was in the 10–20% range versus control for total cell cholesterol and in the 30–60% range versus control for membrane cholesterol, depending on transfection efficiency (Fig. 1). Conversely, SH-SY5Y cells were treated with 10 μM Δ22 for 48 hrs to specifically inhibit the enzymatic activity of seladin-1. This treatment significantly reduced total and membrane cell cholesterol by 10% and 15%versus control, respectively (Fig. 1). The time dependent accumulation of the substrate desmosterol in Δ22-treated cells indicates the selectivity of the inhibitor (Fig. 2A). Cell treatment with the inhibitor and its effect on cell desmosterol content do not affect cell viability as assessed by the MTT assay (Fig. 2B). In another set of experiments carried out by incubating human neuroblastoma cells in the presence of either PEG-choles-terol or β-CD we got similar modifications of membrane cholesterol content independently from seladin-1 modulation (Fig. 1).
Fig 1.
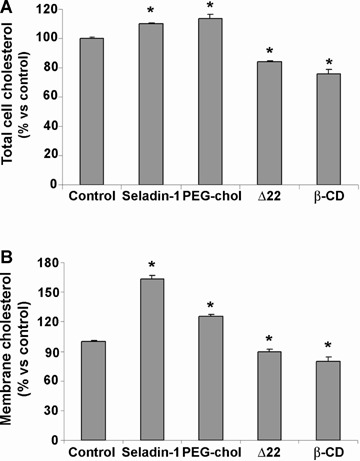
Modulation of total and membrane cholesterol in human SH-SY5Y neuroblastoma cells. (A) Total cholesterol and (B) membrane cholesterol in SH-SY5Y neuroblastoma cells 48 hrs after the transient transfection of seladin-1, 1 hr after incubation with 0.1 mM PEG-chol, 48 hrs after the selective inhibition of the enzyme with 10 μM Δ22 and 30 min. after cell culture supplementation with 1mM β-CD. The amount of cholesterol was determined by gas chromatography-mass spectrometry (GC-MS) using stigmasterol as internal standard as specified under Materials and Methods. The reported values are representative of three independent experiments. *Significant difference (P≤ 0.05) versus control cells.
Fig 2.
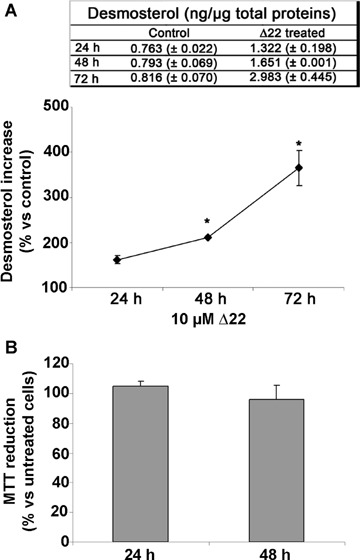
Δ22 inhibits seladin-1 enzymatic activity. (A) Desmosterol accumulation during the treatment of SH-SY5Y cells with 10 μM Δ22 measured in total cell lysates. (B) MTT assay on SH-SY5Y treated for 24 and 48 hrs with 10 μM Δ22.
Increasing cell cholesterol reduces Aβ aggregate interaction with the cell membrane
Cell degeneration in amyloid diseases appears mediated by a toxic mechanism involving some interaction of the aggregated species with the plasma membrane of the affected cells [30, 37–39]. In seladin-1 overexpressing cells and in PEG-cholesterol supplemented cells the increase of plasma membrane cholesterol resulted in a reduced Aβ oligomer binding to the plasma membrane respect to control cells. This is shown by confocal microscopy analysis at different focal lengths of neuroblastoma cells exposed for 10, 20, 30 and 60 min. to Aβ42 pre-fibrillar aggregates (Fig. 3A). Conversely, the same amyloid oligomers added to the cell culture medium appear to accumulate more quickly and to a greater extent at the plasma membrane in Δ22 and β-CD treated cells characterized by a reduced content of cholesterol than in control cells (Fig. 3A). The presence of amyloid aggregates bound to the plasma membrane of exposed cells was detected by immunofluorescence using monoclonal anti-Aβ antibodies after cell membrane permeabilization with glycerol. Notably, the red fluorescence signals related to 6E10 antibody that recognizes also the full length human APP, are negligible in all the analysed conditions before Aβ treatment (see time 0). This evidence let us to rule out the possibility that the differences in the accumulation of exogenous Aβ at the cell membrane were due to different levels of APP expression in these cells. The analysis of SH-SY5Y neuroblastoma cells transfected with an empty expression plasmid without the seladin-1 encoding gene and of ethanol-exposed cells confirmed the specificities of the recorded fluorescence signals (Fig. 3B). This evidence agrees with the Congo Red assay showing that seladin-1 overexpression and cell media supplementation with soluble cholesterol resulted in a significant reduction of rate and extent of Aβ42 aggregate binding to the cell plasma membranes respect to controls (Fig. 3C). Conversely, the loss of membrane cholesterol resulted in a higher rate of aggregate binding to the surface of SH-SY5Y cells treated with the specific seladin-1 inhibitor Δ22 or with β-CD as compared to control cultures. Finally, the ability of exposed cells to bind Aβ aggregates appeared saturable, reaching its limit in the first 10–30 min. (Fig. 3C). No significant difference in Congo Red absorbance values between different groups of Aβ42-untreated cells was observed. Seladin-1 has been shown to affect plasmin activity [29] that is involved in Aβ degradation [20]. Therefore, we measured plasmin activity in lysates of control cells, seladin-1 overexpressing cells and Δ22 treated cells. As shown in Figure 4, transient transfection of seladin-1 in SH-SY5Y resulted in a significantly higher plasmin activity than in control cultures. Conversely, loss in membrane cholesterol resulting from cell treatment with Δ22, led to a significant reduction of plasmin activity respect to control cells. The variation of plasmin activity respect to control is greater for seladin-1 overexpressing cells than for Δ22 treated cells. This effect, as described by Crameri et al. [29], is likely ascribable to the enhanced ability of cholesterol rich membranes to bind plas-minogen. Having assessed the link between seladin-1 expression, membrane cholesterol levels and Aβ aggregate interaction with the plasma membrane of exposed cells, we sought to investigate whether the modulation of seladin-1 expression was associated with any modification of plasma membrane permeability to the external Ca2+ ions.
