

Abstract
Type 2 diabetes mellitus induces a characteristic platelet hyperactivity that might be due to several factors including oxidativ stress and abnormal intracellular Ca2+ homeostasis. Hyperhomocysteinaemia is considered a risk factor in the development of thrombosis although its effect on platelet function and the mechanisms involved are still poorly understood. Here we show tha homocysteine induce a concentration-dependent increase in endogenous production of reactive oxygen species (ROS), which was significantly greater in platelets from diabetic patients than in controls. Platelet treatment with homocysteine resulted in Ca2+ release from the dense tubular system and the acidic stores. Ca2+ mobilization-induced by homocysteine consisted in two components, an initial slow increase in intracellular free Ca + concentration ([Ca +]i) and a rapid and marked increase in [Ca2+]i, th second leading to the activation of platelet aggregation. As well as ROS generation, Ca2+ mobilization and platelet aggregation were significantly greater in platelets from diabetic donors than in controls, which indicate that platelets from diabetic donors are more sensitive to homocysteine. These findings, together with the hyperhomocysteinaemia reported in diabetic patients, strongly suggest that homocysteine might be considered a risk factor in the development of cardiovascular complications associated to type 2 diabetes mellitus.
Keywords: thrombin, homocysteine, aggregation, platelets, calcium signalling, ROS production
Introduction
Homocysteine is a sulphur-containing amino acid produced by conversion of methionine, an essential amino acid that is regularly consumed within the diet. Homocysteine is metabolized by re-methylation or transsulphuration, processes that require either vitamin B12 or B6, respectively, as cofactors [1]. In normal conditions, the plasma levels of homocysteine in humans are low (5-15 μM); however, genetic defects in the enzymes metabolizing homocysteine or environmental factors markedly increase homocysteinaemia. Mutations in cystathione-β-syn-thase or 5,10-methylenetetrahydrofolate reductase are associated with excess levels of homocysteine (>50 μM and up to 400 μm) [1–4]. Environmental factors include reduced folate as well as vitamins B12 and B6 intake, increasing methionine intake, smoking, coffee drinking and the administration of certain drugs, such as steroids or cyclosporine [1].
Hyperhomocysteinaemia is associated with increased risk of atherosclerosis and the development of arterial and venous thrombosis [5]. Studies in animal models have revealed that hyperhomocysteinaemia result in increased oxidant stress [6], impaired endothelial function [7, 8] and increased thrombogenicity [8]. Hyperhomocysteinaemia has also been associated with an increase in platelet activation [9]. Leoncini et al.[10] have reported that homocysteine increase platelet responsiveness to thrombin in a concentration-dependent manner. In addition, acute homocysteine increases in rats have been shown to be associated to an increase in platelet aggregation [11]. Some of the effects of homocysteine on human platelet function have been explained by a reduction in nitric oxide formation or bioavailability [10, 12]. In addition, platelet hyperactivity induced by homocysteine has been shown to involve activation of the p38 mitogen-activated protein kinase (MAPK)/cytosolic phospholipase A (cPLA2) pathway, leading to the synthesis of thromboxane A2 and the activation of integrin aIIbP3 [13]. Homocysteine-induced platelet hyperactivity also involves activation of PLCγ2, which requires signals through oxygen free radicals [14].
Oxidative stress is a phenomenon involved in platelet hyperactivity in patients with type 2 diabetes mellitus [15]. In this type of diabetes mellitus, oxidative stress impairs glucose uptake in muscle cells and adipocytes leading to chronic hyper-glycaemia, which, in turn, causes glucotoxic alterations in many cell types, such as pancreatic β cells, resulting in a decrease in insulin content, secretion and activity [16, 17]. In addition, diabetes mellitus leads to several cardiovascular disorders, including platelet dysfunction associated to an abnormal intracellular Ca2+ homeostasis, which are the major cause of morbidity and mortality in type 2 diabetes mellitus [18–20]. Type 2 diabetic subjects have been reported to show 40% greater plasma levels of homocysteine than healthy subjects [21]; therefore, as homocysteine stimulates oxidative stress and inhibits nitric oxide synthesis, hyperhomocysteinaemia has been presented as a candidate for the generation of oxidative damage in type 2 diabetes.
Thus, in an attempt to elucidate the intracellular mechanisms involved in homocysteine-induced effects in diabetic patients, we have investigated aggregation, intracellular Ca2+ mobilization, and endogenous reactive oxygen species (ROS) generation induced by homocysteine in platelets from type 2 diabetics and healthy subjects.
Materials and methods
Materials
Apyrase (grade VII), ethylene glycol-bis (2-aminoethylether)-N,N,N’,N’,-tetraacetic acid (EGTA), aspirin, bovine serum albumin (BSA), thrombin, adenosine diphosphate (ADP), thapsigargin (TG), 2,5 di-(tertbutyl)-1,4-hydroquinone (TBHQ) and homocysteine (Hcy) were from Sigma (Madrid, Spain). Fura-2 acetoxymethyl ester (fura-2/AM), 5-(and-6)-chloromethyl-2´,7´-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) and calcein AM were from Molecular Probes (Leiden, The Netherlands). All other reagents were of analytical grade and purchased from Panreac (Barcelona, Spain).
Participants
The studied population consisted of 28 type 2 diabetic and 26 age- and gender-matched healthy (control) donors. The clinical and metabolic characteristics of type 2 diabetic patients and healthy individuals are summarized in Table 1. Clinical laboratory parameters were measured by routine laboratory methods. Blood glucose concentration in diabetic patients was in the range of 180 to 240 mg/dl. The glycosylated Hb levels (HbA1c) were used as an index of metabolic control. Only blood of diabetic patients with a level of HbA1c > 6% was selected for experiments. The control participants were age- and gender-matched healthy individuals that had HbA1c levels in the normal range (3.5–5%).
Table 1.
Clinical and metabolic characteristics of type 2 diabetic patients and healthy individuals.
| Healthy individuals | Type 2 diabetics | |
|---|---|---|
| Number | 26 | 28 |
| Age (years) | 57 ± 8 | 58 ± 9 |
| B.M.I. | 26.1 ± 2.9 | 27.7 ± 2.5 |
| Diabetes duration (years) | - | 7.8 ± 2.8 |
| Fasting glycaemia (mg/dl) | 92 ± 5 | 190 ± 12 |
| HbA1C (%) | 4.2 ± 0.5 | 8.5 ± 0.8* |
| Albumin (g/l) | 4.3 ± 0.2 | 4.3 ± 0.4 |
| Cholesterol (mmol/l) | ||
| Total | 5.9 ± 0.5 | 6.0 ± 0.9 |
| HDL | 1.5 ± 0.4 | 1.5 ± 0.6 |
| VLDL | 0.6 ± 0.1 | 0.6 ± 0.3 |
| Low-density lipoprotein (LDL) | 4.0 ± 0.8 | 4.0 ± 0.9 |
| Triglycerides (mmol/l) | 1.2 ± 0.5 | 1.3 ± 0.4 |
| Fibrinogen (mg/dl) | 330 ± 28 | 324 ± 35 |
| Creatinine (mg/dL) | 1.0 ± 0.2 | 1.0 ± 0.3 |
| Blood pressure (mmHg) | ||
| Systolic | 124 ± 8 | 127 ± 7 |
| Diastolic | 82 ± 5 | 83 ± 5 |
Values given are presented as mean ± S.E.M.
