

Abstract
Key points
CaV2.1 channels constitute a dominant Ca2+ entry pathway into brain neurons, triggering downstream Ca2+-dependent processes such as neurotransmitter release.
CaV2.1 is itself modulated by Ca2+, resulting in activity-dependent enhancement of channel opening termed Ca2+-dependent facilitation (CDF).
Real-time Ca2+ imaging and Ca2+ uncaging here reveal that CDF turns out to be strikingly faster, more Ca2+ sensitive, and larger than anticipated on previous grounds.
Robust resolution of the quantitative profile of CDF enables deduction of a realistic biophysical model for this process.
These results suggest that CaV2.1 CDF would figure most prominently in short-term synaptic plasticity and cerebellar Purkinje cell rhythmicity.
Abstract
CaV2.1 (P-type) voltage-gated Ca2+ channels constitute a major source of neuronal Ca2+ current, strongly influencing rhythmicity and triggering neurotransmitter release throughout the central nervous system. Fitting with such stature among Ca2+ entry pathways, CaV2.1 is itself feedback regulated by intracellular Ca2+, acting through calmodulin to facilitate channel opening. The precise neurophysiological role of this calcium-dependent facilitation (CDF) remains uncertain, however, in large measure because the very magnitude, Ca2+ dependence and kinetics of CDF have resisted quantification by conventional means. Here, we utilize the photo-uncaging of Ca2+ with CaV2.1 channels fluxing Li+ currents, so that voltage-dependent activation of channel gating is no longer conflated with Ca2+ entry, and CDF is then driven solely by light-induced increases in Ca2+. By using this strategy, we now find that CDF can be unexpectedly large, enhancing currents by as much as twofold at physiological voltages. CDF is steeply Ca2+ dependent, with a Hill coefficient of approximately two, a half-maximal effect reached by nearly 500 nm Ca2+, and Ca2+ on/off kinetics in the order of milliseconds to tens of milliseconds. These properties were established for both native P-type currents in cerebellar Purkinje neurons, as well as their recombinant channel counterparts under heterologous expression. Such features suggest that CDF of CaV2.1 channels may substantially enhance the regularity of rhythmic firing in cerebellar Purkinje neurons, where regularity is believed crucial for motor coordination. In addition, this degree of extensive CDF would be poised to exert large order-of-magnitude effects on short-term synaptic plasticity via rapid modulation of presynaptic Ca2+ entry.
Introduction
CaV2.1 channels are perhaps the most abundant voltage-gated Ca2+ channel in the mammalian brain (Mori et al. 1991), furnishing a dominant Ca2+ entry pathway into presynaptic release terminals (Hillman et al. 1991; Dunlap et al. 1995; Westenbroek et al. 1995; Craig et al. 1998), as well as into somatic and postsynaptic compartments (Regan, 1991; Usowicz et al. 1992; Indriati et al. 2013). These channels thereby constitute the pre-eminent trigger of neurotransmitter release in the CNS (Uchitel et al. 1992), initiate Ca2+-dependent forms of synaptic and developmental plasticity (Zucker & Regehr, 2002; Finch et al. 2012; Lamont & Weber, 2012; Miyazaki et al. 2012; Wheeler et al. 2012), and support and modulate dendritic Ca2+ spiking important for motor coordination (Womack & Khodakhah, 2002, 2004). In all, dynamic regulation of CaV2.1 activity is probably a significant contributor to the complexity and richness of neurophysiology and neurocomputation.
Early studies in squid reported that the amplitude of voltage-gated Ca2+ currents changed little with altered stimulation patterns, suggesting that other Ca2+-dependent processes were responsible for differing forms of rapid Ca2+-dependent plasticity (Charlton et al. 1982). For example, paired-pulse facilitation of synaptic efficacy has been attributed to the accumulation of residual Ca2+ that primes Ca2+ triggering sites for vesicle release, and short-term synaptic depression has been explained in terms of vesicle depletion (Regehr, 2012). These mechanisms have attracted considerable interest because short-term synaptic plasticity is considered to be essential for the neurocomputational repertoire of the brain (Abbott & Regehr, 2004). That said, if the amplitude of CaV2.1 currents were variably modulated by recent activity, an additional and powerful class of plasticity mechanisms would arise.