Fig 3.
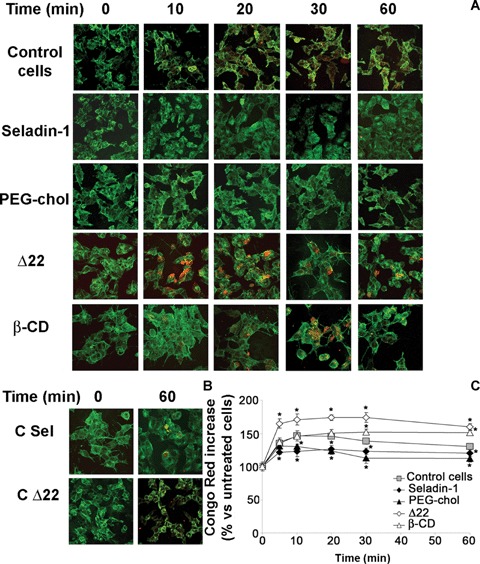
Increasing cell cholesterol reduces amyloid-β peptide (Aβ) aggregate interaction with the cell membrane. (A, B) Representative confo-cal microscopy images showing aggregates in contact with, and penetrating into, the plasma membrane of neuroblastoma cells under different experimental conditions. Cells transfected with empty expression plasmid without seladin-1 gene (C Sel) and cells exposed to ethanol (C Δ22) were used as internal controls. After treatment for 0, 10, 20, 30 and 60 min. with Aβ42 pre-fibrillar aggregates, counterstaining was performed with fluorescein-conjugated wheat germ agglutinin to detect plasma membrane profile (green). The aggregates were labelled with monoclonal mouse 6E10 anti-Aβ antibodies and Texas Red-conjugated anti-mouse secondary antibodies with plasma membrane permeabilization with glycerol, as specified under Materials and methods. (C) Time-course of amyloid aggregate binding to neuroblastoma cells. After the exposure to 1.0 μM Aβ42 pre-fibrillar aggregates for 5, 10, 20, 30, 60 min., cells were washed twice with PBS and the residual aggregate-cell complex was stained with 1.0 μM Congo Red for 20 min. Under these conditions, Congo Red-staining is a measure of the amount of pre-fibrillar aggregates adsorbed to cell membrane surface. *Significant difference (P≤ 0.05) versus control cells.
Fig 4.
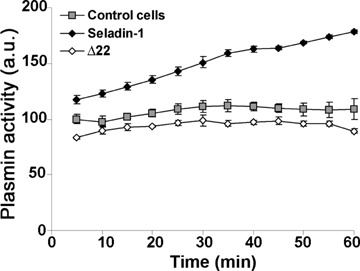
Seladin-1 modulation affects plasmin activity. Plasmin activity, was assayed using the chromogenic substrate S-2251 in total lysates from SH-SY5Y control cells, from cells 48 hrs after the transient trans-fection of seladin-1 and from cells 48 hrs after the selective inhibition of the enzyme with 10 μM Δ22. For details, see under Materials and methods.
