

Abstract
Plaque rupture is the most common type of plaque complication and leads to acute ischaemic events such as myocardial infarction and stroke. Calcification has been suggested as a possible indicator of plaque instability. Although the role of matrix vesicles in the initial stages of arterial calcification has been recognized, no studies have yet been carried out to examine a possible role of matrix vesicles in plaque destabilization. Tissue specimens selected for the present study represented carotid specimens obtained from patients undergoing carotid endarterectomy. Serial frozen cross-sections of the tissue specimens were cut and mounted on glass slides. The thickness of the fibrous cap (FCT) in each advanced atherosclerotic lesion, containing a well developed lipid/necrotic core, was measured at its narrowest sites in sets of serial sections. According to established criteria, atherosclerotic plaque specimens were histologically subdivided into two groups: vulnerable plaques with thin fibrous caps (FCT <100 μm) and presumably stable plaques, in which fibrous caps were thicker than 100 μm. Twenty-four carotid plaques (12 vulnerable and 12 presumably stable plaques) were collected for the present analysis of matrix vesicles in fibrous caps. In order to provide a sufficient number of representative areas from each plaque, laser capture microdissection (LCM) was carried out. The quantification of matrix vesicles in ultrathin sections of vulnerable and stable plaques revealed that the numbers of matrix vesicles were significantly higher in fibrous caps of vulnerable plaques than those in stable plaques (8.908±0.544 versus 6.208±0.467 matrix vesicles per 1.92 μm2 standard area; P= 0.0002). Electron microscopy combined with X-ray elemental microanalysis showed that some matrix vesicles in atherosclerotic plaques were undergoing calcification and were characterized by a high content of calcium and phosphorus. The percentage of calcified matrix vesicles/microcalcifications was significantly higher in fibrous caps in vulnerable plaques compared with that in stable plaques (6.705±0.436 versus 5.322±0A94; P= 0.0474). The findings reinforce a view that the texture of the extracellular matrix in the thinning fibrous cap of atherosclerotic plaque is altered and this might contribute to plaque destabilization.
Keywords: matrix vesicles, calcification, artery, atherosclerosis, pathology
Introduction
Plaque rupture is the most common type of plaque complication [1–6]. Atherosclerotic plaque at the carotid bifurcation is often associated with transient ischaemic attack and ischaemic stroke [7–9]. Over recent years, the features of vulnerable atherosclerotic plaque have been intensely studied [1–10]. The most common type of unstable plaque is the thin-cap fibroatheroma, an eccentric lesion composed of a lipid rich core with a thin fibrous cap (<100 μm) [11–14]. The mechanisms of plaque rupture are not yet well understood, but plaque rupture is defined as the breakdown of the fibrous cap with an intrusion of blood products into the lesion [1–14]. Typical histological features of an unstable plaque include a lipid-rich necrotic core, consisting of dying macrophages and foam cells, and lipid components including free cholesterol, cholesterol esters and cholesterol crystals [1–6]. Unstable atherosclerotic plaque is marked by a reduction in extracellular matrix components, including collagen and proteoglycans, within the fibrous cap as well as by an intensified inflammatory response resulting from an increase in the infiltration of plaque by monocytes/macrophages, lymphocytes and dendritic cells [1–6, 15–21].
The vulnerability of atherosclerotic plaque is critically dependent on many factors including endothelial cell function, inflammatory cells, cytokine production, smooth muscle cell content and cell death, including necrosis and apoptosis [1–6, 22–25]. Calcification has also been suggested as a possible indicator of plaque instability, especially as this feature of plaque can be readily detected by several imaging techniques [26–31]. Studies utilizing various techniques have shown that calcium deposits in carotid and coronary atherosclerotic lesions may occur in various forms [26–32]. Intravascular ultrasound (IVUS) has been used to define sensitivity and specificity of the detection of different grades of histologic calcifications in the plaques [33]. Small ‘spotty’ calcifications in atherosclerotic plaques were found to be often associated with a fibrofatty plaque [34]. A strong inverse correlation has been reported between the extent of carotid plaque calcification and the intensity of plaque fibrous cap inflammation as determined by the degree of macrophage infiltration [34]. Using B-mode ultrasonography, Gorgan and colleagues [35] reported that symptomatic plaques were more echolucent and less calcified than asymptomatic plaques and were associated with a greater degree of histopathologic plaque necrosis. Rao and colleagues [29] reported that a trend towards an increase in plaque instability was seen with low amounts of calcification, but plaques with a high degree of calcification appeared to be more stable. It has been suggested that the distribution of calcified deposits thoughtout plaques, but not the degree of calcification, determines plaque stability [28, 30]. Vengrenyuk and coworkers [28] have hypothesized that fibrous cap microcalcifications, which have gone undetected because they lie below the visibility of current in vivo imaging techniques, cause local stress concentrations that lead to interfacial debonding. A mathematical model predicts that inclusions located in an area of high circumferential stress (>300 kPa) in the fibrous cap can intensify this stress to nearly 600 kPa when the cap thickness is less than 65 μm. The most likely candidates for the inclusions are microcalcifications [28].
Initial stages of the formation of microcalcifications are associated with the presence of matrix vesicles in the arterial wall [36]. Matrix vesicles have been defined as extracellular membrane-invested particles frequently observed at sites of initial calcification [36]. Mineral-associated vesicles were found in human atherosclerotic arteries [37–39] as well as in arteries of rabbits fed cholesterol diets [40–42]. In a previous study, we isolated matrix vesicles from human atherosclerotic plaques and demonstrated that matrix vesicles represent a structural basis for the formation of calcified deposits [43].
Despite obvious interest in the elucidation of a possible role of matrix vesicles in plaque instability, no previous studies have examined the characteristics and numbers of matrix vesicles in unstable plaques. The present study was undertaken to quantify and compare the relative numbers (relative densities) of calcified matrix vesicles in fibrous caps of vulnerable (FCT < 100 μm) and presumably stable atherosclerotic plaques (FCT >100 μm).