P < 0.05 compared to healthy donors.
Platelet preparation
Blood was obtained from type 2 diabetic patients and healthy volunteers in the clinical analysis laboratory, Cáceres, Spain, as approved by Local Ethical Committees and in accordance to the Declaration of Helsinki. Blood was collected at 9:00 a.m. Blood was mixed with one-sixth volume of acid/citrate dextrose anticoagulant containing (in mM): 85 sodium citrate, 78 citric acid and 111 D-glucose. Platelet-rich plasma was then prepared by centrifugation for 5 min. at 700 g and, except for aggregation studies, aspirin (100 μm) and apyrase (40 μg/ml) were added. Cells were then collected by centrifugation at 350 g for 20 min. and re-suspended in HEPES-buffered saline (HBS) containing (in mM): 145 NaCl, 10 4-(2-hydroxyethyl) piper-azine-1-ethanesulfonic acid (HEPES), 10 D-glucose, 5 KCl, 1 MgSO4, pH 7.45 and supplemented with 0.1% w/v BSA and 40 μg/ml apyrase.
Platelets viability
This was assessed by calcein loading of the cells and trypan blue exclusion technique. For calcein loading, platelets were incubated for 30 min. with 5 μm calcein-AM at 37°C, centrifuged at 350 gfor 20 min. and the pellet suspended in HBS. Cells were treated with the agents used in this study for the times indicated, centrifuged again, and re-suspended in HBS. Two millilitre aliquots were used for fluorescence recording (spec-trophotometer Varian Ltd., Madrid, Spain) at (ex: 494 nm and λ em: 535 nm. In this assay ∼95% of cells were viable in our platelet suspensions, a result further confirmed by the trypan blue exclusion technique. No significant effect on cell viability was detected after treatment with Hcy or after incubation in HBS for 6000 sec, the longest period investigated for platelet aggregation.
Platelet aggregation
Platelet aggregation of washed platelets was monitored in a Chronolog (Havertown, PA, U.S.A.) aggregometer. Platelets (300 μl) prepared as described above were suspended in HBS and stimulated in the aggregometer cuvettes at 37°C under stirring at 1200 rpm [15]. The time required to reach the maximum aggregation rate was then determined from the traces recorded.
Intracellular ROS production through the oxidation of CM-H2DCFDA
CM-H2DCFDA is a ROS-sensitive probe that can be used to detect ROS production in living cells. It passively diffuses into cells, where its acetate groups are cleaved by intracellular esterases, releasing the corresponding dichlorodihydrofluorescein derivative. CM-H2DCFDA oxidation yields a fluorescent adduct (DCF) that is trapped inside the cell. Platelet-rich plasma was incubated at 37°C with 10 μm CM-H2DCFDA acetyl ester for 30 min., platelets were then centrifuged and the pellet was re-suspended in HBS. Fluorescence was recorded from 1 ml aliquots using a Shimadzu Spectrophotometer (Shimadzu, Japan) at λ ex: 488 nm and λ em: 530 nm. Traces were fitted to the equation: y = A + Bx where B is the slope.
Measurement of intracellular free calcium concentration ([Ca2+]i)
Cells were incubated at 37°C with 2 μm fura-2 AM for 45 min, collected by centrifugation at 350 g for 20 min. and resuspended in HBS. Fluorescence was recorded from 1 ml aliquots of magnetically stirred platelet suspensions (108 cells/ ml) at 37°C using the fluorescence Spectrophotometer (Varian Ltd., Madrid, Spain) with λ ex: 340 and 380 nm and λ em: 505 nm. Changes in [Ca2+]i were monitored using the fura-2 340/380 fluorescence ratio and calibrated according to the method of Grynkiewicz et al. [22]. Ca2+ release and influx was estimated using the integral of the rise in [Ca2+]i for 2.5 min after addition of agonists or CaCl2, respectively [23].
Statistical analysis
Data are shown as mean±S.E.M. analysis of statistical significance was performed using Student's t-test. P < 0.05 was considered to be significant for a difference. Correlations were calculated with the use of GraphPad Prism software (version 2.0, Graph Pad Software for Science, San Diego, USA).
Results
ROS generation induced by Hcy in platelets from healthy and type 2 diabetic patients
Intracellular ROS production was estimated using CM-H2DCFDA. As shown in Figures 1A–C, treatment of human platelets from healthy donors with Hcy resulted in a slow but sustained increase in the DCF fluorescence. The results of linear regression show that Hcy evoked endogenous ROS generation in a concentration-dependent manner with slopes of 30 ± 1 × 10-5, 51 ± 1 × 10-5and 67 ± 1 × 10-5 at the concentrations 50, 100 and 200 μm, respectively.
Fig 1.
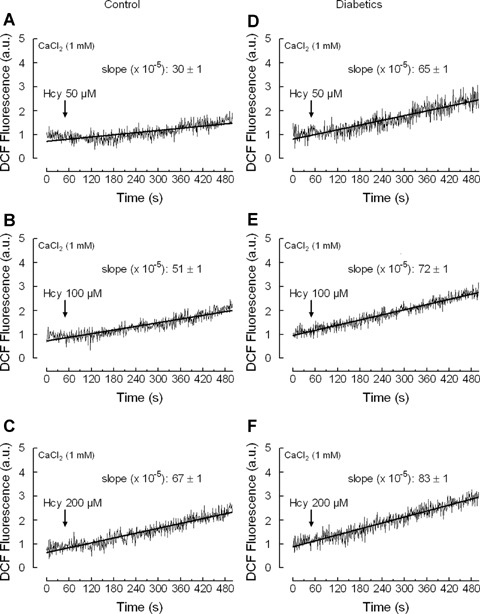
Homocysteine induces reactive oxygen species (ROS) generation in platelets from healthy and diabetic donors. Human platelets from healthy (A–C) or diabetic donors (E-F) were loaded with CM-H2DCFDA and then treated with 50 μm (A), 100 μm (β) or 200 μm (C) homocysteine (Hcy) in a medium containing 1 mM Ca2+. Traces are the average of 10 independent experiments. Kinetic parameters (slopes) were determined by linear regression, which is depicted as the line drawn through the points. Slope values are mean±S.E.M.