Seladin-1 protects against alteration of intracellular Ca2+ levels in cells exposed to Aβ aggregates
Derangement of ion distribution across the plasma membrane is a shared and early biochemical modification displayed by cells exposed to toxic amyloid aggregates [32, 39, 40]. To investigate Ca2+ homeostasis in our cell systems, we performed a confocal microscopy time course analysis of the intracellular Ca2+ content in unfixed viable differently treated SH-SY5Y cells plated on glass coverslips upon exposure to Aβ42 aggregates. A higher membrane cholesterol content prevented the early and sharp increase in cytosolic free Ca2+ levels induced by Aβ aggregates in control cells with basal cholesterol content (Fig. 5A). Actually, both in seladin-1 overexpressing cells and in PEG-cholesterol treated cells the content of cytosolic Ca2+ was substantially the same as that found in cells not exposed to the aggregates and significantly lower than in aggregate-exposed control cultures (Fig. 5C). Similar experiments carried out on SH-SY5Y cells transfected with an empty expression plasmid without the seladin-1 encoding gene or on ethanol-exposed cells confirmed the specificities of the fluorescence signals (Fig. 5B). Conversely, loss in membrane cholesterol, resulting from cell treatment with Δ22 or β-CD, triggered an accelerated and greater increase of cytosolic free Ca2+ (Fig. 5C). Notably, no fluorescence signal decay was observed in the low membrane cholesterol cells during the 20 min. continuous analysis. Moreover, Aβ aggregates failed to induce any modification of the calcein signal in low cholesterol membranes from A22-treated cells such as in control cells (Fig. 6A). Furthermore, to discriminate whether the increase in cytosolic Ca2+ concentration evoked by Aβ aggregates arose from the influx of extracellular Ca2+ across the plasma membrane or from Ca2+ release from the intracellular stores, we conduced matching experiments on cells cultured in Ca2+-free medium. The increase in Ca2+ fluorescence signal observed in cells exposed to Aβ aggregates in Ca2+-containing media was completely abolished when the cells were cultured in a Ca2+-free medium (Fig. 6B). Next, we investigated whether membrane cholesterol modulation following regulation of seladin-1 expression also resulted in modifications of Aβ aggregate toxicity to exposed cells.
Fig 5.
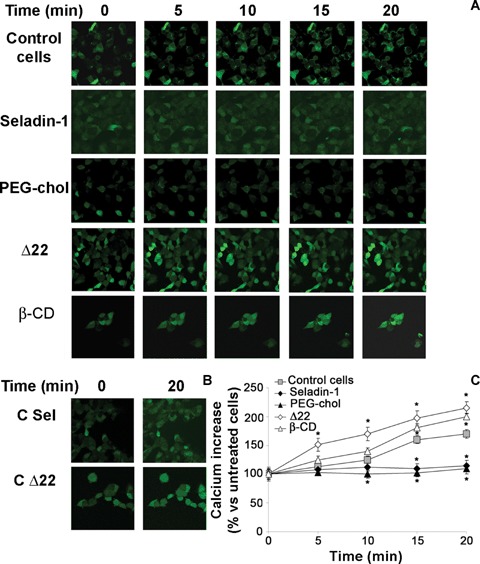
Intracellular free Ca2+ levels in neuroblastoma cells exposed to Aβ aggregates. A, Continuous confocal microscopy analysis of intracellular free Ca2+ levels in neuroblastoma cells during the first 20 min. of exposure to Aβ42 pre-fibrillar aggregates. Cytosolic Ca2+ levels were imaged by confocal microscopy using the fluorescent dye Fluo-3AM as a probe, according to the method described under Materials and methods. (B) Changes in intracellular free Ca2+ levels in empty expression plasmid without seladin-1 gene (C Sel) and cells exposed to ethanol (C Δ22). (C) A variable number of representative living cells ranging from 10 to 22 were analysed in each coverslip in three independent experiments. Fluorescence signals are expressed as average of fractional changes in all cells above the resting baseline, ΔF/F, where F is the average baseline fluorescence before the application of the amyloid aggregates and ΔF is the fluorescence change over the baseline. Values are expressed as% versus untreated cells. *Significant difference (P≤0.05) versus control cells.
Fig 6.
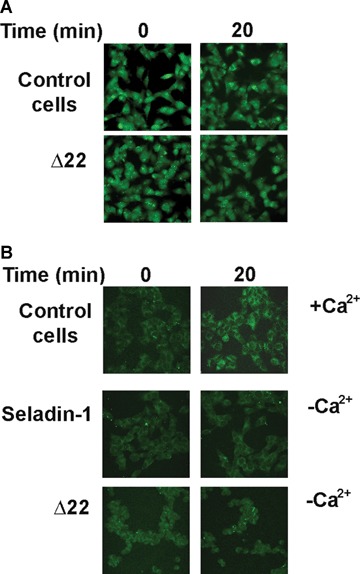
Aβ aggregates induce a rise in intracellular Ca2+ by affecting membrane permeability. (A) Representative confocal microscopy images of Δ22 treated cells loaded with calcein-AM and exposed or not to Aβ aggregates. (B) Confocal microscopy analysis of intracellular free Ca2+ in fixed neuroblastoma cells exposed for 20 min. to Aβ42 aggregates either in Ca2+-containing (+ Ca2+) and in Ca2+-free (−Ca2+) medium. For details see under Materials and methods.