Material and methods
Tissue specimens
The carotid artery specimens were obtained from patients undergoing carotid endarterectomy. Informed consent was obtained from each patient. The investigation conforms with the principles outlined in the Declaration of Helsinki. The protocol of the study was approved by the Human Research Committee of the St Vincent's Hospital Sydney. Pre-operative duplex scanning, magnetic resonance imaging or angiography of the carotid arteries were performed. The indications for endarterectomies were stenosis of internal carotid artery of more than 70% for symptomatic patients and carotid stenosis of more than 80% for asymptomatic patients [44, 45]. Unfixed specimens were immediately embedded in OCT compound, rapidly frozen in liquid nitrogen and stored at −70°C until cryostat sectioning.
For the present investigation, 24 carotid plaques were selected as described below.
Selection of tissue specimens for further analysis
Serial cross-sections were cut (7 μm thick) and mounted on glass slides. To analyse the plaque morphology, representative sections of each plaque were stained with Mayer's haematoxylin. In order to examine the degrees of calcification of atherosclerotic plaques, sections were stained with Alizarin red S and by von Kossa technique. The examined plaques corresponded to Va, Vb and VI type atherosclerotic lesions according to the classification of Stary and colleagues [46]. In each atherosclerotic plaque, the fibrous cap, plaque core and shoulders were histologically identified as previously described [47]. The fibrous cap was defined as the area between the plaque core and the vessel lumen. The plaque shoulders were located at the edges of the fibrous cap, showing the largest increase of plaque thickness over the lumen circumference. The borders between the plaque core and the fibrous cap, as well as the borders between the plaque core and the media, were histologically distinguishable due to the cell-rich and fibrillar structure of the fibrous cap or the media, in contrast to the acellular structure of the plaque lipid/necrotic core. The thickness of the fibrous cap (FCT) was measured at its narrowest site in sets of serial sections. For further analysis, only advanced atherosclerotic plaques with large and well-developed necrotic cores as well as with plexuses of neovascularization in shoulder regions were selected. A number of studies of advanced atherosclerotic lesions have shown that the thickness of the fibrous cap that overlies the necrotic core distinguishes a stable lesion from one that is at high risk for rupture and thromboembolic events [11–14]. The threshold thickness of 100 μm that poses an increased risk for cap rupture has been established in these studies [11–14]. According to this established criteria [11–14], atherosclerotic specimens were histologically subdivided into two groups: relatively stable plaques (FCT >100 μm) (referred to hereafter in the text as ‘stable plaques’) and vulnerable plaques (FCT <100 μm), as used previously [48]. Ninety-three carotid plaques were examined. According to routine histology, amongst the 93 specimens, two specimens from symptomatic patients had intensive haemorrhage, presumably as result of plaque rupture. In these two specimens, the areas between the necrotic cores and the arterial lumen were filled with inflammatory cells and thrombotic masses. It was impossible to identify the borders of the degenerating fibrous caps and, therefore, the analysis of matrix vesicles in these two plaques was impractical. Amongst the remaining 91 specimens, only 12 plaques had fibrous caps with a thickness of less than 100 μm and all these plaques represented tissue specimens obtained from symptomatic patients. These 12 plaques constituted Group A of the present study (vulnerable plaques). For comparison, 12 specimens were randomly taken from the remaining 28 symptomatic patients. No specimens from asymptomatic patients had fibrous caps with a thickness of less than 100 μm and, therefore, the present analysis was limited to a comparison of plaques obtained from symptomatic patients. According to histology, plaques in both groups represented fibroatheromas, in which the necrotic cores exhibited various degrees of calcification. In both groups, the extent of the carotid disease was similar. All patients were receiving various medications. The patients' medical histories are provided in Table 1.
Table 1.
Medical history of patients
| Group A (Vulnerable plaques) (n = 12) | Group B (Stable plaques)(n= 12) | |
|---|---|---|
| Age, years | 69 (63–79) | 71 (65–81) |
| Sex, male, n | 7 | 8 |
| Diabetes, n | 3 | 3 |
| Smoking, n | 5 | 4 |
| Hyperlipidemia, n | 5 | 6 |
| Hypertension, n | 7 | 6 |
The slides with sections of stable and vulnerable plaques were further used in order to microdissect samples of fibrous caps along the border with the necrotic cores.
Laser capture microdissection
In order to collect a sufficient number of representative areas from each plaque, LCM was carried out as described previously [49]. In brief, after being cut on a cryostat, sections were placed onto slides covered with polyethylene-naphtalate (PEN) membranes (PALM Microlaser Technologies; 1440–1000). A PALM Laser-MicroBeam System (PALM Microlaser Technologies), which enabled the contact-free isolation of tissue fragments of fibrous caps, was used for microdissection. At least 10 microdissected fibrous plaque samples from each carotid plaque were catapulted into lids of 0.5-ml reaction tubes, using the laser pressure catapulting technique of the instrument.
Preparation of microdissected tissue samples was carried out according to a modified technique described by Grant and Jerome [50], as used previously [49]. The entire lids of 0.5-ml reaction tubes with microdissected cells were fixed in 1% glutaraldehyde in Phosphate Buffered Saline (PBS), post-fixed in 1% OsO4 and embedded in Araldite. Serial ultrathin sections of the embedded lids containing microdissected tissue areas were cut and placed on formvar-coated grids.
Transmission electron microscopy and statistical analysis
Ultrathin sections were stained with uranyl acetate and lead citrate and examined with the aid of Hitachi H7000 and Morgagni 268D electron microscopes. From each Araldite block, 10 ultrathin sections were used for the quantitative analysis of matrix vesicles. Quantitative analysis was carried out at ×56,000 magnification. In each ultrathin section, matrix vesicles and microcalcifications were calculated in a standard area of 1.92 μm2 (10 random acellular areas in each specimen were analysed). The results for variables were expressed as mean ± SD. Statistical analysis was performed by t-test using Prism® 4. P-values of less than 0.05 were considered as statistically significant.