Treatment of platelets from diabetic donors with increasing concentrations of Hcy resulted also in a concentration-dependent increase in DCF fluorescence (Fig. 1D-F). However, in diabetic platelets the linear regression slopes were 65 ± 1 × 10-5, 72 ± 1 × 10-5 and 83 ± 1 × 10-5 at the Hcy concentrations of 50, 100 and 200 μm, respectively; (P < 0.05). This indicates that in diabetes the rate of endogenous ROS generation induced by Hcy in platelets was significantly greater compared to that observed in platelets from healthy donors. A statistically significant correlation was observed between Hcy concentration and endogenous ROS production both in healthy individuals (y = 0.071x - 2.142, R2 = 0.998, P < 0.05) and diabetic donors (y = 0.125x – 0.301, R2 = 0.994, P < 0.05), which is consistent with previous studies reporting correlation between plasma Hcy concentration and platelet ROS levels [21].
induces Ca2+ mobilization in platelets from healthy and type 2 diabetic patients
The possibility that Hcy-induced endogenous ROS production might be associated to calcium mobilization, as previously reported [15, 24, 25], was tested by examining the effect of Hcy on Ca2+ release from the intracellular stores and Ca2+ entry through the plasma membrane. In human platelets two Ca2+ stores have been described: the TBHQ-insensitive dense tubular system (DTS) and the TBHQ-sensitive acidic stores based on immnunolocalisation studies [26], the different sensitivity to TG and TBHQ [27, 28] and the distinct sensitivity to agonists [29, 30]. We have investigated the effect of Hcy on Ca2+ release from the DTS, by previous depletion of the acidic stores using TBHQ. In a Ca2+-free medium, treatment of platelets from healthy donors with 20 μm TBHQ induced a sustained increase in [Ca2+]i; subsequent addition of Hcy induced a concentration-dependent increase in [Ca2+]i indicative of Ca2+ release from the DTS (Fig. 2A, C, E and G). In platelets from type 2 diabetic patients Hcy evoked a concentration-dependent Ca2+ release from the DTS, which was found to be significantly higher at 50, 100 and 200 μm than those observed in control platelets (Fig. 2B, D, F and G; P < 0.05).
Fig 2.
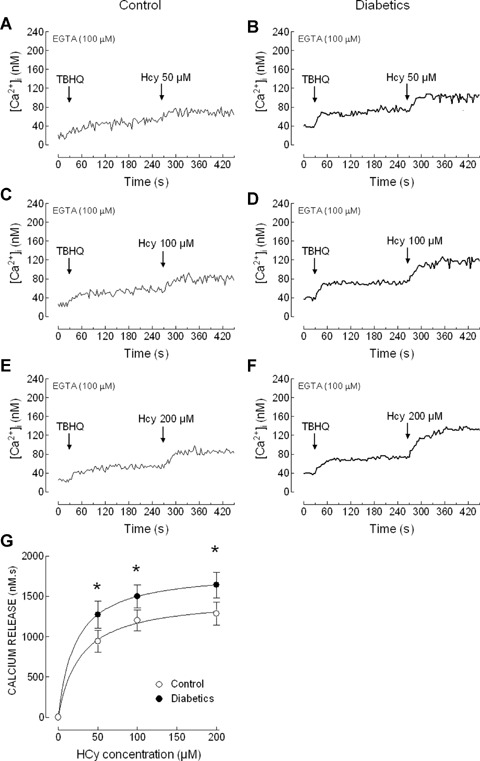
Homocysteine releases Ca2+ from 2,5 di-(tertbutyl)-1,4-hydroquinone (TBHQ)– insensitive stores in platelets from healthy and diabetic donors. Human platelets from healthy individuals (A, C and E) and diabetic patients (B, D and F) were suspended in a Ca2+-free medium (100 μm EGTA was added). Cells were treated with 20 μm TBHQ and 4 min. later homocysteine (Hcy; 50, 100 or 200 μm) was added, as indicated. Traces are representative of 10–21 independent experiments. (G) Values represent Hcy-induced Ca2+ release estimated as described in Material and methods. P < 0.05 compared to Hcy-evoked Ca2+ release in platelets from healthy donors.
In order to investigate Ca2+ release from the acidic stores induced by Hcy we have depleted the DTS, by using a combination of ADP (10 μm) and TG (10 nM), which selectively inhibits SERCA2b present in the DTS [35], prior to the addition of Hcy. Treatment of platelets with ADP + TG resulted in a transient increase in [Ca2+]i indicative of Ca2+ release from the DTS (Fig. 3A-F). Subsequent addition of Hcy induced a sustained increase in [Ca2+]i that was found to be significantly greater in platelets from diabetics than in controls (Fig. 3G; P < 0.05). Hcy-evoked Ca2+ release from the acidic stores was not found to be concentration-dependent. Hcy concentration and Ca2+ release from the DTS were significantly correlated in healthy individuals (y = 4.688x+728, R2= 0.995, P < 0.05) and diabetic donors (y = 2.897x+1141, R2= 0.994, P < 0.05). However, no correlation was found between Hcy concentration and Ca2+ release from the acidic stores in healthy (y = 0.717x + 1272, r2= 0.448,P= 0.53) and diabetic donors (y=-1.127x + 2212, R2= 0.615, P = 0.42).
Fig 3.
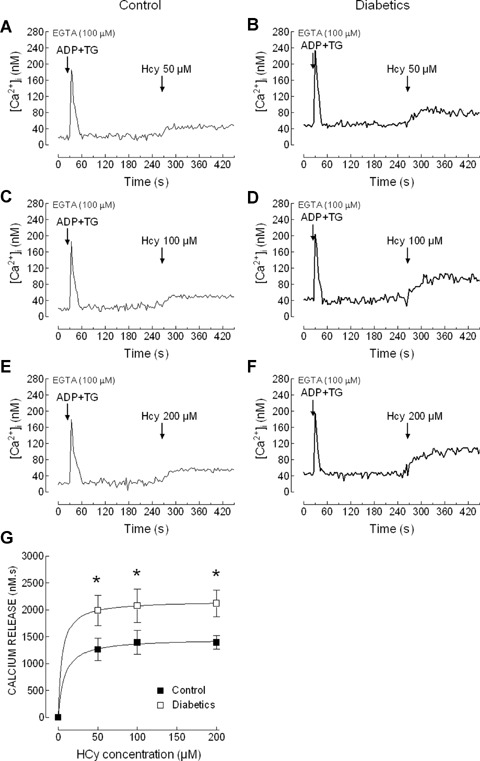
Homocysteine releases Ca2+ from TBHQ-sensitive stores in platelets from healthy and diabetic donors. Human platelets from healthy individuals (A, C and E) and diabetic patients (B, D and F) were suspended in a Ca2+-free medium (100 μm EGTA was added). Cells were treated with 10 μm ADP in combination with 10 nM thapsi-gargin (TG) and 4 min. later homocysteine (Hcy; 50, 100 or 200 μm) was added, as indicated. Traces are representative of 10–14 independent experiments. (G) Values represent Hcy-induced Ca2+ release estimated as described in Material and methods. P < 0.05 compared to Hcy-evoked Ca2+ release in platelets from healthy donors.