High membrane cholesterol prevents Aβ aggregate cytotoxicity
To assess the effect of the regulation of membrane-cholesterol homeostasis on amyloid toxicity, we evaluated the effect of our treatments both on cell morphology and on the mitochondrial status. As shown in Figure 7A, morphological evaluation of SH-SY5Y cells using Hoechst 33342 staining revealed no marked characteristics of apoptosis in cells overexpressing seladin-1 or treated with PEG-cholesterol after their exposure to Aβ aggregates for 24 hrs respect to similarly exposed control cells with basal cholesterol content. Accordingly, as revealed by the MTT assay reported in Figure 7B, cells overexpressing seladin-1 or treated with PEG-cholesterol exposed to Aβ42 aggregates displayed significantly improved viability respect to similarly exposed control cells with basal cholesterol content. Conversely, loss in membrane cholesterol following cell treatment with Δ22 or β-CD resulted in a significant increase in the number of cells showing nuclear condensation (Fig. 7A) and in a significant impairment of cell viability (Fig. 7B) upon exposure to the aggregates, respect to similarly exposed control cells with basal cholesterol content. Finally, the selectivity of the cytoprotective role of seladin-1 was investigated in cells exposed to aggregates other than Aβ42, such as Aβ40 and amylin (Fig. 7C). A reduced susceptibility to the stress induced by Aβ40 was observed in seladin-1 overexpressing cells respect to control cells with basal cholesterol content, confirming the increased resistance of these cells. Reduced vulnerability was also seen when the same cells were exposed to amylin oligomers, further supporting the generality of these effects. On the other hand, an increased vulnerability, though to a different extent, to both Aβ40 and amylin aggregates, was observed in A22-treated cells.
Fig 7.
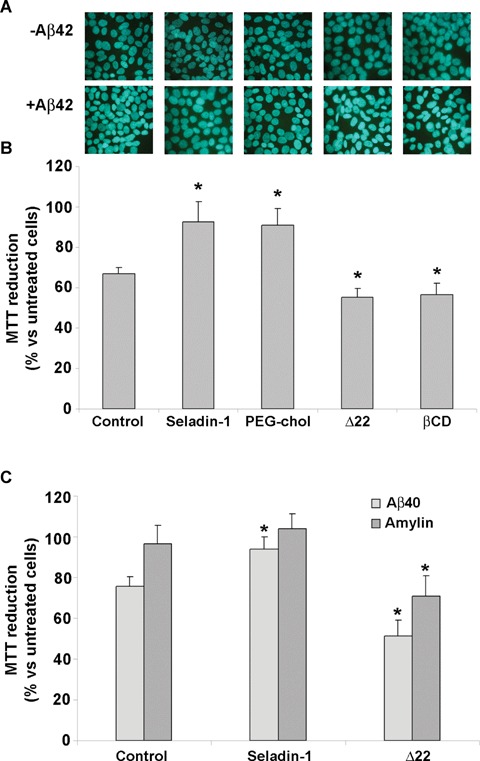
Seladin-1 protects against Aβ aggregate toxicity. (A) The effect of Aβ42 aggregates on neuroblastoma cell morphology was evaluated using the Hoechst 33342 dye staining. Representative blue fluorescence micrographs of untreated (–Aβ42) or exposed SH-SY5Y cells to Aβ42 aggregates for 24 hrs (+Aβ42). For details, see under Materials and methods. (B) Membrane cholesterol enrichment by seladin-1 overexpression or PEG-cholesterol treatment protects neuroblastoma cells against amyloid aggregate toxicity. Cell viability was checked by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) reduction test in cells treated with 1.0 μM Aβ42 pre-fibrillar aggregates for 24 hrs. The reported values are representative of four independent experiments, each performed in triplicate. (C) Cell viability was also checked by the MTT reduction test in control cells, seladin-1 overexpressing cells and Δ22 treated cells exposed to 1.0μM β40 and amylin pre-fibrillar aggregates for 24 hrs. The reported values are representative of three independent experiments, each performed in triplicate.*Significant difference (P≤0,05) versus control cells.
Discussion
The possible relation between cholesterol and neurodegeneration has recently led to focus on the role played by seladin-1, a flavo-protein with desmosterol reductase activity that catalyses the last step of cholesterol synthesis. Seladin-1 appears to be down-regulated in brain areas more severely affected by AD and its overexpression increases the resistance against Aβ-induced neurode-generation [24, 28]. Moreover, reduced brain cholesterol, desmosterol accumulation, displacement of BACE1 from DRM light fractions to APP containing fractions with increased APP β cleavage and Aβ production have been reported in seladin-1 gene knockout mice respect to wild-type mice; conversely, seladin-1 overexpres-sion in SH-SY5Y cells increases cell membrane cholesterol favouring BACE1 recruitment into APP-devoid DRM fractions and reducing both APP processing and Aβ peptide production [29]. These data strongly support the involvement of seladin-1 in the pathway of Aβ production, however they do not address the possibility that cholesterol variations resulting from seladin-1 modulation can affect directly cell resistance to Aβ toxicity.