Electron microscopy combined with X-ray elemental microanalysis
Non-stained ultrathin sections of LR White resin-embedded tissue specimens of plaques were subjected to elemental microanalysis using a LINK QX 200J energy dispersive X-ray microanalysis system attached to a Hitachi H7000 electron microscope, as previously reported [51].
Results
The histological level of examination utilizing haematoxylin staining demonstrated the presence of calcifications in the necrotic core in each specimen containing advanced atherosclerotic plaque (Fig. 1A–G). Staining with Alizarin red S and by von Kossa technique confirmed the presence of calcified deposits of various sizes located in the acellular necrotic core (Fig. 1B-G.). A pale and diffusely distributed Alizarin red S staining was detected in the extracellular matrix of the fibrous caps of some specimens but this staining was unsuitable for a quantitative analysis.
Fig 1.
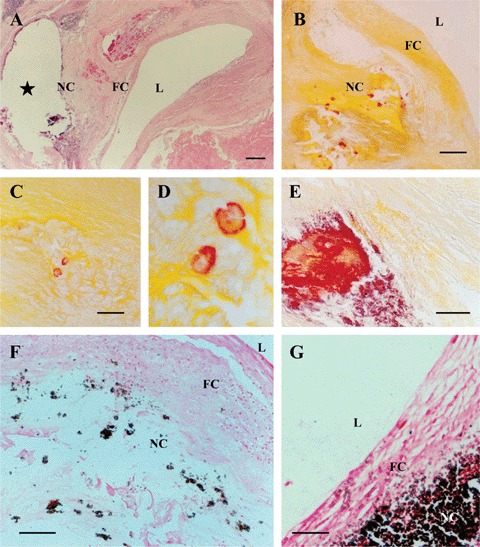
Histologically identifiable large and huge calcified deposits visualized by means of routine histology (A; Haematoxylin & eosin staining) and histo-chemistry using Alizarin red S (B–E) and von-Kossa techniques (F, G). (D) is a detail of (C). In (A, B, F and G), L, arterial lumen; FC, fibrous cap; NC, necrotic core. In (A), star indicates an area where a huge calcified deposit has been formed; this huge calcified deposit was lost during tissue sectioning. Scale bars: 400 μm (A), 250 μm (B, F), 50 μm (C, E), 150 μm (G).
At an electron microscopic level of examination, matrix vesicles were observed in fibrous caps of all plaques selected for further quantitative analysis. In both vulnerable and stable plaques, matrix vesicles were covered by two or several layers of electron-dense material closely opposed to each other and thus, forming concentric membranes (lamellae) (Fig. 2A-H). Matrix vesicles were filled with granular or relatively homogenous material of various electron densities (Fig. 2A-H). No difference in the fine structure of matrix vesicles was detected between specimens of vulnerable and stable plaques. The size of matrix vesicles varied from 90 to 300 nm in both vulnerable and stable plaques. The quantification of matrix vesicles in stable and vulnerable plaques revealed that the numbers (relative densities) of matrix vesicles were significantly higher in fibrous caps of vulnerable plaques than those in stable plaques (8.908±0.544 versus 6.208±0.467 matrix vesicles per 1.92 μm2; P= 0.0002) (Fig. 3A–D).
Fig 2.
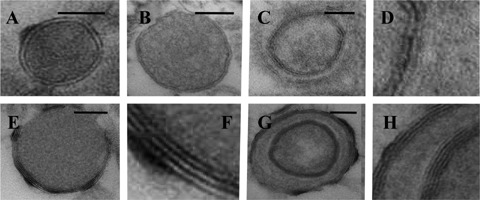
Typical appearance of matrix vesicles in the fibrous cap of atherosclerotic plaque (A-H). (A-B): Matrix vesicles surrounded by two electron-dense layers (lamellae) and filled with granular (A) or homogenous material (B) of medium electron density. (D-H): Matrix vesicles covered by multiple lamellae. (D) is a detail of (C). (F) is a detail of (E). (H) is a detail of (G). Transmission electron microscopy (TEM). Scale bars: 50 nm (A, B, C, EandG).
Fig 3.
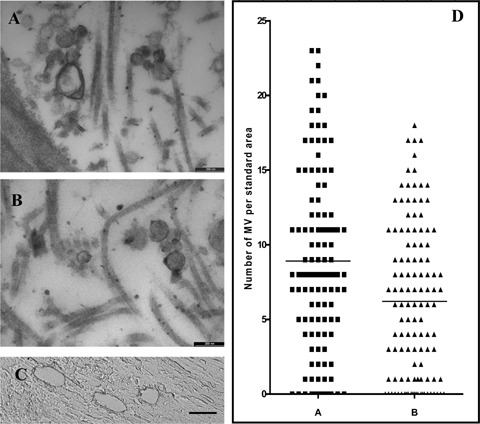
Examples of images of matrix vesicles located in fibrous caps in vulnerable (A) and stable plaques (B), which were used for the quantitative analysis (see Methods section): TEM. Scale bars: 200 nm (A-B). (C): Image showing an example of laser capture microdissection of tissue samples from a fibrous cap. Scale bar: 25 μm (D): Numbers/relative densities of matrix vesicles in fibrous caps in vulnerable (A) and stable plaques (B).
Electron microscopic examination showed that some matrix vesicles in the fibrous caps of both vulnerable and stable plaques displayed signs of calcification (Fig. 4A-G). Elemental microanalysis demonstrated that calcified matrix vesicles had a high content of calcium and phosphorus (Fig. 4H). Some matrix vesicles were completely calcified and displayed a homogeneous, very high electron density throughout while others showed different degrees of calcification (Fig. 4A-G). Calcifying matrix vesicles displayed marked variations in the patterns of calcification but in not yet completely calcified matrix vesicles, the presence of lamellar structures located on the periphery was evident (Fig. 4B and C), indicating that the formation of the microcalcifications had occurred on a structural basis of matrix vesicles. In some matrix vesicles, spicules of calcium salt crystals were associated with lamellar structures (Fig. 4D-F), while in others, spicules of calcium salt crystals were distributed quite irregularly (Fig. 4G). The percentage of calcified matrix vesicles/microcalcifications in the total matrix vesicle population was examined and it was found that the percentage of calcified matrix vesicles/microcalcifications was significantly higher in the tissue specimens of fibrous caps collected from vulnerable plaques than that from stable plaques (6.705±0.436 versus 5.322±0.494; P= 0.0474).