We have further investigated the ability of Hcy to induce Ca2+ entry. As shown in Figure 4A and B, treatment of platelets with increasing concentrations of Hcy (50-200 μm) in a medium containing 1 mM Ca2+ resulted in a rapid, concentration-dependent and sustained increase in [Ca2+]i that was found to be significantly higher to that observed in the absence of extracellular Ca2+ (data not shown). The rise in [Ca2+]i evoked by Hcy was found to be significantly increased in diabetic patients compared to controls (Fig. 4A and B; P < 0.05).
Fig 4.
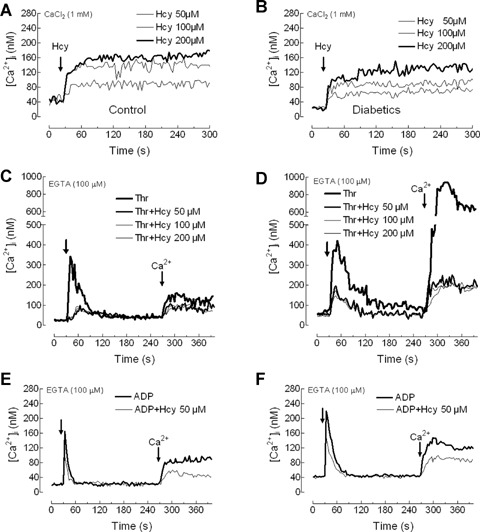
Homocysteine stimulates Ca2+ entry and impairs Ca2+ mobilization by thrombin in platelets from healthy and diabetic donors. (A and B) Human platelets from healthy individuals (A) and diabetic patients (B) were suspended in HEPES-buffered saline (HBS) containing 1 mM Ca2+ and then exposed to increasing concentrations of homocysteine (Hcy; 50–200 μm). (C–F) Human platelets from healthy individuals (C and E) and diabetic patients (D and F) were suspended in a Ca2+-free medium (100 μm EGTA was added). Cells were treated (thick arrow) with either 0.1 U/ml thrombin (Thr; C and D) or with 10 μm ADP (E and F), in the absence or presence of 50, 100 or 200 μm Hcy, as indicated, and 4 min. later CaCl2 (300 μm) was added to the medium to initiate Ca2+ entry. Traces are representative of 8–12 independent experiments.
Subsequently, we questioned whether Hcy impairs Ca2+ mobilization induced by agonists. Treatment of platelets with 0.1 U/ml thrombin resulted in a transient increase of [Ca2+]i. The initial peak [Ca2+]i elevation above basal after thrombin was 297 ± 37 and 392 ± 56 nM in platelets from healthy and diabetic donors (Fig. 4C and D; P < 0.05), which is consistent with previous studies reporting abnormal Ca2+ mobilization in diabetic platelets [19, 20, 32]. The subsequent addition of Ca2+ (300 μm) to the external medium induced a sustained elevation in [Ca2+]i indicative of Ca2+ entry. As shown in Figure 4C and D, Ca2+ entry was significantly higher in platelets from diabetic patients (the integrals of the rise in [Ca2+]i for 2.5 min. after addition of CaCl2 were 14111 ± 2334 and 52108 ± 7560 nM/s in healthy and diabetic patients, respectively; P<0.05). In the presence of 50 μm Hcy thrombin-evoked Ca2+ release was significantly reduced (the initial peak [Ca2+]i elevation above basal after thrombin was 71 ± 6 and 139 ± 10 nM in platelets from healthy and diabetic donors, respectively, and similar results were obtained with 100 and 200 μm Hcy; Fig. 4C and D; P < 0.05). Hcy also impaired thrombin-evoked Ca2+ entry in healthy individuals and diabetic patients (the integrals of the rise in [Ca2+]i for 2.5 min. after addition of CaCl2 were 5850 ± 283, 5389 ±352 and 5203 ± 383 nM/s in platelets from healthy individuals and 8755 ± 996, 8578 ± 235 and 8324 ± 124 nM/s in platelets from diabetic donors, treated in the presence of 50, 100 and 200 μm Hcy; Fig. 4C and D; P < 0.05). Similarly, Hcy significantly reduced Ca + mobilization evoked by 10 μm ADP, including both Ca2+ release from the intracellular stores (the initial peak [Ca2+]i elevation above basal after ADP was 160 ± 7 and 218 ± 11 nM in platelets from healthy and diabetic donors in the absence of Hcy and 82±5 and 129±12 nM in platelets from healthy individuals and diabetic patients in the presence of 50 μm Hcy; Fig. 4E and F; P < 0.05) and Ca2+ entry (the integrals of the rise in [Ca2+]i for 2.5 min. after addition of CaCl2 were 5341 ±470 and 7210±611 nM in platelets from healthy and diabetic donors in the absence of Hcy and 2892±352 and 4125 ± 512 nM in platelets from healthy individuals and diabetic patients in the presence of 50 μm Hcy; Fig. 4E and F; P < 0.05).
Effect of Hcy on platelet aggregation
As shown in Figures 5 and 6(A)–(C) and Table 2, treatment of platelets suspended in a medium containing 1 mM Ca2+ with Hcy was without effect for several minutes, then induced a slow shape change indicated by a decrease in light transmission, followed by a large increase in light transmission as platelets aggregated. The percentage and rate of aggregation was found to be greater in platelets from diabetic donors compared to controls (Table 2), which is consistent with the greater effect of Hcy in ROS production and Ca2+ mobilization in diabetics shown above. In contrast, the lag time was significantly extended in platelets from patients with type 2 diabetes mellitus at all the concentrations of Hcy used (Table 2).
Fig 5.