In the present study, we investigated whether the modulation of membrane cholesterol by seladin-1 overexpression or inhibition could influence Aβ42 aggregate cytotoxicity to human neurotypic SH-SY5Y cells by modulating Aβ aggregate binding to the cell membrane, in most cases a recognized key step in amyloid cytotoxicity, and membrane permeability to the external Ca2+. We found that the levels of both the total and the plasma membrane cholesterol were significantly higher in seladin-1 overexpressing cells than in control cells, in agreement with previous data [29]. Conversely, inhibiting the enzymatic activity of seladin-1 resulted in a significant reduction of total and membrane cell cholesterol. As far as we know, this is the first time that a reduction of the enzymatic activity of seladin-1 is correlated to a decrease of membrane cholesterol and an increase of cell vulnerability to aggregate toxicity. In spite of the limitation of the cell model used, this finding supports the idea that the low seladin-1 levels found in AD brains [24] are responsible for either the reduction of membrane cholesterol described in those brains [21] and the increase of the neuronal vulnerability to Aβ aggregates.
Our data show that neuroblastoma cells enriched in membrane cholesterol following overexpression of seladin-1 or treatment with PEG-cholesterol display higher resistance to Aβ42 aggregate toxicity, in agreement with previous data on human neuroglioma H4 cells and FNC-B4 cells [24, 28]. Conversely, reducing cholesterol levels by seladin-1 inhibition or by cell treatment with β-CD resulted in increased cell vulnerability to Aβ aggregates. Our evidence suggests that seladin-1 is able to modulate plasmin activity that is enhanced in seladin-1 overexpressing cells and reduced, although to a lesser extent, in Δ22 treated cells. This effect, as described by Crameri et al. [29], is probably ascribable to the enhanced ability of cholesterol rich membranes to bind plasminogen. Therefore the seladin-1-induced variation in membrane cholesterol modulates Aβ42 toxicity at least through two different mechanisms: the modification of membrane permeability and the modulation of plasmin activity. These two mechanisms appear to affect in a different way cell susceptibility to Aβ peptides. In particular, the change in membrane permeability induced by Δ22 cell treatment is likely the predominant effect, while both two mechanisms can equally contribute to cell protection when seladin-1 is overexpressed. However, when seladin-1 is overexpressed, other accessory effects of this multifunctional protein associated to its ability to reduce caspase 3 activity [41, 42] and to control the Ras/p53 signalling pathway [43] cannot be excluded. On the other hand our data indicate that, at least in our system, a reduction of seladin-1 enzymatic activity with no modification of the total amount of the protein, is sufficient to increase cell vulnerability by lowering membrane cholesterol. Overall, our data and those previously reported suggest that the higher vulnerability to Aβ aggregates of the AD relevant brain areas where seladin-1 is down-regulated results, at least in part, from the decrease of its enzymatic activity and hence of the content of cholesterol in the neuronal membranes. It is worth noting that this effect is unlikely to result from the build-up of the seladin-1 substrate desmosterol in seladin-1 deficient cells; in fact it has previously been reported that the increase of desmosterol in cultured cells does not affect DMR structure and function [29]. Moreover, our data show that the treatment with Δ22 did not affect cell viability. Conversely, the protective effect demonstrated in cells overexpressing seladin-1 should correspond to a decrease in cell desmosterol for the enhancement of seladin-1 enzymatic activity. The final effect of cholesterol-desmosterol modulation on cell susceptibility to Aβ42 peptides is similar to that obtained with β-CD or PEG-Chol as assessed by the MTT assay. These latter treatments did not affect cell desmosterol only modulating cell cholesterol. These data, although indirectly, support our opinion that cholesterol modulation and not desmosterol modification is responsible of this final effect. Remarkably, Aβ40 and amylin treatments of seladin-1 over-expressing or Δ22 inhibited cells affect cell viability resembling Aβ42 outcome. These data suggest that the seladin-1-dependent modification of membrane cholesterol is generally able to modulate the toxic effect of amyloidogenic peptides independently from their amino acid sequence.