More recently, rapid voltage-dependent inactivation (VDI) has been identified in CaV2.1 channels paired with certain auxiliary β subunits (De Waard & Campbell, 1995). In certain contexts where such subunits predominate, it has been argued that such VDI could underlie forms of short-term synaptic depression (Patil et al. 1998). Apart from VDI, Ca2+–calmodulin (Ca2+–CaM) regulation of CaV2.1 channels has emerged as another potential mechanism supporting activity-dependent plasticity (Lee et al. 2000; DeMaria et al. 2001). Intriguingly, CaM regulation of CaV2.1 exhibits a bipartite format relating to the lobes of an indwelling CaM bound to the intracellular carboxy tail of channels (DeMaria et al. 2001; Erickson et al. 2001). Ca2+ binding to the C-terminal lobe of CaM induces CDF, whereas Ca2+ binding to the N-terminal lobe of CaM induces a Ca2+-dependent inactivation (CDI) (DeMaria et al. 2001). Indeed, there have been hints that such forms of regulation are at play within neuronal preparations (Borst & Sakmann, 1998; Xu et al. 2007; Mochida et al. 2008; Adams et al. 2010; Catterall et al. 2013), and there have been significant efforts to clarify the structure–function underpinnings of such channel regulation (DeMaria et al. 2001; Lee et al. 2003; Kim et al. 2008; Mori et al. 2008).
Nonetheless, progress has been stymied by the inability to quantify CDF and CDI in more than a cursory qualitative manner. Even the coarse Ca2+ sensitivity and magnitude of these forms of regulation remain obscure, complicating efforts to ascribe physiological relevance and decipher structure–function mechanisms. These limitations are particularly acute for CDI of CaV2.1 channels, which can only be robustly induced when large Ca2+ currents are present with minimal levels of Ca2+ buffering (Soong et al. 2002). Thus, the levels of Ca2+ required for CDI may exceed physiological bounds.
To overcome these limitations, we scrutinized the obstacles to determining the Ca2+ sensitivity of CaV2.1 regulation as illustrated in Fig.1A. One typically quantifies channel regulation by measuring whole-cell Ca2+ currents (I), whose magnitude is specified by channel activation gating (top left box) that is modulated by a CaM regulatory system (top right box in red). However, the Ca2+ concentration at the inner mouth of the channel, labelled as nanodomain Ca2+ in Fig.1B, constitutes the actual input to the CaM regulatory module, and this nanodomain Ca2+ is difficult to measure and control. The challenges arise because nanodomain Ca2+ results from the superposition of local Ca2+ fluxing through a ‘home’ channel (Fig.1B, red arrow) and global Ca2+ sources comprising other Ca2+ channels, transport pathways and internal stores. The local Ca2+ input is complex, reflecting stochastic channel gating, as well as local diffusion and Ca2+ buffering near the channel mouth (Fig.1A, ‘local’ pathway). The global Ca2+ input derives from the activity of other Ca2+ channels and sources at a distance, as shaped by cellular Ca2+ diffusion, geometry and buffering (Fig.1A, ‘global’ pathway). There are numerous unknowns among these multiple determinants of local and global Ca2+ inputs. Moreover, the relevant nanodomain Ca2+ concentration cannot be measured in most instances, because commonly used Ca2+ fluorescent dyes only furnish readouts of spatially averaged global Ca2+ (Fig.1B, light-shaded region), and more intricate near-field measurements are currently impractical for the present question (Tay et al. 2012). For CDF, further challenges arise because channel facilitation transpires rapidly over a time scale similar to the growing opening of channels following the onset of a voltage step (Fig.1A, ‘activation gating’). Hence, it is difficult to disambiguate the contributions of CaM-mediated CDF and voltage activation. As a result, previous attempts to quantify CDF have done so indirectly, either by estimating the degree to which channels can be prefacilitated during a preceding voltage step (DeMaria et al. 2001; Chaudhuri et al. 2007; Adams et al. 2010), or by monitoring the gradual increase in currents in response to a train of brief voltage steps (Lee et al. 2000, 2003; Chaudhuri et al. 2007). Though useful for appraising relative changes in CDF, such methods do not quantify the absolute extent of CDF, and can only assess regulation over a narrow voltage range near the midpoint of voltage activation.