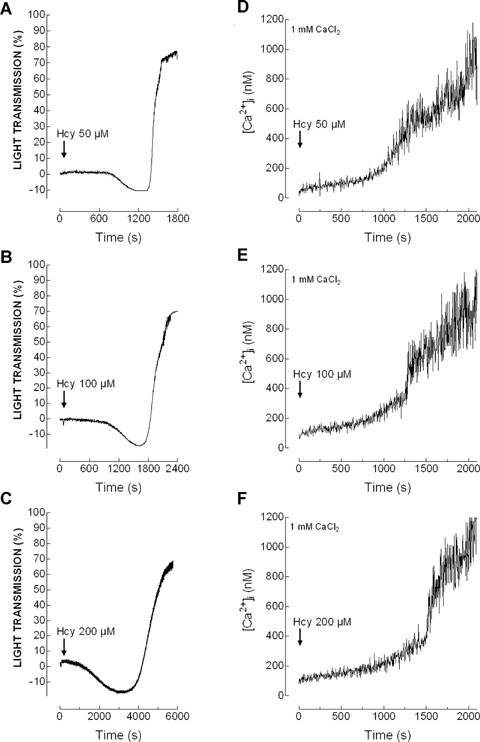
Homocysteine induces shape change and platelet aggregation. Human platelets from healthy donors were suspended in HBS containing 1 mM Ca2+ and then treated with increasing concentrations of homocysteine (Hcy; 50–200 μm), as indicated. Aggregation of human platelets was induced at a shear rate of 1200 rpm at 37°C in a Chronolog aggregometer as described in Material and methods. Elevations in [Ca2+]i were monitored using the 340/380 nm ratio and traces were calibrated in terms of [Ca2+]i. Traces shown are representative of 11 separate experiments.
Fig 6.
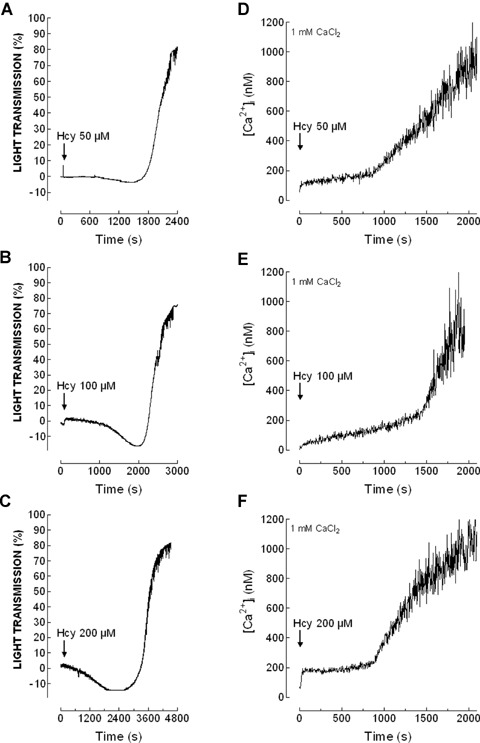
Shape change and aggregation induced by homocysteine in platelet from diabetic patients. Human platelets from type 2 diabetics were suspended in HBS containing 1 mM Ca2+ and then treated with increasing concentrations of homocysteine (Hcy; 50-200 μm), as indicated. Aggregation of human platelets was induced at a shear rate of 1200 rpm at 37°C in a Chronolog aggregometer as described in Material and methods. Elevations in [Ca2+]i were monitored using the 340/380 nm ratio and traces were calibrated in terms of [Ca2+]i. Traces shown are representative of 11 separate experiments.
Table 2.
Homocysteine induces platelet aggregation
| Hey concentration (μm) | Lag-time (sec.) | % rate | % aggregation |
|---|---|---|---|
| Platelets from healthy donors | |||
| 50 | 689.8 ± 141.5 | 52.6 ± 4.0 | 79.0 ± 3.6 |
| 100 | 715.1 ± 116.2 | 62.0 ± 6.9 | 67.2 ± 4.1 |
| 200 | 990.2 ± 168.1 | 41.0 ± 1.9 | 60.2 ± 5.1 |
| Platelets from type 2 diabetic patients | |||
| 50 | 1054.9 ± 220.7* | 48.0 ± 3.5 | 86.8 ± 1.5* |
| 100 | 1316.3 ± 223.9* | 51.5 ± 7.6 | 76.4 ± 3.7* |
| 200 | 1593.8 ± 307.4* | 45.8 ± 2.4* | 71.8 ± 4.8* |
Human platelets were suspended in a HBS containing 1 mM Ca2+ and then were treated at 37°C with increasing concentrations of homocys-teine and platelet aggregation was induced at a shear rate of 1200 rpm at 37°C. Values given are presented as mean ± S.E.M. of 11 separate determinations. *P < 0.05 compared to the response in platelets from healthy donors.
Further examination of Ca2+ mobilization by Hcy in the same cell preparation used to determined platelet aggregation reported an initial slow and sustained increase in [Ca2+]i followed by a rise in the rate of increase of [Ca2+]i indicative of Ca2+ influx (Figs. 5 and 6D–F). The change in the rate of Ca2+ entry coincides with the initiation of shape change during platelet aggregation, which suggest that the rise in [Ca2+]i might be responsible for Hcy-evoked platelet aggregation.
Despite we found that Hcy induces platelet aggregation and Ca2+ mobilization, we did not find a satisfactory correlation between Hcy concentrations and the percentage of platelet aggregation either in healthy (y = (0.125x + 83, R2= 0.909, P= 0.19) or diabetic donors (y = (0.091x + 89, R2= 0.802, P= 0.28), as well as between Hcy concentration and Ca2+ entry in healthy (y = 2320x + 220,800, R2= 0.729, P= 0.34) or diabetic donors (y = 3676x + 524500, R2= 0.949, P= 0.14).
Discussion
Circulating platelets are activated in response to a variety of physiological agonists, such as thrombin, that become available after the vascular injury. Platelet responsiveness might be altered by a number of pathological situations, including hypertension [33], myeloproliferative disorders [34] or diabetes mellitus [19, 32]. Oxidative stress has been shown to be involved in the development of abnormal signalling pathways leading to platelet hyperaggregability [35, 36].
Hyperhomocysteinaemia is considered an independent risk factor for vascular thrombosis; however the mechanisms underlying these alterations remains poorly understood. The role of oxidative stress in hyperhomocysteinaemia-induced cardiovascular disorders is plausible since Hcy promotes oxidant injury to vascular and blood cells [13, 14, 22]. Platelets from type 2 diabetic patients are chronically exposed to high plasma levels of Hcy [21], which might be involved in the development of cardiovascular complications in these patients. Using preparations of washed platelets, we have found here that Hcy was able to induce endogenous generation of ROS that was markedly greater in cells from diabetic patients. This fact, together with the continuous exposure to hyperhomocysteinaemia [21], could increase the risk of thrombosis. The greater endogenous ROS production in platelets from diabetic donors compared to controls is consistent with previous observations using platelet agonists [36–38].