We previously reported that amyloid cytotoxicity can be related, among others, to the ability of the aggregates to interact with the plasma membrane [32]; in addition, disruption of cholesterol homeostasis can be detrimental to cells because toxic Aβ aggregates interact more easily with cholesterol-depleted membranes [16, 17]. Therefore, we investigated whether membrane content of cholesterol could affect the rate and extent of Aβ42 aggregate interaction with the plasma membrane of our neuronal cell model. We found by confocal microscopy and Congo red assay that the binding rate of the Aβ42 assemblies to the plasma membranes of Aβ42-exposed cells was significantly faster and increased in cholesterol-poor cells treated with Δ22 and β-CD than in cholesterol-enriched cells overexpressing seladin-1 or treated with PEG-cholesterol. These findings confirm previous results [32] and the idea that the ability of Aβ42 aggregates to bind to the plasma membrane is significantly affected by its content in cholesterol. In particular, our data agree with several reports indicating that cholesterol content affects membrane physical features, such as fluidity and density of lipid packing resulting in alterations of aggregate recruitment to the membrane and membrane permeabilization [17]. Under our experimental conditions, the differences found between Δ22-and β-CD-treated cells in Aβ binding to the cell membrane and in the intracellular calcium spikes cannot be explained by a significant variation in cholesterol membrane content. An in depth investigation of membrane distribution of cholesterol in Δ22-and β-CD-treated cells appear useful to substantiate this different behaviour. It has previously been reported that Aβ binding and aggregation occurs in the lipid raft and is mediated by clusters of the ganglioside GM1, as detected by ThT or Congo Red [44]. It was hypothesized that Aβ adopts an altered conformation through its binding to GM1 acting as a seed for Aβ fibrillo-genesis in AD brain [44]. It has also been reported that GM1 clusters are susceptible to membrane cholesterol depletion [45]. Recent evidence suggests that Aβ interacts with the APP present at the cell surface and acts as a ligand of its own precursor resulting in a cell death-related signal [46]. We cannot exclude that different APP distribution and/or accessibility in the plasma membranes, likely resulting from lipid raft reorganization at our different experimental conditions, may contribute to the interaction of the Aβ aggregates with the cell membranes.
It has previously been shown that enriching with cholesterol the plasma membranes of PC12, SH-SY5Y and GT1-7 cells modifies membrane fluidity preventing Aβ incorporation into, and permeabilization of, the cell membrane [16, 17, 31]. A leading theory on the molecular basis of amyloid toxicity is that pore-like pre-fib-rillar aggregates interact with the cell membranes leading to membrane permeabilization and free Ca2+ imbalance [37, 47–51]. Our confocal images show that in our exposed cells an early, sharp increase of free cytosolic Ca2+ does occur, in agreement with previously reported findings [40]. Actually, our cholesterol-poor cells displayed a prompt and enhanced Ca2+ increase upon aggregate binding to the cell membrane respect to control cells whereas cholesterol-enriched cells displayed slower and reduced Aβ aggregate binding to the plasma membrane resulting in a delayed and significantly reduced rise of free Ca2+ content respect to control cells. The Ca + signals evoked by Aβ42 oligomers were abolished in cells cultured in Ca2+-free medium, indicating a main contribution from extracellular, rather than from the intracellular, Ca2+ sources. We also sought to provide information on weather intracellular free Ca2+ dyshomeostasis in seladin-1 down-regulated neurons of AD-affected brain areas could result from a generalized increase in membrane permeability [39, 52] or from the formation of membrane pores [30, 49–51]. In our cell model, we did not observe any rapid fluorescence signal decay in low membrane cholesterol cells, suggesting the absence of a fluorescent dye leakage invoked by a generalized increase in membrane permeability induced by Aβ aggregates. On the contrary, our calcein data suggest a selective-specific pore-like mechanism for oligomer-mediated toxicity, even though minor changes of membrane permeability cannot be ruled out. The relevance of these finding is stressed by the well known association between alterations of intracellular free Ca2+ and either oxidative stress [53] and cell death [54].
Overall, our results provide information useful to depict a mechanism of cell impairment and death that can be common to pre-fibrillar aggregates of most peptides and proteins. It apparently starts with aggregate binding to the cell membrane resulting in membrane permeabilization, subsequent alteration of ion distribution inside exposed cells and oxidative stress [32, 55]. Seladin-1 down-regulation or inhibition can contribute to such a chain of events, indicating the importance, among the different activities of seladin-1, of its regulatory effect on membrane cholesterol levels. Conversely, seladin-1 overexpression and the resulting membrane cholesterol increase provide a mechanistic explanation of the cytoprotection by seladin-1 against aggregate toxicity. Our data, together with many others, suggest that neuronal integrity requires the maintenance of a proper steady-state level of brain cholesterol and that its reduction, even a moderate one such as that obtained by seladin-1 inhibition or by β-CD treatment, results in severe impairment of cell viability. These results implement the recently published data on the role of a moderate reduction of membrane cholesterol in AD pathogenesis [12]. In particular, they suggest that a mild loss of neuronal membrane cholesterol results not only in altered APP processing with increased Aβ production [15, 29] but also in a quicker and increased binding of secreted Aβ oligomers to the neuronal membrane with subsequent alteration of Ca2+ distribution and cell damage. The recently reported seladin-1 down-regulation in AD patients is likely to contribute to this mechanism by a reduced cholesterol synthesis. Taken together these and other recently reported data highlight Seladin-1/DHCR24 as a possible new susceptibility gene and pharmacological target for sporadic AD. Further investigation is needed to confirm such possibility.