Fig 4.
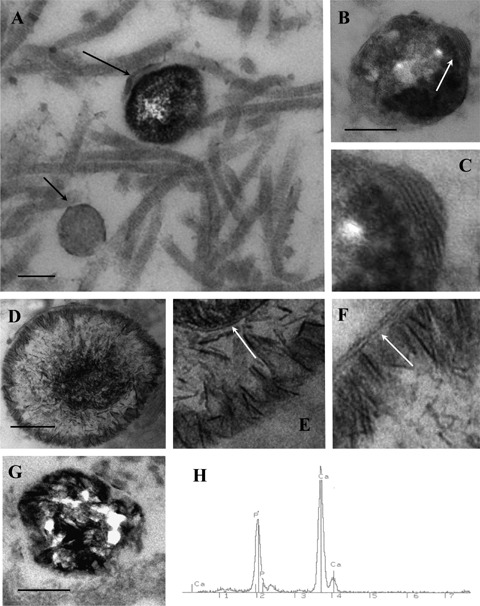
Typical appearance of matrix vesicles undergoing calcification (A-G). In (A), the large arrow shows a calcifying matrix vesicle that is characterized by a very high electron density while the small arrow shows a non-calcified matrix vesicle that displays a medium electron density. In (B), the arrow shows the zone of a calcifying matrix vesicle where the association of calcified deposits with the multilamellar structure is evident. Note that calcification occurs inside the matrix vesicle as well as along the vesicle contour. (C) is a detail of (B). In (D-F), spicules of calcium salt crystals are distributed along the contour of a matrix vesicle where they are associated with lamellar structures (shown by arrows in Fig. E and F). (E) and (F) are details of (D). In (G), spicules of calcium salt crystals are distributed irregularly throughout the matrix vesicle. TEM. Scale bars: 100 nm (A, B, D, G). (H): X-ray elemental microanalysis showing the presence of calcium and phosphorus in a calcifying matrix vesicle.
Discussion
Plaque ruptures most often in thinning fibrous caps [1–6, 11–14]. Analysis of alterations occurring during the thinning of the fibrous cap is important [1–6, 11–14]. During recent years, a number of studies focused on the elucidation of the possible contribution of the extracellular matrix to plaque destabilization [1–6, 18, 19] but no previous work has examined the possible contribution of matrix vesicles to plaque destabilization.
Matrix vesicles have been isolated from human atherosclerotic lesions and arteries of experimental animals [39, 41, 43]. While there has been little study of the lipid composition of human vascular matrix vesicles, it is known that vascular matrix vesicles contain approximately equimolar amounts of phospholipids and sterols, of which cholesteryl arachidonate comprises 2.3%[43]. Matrix vesicles contain bone morphogenic proteins and non-collagenous bone matrix proteins including osteopontin, osteonectin and matrix Gla protein in atherosclerotic lesions [36, 52, 53]. Annexins are the main group of proteins in matrix vesicles [36]. The origin of matrix vesicles in atherosclerotic lesions is not yet well understood. In other calcified tissues, matrix vesicle biogenesis occurs by polarized budding and pinching-off of vesicles from specific regions of the outer plasma membranes of differentiating growth plate chondrocytes, osteoblasts and odontoblasts [36]. Shedding of microvesicles from the surface of structurally intact smooth muscle cells has been demonstrated in atherosclerotic lesions [38] but the release of a large number of microvesicular structures into the extracellular space occurring during cell death has also been reported [54]. Bauriedel and colleagues [55] have reported that the presence of membrane surrounded cytoplasmic remnants of apoptotic smooth muscle cells, which might represent matrix vesicles, were markedly increased in unstable angina lesions. It is impossible to exclude that some matrix vesicles in the arterial wall may form simply as a result of physicochemical processes, similarly as this occurs in vitro when liposomes are produced [56, 57].
The present study revealed that in vulnerable plaque where the fibrous caps were thinner than 100 μm, the relative numbers (relative densities) of matrix vesicles were significantly higher than those in presumably stable plaque with a thicker fibrous cap. The increased relative density of matrix vesicles could alter the texture of the connective stroma of fibrous plaque, rendering the plaque prone to rupture. The mechanisms leading to an increased number of matrix vesicles in the thinning fibrous cap require further investigation. No unique features of the populations of matrix vesicles in vulnerable plaque were detected and this might suggest that the difference in matrix vesicles between more and less stable plaques is of a quantitative nature. However, our study was limited to morphological analysis and, therefore, it cannot be excluded that future biochemical analysis might reveal unique features of matrix vesicles in vulnerable plaques.