Several mechanisms have been reported to underlie the effect of Hcy on platelet function, including activation of cPLA2 and PLCγ2 [12, 13]. A recent study has reported that Hcy induces Ca2+ release from intracellular pools in washed platelets [13], although the authors did not discriminate the identity of the Ca2+ stores. Human platelets present two separate Ca2+ stores differentiated by the distinct sensitivity to TG and TBHQ. The main store, expressing SERCA2b, which is inhibited by low concentrations of TG and is insensitive to TBHQ [28], has long been identified as the DTS, and the TBHQ-sensitive store has recently been identified as an acidic organelle [29] expressing SERCA3 [26]. Here we show that Hcy releases Ca2+ from both stores. Our results indicate that depletion of the DTS by Hcy occurs in a concentration-dependent fashion while that of the acidic stores was not found to be concentration-dependent; the latter result is consistent with our previous observations using the physiological agonist thrombin [34]. We have found that Hcy, at concentrations between 50 and 200 μm, impairs throm-bin-evoked Ca2+ release and Ca2+ entry, and Ca2+ mobilization by ADP was also found to be reduced by 50 μm Hcy. Similar findings have been reported in a human umbilical cord endothelial cell line [34]. The effect of Hcy inducing Ca2+ mobilization per se but impairing agonist-induced Ca2+ mobilization resembles that previously reported for H2O2 in platelets, which plays a dual role in Ca2+ signalling, both stimulating Ca2+ mobilization by itself but also impairing agonist-evoked Ca2+ fluxes [24]. The inhibitory effect of H2O2 on agonist-evoked Ca2+ mobilization has been attributed to the impairment of the dynamics of actin cytoskeleton polymerizationdepolymerization induced by agonists [41, 42]. Actin re-organization, as well as microtubular remodelling, has been widely demonstrated to be required for the activation of store-operated Ca2+ entry and secretion in platelets and other non-excitable cells [43–46]. In umbilical endothelial cells, the inhibitory effect of Hcy on store-operated Ca2+ entry has been reported to be mediated through inhibition of actin polymerization [40], which, as for H2O2, might be responsible for the effect of Hcy on agonist-evoked Ca2+ mobilization in human platelets.
Store-operated Ca2+ entry is a mechanism regulated by the filling state of the intracellular Ca2+ stores that has been presented as a major Ca2+ entry mechanism involved in the activation of a number of cellular processes and is required for full platelet activation [47–49]. The results in this study show that Hcy per se activates Ca2+entry which consists of two phases: an initial stage consisting in a small but sustained increase in [Ca2+]i followed by a rapid increase in [Ca2+]i. Interestingly, the pattern of Ca2+ mobilization parallels that of platelet aggregation by Hcy. During the initial phase of Ca2+ mobilization we did not detect activation of platelet aggregation or cell shape change, probably due to the low [Ca2+]i reached at this stage, but the initiation of the second phase, which induces a remarkable increase in [Ca2+]i seems to induce the beginning of platelets shape change and aggregation. Although speculative, the rapid Ca2+ mobilization observed during the second phase might be mediated by direct effects of Hcy on the Ca2+ handling mechanisms, perhaps by ROS production [24, 50, 51], or through the activation of secretory pathways leading to the release of platelet agonists. Taking together, the findings in this study suggest that Hcy-induced Ca2+ mobilization is involved in the activation of platelet aggregation.
No significant correlation was found between Hcy concentration and either the second phase of Ca2+ entry or the percentage of aggregation. Despite we have suggested that ROS production might be involved in Hcy-induced Ca2+ mobilization, the oxidative stress induced by high concentrations of Hcy (ROS production is enhanced as cells are exposed to greater concentrations of Hcy (see Fig. 1)) might exert inhibitory effects on Ca2+ mobilization as previously reported [24, 52], thus preventing a satisfactory correlation between Hcy concentrations and Ca2+ mobilization.
In summary, our results demonstrate that Hcy induces endogenous ROS generation in human platelets, which might lead to Ca2+ mobilization, as previously reported [50, 51], and, subsequently, to platelet aggregation. All the parameters tested in the present study, ROS generation, Ca2+ mobilization and platelet aggregation are more elevated in platelets from diabetic donors than in controls, even though they were exposed to the same concentrations of Hcy, which indicates that platelets from diabetic donors are more sensitive to plasma Hcy levels. These findings, together with the reported hyperhomocysteinaemia in patients with type 2 diabetes mellitus [21, 53], which has been associated with the development of atherosclerosis, thrombosis and other cardiovascular disorders [5, 9, 54], strongly suggest that Hcy might be considered a risk factor in the development of cardiovascular disorders associated to type 2 diabetes mellitus.
Acknowledgments
This work was supported by M.E.C.–FEDER (BFU2007-60104/BFI), Junta de Extremadura-Consejería de Educación, Ciencia y Tecnología & FEDER (2PR04A009) and Consejería de Sanidad y Consumo. N. Alexandru was the recipient of 3 month training at University Extremadura, Spain financially supported by a grant from the FP6 European Community SSA-SERA Project 016873. We thank Dr. Hernandez-Cruz for providing blood samples and Mercedes Gómez Blázquez for her technical assistance.