Acknowledgments
This study was supported by grants from the Italian MIUR (project numbers 2005054147_001; 2005053998_001 and 2006069900_001); from the Regione Toscana (TRESOR project, principal investigator Prof. Mario Serio) and from Ente Cassa di Risparmio di Firenze (grant n. 2005.07.01).
References
- 1.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 3.Takeda K, Araki W, Tabira T. Enhanced generation of intracellular Abeta42 amyloid peptide by mutation of presenilins PS1 and PS2. Eur J Neurosci. 2004;19:258–64. doi: 10.1111/j.0953-816x.2003.03135.x. [DOI] [PubMed] [Google Scholar]
- 4.Herman GE. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet. 2003;12 Spec No 1:R75–88. doi: 10.1093/hmg/ddg072. [DOI] [PubMed] [Google Scholar]
- 5.Ledesma MD, Dotti CG. The conflicting role of brain cholesterol in Alzheimer's disease: lessons from the brain plasminogen system. Biochem Soc Symp. 2005;72:129–38. doi: 10.1042/bss0720129. [DOI] [PubMed] [Google Scholar]
- 6.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–4. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 8.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–43. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 9.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–61. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Austen B, Christodoulou G, Terry JE. Relation between cholesterol levels, statins and Alzheimer's disease in the human population. J Nutr Health Aging. 2002;6:377–82. [PubMed] [Google Scholar]
- 11.Svennerholm L, Bostrom K, Jungbjer B, Olsson L. Membrane lipids of adult human brain: lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J Neurochem. 1994;63:1802–11. doi: 10.1046/j.1471-4159.1994.63051802.x. [DOI] [PubMed] [Google Scholar]
- 12.Shabab LA, Hsiung G-YR, Feldman H. Cholesterol in Alzheimer's disease. Lancet Neurol. 2005;4:841–52. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- 13.Kaether C, Haass C. A lipid boundary separates APP and secretases and limits amyloid beta-peptide generation. J Cell Biol. 2004;167:809–12. doi: 10.1083/jcb.200410090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vetrivel KS, Thinakaran G. Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology. 2006;66:69–73. doi: 10.1212/01.wnl.0000192107.17175.39. [DOI] [PubMed] [Google Scholar]
- 15.Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167:953–60. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yip CM, Elton EA, Darabie AA, Morrison MR, McLaurin J. Cholesterol, a modulator of membrane-associated Abeta-fibrillogenesis and neurotoxicity. J Mol Biol. 2001;311:723–34. doi: 10.1006/jmbi.2001.4881. [DOI] [PubMed] [Google Scholar]
- 17.Arispe N, Doh M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer's disease Abeta(1-40) and (1-42) peptides. FASEB J. 2002;16:1526–36. doi: 10.1096/fj.02-0829com. [DOI] [PubMed] [Google Scholar]
- 18.Kawahara M, Kuroda Y. Intracellular calcium changes in neuronal cells induced by Alzheimer's beta-amyloid protein are blocked by estradiol and cholesterol. Cell Mol Neurobiol. 2001;21:1–13. doi: 10.1023/A:1007168910582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sponne I, Fifre A, Koziel V, Oster T, Olivier JL, Pillot T. Membrane cholesterol interferes with neuronal apoptosis induced by soluble oligomers but not fibrils of the amyloid β peptide. FASEB J. 2004;18:836–8. doi: 10.1096/fj.03-0372fje. [DOI] [PubMed] [Google Scholar]
- 20.Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep. 2000;1:530–5. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ledesma MD, Abad-Rodriguez J, Galvan C, Biondi E, Navarro P, Delacourte A, Dingwall C, Dotti CG. Raft disorganization leads to reduced plasmin activity in Alzheimer's disease brains. EMBO Rep. 2003;4:1190–6. doi: 10.1038/sj.embor.7400021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ledesma MD, Da Silva JS, Schevchenko A, Wilm M, Dotti CG. Proteomic characterisation of neuronal sphingolipid-cholesterol microdomains: role in plasminogen activation. Brain Res. 2003;987:107–116. doi: 10.1016/s0006-8993(03)03296-7. [DOI] [PubMed] [Google Scholar]
- 23.Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I. Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637–43. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 24.Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-lsla T, Behl C, Levkau B, Nitsch RM. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer's disease-associated neurodegeneration and oxidative stress. J Neurosci. 2000;20:7345–52. doi: 10.1523/JNEUROSCI.20-19-07345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, FitzPatrick DR, Kelley Rl, Wanders RJ. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–94. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benvenuti S, Saccardi R, Luciani P, Urbani S, Deledda C, Cellai I, Francini F, Squecco R, Rosati F, Danza G, Gelmini S, Greeve I, Rossi M, Maggi R, Serio M, Peri A. Neuronal differentiation of human mesenchymal stem cells: changes in the expression of the Alzheimer's disease-related gene seladin-1. Exp Cell Res. 2006;312:2592–604. doi: 10.1016/j.yexcr.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 27.Luciani P, Ferruzzi P, Arnaldi G, Crescioli C, Benvenuti S, Valeri A, Greeve I, Serio M, Mannelli M, Peri A. Expression of the novel adrenocorticotropin-responsive gene selective Alzheimer's disease indicator-1 in the normal adrenal cortex and in adrenocortical adenomas and carcinomas. J Clin Endocrinol Metab. 2004;89:1332–9. doi: 10.1210/jc.2003-031065. [DOI] [PubMed] [Google Scholar]