Figure 1. Ca2+ sources influencing CaV2.1 opening.
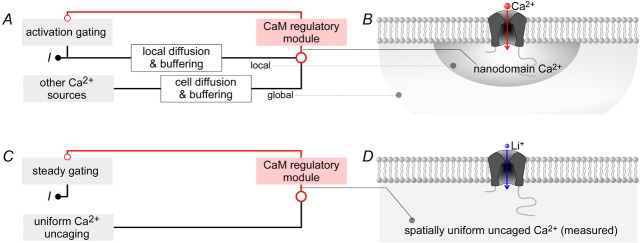
A and B, Ca2+–CaM regulation of channels arises from a superposition of local and global Ca2+ sources. CaV2.1 channels stimulated by a voltage step give rise to Ca2+ current I, which is proportional to channel activation gating. The Ca2+ current, shaped by local diffusion and buffering, creates a local nanodomain Ca2+ signal that feeds into the CaM regulatory module, which bestows feedback modulation on activation gating. Concurrently, Ca2+ from distant channels shaped by cell diffusion and buffering creates a global Ca2+ signal which is also an input into the CaM regulatory module. C and D, simplified scheme employing Li+ as charge carrier. Stimulating CaV2.1 here gives rise to Li+ current I, which is allowed to reach steady state (steady gating) before Ca2+ is uncaged to influence the CaM regulatory module. Importantly, in this system, Ca2+ uncaging generates the only Ca2+ signal, which is spatially uniform and measurable.
Thus appraised, we here devised strategies to directly measure and control nanodomain Ca2+ while recording CaV2.1 whole-cell currents at steady state with respect to activation gating. Figure1C and D schematizes the overall approach, where Li+ is substituted for Ca2+ as charge carrier through these channels (Fig.1D). In so doing, Ca2+ influx through channels no longer triggers CaM regulation, and channel gating is uncoupled from induction of the CaM regulatory module (Fig.1C). Instead, uniformly dialysed DM-nitrophen (caged Ca2+) is photolysed to abruptly produce spatially uniform Ca2+ elevations throughout the cytoplasmic compartment, including the channel nanodomain (Fig.1D). Furthermore, ratiometric Ca2+ fluorescent dyes permit real-time measurement of Ca2+ concentration. Accordingly, we directly control and measure the Ca2+ input to the CaM regulatory module (Fig.1C) (Tadross et al. 2013; Ben-Johny et al. 2014). Importantly, photo-uncaging can be produced well after channels have achieved steady state with respect to voltage activation, so that the changes in CaV2.1 current directly reflect the effects of CaM regulation alone (Fig.1C, steady gating).
Using this approach, we now find that CDF of CaV2.1 is larger, faster and more Ca2+ sensitive than expected. Importantly, the properties of CDF render Ca2+ current modulation particularly strong during physiological spike-like voltage stimuli. By contrast, CDI turns out to be more than 50-fold less Ca2+ sensitive, such that CDF can, in many instances, develop in isolation from CDI. Our results pertain to both recombinant CaV2.1 channels and native CaV2.1 currents within cerebellar Purkinje neurons. Moreover, CaV2.1 behaviour in both contexts could be quantitatively predicted by a common biophysical model of CaV2.1 gating and CDF, gleaned from extensive experimental constraints. Overall, CDF of CaV2.1 channels is well positioned to influence activity-dependent processes throughout the CNS.