References
- 1.Undas A, Brozek J, Szczeklik A. Homocysteine and thrombosis: from basic science to clinical evidence. Thromb Haemost. 2005;94:907–15. doi: 10.1160/TH05-05-0313. [DOI] [PubMed] [Google Scholar]
- 2.Frantzen F, Faaren AL, Alfheim I, Nordhei AK. Enzyme conversion immunoassay for determining total homocysteine in plasma or serum. Clin Chem. 1998;44:311–16. [PubMed] [Google Scholar]
- 3.Selhub J. Homocysteine metabolism. Ann Rev Nutr. 1999;19:217–46. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- 4.Wierzbicki AS. Homocysteine and cardiovascular disease: a review of the evidence. Diab Vasc Dis Res. 2007;4:143–50. doi: 10.3132/dvdr.2007.033. [DOI] [PubMed] [Google Scholar]
- 5.Cattaneo M. Heperhomocysteinemia, atherosclerosis and thrombosis. Thromb. Haemost. 1999;81:165–76. [PubMed] [Google Scholar]
- 6.Kanani PM, Sinkey CA, Browning RL, Allaman M, Knapp HR, Haynes WG. Role of oxidant stress in endothelial dysfunction produced by experimental hyper-homocysteinemia in humans. Circulation. 1999;100:1161–8. doi: 10.1161/01.cir.100.11.1161. [DOI] [PubMed] [Google Scholar]
- 7.Stamler JS, Osborne JA, Jaraki O, Rabbani LE, Mullins M, Singel D, Loscalzo J. Adverse vascular effects of homocysteine are modulated by endothe-lium-derived relaxing factor and related oxides of nitrogen. J Clin Invest. 1993;91:308–18. doi: 10.1172/JCI116187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lentz SR. Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost. 2005;3:1646–54. doi: 10.1111/j.1538-7836.2005.01364.x. [DOI] [PubMed] [Google Scholar]
- 9.Coppola A, Davi G, De Stefano V, Mancini FP, Cerbone AM, Di Minno G. Homocysteine, coagulation, platelet function, and thrombosis. Semin Thromb Hemost. 2000;26:243–54. doi: 10.1055/s-2000-8469. [DOI] [PubMed] [Google Scholar]
- 10.Leoncini G, Pascale R, Signorello MG. Effects of homocysteine on L-arginine transport and nitric oxide formation in human platelets. Eur J Clin Invest. 2003;33:713–9. doi: 10.1046/j.1365-2362.2003.01203.x. [DOI] [PubMed] [Google Scholar]
- 11.Durand P, Lussier-Cacan S, Blache D. Acute methionine load-induced hyper-homocysteinemia enhances platelet aggregation, thromboxane biosynthesis, and macrophage-derived tissue factor activity in rats. FASEB J. 1997;11:1157–68. [PubMed] [Google Scholar]
- 12.Upchurch GR, Jr, Welch GN, Fabian AJ, Freedman JE, Johnson JL, Keaney JF, Jr, Loscalzo J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J Biol Chem. 1997;272:17012–7. doi: 10.1074/jbc.272.27.17012. [DOI] [PubMed] [Google Scholar]
- 13.Leoncini G, Bruzzese D, Signorello MG. Activation of p38 MAPKinase/cPLA2 pathway in homocysteine-treated platelets. J Thromb Haemost. 2006;4:209–16. doi: 10.1111/j.1538-7836.2005.01708.x. [DOI] [PubMed] [Google Scholar]
- 14.Leoncini G, Bruzzese D, Signorello MG. A role for PLCgamma2 in platelet activation by homocysteine. J Cell Biochem. 2007;100:1255–65. doi: 10.1002/jcb.21123. [DOI] [PubMed] [Google Scholar]
- 15.Redondo PC, Jardin I, Hernandez-Cruz JM, Pariente JA, Salido GM, Rosado JA. Hydrogen peroxide and peroxynitrite enhance Ca2+ mobilization and aggregation in platelets from type 2 diabetic patients. Biochem Biophys Res Commun. 2005;333:794–802. doi: 10.1016/j.bbrc.2005.05.178. [DOI] [PubMed] [Google Scholar]
- 16.Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes. 1998;47:1562–9. doi: 10.2337/diabetes.47.10.1562. [DOI] [PubMed] [Google Scholar]
- 17.Cooksey RC, Jouihan HA, Ajioka RS, Hazel MW, Jones DL, Kushner JP, McClain DA. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology. 2004;145:5305–12. doi: 10.1210/en.2004-0392. [DOI] [PubMed] [Google Scholar]
- 18.Cimminiello C, Milani M. Diabetes mellitus and peripheral vascular disease: is aspirin effective in preventing vascular events? Diabetologia. 1996;39:1402–4. doi: 10.1007/s001250050590. [DOI] [PubMed] [Google Scholar]
- 19.Saavedra FR, Redondo PC, Hernandez-Cruz JM, Salido GM, Pariente JA, Rosado JA. Store-operated Ca2+ entry and tyrosine kinase pp60src hyperactivity are modulated by hyperglycemia in platelets from patients with non insulin-dependent diabetes mellitus. Arch Biochem Biophys. 2004;432:261–8. doi: 10.1016/j.abb.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 20.Rosado JA, Saavedra FR, Redondo PC, Hernandez-Cruz JM, Salido GM, Pariente JA. Reduced plasma membrane Ca2+-ATPase function in platelets from patients with non-insulin-dependent diabetes mellitus. Haematologica. 2004;89:1142–4. [PubMed] [Google Scholar]
- 21.Signorello MG, Viviani GL, Armani U, Cerone R, Minniti G, Piana A, Leoncini G. Homocysteine, reactive oxygen species and nitric oxide in type 2 diabetes mellitus. Thromb Res. 2007;120:607–13. doi: 10.1016/j.thromres.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 23.Rosado JA, Sage SO. Farnesylcysteine analogues inhibit store-regulated Ca2+ entry in human platelets: evidence for involvement of small GTP-binding proteins and actin cytoskeleton. Biochem J. 2000;347:183–92. [PMC free article] [PubMed] [Google Scholar]
- 24.Redondo PC, Salido GM, Pariente JA, Rosado JA. Dual effect of hydrogen peroxide on store-mediated calcium entry in human platelets. Biochem Pharmacol. 2004;67:1065–76. doi: 10.1016/j.bcp.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 25.Tintinger GR, Theron AJ, Potjo M, Anderson R. Reactive oxidants regulate membrane repolarization and store-operated uptake of calcium by formyl peptide-activated human neutrophils. Free Radic Biol Med. 2007;42:1851–7. doi: 10.1016/j.freeradbiomed.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Kovacs T, Berger G, Corvazier E, Paszty K, Brown A, Bobe R, Papp B, Wuytack F, Cramer EM, Enouf J. Immunolocalization of the multi-sarco/endoplasmic reticulum Ca2+ ATPase system in human platelets. Br J Haematol. 1997;97:192–203. doi: 10.1046/j.1365-2141.1997.9982639.x. [DOI] [PubMed] [Google Scholar]
- 27.Bobe R, Bredoux R, Wuytack F, Quarck R, Kovacs T, Papp B, Corvazier E, Magnier C, Enouf J. The rat platelet 97-kDa Ca2+ ATPase isoform is the sarcoendoplasmic reticulum Ca2+ ATPase 3 protein. J Biol Chem. 1994;269:1417–24. [PubMed] [Google Scholar]