- 28.Benvenuti S, Luciani P, Vannelli GB, Gelmini S, Franceschi E, Serio M, Peri A. Estrogen and SERMs exert neuroprotective effects and stimulate the expression of seladin-1, a recently discovered anti-apoptotic gene, in human neuroblast long-term cell cultures. J Clin Endocrinol Metab. 2005;90:1775–82. doi: 10.1210/jc.2004-0066. [DOI] [PubMed] [Google Scholar]
- 29.Crameri A, Biondi E, Kuehnle K, Lutjohann D, Thelen KM, Perga S, Dotti CG, Nitsch RM, Ledesma MD, Mohajeri MH. The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J. 2006;25:432–43. doi: 10.1038/sj.emboj.7600938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA. 1993;90:567–71. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275:14077–83. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- 32.Cecchi C, Baglioni S, Fiorillo C, Pensalfini A, Liguri G, Nosi D, Rigacci S, Bucciantini M, Stefani M. Insights into the molecular basis of the differing susceptibility of varying cell types to the toxicity of amyloid aggregates. J Cell Sci. 2005;118:3459–70. doi: 10.1242/jcs.02473. [DOI] [PubMed] [Google Scholar]
- 33.Cecchi C, Fiorillo C, Baglioni S, Pensalfini A, Bagnoli S, Nacmias B, Sorbi S, Nosi D, Relini A, Liguri G. Increased susceptibility to amyloid toxicity in familial Alzheimer's fibroblasts. Neurobiol Aging. 2007;28:863–76. doi: 10.1016/j.neurobiolaging.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Cecchi C, Pensalfini A, Stefani M, Baglioni S, Fiorillo C, Cappadona S, Caporale R, Nosi D, Ruggiero M, Liguri G. Replicating neuroblastoma cells in different cell cycle phases display different vulnerability to amyloid toxicity. J Mol Med. 2008;86:197–209. doi: 10.1007/s00109-007-0265-3. [DOI] [PubMed] [Google Scholar]
- 35.Bradford MM. A rapid sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Datki Z, Papp R, Zadori D, Soos K, Fulop L, Juhasz A, Laskay G, Hetenyi C, Mihalik E, Zarandi M, Penke B. In vitro model of neurotoxicity of Abeta 1-42 and neuroprotection by a pentapeptide: irreversible events during the first hour. Neurobiol Dis. 2004;17:507–15. doi: 10.1016/j.nbd.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–99. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 38.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–6. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 39.Demuro A, Mina E, Kayed R, Milton S, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 40.Bhatia R, Lin H, Lal R. Fresh and globular amyloid beta protein (1-42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. FASEB J. 2000;14:1233–43. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- 41.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 42.Mohajeri MH, Saini K, Schultz JG, Wollmer MA, Hock C, Nitsch RM. Passive immunization against beta-amyloid pep-tide protects central nervous system (CNS) neurons from increased vulnerability associated with an Alzheimer's disease-causing mutation. J Biol Chem. 2002;277:33012–7. doi: 10.1074/jbc.M203193200. [DOI] [PubMed] [Google Scholar]
- 43.Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxida-tive stress by Seladin-1. Nature. 2004;432:640–5. doi: 10.1038/nature03173. [DOI] [PubMed] [Google Scholar]
- 44.Yanagisawa K. Role of gangliosides in Alzheimer's disease. Biochim et Biophys Acta. 2007;1768:1943–51. doi: 10.1016/j.bbamem.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Fujita A, Cheng J, Hirakawa M, Furukawa K, Kusunoki S, Fujimoto T. Gangliosides GM1 and GM3 in the Living Cell Membrane Form Clusters Susceptible to Cholesterol Depletion and Chilling. Mol Biol Cell. 2007;18:2112–22. doi: 10.1091/mbc.E07-01-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597-624) FASEB J. 2006;20:1254–6. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–90. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 48.Orrenius S, Zhovotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 49.Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci USA. 2005;102:10427–32. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arispe N, Diaz JC, Simakova O. Abeta ion channels. Prospects for treating Alzheimer's disease with Abeta channel blockers. Biochim Biophys Acta. 2007;1768:1952–65. doi: 10.1016/j.bbamem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 51.Lal R, Lin H, Quist AP. Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim Biophys Acta. 2007;1768:1966–75. doi: 10.1016/j.bbamem.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 53.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. 2002;38:713–21. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 54.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–3. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 55.Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D, Dobson CM, Stefani M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem. 2004;279:31374–82. doi: 10.1074/jbc.M400348200. [DOI] [PubMed] [Google Scholar]