Methods
Molecular biology
The parental human CaV2.1 clones were gifts from Dr Terry Snutch (University of British Columbia). The exact splice background (Soong et al. 2002) used was: Δ10A (+G); 16+/17+; Δ17A (−VEA); −31* (−NP); 37a (EFa) or 37b (EFb); 43+/44+; 47− or 47+. Henceforth, for compactness, we will refer to the main constructs as EFa −47, EFb +47 and EFb −47. The entire channel ORF was transferred into the pEGFP-N3 vector (Clontech, Mountain View CA) with restriction enzymes Nhe1/XbaI for ease of mutagenesis (enhanced green fluorescent protein (eGFP) coding sequence replaced). The EFb variant in this same background was generated by transferring the relevant region from our CaV2.1 EFb clone (Soong et al. 2002) with XhoI/BglII. The pore mutants were all generated by overlap extension PCR, as previously described (Ho et al. 1989). The primers used to generate E1A were: CCCGTCCGACAGAACTGCCTC (flank1_F), GATCAGTCCACCCTgCCATGGTTATGC (E1A_R), GCATAACCATGGcAGGGTGGACTGATC (E1A_F) and TAGGGTCCCTCCCGGCTCAG (flank1_R). The PCR product containing the mutation was then transferred back into our channel backbone with AgeI/PvuI. E2A was generated with the same flanking primers and restriction enzymes, with these overlap primers: CCTGACGGGCGcAGACTGGAAC (E2A_F) and GTTCCAGTCTgCGCCCGTCAGG (E2A_R). The primers for E3A were AACAGGCCCGCTACCACG (flank3_F), CCAGCCTgCTCCCGTGGAC (E3A_R), GTCCACGGGAGcAGGCTGG (E3A_F) and GAAGTTATTCCCAAACTCAGTCACGAGG (flank3_R), and transferred with enzymes PvuI/EcoRV. The primers for E4A were TGTTCTGGGCAGCATCACCG (flank4_F), GCCAAGCTgCCCCGG TGG (E4A_R), CCACCGGGGcAGCTTGGC (E4A_F) and CAGCTTCTTGGCCTTGCTCTGC (flank4_R), and inserted with enzymes EcoRV/BglII. All mutants were verified by Sanger sequencing.
Cell culture and transfection
Human embryonic kidney 293 (HEK 293, ATCC) cells were cultured under sterile conditions on glass coverslips in 60 mm plates at 37°C with 5% CO2, and transiently transfected with a calcium phosphate protocol (Brody et al. 1997). We used 2–5 μg of cDNA encoding the desired channel α1 subunit, with 4 μg each of rat brain β2a and α2δ subunits. SV40 T-antigen, 1 μg, and cyan fluorescent protein (CFP), 1 μg, were also co-transfected to enhance expression and identify transfected cells. For experiments not involving Ca2+ uncaging, β2a-IRES2-eGFP was used in place of β2a plus CFP to identify transfected cells. All constructs were driven by a CMV promoter, and cells were used for electrophysiology 1–3 days after transfection.
Dissociated Purkinje neurons
Cerebellar Purkinje neurons were isolated from postnatal day (P) 14–18 C57/B6 mice (Jackson Labs, Bar Harbor ME) as previously described (Adams et al. 2010). Animals were handled according to protocols approved by the Johns Hopkins University's Animal Care and Use Committee. Briefly, mice were anesthesized with isoflurane, and swiftly decapitated with a guillotine. Purkinje neurons were then enzymatically dissociated from cerebellar cortex and plated on poly-d-lysine-coated glass-bottom dishes (In Vitro Scientific, Sunnyvale CA). Cells were allowed to recover for 1 h at room temperature before experimentation.
Electrophysiology
For both HEK 293 cells and Purkinje neurons, whole-cell recordings were obtained using Axopatch 200 or 200A amplifiers (Molecular Devices, Sunnyvale CA) at room temperature. Electrodes were pulled (model P-97, Sutter Instruments, Novato CA) from borosilicate glass capillaries (MTW 150-F4, World Precision Instruments, Sarasota FL) and fire-polished with a microforge (Narishige, Tokyo) to achieve resistances of 1–3 MΩ before series resistance compensation of 70–80%. Currents were low-pass filtered at 5 kHz, and leak-subtracted with a P/4–P/8 protocol. Data acquisition (digitized with ITC-18 (Instrutech, Longmont CO)) and analysis of electrophysiological data were done with custom software written in MATLAB (MathWorks, Natick MA).