The important role of matrix vesicles in the initiation of calcification is recognized [36–43]. In some circumstances, vascular smooth muscle cells promote calcification by the mechanisms similar to those occurring during growth plate mineralization [36–43]. According to the current concept, the initiation of the formation of mineral crystals within matrix vesicles is augmented by the activity of matrix vesicle phosphatases and calcium-binding molecules, all of which are concentrated in or near the matrix vesicle membrane (lamellae) [36]. Membranes of matrix vesicles contain alkaline phosphatase, which hydrolyses a variety of substrates leading to the increase in Pi local levels [58, 59]. Alkaline phosphatase activity facilitates inactivation of mineralization inhibitors such as adenosine triphosphate (ATP) and pyrophos-phate ions [36, 59]. An increase in intravesicular Pi levels is thought to be partly mediated by NaPiTs, which include type III transporter glvr-1 [59–61]. Mineralization-competent matrix vesicles have been shown to contain a nucleational core composed of amorphous calcium phosphate, phosphatidylserine and annexins [36, 59–61] while mineralization-incompetent matrix vesicles lack these components [59–61]. Mineralization-competent matrix vesicles are able to locally increase calcium and Pi levels, thereby permitting crystal nucleation [59–61]. Members of the annexin family mediate calcium uptake into matrix vesicles [36, 59–61]. In the presence of elevated levels of calcium and phosphate ions, the release of mineralization-competent matrix vesicles occurs [36, 59–61]. The mineralization begins with crystal release through the matrix vesicle membrane exposing preformed hydroxyapatite crystals to the extracellular fluid [36]. The extracellular fluid contains sufficient Ca2+ and PO43− to support continuous crystal proliferation, with preformed crystals serving as templates for the formation of new crystals by a process of homologous nucleation [36]. The membrane-invested internal microenvironment protects the pre-current mineral nuclei in a pre-crystalline state, before conversion to hydroxyapatite [36]. It is thought that once the first calcium phosphate crystals have coalesced to form a mineralization front, crystal multiplication then occurs independently of matrix vesicles [36, 59]. The present study revealed that the percentage of matrix vesicles undergoing calcification is higher in plaques with thinner fibrous caps. This suggests that the rate of calcification of matrix vesicles might be higher in fibrous caps of vulnerable plaques. The increase in numbers/relative density of calcifying matrix vesicles could alter the texture of the connective stroma of fibrous plaque, rendering the plaque prone to rupture.
An increased calcification of matrix vesicles in the fibrous cap of vulnerable plaque might depend on many factors including endothelial cell function, inflammatory cells, cytokine production, smooth muscle cell content and cell death, all of which are considered as determinants of plaque rupture [1–8, 62, 63]. Ectopic calcifications have been often associated with apoptotic or necrotic processes, both of which lead to the release of a large number of membrane invested microfragments of the cytoplasm in the extracellular space [64]. It has been suggested that apoptotic bodies may proceed to calcify, depending on the local microenvironment [65, 66]. In advanced atherosclerotic plaques, matrix vesicles derived from smooth muscle cells contain BAX protein, indicating that they may be remnants of apoptotic cells [67]. During programmed cell death, calcium and Pi ions stored in mitochondria and sarcoplasm can be incorporated in apoptotic bodies contributing to the formation of calcium phosphate crystals. In vitro studies have shown that induction of apoptosis in vascular smooth muscle cells is accompanied by the release of calcium-rich structures [65, 68]. Inhibition of apoptosis by the broad-range caspase inhibitor ZVAD-FMK reduces the extent of mineralization in culture of vascular smooth muscle cells [65]. It has been suggested that ZVAD-FMK inhibits mineralization at the level of apoptotic body release [65].
The present study is limited to the comparison of the spatial densities of matrix vesicles between thick (>100 μm) and thin (<100 μm) fibrous caps. Apart from plaque morphology [1–8], other factors such as haemodynamic forces and pharmacological modulation have been recognized as determinants of plaque stability [69–73]. Carotid atherosclerosis is thought to be locally modulated by haemodynamic forces of wall shear stress and circumferential wall tension, respectively representing the frictional and the perpendicular force of the blood flowing on the vascular wall [69–71]. The impact of haemodynamic forces on vulnerable plaque rupture is not well understood especially as ruptures most frequently occur in regions where computational finite element (FEM) and fluid structure interaction (FSI) models do not predict maximal stress [74–76]. Plaques rupture in the central part of the fibrous cap rather than in regions of high curvature at plaque shoulders where the models predict maximum tissue stresses [74–76]. Analysing 3D MRI images of plaques using a FSI model, Tang et al. [77] have shown that maximal stress appears at healthy parts of the vessel where the vessel wall is thinner than the wall on the diseased plaque side or where vessel wall curvature is large.
Conclusion
The results of the present study indicate that the accumulation and calcification of matrix vesicles might be involved in the process of plaque destabilization, possibly through the alteration of the texture of connective stroma in plaque fibrous caps. Further studies are needed to analyse the mechanisms responsible for the increased relative density of matrix vesicles and microcalcifications in the thinning fibrous cap and the effects of these events on plaque destabilization. In particular, investigating the cell composition, cell density and the occurrence and peculiarities of cell death might help understand the increase in the density of matrix vesicles in the fibrous cap of vulnerable plaque.
Acknowledgments
This work was supported by grants from the St Vincent's Clinic Foundation, Sydney, and from the School of Medicine, University of Western Sydney.
References
- 1.Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N EnglJMed. 1997;336:1276–82. doi: 10.1056/NEJM199705013361802. [DOI] [PubMed] [Google Scholar]
- 2.Shah PK. Mechanisms of plaque vulnerability and rupture. J Am Coll Cardiol. 2003;41:S15–22. doi: 10.1016/s0735-1097(02)02834-6. [DOI] [PubMed] [Google Scholar]
- 3.Mitra AK, Dhume AS, Agrawal DK. “Vulnerable plaques’-ticking of the time bomb. Can J Physiol Pharmacol. 2004;82:860–71. doi: 10.1139/y04-095. [DOI] [PubMed] [Google Scholar]
- 4.Hennerici MG. The unstable plaque. Cerebrovasc Dis. 2004;17(Suppl 3):17–22. doi: 10.1159/000075300. [DOI] [PubMed] [Google Scholar]
- 5.Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. 2005;46:937–54. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- 6.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13–8. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 7.Carr S, Farb A, Pearce WH, Virmani R, Yao JS. Atherosclerotic plaque rupture in symptomatic carotid artery stenosis. J Vase Surg. 1996;23:755–65. doi: 10.1016/s0741-5214(96)70237-9. [DOI] [PubMed] [Google Scholar]
- 8.Virmani R, Ladich ER, Burke AP, Kolodgie FD. Histopathology of carotid atherosclerotic disease. Neurosurgery. 2006;59(Suppl 3):S219–27. doi: 10.1227/01.NEU.0000239895.00373.E4. [DOI] [PubMed] [Google Scholar]
- 9.Redgrave JN, Rothwell PM. Asymptomatic carotid stenosis: what to do. Curr Opin Neurol. 2007;20:58–64. doi: 10.1097/WCO.0b013e328012da60. [DOI] [PubMed] [Google Scholar]
- 10.Redgrave JN, Lovett JK, Gallagher PJ, Rothwell PM. Histological assessment of 526 symptomatic carotid plaques in relation to the nature and timing of ischemic symptoms: the Oxford plaque study. Circulation. 2006;113:2320–8. doi: 10.1161/CIRCULATIONAHA.105.589044. [DOI] [PubMed] [Google Scholar]
- 11.Kolodgie FD, Burke AP, Farb A, Gold HK, Yuan J, Narula J, Finn AV, Virmani R. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16:285–92. doi: 10.1097/00001573-200109000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Virmani R, Burke AP, Kolodgie FD, Farb A. Pathology of the thin-cap fibroatheroma: a type of vulnerable plaque. J Interv Cardiol. 2003;16:267–72. doi: 10.1034/j.1600-0854.2003.8042.x. [DOI] [PubMed] [Google Scholar]
- 13.Mehta AB, Shah S. Unstable or High Risk Plaque: How Do We Approach It? MJAFI. 2006;62:2–7. doi: 10.1016/S0377-1237(06)80141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W, Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation. 2003;108:1664–72. doi: 10.1161/01.CIR.0000087480.94275.97. [DOI] [PubMed] [Google Scholar]
- 15.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the unstable plaque. Prog Cardiovascular Dis. 2002;44:349–56. doi: 10.1053/pcad.2002.122475. [DOI] [PubMed] [Google Scholar]
- 16.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb VascBiol. 2000;20:1262–75. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Ortiz A, Badimon JJ, Falk E, Fuster V, Meyer B, Mailhac A, Weng D, Shah PK, Badimon L. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23:1562–9. doi: 10.1016/0735-1097(94)90657-2. [DOI] [PubMed] [Google Scholar]
- 18.Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- 19.Burleigh MC, Briggs AD, Lendon CL, Davies MJ, Born GV, Richardson PD. Collagen types I and III, collagen content, GAGs and mechanical strength of human atherosclerotic plaque caps: span-wise variations. Atherosclerosis. 1992;96:71–1. doi: 10.1016/0021-9150(92)90039-j. [DOI] [PubMed] [Google Scholar]
- 20.Yilmaz A, Lochno M, Traeg F, Cicha I, Reiss C, Stumpf C, Raaz D, Anger T, Amann K, Probst T, Ludwig J, Daniel WG, Garlichs CD. Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques. Atherosclerosis. 2004;176:101–10. doi: 10.1016/j.atherosclerosis.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 21.Erbel C, Sato K, Meyer FB, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. Functional profile of activated dendritic cells in unstable atherosclerotic plaque. Basic Res Cardiol. 2007;102:123–32. doi: 10.1007/s00395-006-0636-x. [DOI] [PubMed] [Google Scholar]
- 22.Kockx MM, Herman AG. Apoptosis in atherosclerosis: beneficial or detrimental? Cardiovasc Res. 2000;45:736–46. doi: 10.1016/s0008-6363(99)00235-7. [DOI] [PubMed] [Google Scholar]
- 23.Kolodgie FD, Narula J, Haider N, Virmani R. Apoptosis in atherosclerosis. Does it contribute to plaque instability? Cardiol Clin. 2001;19:127–39. doi: 10.1016/s0733-8651(05)70199-5. [DOI] [PubMed] [Google Scholar]
- 24.Rossi ML, Marziliano N, Merlini PA, Bramucci E, Canosi U, Belli G, Parenti DZ, Mannucci PM, Ardissino D. Different quantitative apoptotic traits in coronary atherosclerotic plaques from patients with stable angina pectoris and acute coronary syndromes. Circulation. 2004;110:1767–73. doi: 10.1161/01.CIR.0000142865.04816.89. [DOI] [PubMed] [Google Scholar]
- 25.Clarke M, Bennett M. The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am JNephrol. 2006;26:531–5. doi: 10.1159/000097815. [DOI] [PubMed] [Google Scholar]
- 26.Pham PH, Rao DS, Vasunilashorn F, Fishbein MC, Goldin JG. Computed tomography calcium quantification as a measure of atherosclerotic plaque morphology and stability. Invest Radiol. 2006;41:674–80. doi: 10.1097/01.rli.0000233325.42572.08. [DOI] [PubMed] [Google Scholar]
- 27.Cola C, Clementi E, Biondi-Zoccai G, Sangiorgi G. From carotid plaque biology to serologic markers of vulnerability to predict the risk of cerebrovascular events. Ada ChirBelg. 2007;107:129–42. [PubMed] [Google Scholar]
- 28.Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L, Weinbaum S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci USA. 2006;103:14678–83. doi: 10.1073/pnas.0606310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao DS, Goldin JG, Fishbein MC. Determinants of plaque instability in atherosclerotic vascular disease. Cardiovasc Pathol. 2005;14:285–93. doi: 10.1016/j.carpath.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Li ZY, Howarth S, U-King-lm J, Gillard J. Atheroma: is Calcium Important or Not? A Modelling Study of Stress Within the Atheromatous Fibrous Cap in Relation to Position and Size of Calcium Deposits. ConfProc IEEE Eng Med Biol Soc. 2005;3:2236–9. doi: 10.1109/IEMBS.2005.1616908. [DOI] [PubMed] [Google Scholar]
- 31.Lin TC, Tintut Y, Lyman A, Mack W, Demer LL, Hsiai TK. Mechanical response of a calcified plaque model to fluid shear force. Ann Biomed Eng. 2006;34:1535–41. doi: 10.1007/s10439-006-9182-9. [DOI] [PubMed] [Google Scholar]
- 32.Higgins CL, Marvel SA, Morrisett JD. Quantification of calcification in atherosclerotic lesions. Arterioscler Thromb VascBiol. 2005;25:1567–76. doi: 10.1161/01.ATV.0000172017.79441.73. [DOI] [PubMed] [Google Scholar]
- 33.Ehara S, Kobayashi Y, Yoshiyama M, Ueda M, Yoshikawa J. Coronary Artery Calcification Revisited. J Atheroscler Thrombosis. 2006;13:31–7. doi: 10.5551/jat.13.31. [DOI] [PubMed] [Google Scholar]
- 34.Shaalan WE, Cheng H, Gewertz B, McKinsey JF, Schwartz LB, Katz D, Cao D, Desai T, Glagov S, Bassiouny HS. Degree of carotid plaque calcification in relation to symptomatic outcome and plaque inflammation. J Vase Surg. 2004;40:262–9. doi: 10.1016/j.jvs.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 35.Grogan JK, Shaalan WE, Cheng H, Gewertz B, Desai T, Schwarze G, Glagov S, Lozanski L, Griffin A, Castilla M, Bassiouny HS. B-mode ultrasonographic characterization of carotid atherosclerotic plaques in symptomatic and asymptomatic patients. J Vase Surg. 2005;42:435–41. doi: 10.1016/j.jvs.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 36.Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–6. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- 37.Tanimura A, McGregor DH, Anderson HC. Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med. 1983;172:173–7. doi: 10.3181/00379727-172-41542. [DOI] [PubMed] [Google Scholar]
- 38.Bobryshev YV, Lord RS, Warren BA. Calcified deposit formation in intimal thickenings of the human aorta. Atherosclerosis. 1995;118:9–21. doi: 10.1016/0021-9150(95)05588-n. [DOI] [PubMed] [Google Scholar]
- 39.Hsu HH, Camacho NP. Isolation of calcifiable vesicles from human atherosclerotic aortas. Atherosclerosis. 1999;143:53–62. doi: 10.1016/s0021-9150(98)00322-0. [DOI] [PubMed] [Google Scholar]
- 40.Hsu HH, Camacho NC, Tawfik O, Sun F. Induction of calcification in rabbit aortas by high cholesterol diets: roles of calcifi-able vesicles in dystrophic calcification. Atherosclerosis. 2002;161:85–94. doi: 10.1016/s0021-9150(01)00623-2. [DOI] [PubMed] [Google Scholar]
- 41.Hsu HH, Tawfik O, Sun F. Mechanisms of calcification by vesicles isolated from atherosclerotic rabbit aortas. Biochim BiophysActa. 2002;1563:18–22. doi: 10.1016/s0005-2736(02)00371-1. [DOI] [PubMed] [Google Scholar]
- 42.Hsu HH, Tawfik O, Sun F. Mechanism of dystrophic calcification in rabbit aortas: temporal and spatial distributions of calcifying vesicles and calcification-related structural proteins. Cardiovasc Pathol. 2004;13:3–10. doi: 10.1016/S1054-8807(03)00093-0. [DOI] [PubMed] [Google Scholar]
- 43.McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, Lord RS, Geczy CL. S100A8 and S100A9 in human arterial wall. Implications foratherogenesis. J Biol Chem. 2005;280:41521–9. doi: 10.1074/jbc.M509442200. [DOI] [PubMed] [Google Scholar]
- 44.Lord RSA. Surgery of occlusive cere-brovascular disease. CV Mosby; 1986. St Louis: Mo: [Google Scholar]
- 45.Lord RSA. Carotid endarterectomy: options and outcomes. Aust N Z J Surg. 1995;65:151–9. doi: 10.1111/j.1445-2197.1995.tb00598.x. [DOI] [PubMed] [Google Scholar]
- 46.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–74. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 47.Bobryshev YV, Lord RSA. Mapping of vascular dendritic cells in atherosclerotic arteries suggests their involvement in local immune-inflammatory reactions. Cardiovasc Res. 1998;37:799–810. doi: 10.1016/s0008-6363(97)00229-0. [DOI] [PubMed] [Google Scholar]
- 48.Bobryshev YV, Lord RSA. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J Histochem Cytochem. 2005;53:781–5. doi: 10.1369/jhc.4B6570.2005. [DOI] [PubMed] [Google Scholar]
- 49.Bobryshev YV. Intracellular localization of oxidized low-density lipoproteins in atherosclerotic plaque cells revealed by electron microscopy combined with laser capture microdissection. J Histochem Cytochem. 2005;53:793–7. doi: 10.1369/jhc.4B6602.2005. [DOI] [PubMed] [Google Scholar]
- 50.Grant K, Jerome WG. Laser capture microdissection as an aid to ultrastructural analysis. Microsc Microanal. 2002;8:170–5. doi: 10.1017/s143192760202010x. [DOI] [PubMed] [Google Scholar]
- 51.Bobryshev YV, Lord RSA. Detection of vascular dendritic cells accumulating calcified deposits in the cytoplasm. Tissue Cell. 1998;30:383–8. doi: 10.1016/s0040-8166(98)80052-9. [DOI] [PubMed] [Google Scholar]
- 52.Canfield AE, Farrington C, Dziobon MD, Boot-Handford RP, Heagerty AM, Kumar SN, Roberts IS. The involvement of matrix glycoproteins in vascular calcification and fibrosis: an immunohistochemical study. J Pathol. 2002;196:228–34. doi: 10.1002/path.1020. [DOI] [PubMed] [Google Scholar]
- 53.Dhore CR, Cleutjens JP, Lutgens E, Cleutjens KB, Geusens PP, Kitslaar PJ, Tordoir JH, Spronk HM, Vermeer C, Daemen MJ. Differential Expression of Bone Matrix Regulatory Proteins in Human Atherosclerotic Plaques. Arterioscler, Thromb Vase Biol. 2001;21:1998–2003. doi: 10.1161/hq1201.100229. [DOI] [PubMed] [Google Scholar]
- 54.Proudfoot D, Skepper JN, Hegyi L, Farzaneh-Far A, Shanahan CM, Weissberg PL. The role of apoptosis in the initiation of vascular calcification. Z Kardiol. 2001;90:43–6. doi: 10.1007/s003920170041. [DOI] [PubMed] [Google Scholar]
- 55.Bauriedel G, Hutter R, Welsch U, Bach R, Sievert H, Luderitz B. Role of smooth muscle cell death in advanced coronary primary lesions: implications for plaque instability. Cardiovasc Res. 1999;41:480–8. doi: 10.1016/s0008-6363(98)00318-6. [DOI] [PubMed] [Google Scholar]
- 56.Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ, Valenzuela SM. Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic ResCardiol. 2007;102:133–43. doi: 10.1007/s00395-006-0637-9. [DOI] [PubMed] [Google Scholar]
- 57.Skrtic D, Eanes ED. Effect of different phospholipid-cholesterol membrane compositions on liposome-mediated formation of calcium phosphates. Calcif Tissue Int. 1992;50:253–60. doi: 10.1007/BF00296290. [DOI] [PubMed] [Google Scholar]
- 58.Boskey AL. Mineral-matrix interactions in bone and cartilage. Clin Orthop. 1992;281:244–24. [PubMed] [Google Scholar]
- 59.Magne D, Julien M, Vinatier C, Merhi-Soussi F, Weiss P, Guicheux J. Cartilage formation in growth plate and arteries: from physiology to pathology. Bioessays. 2005;27:708–16. doi: 10.1002/bies.20254. [DOI] [PubMed] [Google Scholar]
- 60.Wu LN, Genge BR, Dunkelberger DG, LeGeros RZ, Concannon B, Wuthier RE. Physicochemical characterization of the nucleational core of matrix vesicles. J Biol Chem. 1997;272:4404–11. doi: 10.1074/jbc.272.7.4404. [DOI] [PubMed] [Google Scholar]
- 61.Kirsch T, Nah HD, Shapiro IM, Pacifici M. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J Cell Biol. 1997;137:1149–60. doi: 10.1083/jcb.137.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feeley TM, Leen EJ, Colgan MP, Moore DJ, Hourihane DO, Shanik GD. Histologic characteristics of carotid artery plaque. J Vase Surg. 1991;13:719–24. [PubMed] [Google Scholar]
- 63.Bassiouny HS, Sakaguchi Y, Mikucki SA, McKinsey JF, Piano G, Gewertz BL, Glagov S. Juxtalumenal location of plaque necrosis and neoformation in symptomatic carotid stenosis. J Vase Surg. 1997;26:585–94. doi: 10.1016/s0741-5214(97)70056-9. [DOI] [PubMed] [Google Scholar]
- 64.Kim KM. Apoptosis and calcification. Scanning Microsc. 1995;9:1137–75. [PubMed] [Google Scholar]
- 65.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–62. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- 66.Proudfoot D, Shanahan CM. Biology of calcification in vascular cells: intima versus media. Herz. 2001;26:245–51. doi: 10.1007/pl00002027. [DOI] [PubMed] [Google Scholar]
- 67.Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97:2307–15. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- 68.Anderson HC. Mechanisms of pathologic calcification. Rheum Dis Clin North Am. 1988;14:303–19. [PubMed] [Google Scholar]
- 69.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53:502–14. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 70.Carallo C, Irace C, Pujia A, De Franceschi MS, Crescenzo A, Motti C, Cortese C, Mattioli PL, Gnasso A. Evaluation of common carotid hemodynamic forces. Relations with wall thickening. Hypertension. 1999;34:217–21. doi: 10.1161/01.hyp.34.2.217. [DOI] [PubMed] [Google Scholar]
- 71.Grogan JK, Shaalan WE, Cheng H, Gewertz B, Desai T, Schwarze G, Glagov S, Lozanski L, Griffin A, Castilla M, Bassiouny HS. B-mode ultrasonographic characterization of carotid atherosclerotic plaques in symptomatic and asymptomatic patients. J Vase Surg. 2005;42:435–41. doi: 10.1016/j.jvs.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 72.Mezzetti A. Pharmacological modulation of plaque instability. Lupus. 2005;14:769–72. doi: 10.1191/0961203305lu2218oa. [DOI] [PubMed] [Google Scholar]
- 73.Wilensky RL, Hamamdzic D. The molecular basis of vulnerable plaque: potential therapeutic role for immunomodulation. Curr Opin Cardiol. 2007;22:545–51. doi: 10.1097/HCO.0b013e3282f028fe. [DOI] [PubMed] [Google Scholar]
- 74.Cheng GC, Loree HM, Kamm RD, Fishbein MC, Lee RT. Distribution of circumferential stress in ruptured and stable atherosclerotic lesions. A structural analysis with histopathological correlation. Circulation. 1993;87:1179–87. doi: 10.1161/01.cir.87.4.1179. [DOI] [PubMed] [Google Scholar]
- 75.Lee RT, Loree HM, Cheng GC, Lieberman EH, Jaramillo N, Schoen FJ. Computational structural analysis based on intravascular ultrasound imaging before in vitro angioplasty: prediction of plaque fracture locations. JAm Coll Cardiol. 1993;21:777–82. doi: 10.1016/0735-1097(93)90112-e. [DOI] [PubMed] [Google Scholar]
- 76.Loree HM, Kamm RD, Stringfellow RG, Lee RT. Effects of fibrous cap thickness on peak circumferential stress in model atherosclerotic vessels. Circ Res. 1992;71:850–8. doi: 10.1161/01.res.71.4.850. [DOI] [PubMed] [Google Scholar]
- 77.Tang D, Yang C, Zheng J, Woodard PK, Saffitz JE, Petruccelli JD, Sicard GA, Yuan C. Local maximal stress hypothesis and computational plaque vulnerability index for atherosclerotic plaque assessment. Ann Biomed Eng. 2005;33:1789–801. doi: 10.1007/s10439-005-8267-1. [DOI] [PMC free article] [PubMed] [Google Scholar]