- 28.Enyedi A, Paszty K, Kovacs T, Sarkadi B, Gardos G, Magnier C, Wuytack F, Enouf J. Simultaneous presence of two distinct endoplasmic-reticulum-type calcium-pump isoforms in human cells. Characterization by radio-immunoblotting and inhibition by 2,5-di-(t-butyl)-1,4-ben-zohydroquinone. Biochem J. 1992;288:297–302. doi: 10.1042/bj2880297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez JJ, Camello-Almaraz C, Pariente JA, Salido GM, Rosado JA. Ca2+ accumulation into acidic organelles mediated by Ca2+- and vacuolar H+-ATPases in human platelets. Biochem J. 2005;390:243–52. doi: 10.1042/BJ20050168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez JJ, Redondo PC, Salido GM, Pariente JA, Rosado JA. Two distinct Ca2+ compartments show differential sensitivity to thrombin, ADP and vasopressin in human platelets. Cell Signal. 2006;18:373–81. doi: 10.1016/j.cellsig.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Jardin I, Redondo PC, Salido GM, Rosado JA. Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim Biophys Acta. 2008;1783:84–97. doi: 10.1016/j.bbamcr.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Chaabane C, Dally S, Corvazier E, Bredoux R, Bobe R, Ftouhi B, Raies A, Enouf J. Platelet PMCA- and SERCA-type Ca2+ ATPase expression in diabetes: a novel signature of abnormal megakary-ocytopoiesis. J Thromb Haemost. 2007;5:2127–35. doi: 10.1111/j.1538-7836.2007.02709.x. [DOI] [PubMed] [Google Scholar]
- 33.Blankenship KA, Dawson CB, Aronoff GR, Dean WL. Tyrosine phosphorylation of human platelet plasma membrane Ca2+-ATPase in hypertension. Hypertension. 2000;35:103–7. doi: 10.1161/01.hyp.35.1.103. [DOI] [PubMed] [Google Scholar]
- 34.Avram S, Lupu A, Angelescu S, Olteanu N, Mut-Popescu D. Abnormalities of platelet aggregation in chronic myelopro-liferative disorders. J Cell Mol Med. 2001;5:79–87. doi: 10.1111/j.1582-4934.2001.tb00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jardin I, Redondo PC, Salido GM, Pariente JA, Rosado JA. Endogenously generated reactive oxygen species reduce PMCA activity in platelets from patients with non-insulin-dependent diabetes mellitus. Platelets. 2006;17:283–8. doi: 10.1080/09537100600745187. [DOI] [PubMed] [Google Scholar]
- 36.Leoncini G, Signorello MG, Piana A, Carrubba M, Armani U. Hyperactivity and increased hydrogen peroxide formation in platelets of NIDDM patients. Thromb Res. 1997;86:153–60. doi: 10.1016/s0049-3848(97)00058-3. [DOI] [PubMed] [Google Scholar]
- 37.Schaeffer G, Wascher TC, Kostner GM, Graier WF. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia. 1999;42:167–76. doi: 10.1007/s001250051135. [DOI] [PubMed] [Google Scholar]
- 38.Bouaziz A, Salido S, Linares-Palomino PJ, Sanchez A, Altarejos J, Bartegi A, Salido GM, Rosado JA. Cinnamtannin B-1 from bay wood reduces abnormal intra-cellular Ca2+ homeostasis and platelet hyperaggregability in type 2 diabetes mellitus patients. Arch Biochem Biophys. 2007;457:235–42. doi: 10.1016/j.abb.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 39.Jardin I, Ben Amor N, Bartegi A, Pariente JA, Salido GM, Rosado JA. Differential involvement of thrombin receptors in Ca2+ release from two different intracellular stores in human platelets. Biochem J. 2007;401:167–74. doi: 10.1042/BJ20060888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang HS, Xiao JH, Cao EH, Qin JF. Homocysteine inhibits store-mediated calcium entry in human endothelial cells: evidence for involvement of membrane potential and actin cytoskeleton. Mol Cell Biochem. 2005;269:37–47. doi: 10.1007/s11010-005-3168-z. [DOI] [PubMed] [Google Scholar]
- 41.Rosado JA, González A, Salido GM, Pariente JA. Effects of reactive oxygen species on actin filament polymerisation and amylase secretion in mouse pancreatic acinar cells. Cell Signal. 2002;14:547–56. doi: 10.1016/s0898-6568(01)00273-x. [DOI] [PubMed] [Google Scholar]
- 42.Zhu D, Tan KS, Zhang X, Sun AY, Sun GY, Lee JC. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J Cell Sci. 2005;118:3695–703. doi: 10.1242/jcs.02507. [DOI] [PubMed] [Google Scholar]
- 43.Rosado JA, Salido GM, Jensen RT, Garcia LJ. Are tyrosine phosphorylation of p125(FAK) and paxillin or the small GTP binding protein, rho, needed for CCK-stimulated pancreatic amylase secretion? Biochim Biophys Acta. 1998;1404:412–26. doi: 10.1016/s0167-4889(98)00072-x. [DOI] [PubMed] [Google Scholar]
- 44.Patterson RL, van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–99. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- 45.Rosado JA, Sage SO. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Trends Cardiovasc Med. 2000;10:327–32. doi: 10.1016/s1050-1738(01)00073-1. [DOI] [PubMed] [Google Scholar]
- 46.Bouaziz A, Amor NB, Woodard GE, Zibidi H, Lopez JJ, Bartegi A, Salido GM, Rosado JA. Tyrosine phosphorylation / dephosphorylation balance is involved in thrombin-evoked microtubular reorganisation in human platelets. Thromb Haemost. 2007;98:375–84. [PubMed] [Google Scholar]
- 47.Parekh AB. Store-operated Ca2+ entry: Dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol. 2003;547:333–48. doi: 10.1113/jphysiol.2002.034140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Putney JW., Jr Store-operated calcium channels: How do we measure them, and why do we care? Sci STKE. 2004;243:1–3. doi: 10.1126/stke.2432004pe37. [DOI] [PubMed] [Google Scholar]
- 49.Rosado JA, Redondo PC, Sage SO, Pariente JA, Salido GM. Store-operated Ca2+ entry: vesicle fusion or reversible trafficking and de novo conformational coupling? J Cell Physiol. 2005;205:262–9. doi: 10.1002/jcp.20399. [DOI] [PubMed] [Google Scholar]
- 50.Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Shea TB. Homocysteine potentiates beta-amyloid neurotoxicity: role of oxidative stress. J Neurochem. 2001;78:249–53. doi: 10.1046/j.1471-4159.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- 51.Redondo PC, Salido GM, Rosado JA, Pariente JA. Effect of hydrogen peroxide on Ca2+ mobilisation in human platelets through sulphydryl oxidation dependent and independent mechanisms. Biochem Pharmacol. 2004;67:491–502. doi: 10.1016/j.bcp.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 52.Elliott SJ, Schilling WP. Oxidative stress inhibits bradykinin-stimulated 45Ca2+ flux in pulmonary vascular endothelial cells. Am J Physiol. 1991;260:H549–56. doi: 10.1152/ajpheart.1991.260.2.H549. [DOI] [PubMed] [Google Scholar]
- 53.Koubaa N, Nakbi A, Smaoui M, Abid N, Chaaba R, Abid M, Hammami M. Hyperhomocysteinemia and elevated ox-LDL in Tunisian type 2 diabetic patients: Role of genetic and dietary factors. Clin Biochem. 2007;40:1007–14. doi: 10.1016/j.clinbiochem.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 54.Rasouli ML, Nasir K, Blumenthal RS, Park R, Aziz DC, Budoff MJ. Plasma homocysteine predicts progression of atherosclerosis. Atherosclerosis. 2005;181:159–65. doi: 10.1016/j.atherosclerosis.2005.01.001. [DOI] [PubMed] [Google Scholar]