Whole-cell solutions
Internal solutions
For the Ca2+ block and pre-pulse CDF characterizations, the internal solution contained (in mm): 135 CsMeSO3, 5 CsCl, 1 MgCl2, 4 MgATP, 1 EGTA, 10 Hepes, with pH titrated to 7.4 with CsOH, and adjusted to 295 mosmol l−1 with glucose. For Ca2+-uncaging experiments the internal solution contained: 135 mm CsCl, 40 mm Hepes (pH 7.4), 1–2 mm citrate, 5 μm Fluo-2 (TefLabs, Austin TX) + 5 μm Fluo-2 Low Affinity (TefLabs), 2.5 μm Alexa568 (Invitrogen, Waltham MA), 2 mm DM-Nitrophen (DMNP-EDTA) (Invitrogen) and 0.25–1 mm CaCl2. The two Ca2+ dyes with different Ca2+ sensitivities were combined with Alexa568 for increased dynamic range of ratiometric Ca2+ detection. Calibration of premixed stocks of Fluo/Alexa was performed both in vitro with droplets and also in vivo by patching HEK 293 cells in the whole-cell configuration with internal solutions containing different free Ca2+ concentrations, buffered by either 5 mm EGTA, 5 mm HEDTA, or 5 mm NTA. Free Ca2+ concentrations were calculated with MaxChelator (Stanford).
External solutions
For characterizations of CDF with the pre-pulse protocol, the bath solution contained (in mm): 140 TEA-MeSO3, 10 Hepes (pH 7.4 with TEA-OH) and 5 CaCl2 or 5 BaCl2, adjusted to 295 mosmol l−1 with glucose. For Ca2+ block experiments, bath solution contained (in mm): 80 TEA-MeSO3, 10 Hepes (pH 7.4), 80 LiCl and 0.5–2.5 CaCl2 buffered by 5 EGTA, 5 HEDTA or 5 NTA depending on the desired concentration of free Ca2+ (295 mosmol l−1 with glucose). Free Ca2+ concentrations of these solutions were calculated using MaxChelator (Stanford). For Ca2+-uncaging experiments, bath solution contained (in mm): 80 TEA-MeSO3, 10 Hepes (pH 7.4), 80 LiCl and 2 EGTA (295 mosmol l−1 with glucose). Unless otherwise specified, all reagents were obtained from Sigma-Aldrich.
Ca2+ imaging and uncaging
All Ca2+-uncaging experiments were done on a Nikon TE2000-U inverted microscope with a 40×/1.3 Plan Fluor objective as previously described (Tadross et al. 2013). Briefly, dyes are excited by a 514 nm argon laser, with the resulting fluorescence obtained at two channels after splitting by a 545 DCLP dichroic mirror, and filtering either with 545/40BP (Fluo) or 580LP (Alexa568). Brief UV pulses were generated by a UV flash photolysis system (Cairn Research, Kent, England), with varying UV intensities achieved by adjusting the voltage (50–200 V) and total capacitance (500–4000 μF) of the capacitor bank. The fluorescence of our two dyes as a function of Ca2+ is a superposition of the Ca2+ response of each dye, and exhibits the following relation:
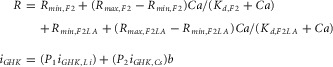 |
where R is the measured green/red fluorescence ratio. We determined Rmin and Rmax of Fluo-2 (F2) and Fluo-2 Low Affinity (F2LA) dyes by whole-cell dialysis of HEK 293 cells with carefully buffered Ca2+ solutions, containing each dye and verified that the in vivo Kd values for both dyes (0.39 and 6.7 μm) were the same as the in vitro Kd values provided by the manufacturer (TEFLabs). Intracellular Ca2+ concentration (Ca) was then determined from the measured R by numerically inverting the above equation.
Simulations: CaV2.1 channel models
The basic unfacilitated channel model consists of six states, with four transitions amongst five closed states before a final transition to the open state. The parameters of these rate constants were chosen such that they fit both the steady-state open probability (Po) versus voltage relations (Fig.5E) as well as the kinetics of channel activation and deactivation in response to voltage steps (Fig.5B).
Figure 5. Contrasts between facilitated and non-facilitated channels at various potentials.

A and B, protocol to define conductance–voltage (G–V) curves of unfacilitated and facilitated channels. Li+ tail currents from CaV2.1 EFa E3A are elicited at −50 mV after 10 ms step depolarizations ranging from −30 to 50 mV. The peak tail currents before (blue) and after (red) Ca2+ uncaging are marked with filled circles. Ca2+ is uncaged during a 40 ms voltage step at −10 mV to monitor progression of CDF. A, expanded view of tail currents (black) after voltage steps to −10 mV before (left) and after (right) Ca2+ uncaging. A kinetic model of CaV2.1 channel gating is illustrated (middle), with simulation results (red, left and right) overlaid on the data. Parameters for model shown in Fig.6C, exemplar G–V from B. The peak of the tail currents, normalized to the peak currents after steps to 40 and 50 mV at baseline Ca2+ (blue), provides an assessment of how the voltage activation curve changes in response to Ca2+ (red). Two effects are seen: an increase in Gmax (a), and a left shift of ∼5 mV (b). D, CDF vs. voltage from B. E and F, similarly, population data (mean ± SEM) with n = 7 cells where Ca2+ was raised > 3 μm. Smooth fit lines are from simulations of the channel model illustrated in A (middle). G and H, population data for CaV2.1 EFb E3A (mean ± SEM) with Ca2+ uncaged to > 3 μm for n = 7 cells.
The full channel model with CDF consists of 3 × 6 states, as illustrated in Fig.8B. The horizontal transitions within the first two rows of states are identical to that of the basic unfacilitated model. The last row of states, however, represents the facilitated mode, with 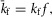 and
and 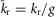 , such that the facilitated channel has a larger maximal Po and increased open times. Additionally, the rate of transitions between the closed states were boosted slightly (
, such that the facilitated channel has a larger maximal Po and increased open times. Additionally, the rate of transitions between the closed states were boosted slightly (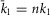 ;
; 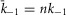 ; n = 1.1) to match the kinetics of voltage activation when channels are fully facilitated (Fig.5B, right). The vertical transitions between states of the three rows are determined by the kinetics of CDF (Fig.8). As there was no discernible voltage dependence to CDF (Fig.7F), the vertical transitions across each of the columns of states are identical, with the exception of the lower-right transition between the open states of rows 2 and 3 where
; n = 1.1) to match the kinetics of voltage activation when channels are fully facilitated (Fig.5B, right). The vertical transitions between states of the three rows are determined by the kinetics of CDF (Fig.8). As there was no discernible voltage dependence to CDF (Fig.7F), the vertical transitions across each of the columns of states are identical, with the exception of the lower-right transition between the open states of rows 2 and 3 where 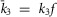 and
and 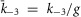 to maintain the necessary path independence. This model is easily represented as a system of differential equations, which was numerically integrated in MATLAB with a stiff ODE solver (ode23s). The channel open probability Po is the probability of residing in any of the three open states, and the simulated macroscopic current is then:
to maintain the necessary path independence. This model is easily represented as a system of differential equations, which was numerically integrated in MATLAB with a stiff ODE solver (ode23s). The channel open probability Po is the probability of residing in any of the three open states, and the simulated macroscopic current is then: 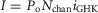 , where iGHK is the voltage-dependent unitary current and Nchan represents the number of channels.
, where iGHK is the voltage-dependent unitary current and Nchan represents the number of channels.
Figure 8. Dynamic Ca2+ responsiveness of CDF.

A, saturating on-kinetics of CDF. Individual normalized current records (grey) from 10 cells, where intracellular Ca2+ was uncaged to >3 μm, are shown overlaid together with their average (black line), fitted by a single exponential with τ = 2 ms. B, CaV2.1 channel model, where the vertical Ca2+-dependent transitions have 3 states (rows), with transitions between 2nd and 3rd rows having no Ca2+ dependence. The correspondence of these three gating modes to previously described conformations underlying CaM regulation of Ca2+ channels (Tadross et al. 2008) is denoted by the numbers within ovoids at the lower right edge of each gating mode. Conformation 1 corresponds to a channel in which the C-terminal lobe of Ca2+-free CaM (apoCaM) is bound to a channel pre-association site; conformation 2 depicts a channel wherein this lobe of apoCaM transiently unbinds from the pre-association site; conformation 3 displays a channel where this transiently unbound lobe of CaM complexes with Ca2+, and conformation 4 represents the channel after the Ca2+-saturated lobe of CaM has interacted with a channel effector site to produce facilitation. For simplicity, conformations 1 and 2 have been combined in simulations used in the present study. C, examplar traces from a single cell, where a family of time-varying Ca2+ inputs by Ca2+ uncaging (second row) trigger a corresponding family of kinetically distinct CDF records (last row). The continuous black lines on the Ca2+ and current axes represent the fit to the Ca2+ waveforms used as the input and the output of the channel model, respectively. