

Abstract
Transforming growth factor-β (TGF-β) signalling is induced in liver as a consequence of damage and contributes to wound healing with transient activation, whereas it mediates fibrogenesis with long-term up-regulation in chronic disease. Smad-dependent TGF-β effects are blunted by antagonistic Smad7, which is transcriptionally activated as an immediate early response upon initiation of TGF-β signalling in most cell types, thereby providing negative feedback regulation. Smad7 can be induced by other cytokines, e.g. IFN-γ, leading to a crosstalk of these signalling pathways. Here we report on a novel mouse strain, denoted S7ΔE1, with a deletion of exon I from the endogenous smad7 gene. The mice were viable and exhibited normal adult liver architecture. To obtain insight into Smad7-depend-ent protective effects, chronic liver damage was induced in mice by carbon tetrachloride (CCI4) administration. Subsequent treatment, elevated serum liver enzymes indicated enhanced liver damage in mice lacking functional Smad7. CCI4-dependent Smad2 phosphoryla-tion was pronounced in S7ΔE1 mice and accompanied by increased numbers of α-smooth muscle actin positive ‘activated’ HSCs. There was evidence for matrix accumulation, with elevated collagen deposition as assessed morphometrically in Sirius red stained tissue and confirmed with higher levels of hydroxyproline in S7ΔE1 mice. In addition, the number of CD43 positive infiltrating lymphocytes as well as of apoptotic hepatocytes was increased. Studies with primary hepatocytes from S7ΔE1 and wild-type mice indicate that in the absence of functional Smad7 protein, hepatocytes are more sensitive for TGF-β effects resulting in enhanced cell death. Furthermore, S7ΔE1 hepatocytes display increased oxidative stress and cell damage in response to CCI4, as measured by reactive oxygen species production, glutathione depletion, lactate dehydrogenase release and lipid peroxidation. Using an ALK-5 inhibitor all investigated CCI4 effects on hepatocytes were blunted, confirming participation of TGF-β signalling. We conclude that Smad7 mediates a protective effect from adverse TGF-β signalling in damaged liver, re-iterating its negative regulatory loop on signalling.
Keywords: TGF-β, Smad7, hepatocytes, liver fibrosis, oxidative stress, mouse model
Introduction
TGF-β isoforms, activins and BMPs, constitute a large family with more than 33 members in human beings. Characteristically, members transmit signals via cell surface serine/threonine kinase receptors and intracellular Smad transcription factors [10]. Smads are segregated into three functional groups. Receptor Smads (R-Smads; Smadsl,2,3,5 and 8) are directly phosphorylated and activated by the TGF-β receptor complex. Co-Smad (Smad4) associates with activated R-Smads and these heteromeric proteins are translocated to the nucleus where they regulate gene transcription by either association with DNA binding proteins or direct binding to promoter sequences in target genes. Smad6 and Smad7 are inhibitory Smads (l-Smads) that down-regulate TGF-β superfamily signalling and thereby modulate biological responses. I-Smad activity is affected, in part, by a feedback mechanism that involves TGF-β-dependent transcriptional regulation of Smad7 [11], thus assuring termination or reducing the strength of TGF-β signalling.
TGF-β is a master mediator in liver fibrogenesis [12], TGF-β favours transition of HSCs to myofibroblasts, stimulates synthesis of ECM proteins, and inhibits their degradation. Strategies aimed at disrupting TGF-β synthesis and/or signalling pathways markedly decrease fibrosis in experimental models [13]. In line with this, we recently used an adenovirus-based gene therapeutic approach for ectopic Smad7 expression in liver cells and thus abrogated fibrogenesis in bile duct ligated rats [4].
The physiological role of Smad7 during liver damage is not clear. We found that quiescent HSCs in culture display a functional negative feedback comprising TGFβ-dependent transient activation of Smad7 expression, which is lost in myofibroblasts [14, 15]. Studies analysing PAI-1 and Col1A2 expression in HSCs further support a model in which endogenous TGF-β-mediated Smad7 terminates fibrotic signals, thus providing a transient TGF-β response after acute liver damage. In contrast, constitutive Smad2 activation and lack of Smad7 expression were present in MFBs throughout chronic liver injury [16]. These results imply that low Smad7 levels are involved in progression of liver fibrosis.
Recently, we generated a novel mouse model, denoted S7ΔE1, with a deletion of exonl from the endogenous Smad7 gene, by replacing the coding region of Smad7 including the starting ATG codon and a part of intron I with a neomycin selection cassette via homologous recombination. This resulted in a deletion of roughly the first half of the Smad7 protein, i.e. the N-terminal 204 aa. Mice heterozygous or homozygous for this deletion were viable and fertile. Mutant B cells showed an over-active TGF-β signalling measured as increase of phosphorylated Smad2-positive B cells leading to increased Ig class switch recombination to IgA, enhanced B cell apoptosis, highlighting a prominent role for Smad7 in regulating the immune system's response to TGF-β[1].
To get further insight into Smad7 function during liver fibrogenesis in vivo, we examined these S7ΔE1 mice in a carbon tetrachloride (CCI4) mouse model of liver fibrosis. We observed increased liver damage, enhanced collagen deposition and a higher number of α-SMA positive cells in mice lacking a functional Smad7 gene. Furthermore, CCI4-dependent inflammation and apoptosis were up-regulated in Smad7 deficient mice. As a consequence, primary hepatocytes from S7ΔE1 mice displayed enhanced and prolonged signalling in response to TGF-β. Our observations confirmed that the hypothesized negative feedback regulation of TGF-β signalling via Smad7 modulates the strength of the pro-fibrogenic response in a model of chronic liver disease.
Materials and methods
Materials
Recombinant active human transforming growth factor-β1(TGF-β1) was obtained from Peprotec (London, UK). Chloramine T solution, perchloric acid, p-dimethylaminobenzaldehyde, Triton-X-100, 2’7’-dichlorofluores-cein diacetate (DCFH-DA), DMSO, William's Medium E, monochlorobimane (MCB), 2-thiobarbituric acid (TBA), acetic acid, 2,6-di-tert-butyl-4-methylphenol, carbon tetrachloride and mineral oil were obtained from Sigma (Miinchen, Germany). Ethanol and methanol were obtained from Otto Fischar GmbH (Saarbriicken, Germany).
Generation of Smad7 mutant mice
Smad7 exonl deleted mice were generated as previously described [1]. Briefly, the Smad7 locus was isolated from a 129Sv genomic library [2]. The targeting vector consisted of a 5.1-kb Pvull-EcoRI genomic 5’ fragment containing the Smad7 promoter and 5’ untranslated leader sequence, followed by a PGKneobpA expression cassette replacing the translated part of exon 1 and theexon l/intron I boundary, a 1 -kb Hindlll-Notl genomic 3’ fragment, and α herpes simplex virus (HSV) thymidine kinase expression cassette in pBluescript SK+. The construct was linearized for electroporation into 129Sv-derived R1-ES cells, and colonies were selected with G418 and gancyclovir. Homologous recombination events were screened by Southern blotting using a Smad7 genomic 5’ probe (1 -kb Pvull-Hindl 11) or a genomic 3’ probe (700-bp Notl-Hindlll), both situated outside the genomic targeting vector sequences. Mice were genotyped by PCR using a common 5’ primer (5’-GCGGGGGAGGGAGGGGTAGAGG-3’) and an exonl antisense primer (5’-GGGGCGAGGAGGCGAGGAGAAAAT-3’) for the wild-type allele (1.7 kb) or a neo antisense primer (5’-GCTACCGGTGGATGTGGAATGTGTGC-3’) for the mutant allele (1.1 kb). Heterozygous mutant mice were backcrossed five generations onto CD-1 mice, obtained from Charles River Laboratories (MA, USA). The mice were used between 4 and 8 months of age, if not stated otherwise. All the in vitro and in vivo experiments were performed with homozygous Smad7 mutant mice. The animal experimentation was approved by the local ethical committee.
Chronic liver injury
Liver fibrosis was induced in Smad7 mutant (n= 8) and CD-1 wild-type (n = 8) mice by intraperitoneal injection of CCI4 (50 μl of 25% CCI4 in mineral oil per 25 g; 2 μl/g body weight) twice weekly. Sham-treated mice (three Smad7 mutant and three wild-type mice) received mineral oil only. After 8 weeks, animals were starved overnight and killed 48 hrs after the last CCI4 injection. We obtained liver samples from several lobes and either fixed them in buffered formalin or snap froze them in liquid nitrogen and stored them at −80°C until use.
Hepatic fibrosis indices
To measure serum values of liver enzymes, blood was taken from the retrobulbary venous complex of anaesthesized mice. Asparate aminotrans-ferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase (AP) values were determined with the VITROS Chemistry Products Calibrator Kit 3 and VITROS Chemistry Systems (Ortho-Clinical Diagnostics, Buckinghamshire, UK).
Hyaluronic acid assay
Concentration of hyaluronic acid in serum was determined with a hyaluronic acid (HA) quantitative test kit (Corgenix, CO, USA) at an absorbance of 450 nm using a Victor 1420 multilable counter spectrometer (Wallac, MA, USA).
Hydroxyproline measurement
Frozen liver tissue was hydrolyzed in 2.0 ml 6 N HCI for 3 hrs at 130°C. The solution was neutralized to pH 7.0 with 2.5 N NaOH. Two millilitres diluted solution (1:40 deionized H20) and 1.0 ml 0.05 M chloramine T solution were mixed and incubated for 20 min. at room temperature. One millilitre 3.15 M perchloric acid was added and the solution was incubated for 5 min. at room temperature. One millilitre 20% p-dimethylaminobenzaldehyde was added and the solution was incubated for 20 min. at 60°C. Absorbance was determined at 557 nm, and hydroxyproline was calculated by comparison to a standard curve as described [3].
Liver histology
Liver specimen were fixed in 10% formalin and embedded them in paraffin. Tissue sections (4 μm) were stained with haematoxylin and eosin for routine examination, or with picro-Sirius red for visualization of hepatic collagen deposition as described [4]. Histological grading (necrosis and inflammatory infiltration) was assessed by a pathologist blinded to the study conditions on at least four fragments from distinct areas of each liver. All samples were scored simultaneously. Blinded to the study conditions, we quantified fibrosis from Sirius red-stained liver tissue sections by morphometric analysis with LEICA QWIN software (Solms, Germany). Presented values were the mean of ten fields (100-fold magnification) taken from four to five liver sections per mouse.
Real time RT-PCR analysis
Total RNA was purified from liver tissue after homogenization in TRIZOL reagent (GIBCO BRL, Eggenstein, Germany; 1 ml/100 mg tissue) using an UltraTurrax (IKA, Staufen, Germany). Samples were normalized by total RNA measurements (260/280 nm) and RNA gel electrophoresis. Liver RNA samples from two or three animals were pooled and cDNA was prepared using the One step RT-PCR Kit from Qiagen (Hilden, Germany). Quantitative real time PCR for α2 type I procollagen was undertaken with SYBR-Green following the manufacturers protocol (Roche, Mannheim, Germany) as previously described [5]. For calibration, cDNA corresponding to 25, 50 or 100 ng total RNA of one sample was used. Subsequently sample cDNAs were analysed, each generated from 100 ng reverse transcribed total RNA from the pooled samples and related to the calibration curve. Linear calibration was performed during each experiment. Mouse procollagen type I primer sequence: 5’-ACCTGTGTGTTCCCTACTCA-3’ (sense) and 5’-GACTGTTGCCTTCGCCTCTG-3’ (antisense).
Immunoblot analysis
Briefly, hepatocytes were lysed in ice cold RIPA buffer (1x Tris-buffered saline, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) as described [6]. Protein concentration was measured with DC protein assay (Biorad, Msnchen, Germany). Thirty micrograms protein were separated by SDS-PAGE using NuPAGE Bis-Tris gels (Invitrogen, Karlsruhe, Germany) and transferred to nitrocellulose membranes. Immunoblotting proceeded as described [7], using the following antibodies: anti-β-actin, -α-smooth muscle actin (SMA) and -proliferating cell nuclear antigen (PCNA) (all from Sigma), -MADH7 (Abeam, Cambridge, UK), -phospho-Smad2, -cleaved cas-pase 3 and horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling, Danvers, USA), -collagen type I (Santa Cruz Biotechnology Santa Cruz, USA) and -CD43 (BD Pharmingen, San Diego, USA).
Immunohistochemistry
Immunohistochemical analyses were performed in liver tissue fixed in formalin and embedded in paraffin. Primary antibodies were used in the following dilutions: anti-α-SMA (1:300), -phospho-Smad2 (1:100), -cleaved cas-pase 3 (1:75) and PCNA (1:500). Endogenous peroxidase was blocked with peroxidase blocking reagent’ (DAKO, Hamburg, Germany) for 30 min. We processed the sections with standard HRP-conjugated antibodies. We showed peroxidase activity using metal-enhanced diaminobenzidine (DAB) substrate (DAKO). We observed no staining when primary antibody was omitted. Slides were counterstained with aqueous Mayers Hamalaun (Merck, Darmstadt, Germany). For quantification, we counted positive stained cells per observation field (10 fields per section, 200-fold magnification).
Isolation of primary hepatocytes and treatment
In vitro studies were performed with primary hepatocytes isolated from livers of male wild-type or S7ΔE1 CD-1 mice (30–40 g, 8–12 weeks of age) using collagenase perfusion as described previously [8]. Cells were plated on collagen-coated plates at a density of 4 × 104 cells/cm2 in Williams' medium E (10% FBS, 2 mM L-glutamine, 1% penicillin/streptomycin and 100 nM dexamethasone). After 4 hrs attachment the medium was changed to Williams medium E (2 mM L-glutamine and 1% penicillin/streptomycin). Different substances for treatment were added to serum-free culture medium for the indicated times and concentrations. Control conditions included cells maintained for the same period in serum-free medium supplemented with the solvent chemical.
The in vitro experiments were done in duplicates, using hepatocytes from two independent preparations. Cell lysates were collected from one well (Western blot analysis) and from two wells of a six-well plate for DNA laddering.
DNA laddering
Genomic DNA fragmentation was analysed as described [9]. Briefly, after 48 hrs treatment primary hepatocytes were collected in lysis buffer and incubated with proteinase K for 4 hrs at 37°C. Proteins were precipitated by adding 4 M NaCI at 4°C for 1 hr and centrifuged at 14.000 rpm for 60 min. at 4°C. DNA was isolated with phenol/cholophorm/isopropanol (Roth). For visualization 10 |xg of DNA was resolved in 1.5% agarose gels and stained with ethidium bromide.
Transient cell infections and reporter gene assays
Immediately after attachment, primary hepatocytes were seeded into 12 well plates and infected for 1 hr with 100 moi of the Smad2/3/4 reporter-adenovirus vector Ad(CAGA)9-MLP-Luc (AdCAGA) as previously described [8]. Cells were serum-starved overnight and stimulated with 1 ng/ml TGF-β for 16 hrs. Cell lysates and luciferase activity measurements were performed with the Steady-Glo Luciferase Assay System (Promega, Wl USA) and values were normalized to protein concentrations. Three wells were analysed per condition to generate the average value.
Measurement of lactate dehydrogenase release
Lactate dehydrogenase (LDH) activity was measured in culture super-natants and cell lysates (0.1% Triton-X-100 in medium) using the cytotoxicity detection kit (LDH; Roche) according to the manufacturers' instructions. The OD (λ= 492 nm/ref = 600 nm) was measured with a rainbow thermo ELISA reader (SLT Labinstruments, Auhof, Germany). Results are given as percentage of total LDH released. All experiments were performed in triplicates from three independent preparations of hepatocytes.
Measurement of reactive oxygen species (R0S), glutathione (GSH) and lipid peroxidation
After stimulation with CCI4 or TGF-β, cells were washed three times with HBSS (Hank's balanced salt solution) followed by incubation with 10 μM DCFH-DA (ROS) or 10 μM MCB (GSH) in medium for 30 min. at 37°C. The residual DCFH-DA and MCB was removed by washing three times with HBSS. Cells were lysed immediately with 0.1% Triton-X-100. Cell lysates were transferred to a white 96-well plate (Nunc, Wiesbaden, Germany) and the fluorescence was determined (ex/em = 485/527 or 355/460 nm: respectively) with a Fluoroskan Ascent fluorescence microplate reader (ThermoLabsystems, Engelsbach, Germany).
For measuring lipid peroxidation, cells were treated for 72 hrs. Cell lysates were collected in ice-cold HBSS supplemented with 1/50 volume of butylated alcohol (0.2% 2,6-di-tert-butyl-4-methylphenol in ethanol) by sonication. One volume TBA (5 mM in acetic acid) solution was added before boiling the reaction mixture for 60 min. Samples were chilled on ice and half volume methanol was added. Samples were centrifuged (13.000 x g, 5 min., RT) and the supernatant was transferred to a white 96-well plate to measure fluorescence (ex/em = 530/590 nm). All experiments were done in triplicates from three independent hepatocyte preparations.
Statistics
Results are expressed as mean ± standard error of the mean (S.E.M.) and were analysed either by Mann-Whitney test or by ANOVA followed by paired comparison, as appropriate. Statistical analysis of quantitative RT-PCR data was performed with Student's t-test for paired data. P< 0.05 was taken as the minimum level of significance.
Results
Lack of a functional Smad7 gene increases liver damage and fibrogenesis in a murine chronic liver disease model
CCI4 treatment of mice incites general hepatotoxicity and causes necroinflammatory liver injury followed by a chronic repair response. To get further insight into the physiological role of Smad7 during liver damage, we treated Smad7 deficient mice, carrying a targeted deletion of exonl corresponding to the N-terminal half of the Smad7 protein, for 8 weeks with CCI4. These mice are denoted S7ΔE1 mice. CCI4 intoxication leads to extensive formation of septa with some nodules in S7ΔE1 mice and the CD-1 control strain. The extent of liver injury after CCI4 treatment, assessed by the release of enzymes ALT, AST, HA and AP into serum and by histological analysis was more pronounced in S7ΔE1 mice compared to CD-1 wild-type mice (AST, 262.8 ± 16.6 versus 121.3 ± 16.8; ALT, 605.5 ± 22.3 versus 362.7 ± 15.4; HA, 562 ± 30.2 versus 355 ± 22.5; AP, 247.3 ± 9.8 versus 191.3 ± 13.3; Fig. 1a-d). Furthermore, liver hydroxyproline content (Fig. 2a), a2 type I procol-lagen mRNA levels (Fig. 2b) and collagen type I protein levels (Fig. 2c) were more elevated in S7ΔE1 mice than in CD-1 wild-type animals. In line with this, there was a difference in the amount of collagen fibres; CD-1 wild-type mice had about 2.5-fold less interstitial collagen deposition compared with S7ΔE1 mice after eight weeks of CCI4 application, as determined by Sirius red staining and morphometric quantification (Fig. 3a and b). Finally, the number of a-SMA positive cells was also higher in S7ΔE1 mice (Fig. 3c and d). Western blot analysis of liver lysates from these mice accordingly display the strongest α-SMA signal (Fig. 2c).
Fig 1.
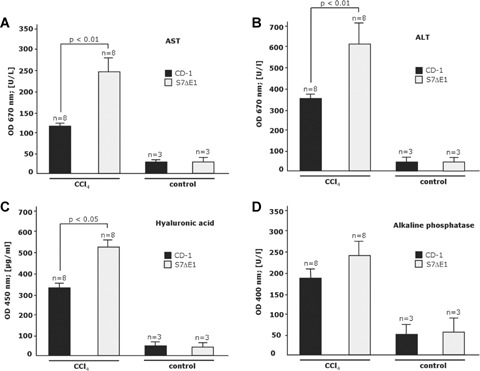
Loss of Smad7 function increases sensitivity for liver damage after CCI4 intoxication in mice. Evaluation of liver enzymes (a–c; VITROS Chemistry Products) and hyaluronic acid (d; Hyaluronic Acid Test Kit from Corgenix) in serum samples after 8 weeks CCI4 application; (a) asparate aminotransferase (AST); (b) alanine aminotransferase (ALT) and (c) alkaline phosphatase (AP). n, number of animals investigated in triplicate measurements.
Fig 2.
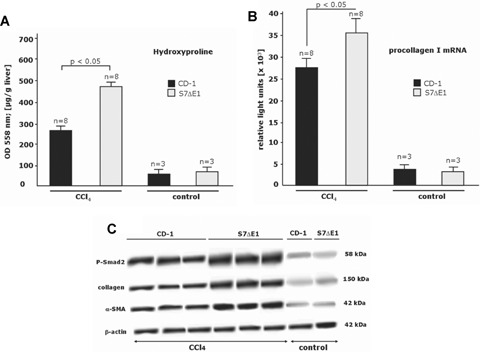
Loss of Smad7 function increases fibrogenesis after CCI4 intoxication in mice, (a) Spectrophotometrical analysis (Pharmacia Biotech, Ultraspec 2000; OD 558 nm) of hydroxyproline concentration, as a measure for the degree of fibrosis in liver tissue samples after 8 weeks of CCI4 application (Jamall et al., 1981; Boigk etal., 1997); (b) RT qRT-PCR for a2 type I procollagen mRNA was undertaken as described in materials and methods. Pooled samples from two different animals were analysed twice, giving similar results in both analyses. Oligonucleotide primers for mouse procollagen type I were as follows: 5’-ACCTGTGTGTTCCCTACTCA-3’ (sense) and 5’-GACTGTTGCCTTCGCCTCTG-3’ (antisense); n indicates the total number of animals investigated; c, Liver protein lysates (20 μg) were examined for Smad2 phosphorylation, α-SMA and type I collagen expression in Western blots as indicated. Representative photomicrographs of results obtained with eight S7ΔE1 and eight wild-type mice after 8 weeks of CCI4 intoxication are shown. Blots were reprobed with a monoclonal anti-β-actin antibody to confirm equal protein loading.
Fig 3.
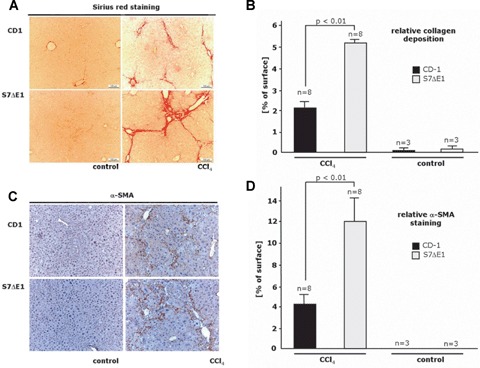
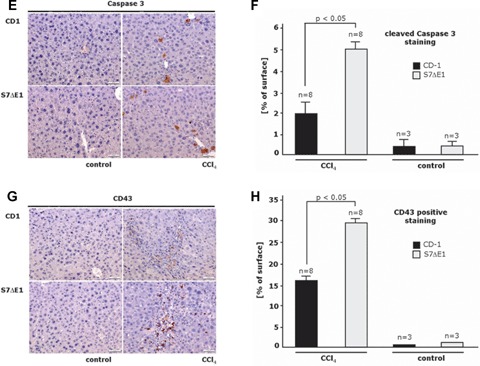
Loss of Smad7 protein function leads to enhanced inflammation and fibrogenesis as well as hepatocyte apopto-sis in a CCI4 model of liver damage in mice. Representative photomicrographs of liver sections from CD-1 and S7ΔE1 strains treated or not (control) for 8 weeks with CCI4: (a) Sirius red staining to semi-quantify collagen deposition; (c) α-smooth muscle actin immunostaining to detect activated HSCs; (e) immunostaining for cleaved caspase 3 to identify apoptotic cells; (g) immunostaining for CD43 to detect inflammatory cells; (b, d, f, h) Morphometric quantification of histochemical and immunohistochemical stagings. Ten fields were selected randomly from each section of different groups (8 animals/ group); for (b), Sirius red staining, LEICA QWIN software (Germany) was used. The graph shows the percentage of positive staining related to the total area investigated; for all other immunostainings (d, f, h), about 200 cells were evaluated per observation field, immunopositive cells were counted and data are expressed as percentage of total cell numbers investigated.
Damage-induced increased levels of active TGF-β in the liver are described to act on hepatocytes by inducing apoptosis [17]. Disrupting the Smad7 gene lead to a moderate, but significant increase of apoptotic cell number in the liver, as determined by IHC using an antibody to cleaved Caspase-3 (Fig. 3e and f).
Further, liver inflammation was analysed in CCI4 intoxicated wild-type and S7ΔE1 mice by identifying CD43-positive cells, indicating infiltrating mononuclear cells and lymphocytes. Lack of a functional Smad7 gene markedly increased the number of CD43-positive cells in the hepatic parenchyma, which were mainly observed in pericentral areas (30 ± 6 versus 14 ± 3 positive cells per field; Fig. 3g and h).
Increased sensitivity to TGF-β-induced apoptosis in S7ΔE1 hepatocytes
Given the higher ratio of apoptotic cells after CCI4-induced damage in the livers of S7ΔE1 mice compared to wild-type animals, we speculated if this was due to a higher sensitivity of hepatocytes to TGF-β. Primary hepatocytes from wild-type and S7ΔE1 mice were isolated and cultured onto collagen-coated plates. Phase contrast microscopy 24 hrs after plating (day 1) showed no differences in cell morphology from either background (Fig. 4a). A characteristic honeycomb shape and predominantly bi-nucleated cells were observed. With increasing culture time, hepatocytes lose these morphological features in a TGF-β-dependent process of epithelial to mesenchymal transition (EMT) [18]. These changes were observed in untreated cells from both genetic backgrounds, but were more pronounced in S7ΔE1 hepatocytes. Stimulation with 5 ng/ml TGF-β for 48 hrs resulted in enhanced dedifferentiation and apoptosis, as judged by cell shape and refringent apoptotic bodies. Again, both features were stronger in S7ΔE1 cells, suggesting an enhanced sensitivity to TGF-β (Fig. 4a).
Fig 4.

Smad7 mutation results in enhanced sensitivity to TGF-p-induced apoptosis in hepatocytes. (a) Phase contrast microscopy of cultured hepa-tocytes from wild-type and S7ΔE1 mice. Day 1 hepatocytes show typical honeycomb shape. Cultures on day 3 present morphological features of EMT and apoptosis, which are more prominent with S7ΔE1 genetic background (see inserts); (b) DNA fragmentation and (c) immunoblot of active cas-pase-3 and PARP (full length and cleaved) as markers of apoptosis. Both methods show increased apoptosis rates in S7ΔE1 compared to wild-type mice; (d) CD1 wild-type and S7ΔE1 hepatocytes were treated for 48 hrs with different concentrations of TGF-β as indicated. Cellular damage was measured by LDH determinations with the culture supernatant. The resulting EC50 value (GraphPad Prism, San Diego, USA) was significantly higher for CD1 wild-type hepatocytes, with P< 0.01 for treatment of cells with 1.25, 2.5 and 5 ng/ml TGF-β.
TGFβ-dependent DNA fragmentation results confirmed the microscopic features and display higher apoptosis rates in S7ΔE1 compared to wild-type cells (Fig. 4b). Western analyses were also in agreement with higher TGFβ-induced apoptosis in S7ΔE1 mice, showing clear poly-ADP-ribose-polymerase (PARP) degradation and stronger Caspase-3 activation in the Smad7 mutant background (Fig. 4c). Remarkably, untreated S7ΔE1 cells are to some extent spontaneously undergoing apoptosis, as indicated by detection of active Caspase-3 protein (Fig. 4c).
To further proof and compare cellular damage by TGF-β, we measured LDH secretion into the culture medium. After 24 hrs, no significant difference in LDH release between S7ΔE1 and wild-type hepatocytes was apparent (data not shown), whereas after 48 hrs damage in S7ΔE1 cells was evident (Fig. 4d). At this time-point, EC50 values of wild-type hepatocytes (∼2.583 ng/ml; R2= 0.9369) were approximately twofold increased compared to S7ΔE1 hepatocytes (∼1.249 ng/ml; R2= 0.9655); P < 0.01 for treatment of cells with 1.25, 2.5 and 5 ng/ml TGF-β. Control conditions and plateau values did not vary significantly between the two cell types.
Smacl7 deficiency does not result in enhanced sensitivity to chemical damage in hepatocytes
The in vivo data indicate that S7ΔE1 mice are more sensitive to CCI4-induced liver damage. To rule out an enhanced sensitivity of hepatocytes to direct chemical damage, we compared the sensitivity of cells from both genetic backgrounds to known hepato-toxic chemicals. Primary cultures were treated for 24 hrs with CCI4, acetaminophen, verapamil, digoxin, tetracycline and DMS0 at varying concentrations. EC50 values indicated no significant difference between S7ΔE1 and wild-type hepatocytes treated with these chemicals (Fig. 5a-f), supporting the notion that the differences in cellular damage by CCI4in vivo are mediated by TGF-β signalling.
Fig 5.
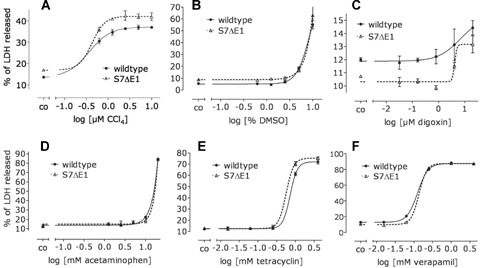
Increased sensitivity to cell damage of S7ΔE1 hepatocytes is not related to direct chemical effects. CD1 wild-type and S7ΔE1 hepatocytes were treated with five different concentrations of CCI4 (a), DMSO (b), digoxin (c), acetaminophen (d), tetracycline (e) and verapamil (f). After 24 h, LDH activity in culture supernatant and cell lysate was measured. The cellular damage is given as% of total LDH released into the culture supernatant. The resulting EC50 values did not vary significantly between the two backgrounds. CCI4 (wild-type: R2= 0.9771, EC50= 0.3700; S7ΔE1: R2= 0.9579, EC50= 0.4253), DMSO (wild-type: R2= 0.9823, EC50= 8.422; S7ΔE1: R2= 0.9213, EC50= 8.220), digoxin (wild-type: R2= 0.5253, EC50= nd; S7ΔE1: R2= 0.8020, EC50= nd), acetaminophen (wild-type: R2= 0.9974, EC50= 58.71; S7ΔE1: R2= 0.9865, EC50= 56.61), tetracycline (wild-type: R2= 0.9948, EC50= 0.7222; S7ΔE1: R2= 0.9970, EC50= 0.5667) and verapamil (wild-type: R2= 0.9993, EC50= 0.1247; S7ΔE1: R2= 0.9997, EC50= 0.1364). **P< 0.01 between wild-type and S7ΔE1 with the same treatment.
S7ΔE1 hepatocytes are more sensitive to TGF-β and CCI4-induced oxidative stress
TGF-β and CCI4 are known inducers of oxidative stress in hepatocytes. Primary hepatocytes were stimulated with TGF-β or CCI4 and as direct measures of oxidative stress, ROS production and GSH depletion were determined. TGF-β stimulation resulted in rapid and transient production of ROS in cells from both backgrounds that peaked between 4 and 8 hrs. However, in S7ΔE1 hepatocytes TGFβ-induced increase in ROS was faster and more prolonged (Fig. 6a). In line with this, cellular GSH showed a more rapid decrease in S7ΔE1 hepatocytes (Fig. 6b) and wild-type hepatocytes recovered much faster from TGFβ-dependent oxidative stress. After 24 hrs, wild-type cells almost reached basal levels of GSH and ROS, whereas S7ΔE1 cells were still stressed, even after 48 hrs.
Fig 6.
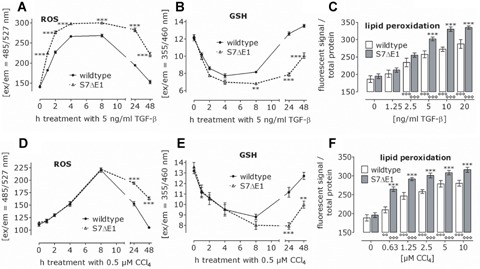
Loss of Smad7 function leads to increased oxidative stress by TGF-β and CCI4. CD1 wild-type and S7ΔE1 hepatocytes were treated with TGF-β (a–c) or CCI4 (d–f). During the first 48 hrs after treatment, levels of ROS (a, d) and cellular glutathione (b, e) were measured. After 72 hrs lipid peroxidation (c, f) was determined. **P< 0.01 and ***P< 0.001 when comparing wild-type and S7ΔE1 hepatocytes under the same conditions. 00P< 0.01 and 00°P< 0.001 when comparing the different treatments to the control condition in wild-type hepatocytes. ***p< 0.001 when comparing the different treatments to the negative control in S7ΔE1 hepatocytes.
After stimulation with 0.5 μM CCI4, no difference in oxidative stress generation was observed between S7ΔE1 and wild-type hepatocytes during the first 8 hrs, whereas after 24 hrs, an enhanced recovery rate was found in wild-type hepatocytes (Fig. 6c and d). Finally, lipid peroxidation increased upon TGF-β and CCI4 treatment in a dose-dependent manner and was higher in S7ΔE1 compared to wild-type hepatocytes (Fig. 6e and f).
It has previously been reported that CCI4 induces an increase in serum levels of TGFβ1[18]. This observation points to the possibility that CCI4incites damage due to modified TGF-β signalling instead of direct effects from the chemical itself.
To proof this assumption, we repeated the experiments with primary hepatocyte cultures in presence or absence of the ALK-5 inhibitor SB431542. Cells were stimulated for 16 hrs with TGF-β and CCI4. The ALK-5 inhibitor indeed antagonized ROS production, GSH decrease and lipid peroxidation to a similar extent in both groups (Fig. 7a-c). SB431542 addition obliviated the differences between S7ΔE1 and wild-type hepatocytes in the assays. We conclude that increased oxidative stress in S7ΔE1 hepatocytes compared to wild-type mice resulted in enhanced cellular damage as observed by LDH release after 48 and 72 hrs treatment with TGF-β and CCI4. Thereby, TGFβ-induced LDH release was comparable to the effect observed with CCI4, indicating direct participation of TGF-β signalling in CCI4-dependent liver damage.
Fig 7.
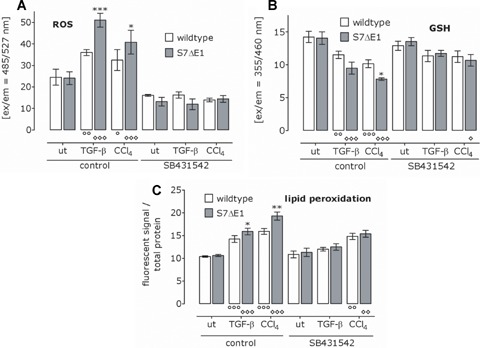
Blocking ALK-5 reduces TGF-β and CCI4-induced oxidative stress and abrogates the differences between CD1 wild-type and S7ΔE1 hepato-cytes. Where indicated, TGF-β/CCI4-treated hepatocytes obtained in addition SB431542. After 16 hrs ROS (a) and GSH (b) levels were determined. After 72 hrs lipid peroxidation was measured (c). *P< 0.05, **P< 0.01 and ***p< 0.001 when comparing wild-type and S7ΔE1 hepatocytes under the same conditions. °P< 0.05, 00P< 0.01 and 000P< 0.001 when comparing the different treatments to the negative control in wild-type hepatocytes. °P< 0.05, P< 0.01 and *p< 0.001 when comparing the different treatments to the control condition in S7ΔE1 hepatocytes.
Increased sensitivity to TGF-β-induced signal transduction in S7ΔE1 hepatocytes
Analysing Smad2 phosphorylation in wild-type and S7ΔE1 hepatocytes 48 hrs after stimulation with 5 ng/ml TGF-β by Western blot indicates that disruption of Smad7 exon 1 results in a higher level of phospho-Smad2 (Fig. 8a). Of note, the endogenous secreted TGF-β activity was biologically relevant, particularly for S7ΔE1 cells, because under basal conditions there was a clear P-Smad2 band in S7ΔE1 but not wild-type cells. To determine TGF-β-induced transcriptional activity, hepatocytes were infected with an adenoviral vector containing a reporter construct for Smad3/4 (AdCAGA) (Fig. 8b). TGF^-induced luciferase activity in S7ΔE1 cells almost fourfold more than in wild-type cells, indicating a stronger sensitivity to TGF-β due to the lack of an effective negative feedback regulation by the disrupted Smad7 gene. Under basal conditions, low luciferase activity was observed that, although minute compared to the levels after TGF-β stimulation, was higher in S7ΔE1 than wild-type cells.
Fig 8.
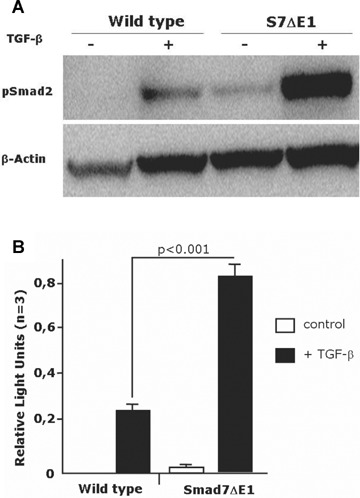
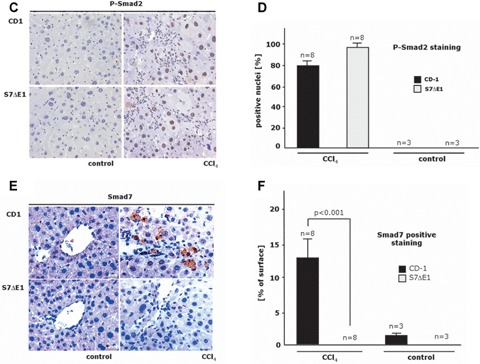
TGF^-induced signalling is enhanced in S7ΔE1 hepatocytes. (a) immunoblot analysis of Smad2 phosphorylation after 48 hrs of stimulation with 5 ng/ml TGF-β. A stronger band is observed in S7ΔE1 hepatocytes compared to wild-type cells. Notice that even under non-stimulated conditions S7ΔE1 cells yield a clear pSmad2 band that is not found with wild-type cells; (b) Smad3/4-luciferase reporter assays indicate that the stronger Smad phosphorylation observed in S7ΔE1 hepatocytes correlates with enhanced transcriptional activity of Smad complexes. Loss of Smad7 function leads to enhanced TGF-β signalling in a CCI4 model of liver damage (c-f). Representative photomicrographs of liver sections from CD-1 and S7ΔE1 strains treated or not (control) for 8 weeks with CCI4 are shown; (c) phospho-Smad2 immunostaining as a quantitative measurement for TGF-β signalling; (e) immunostaining for Smad7; (d, f) morphometric quantification of immunohistochemical stainings. Ten fields were selected randomly from each section of different groups (8 animals/group); about 200 cells were evaluated per observation field, immunopositive cells were counted and expressed as percentage of total cells investigated.
To analyse TGF-β signalling in vivo, we determined Smad2 C-terminal serine phosphorylation with immunohistochemistry. Application of CCI4 for 8 weeks strongly induced TGF-β signalling in liver cells of CD-1 wild-type mice. In line with the in vitro data, the number of phospho-Smad2 positive nuclei was further increased in S7ΔE1 mice (Fig. 8c and d). Similarly, Western blot results with whole liver protein lysates of the same animals display a stronger P-Smad2 band in S7ΔE1 mice (Fig. 2c). As expected, S7ΔE1 mice did not show positive staining for Smad7 in CCI4-treated liver samples. The wild-type mice instead, display a significant number of Smad7 immunopositive cells, indicating physiological presence of negative Smad7 feedback regulation for TGF-β signalling during liver damage in vivo (Fig. 8e and f; supplement Figs 1 and 2). We conclude from these data that the increased sensitivity of S7ΔE1 mice to CCI4-dependent damage is due to enhanced TGF-β signalling.
Discussion
Production of active TGF-β and subsequent signal transduction are strongly induced upon liver damage. Research from the last decade dedicated TGF-β as master pro-fibrogenic cytokine in chronic liver disease and various approaches have been used to block TGF-β effects in animal models, which were successful in abrogating progression of liver fibrosis from different etiologies [19–22]. TGF-β signalling is negatively regulated by Smad7 in most cell types. In the present report we investigated the impact of disrupting this negative control mechanism on developing CCI4-induced liver disease in mice.
After damage with CCI4 the lack of a functional Smad7 gene and protein lead to enhanced TGF-β signalling in different types of liver cells, including HSCs and hepatocytes. Further, we found that liver damage is enhanced in S7ΔE1 mice, as indicated by increased levels of liver enzymes in serum samples and our results implicate participation of TGF-β signalling/Smad7 action in CCI4-dependent hepatocyte damage.
The role of TGF-β signalling in HSCs has been extensively studied [23] and it was found that negative feedback regulation via Smad7 only occurs in quiescent cells, whereas such a regulatory loop is absent in activated HSCs [15]. In line with this, during acute liver injury, profibrogenic TGF-β signalling in HSCs was terminated by Smad7, thus providing a transient response to TGF-β, whereas Smad7 was not induced in MFBs in vivo throughout chronic liver injury, thus allowing constitutive R-Smad activation and subsequent progression of liver fibrosis [16]. On the other hand, Smad7 expression is physiologically induced and was continuously increasing with duration of cholestasis during liver damage in bile duct ligated rats, when investigating whole liver lysates at RNA and protein level [24]. These conflicting results indicate that beside the general picture about Smad7 expression and negative regulation of TGF-β signalling cell type specific differences need to be taken into account.
In the diverse fibroproliferative diseases, TGF-β is mainly profi-brogenic, whereas its anti-inflammatory role seems to be of minor relevance. However, in chronic inflammatory bowel disease (IBD), although active TGF-β is present, an anti-inflammatory role in the mucosa of inflamed regions and isolated mucosal T cells is abrogated by pathophysiological Smad7 over-expression [25]. An anti-sense approach targeting Smad7 mRNA restored TGF-β signalling in inflammatory cells and released the inhibitory effects of pro-inflammatory cytokine synthesis, e.g. IFN-7 or tumour necrosis factor-α (TNF-β). This indicates that Smad7 blunts TGF-β signalling in chronic bowel disease and thus helps to maintain chronic production of pro-inflammatory cytokines in the presence of TGF-β[26]. In a chronic liver disease mouse model, we found increased inflammation in Smad7-deficient animals after CCI4 intoxication, suggesting that the mechanism occurring in chronic IBD is not relevant in chronic liver disease.
Because the number of α-SMA-positive cells, collagen deposition/expression and hydroxyproline content are increased in S7ΔE1 compared to wild-type mice after CCI4 treatment, we conclude that lack of a functional Smad7 gene leads to enhanced TGF-β activation of HSCs.
We have previously shown [27, 28] that beside TGF-β, IFN-7, TNF-α and EGF have been described as potential inducer of Smad7 expression. All of these are up-regulated during the course of liver fibrogenesis and may represent potential mediators of Smad7 dependent negative control of profibrogenic TGF-β signalling, which is now lacking in S7ΔE1 mice.
Given the observed excess in liver damage following CCI4 application and up-regulated apoptosis rate in S7ΔE1 mice, we speculated if this was a consequence of a higher sensitivity of hepatocytes to TGF-βper se, due to the lack of a fully functional Smad7 gene.
We therefore determined if primary hepatocytes from S7ΔE1 mice are more sensitive to TGF-βin vitro. Our studies showed that hepatocytes from wild-type and Smad7 mutant genetic background did not differ in their morphology during the first hours after isolation. However, the typical progressive dedifferentiation process observed in this culture system was more pronounced in cells derived from the S7ΔE1 background. We recently elucidated these changes and characterized them as EMT, which is in part stimulated by autocrine TGF-β secretion from hepatocytes. It is therefore reasonable to expect that a lack of a negative feedback loop by Smad7 deficiency would result in enhanced features induced by TGF-β in culture. In line with this, deletion of exon 1 in Smad7 resulted in enhanced TGF-β signalling in primary hepatocytes. Both, Smad2 phosphorylation and a transcriptional reporter assay for Smad3/4, revealed that initial signal transduction events and transcriptional regulation by Smad complexes are increased in S7ΔE1 hepatocytes.
Controversial data exist about the role of Smad7 in TGF-β-induced apoptosis. Several studies have shown that Smad7 can inhibit TGF-β-induced apoptosis due to its well know role as inhibitor of the Smad pathway. For example, Smad7 is induced by CD40 engagement in B-lymphocytes, resulting in inhibition of TGF-β-dependent growth arrest and apoptosis [29]. Similarly, Smad7 impaired TGFβ-induced apoptosis in Hep3B cells [30]. Conversely, nuclear translocation of the cell survival factor NF-?B is inhibited by Smad7 in podocytes, thereby sensitizing cells to the pro-apoptotic effect of TGF-β, which occurs in part via p38 [31]. Similarly, in prostate carcinoma cells, Smad7 is required for TGF-β-induced apoptosis, acting as stabilizer of a MAPK complex that leads to activation of p38 [32].
In hepatocytes and in the non-differentiated hepatoma cell line AML12, p38 plays also a crucial role in apoptosis. However in this cell-type it was shown that p38 activation relies on GADD45β expression, which is directly induced by Smad3 33]. Recent studies have highlighted the relevance of the Bel family members Bcl-2 and Bim as targets of TGF-β, mediating its pro-apoptotic effects in hepatocytes. The anti-apoptotic member Bcl-2 is down-regulated by TGF-βvia Smad3 [34] leading to the sen-sitization of hepatocytes to pro-apoptotic stimuli. On the other hand, expression of the pro-apoptotic members Bim and Bmf is induced by TGF-β[35]. These effects are dependent on Smad and p38 pathways, but also on ROS generation. We have shown that TGF-β down-regulates the expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL in hepatocytes. In this context, it is therefore reasonable to expect that the lack of a fully functional inhibitory feedback loop would result in increased levels of GADD45β and more vigorous down-regulation of Bcl-2, and possibly a more pronounced increase in Bim or Bmf. Deletion of Exon 1 in the Smad7 gene resulted in increased apoptosis rates, which is demonstrated by cell morphology and molecular markers of apoptosis, such as PARP degradation, Caspase-3 activation and DNA fragmentation. Given that transcriptional responses are enhanced in S7ΔE1 hepatocytes, it is likely that the regulation of the pro- and anti-apoptotic components by TGF-β are stronger in S7ΔE1 hepatocytes due to enhanced signalling. This indicates that Smad7 rather represents an inhibitor than an inducer for TGF^-mediated apoptosis in hepatocytes. This is further supported from studies with B cells from these mutant mice, displaying increased Smad2 phosphorylation and enhanced spontaneous apoptosis [1].
S7ΔE1 hepatocytes are highly sensitive to oxidative stress. Upon addition of TGF-β, the increase in ROS generation and decrease in GSH is pronounced in S7ΔE1 hepatocytes compared to wild-type background. Furthermore, the CD1 wild-type hepatocytes ‘recover’ faster from oxidative stress than S7ΔE1 hepatocytes. Consequently, lipid peroxidation levels were also higher in S7ΔE1 hepatocytes. Two different mechanisms may exert TGF-β mediated oxidative stress [36], that is induction of the NADPH oxidase-like system, accumulating ROS in the mitochondria and down-regulation of antioxidant genes, increasing ROS in the cytoplasm. Thus, lack of negative feedback regulation of TGF-β signalling in S7ΔE1 hepatocytes might result in continuous ROS production by up-regulated NADPH oxidase, the major source of superoxide 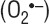 production, which cannot be buffered by the cells due to the lack of antioxidant genes, e.g. GSH-S-transferase, catalase and superoxide dismutase [37], decelerating the recovery of the cell. Thus lack of functional Smad7 leads to ROS accumulation, mitochondrial damage and Cytochrome c release into the cytosol. This concomitantly induces translocation of truncated Bid (tBid) and Bax into the mitochondria [38], triggering apoptosis by activation of execution Caspases 3 and 7. Antioxidants ascorbic acid and superoxide dismutase are able to prevent activation of initiator Caspase 8 and Bid cleavage, thus playing an important role in reduction of apoptosis [39]. Thus, enhanced TGF-β action might reduce these antioxidants, leading to increased levels of ROS and subsequently enhanced apoptosis and lipid peroxidation in hepatocytes.
production, which cannot be buffered by the cells due to the lack of antioxidant genes, e.g. GSH-S-transferase, catalase and superoxide dismutase [37], decelerating the recovery of the cell. Thus lack of functional Smad7 leads to ROS accumulation, mitochondrial damage and Cytochrome c release into the cytosol. This concomitantly induces translocation of truncated Bid (tBid) and Bax into the mitochondria [38], triggering apoptosis by activation of execution Caspases 3 and 7. Antioxidants ascorbic acid and superoxide dismutase are able to prevent activation of initiator Caspase 8 and Bid cleavage, thus playing an important role in reduction of apoptosis [39]. Thus, enhanced TGF-β action might reduce these antioxidants, leading to increased levels of ROS and subsequently enhanced apoptosis and lipid peroxidation in hepatocytes.
Hepatotoxic damages by CCI4 are mostly attributed to its toxic metabolite, trichloromethyl radical (CCI3·), formed by action of various cytochrome P450 isoforms, especially by CYP2E1 [40]. To compare direct chemical effects on S7ΔE1 versus wild-type hepatocytes in vitro, other chemicals were chosen in addition, whose hepatotoxic effects also depend on activity of Cytochrome P450 isoforms. The EC50 values determined by LDH release after 24 hrs treatment with CCI4, acetaminophen, DMSO, digoxin, tetracycline and verapamil did not vary significantly among the different mouse strains, suggesting that the higher sensitivity to CCI4-dependent cellular damage, seen after 48 hrs treatment in S7ΔE1 mice is not due to the pure chemical effect. In line with this, CCI4 treatment does not show differences in oxidative stress measurements in the first 8 hrs after addition. Longer treatment however, increased ROS levels and reduced GSH levels, and this was stronger in S7ΔE1 hepatocytes compared to CD1 wild-type, suggesting a participation of Smad7 in the recovery of the cells from oxidative stress induced by CCI4. The difference became more pronounced after 72 hrs when lipid peroxidation was measured.
Date et al. showed that CCI4 treatment in rats resulted in increased TGF-β serum levels [18]. Thus, we hypothesize that the later-occurring differences in oxidative stress are due to up-coming TGF-β signalling and lack of negative feedback regulation in S7ΔE1 hepatocytes.
This hypothesis, that ROS production after CCI4 is linked to TGF-β/Smad2/Smad3 signalling, was proven with a specific ALK5 inhibitor SB-431542 [41]. CCI4-dependent oxidative stress is reduced in both CD1 wild-type and S7ΔE1 hepatocytes in the presence of the ALK-5 inhibitor (SB431542). Furthermore, the ALK-5 inhibitor was able to diminish the differences among the two backgrounds, which strengthens the idea that active TGF-β signalling is involved in the increased sensitivity of S7ΔE1 hepatocytes to CCI4-dependent cellular damage.
In summary, we present further evidence that Smad7 is critically involved in establishing and progression of chronic liver disease. Smad7 is efficient in blunting TGF-β signalling in the different cell types of the liver and thus inhibits profibrogenic effects. In contrast to quiescent HSCs, activated myofibroblasts lack TGF-β-dependent induction of Smad7 in vitro and in vivo, and thus may contribute to chronic TGF-β signalling and fibrogenesis [5–42]. We now show that mice lacking a functional Smad7 gene display a higher sensitivity for damage and more severely progress to chronic liver disease. The hepatocyte damaging effect of CCI4 is provided the TGFβ/Smad pathway inducing oxidative stress, lipid peroxidation and apoptosis and is under negative control via Smad7. Lack of a functional Smad7 gene results in prolonged and enhanced TGF-β signalling and thus more severe damage. We conclude that Smad7 is physiologically induced in hepatocytes during liver damage and represents an important control for the strength of TGF-β effects. As such, it can be envisaged as an important component of an antifibrotic therapy approach. In long-term studies (about 12 months), we will now investigate if Smad7 deficiency may spontaneously lead to liver damage without an additional toxic challenge.
Acknowledgments
The study was financially supported by the Deutsche Forschungsgemeinschaft, DFG grant DO 373/6–1, SFB542 project C4, the Dietmar Hopp Foundation, Walldorf, Germany, the Stiftung Pathobiochemie, and the BMBF program HepatoSys. We appreciate the excellent technical assistance from A. M Jler. Our thanks also go to JDDW and APDW, who enabled the presentation of our results at the 2nd International Symposium on Alcoholic Liver and Pancreatic Diseases and Cirrhosis at Kobe, October 2007.
References
- 1.Li R, Rosendahl A, Brodin G, Cheng AM, Ahgren A, Sundquist C, Kulkarni S, Pawson T, Heldin CH, Heuchel RL. Deletion of exon I of SMAD7 in mice results in altered B cell responses. J Immunol. 2006;176:6777–84. doi: 10.4049/jimmunol.176.11.6777. [DOI] [PubMed] [Google Scholar]
- 2.Zilian O, Saner C, Hagedorn L, Lee HY, Sauberli E, Suter U, Sommer L, Aguet M. Multiple roles of mouse Numb in tuning developmental cell fates. Curr Biol. 2001;11:494–501. doi: 10.1016/s0960-9822(01)00149-x. [DOI] [PubMed] [Google Scholar]
- 3.Jamall IS, Finelli VN, Que Hee SS. A simple method to determine nanogram levels of 4-hydroxyproline in biological tissues. Anall. Bioch. 1981;112:70–75. doi: 10.1016/0003-2697(81)90261-x. [DOI] [PubMed] [Google Scholar]
- 4.Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, Ten Dijke P, Gressner AM. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–91. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- 5.Dooley S, Delvoux B, Lahme B, Mangasser-Stephan K, Gressner AM. Modulation of transforming growth factor beta response and signaling during transd-ifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology. 2000;31:1094–106. doi: 10.1053/he.2000.6126. [DOI] [PubMed] [Google Scholar]
- 6.Wiercinska E, Wlckert L, Denecke B, Said HM, Hamzavi J, Gressner AM, Thorlkay M, ten Dijke P, Mertens PR, Breitkopf K, Dooley S. Id1 is a critical mediator in TGF-beta-induced transdiffer-entiation of rat hepatic stellate cells. Hepatology. 2006;43:1032–41. doi: 10.1002/hep.21135. [DOI] [PubMed] [Google Scholar]
- 7.Weng HL, Ciuclan L, Liu Y, Hamzavi J, Gotloy P, Gaitantzi H, Kanzler S, Heuchel R, Ueberham U, Gebhardt R, Breitkopf K, Dooley S. Profibrogenic transforming growth factor-beta/activin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology. 2007;46:1257–70. doi: 10.1002/hep.21806. [DOI] [PubMed] [Google Scholar]
- 8.Klingmuller U, Bauer A, Bohl S, Nickel PJ, Breitkopf K, Dooley S, Zellmer S, Kern C, Merfort I, Sparna T, Donauer J, Walz G, Geyer M, Kreutz C, Hermes M, Gotschel F, Hecht A, Walter D, Egger L, Neubert K, Borner C, Brulport M, Schormann W, Sauer C, Baumann F, Preiss R, MacNelly S, Godoy P, Wiercinska E, Ciuclan L, Edelmann J, Zeilinger K, Heinrich M, Zanger UM, Gebhardt R, Maiwald T, Heinrich R, Timmer J, von Weizsacker F, Hengstler JG. Primary mouse hepatocytes for systems biology approaches: a standardized in vitro system for modelling of signal transduction pathways. Syst. Biol. 2006;153:433–47. doi: 10.1049/ip-syb:20050067. [DOI] [PubMed] [Google Scholar]
- 9.Ju W, Ogawa A, Heyer J, Nierhof D, Yu L, Kucherlapati R, Shafritz DA, Bottinger EP. Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol. 2006;26:654–67. doi: 10.1128/MCB.26.2.654-667.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 11.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Bataller R, Brenner DA. Liver fibrosis. JCI. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breitkopf K, Haas S, Wiercinska E, Singer MV, Dooley S. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res. 2005;29:121S–131S. doi: 10.1097/01.alc.0000189284.98684.22. [DOI] [PubMed] [Google Scholar]
- 14.Stopa M, Anhuf D, Terstegen L, Gatsios P, Gressner AM, Dooley S. Participation of Smad2, Smad3, and Smad4 in transforming growth factor beta (TGF-beta)-induced activation of Smad7. The TGF-beta response element of the promoter requires functional Smad binding element and E-box sequences for transcriptional regulation. J Biol Chem. 2000;275:29308–17. doi: 10.1074/jbc.M003282200. [DOI] [PubMed] [Google Scholar]
- 15.Dooley S, Streckert M, Delvoux B, Gressner AM. Expression of Smads during in vitro transdifferentiation of hepatic stellate cells to myofibroblasts. BBRC. 2001;283:554–62. doi: 10.1006/bbrc.2001.4811. [DOI] [PubMed] [Google Scholar]
- 16.Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology. 2002;35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- 17.Ueberham E, Low R, Ueberham U, Schonig K, Bujard H, Gebhardt R. Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibro-sis. Hepatology. 2003;37:1067–78. doi: 10.1053/jhep.2003.50196. [DOI] [PubMed] [Google Scholar]
- 18.Date M, Matsuzaki K, Matsushita M, Tahashi Y, Furukawa F, Inoue K. Modulation of transforming growth factor beta function in hepatocytes and hepatic stellate cells in rat liver injury. Gut. 2000;46:719–24. doi: 10.1136/gut.46.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. Faseb J. 2004;18:816–27. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 20.de Gouville AC, Huet S. Inhibition of ALK5 as a new approach to treat liver fibrotic diseases. Drug news perspect. 2006;19:85–90. doi: 10.1358/dnp.2006.19.2.977444. [DOI] [PubMed] [Google Scholar]
- 21.Kim KH, Kim HC, Hwang MY, Oh HK, Lee TS, Chang YC, Song HJ, Won NH, Park KK. The antifibrotic effect of TGF-beta1 siRNAs in murine model of liver cirrhosis. BBRC. 2006;343:1072–78. doi: 10.1016/j.bbrc.2006.03.087. [DOI] [PubMed] [Google Scholar]
- 22.George J, Roulot D, Koteliansky VE, Bissell DM. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. PNAS. 1999;96:12719–24. doi: 10.1073/pnas.96.22.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seyhan H, Hamzavi J, Wiercinska E, Gressner AM, Mertens PR, Kopp J, Horch RE, Breitkopf K, Dooley S. Liver fibrogen-esis due to cholestasis is associated with increased Smad7 expression and Smad3 signaling. J Cell Mol Med. 2006;10:922–32. doi: 10.1111/j.1582-4934.2006.tb00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakao A, Okumura K, Ogawa H. Smad7: a new key player in TGF-beta-associated disease. Trends in Mol Med. 2002;8:361–63. doi: 10.1016/s1471-4914(02)02376-6. [DOI] [PubMed] [Google Scholar]
- 26.Boirivant M, Pallone F, Di Giacinto C, Fina D, Monteleone I, Marinaro M, Caruso R, Colantoni A, Palmieri G, Sanchez M, Strober W, MacDonald TT, Monteleone G. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-betal-mediated suppression of colitis. Gastroenterology. 2006;131:1786–98. doi: 10.1053/j.gastro.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 27.Stopa M, Benes V, Ansorge W, Gressner AM, Dooley S. Genomic locus and promoter region of rat Smad7, an important antagonist of TGFbeta signaling. Mamm Genome. 2000;11:169–76. doi: 10.1007/s003350010032. [DOI] [PubMed] [Google Scholar]
- 28.Weng H, Mertens PR, Gressner AM, Dooley S. IFN-gamma abrogates profibro-genic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J Hepatol. 2007;46:295–303. doi: 10.1016/j.jhep.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Patil S, Wildey GM, Brown TL, Choy L, Derynck R, Howe PH. Smad7 is induced by CD40 and protects WEHI 231 B-lym-phocytes from transforming growth factor-beta -induced growth inhibition and apop-tosis. J Biol Chem. 2000;275:38363–70. doi: 10.1074/jbc.M004861200. [DOI] [PubMed] [Google Scholar]
- 30.Yamamura Y, Hua X, Bergelson S, Lodish HF. Critical role of Smads and AP-1 complex in transforming growth factor-beta -dependent apoptosis. J Biol Chem. 2000;275:36295–302. doi: 10.1074/jbc.M006023200. [DOI] [PubMed] [Google Scholar]
- 31.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Bottinger EP. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–16. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edlund S, Bu S, Schuster N, Aspenstrom P, Heuchel R, Heldin NE, ten Dijke P, Heldin CH, Landstrom M. Transforming growth factor-betal (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol Biol Cell. 2003;14:529–44. doi: 10.1091/mbc.02-03-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ., Jr Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278:43001–07. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 34.Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell. 2006;9:445–57. doi: 10.1016/j.ccr.2006.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramjaun AR, Tomlinson S, Eddaoudi A, Downward J. Upregulation of two BH3-only proteins, Bmf and Bim, during TGF beta-induced apoptosis. Oncogene. 2007;26:970–81. doi: 10.1038/sj.onc.1209852. [DOI] [PubMed] [Google Scholar]
- 36.Herrera B, Murillo MM, Alvarez-Barrientos A, Beltran J, Fernandez M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic Biol Med. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 37.Kayanoki Y, Fujii J, Suzuki K, Kawata S, Matsuzawa Y, Taniguchi N. Suppression of antioxidative enzyme expression by transforming growth factor-beta 1 in rat hepatocytes. J. Biol. Chem. 1994;269:15488–92. [PubMed] [Google Scholar]
- 38.El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, Lee PC, Hinton RH, Price SC, Kass GE. Involvement of mitochondria in acetamino-phen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol Appl Pharmacol. 2003;191:118–29. doi: 10.1016/s0041-008x(03)00240-0. [DOI] [PubMed] [Google Scholar]
- 39.Porras A, Zuluaga S, Valladares A, Alvarez AM, Herrera B, Fabregat I, Benito M. Long-term treatment with insulin induces apoptosis in brown adipocytes: role of oxidative stress. Endocrinology. 2003;144:5390–401. doi: 10.1210/en.2003-0622. [DOI] [PubMed] [Google Scholar]
- 40.Castillo T, Koop DR, Kamimura S, Triadafilopoulos G, Tsukamoto H. Role of cytochrome P-450 2E1 in ethanol-, carbon tetrachloride- and iron-dependent micro-somal lipid peroxidation. Hepatology. 1992;16:992–6. doi: 10.1002/hep.1840160423. [DOI] [PubMed] [Google Scholar]
- 41.Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C, Martin W, Fornwald J, Lehr R, Harling J, Gaster L, Callahan JF, Olson BA. Inhibition of transforming growth factor (TGF)-betal-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Molec Pharmac. 2002;62:58–64. doi: 10.1124/mol.62.1.58. [DOI] [PubMed] [Google Scholar]
- 42.Dooley S, Delvoux B, Streckert M, Bonzel L, Stopa M, ten Dijke P, Gressner AM. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFbeta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001;502:4–10. doi: 10.1016/s0014-5793(01)02656-4. [DOI] [PubMed] [Google Scholar]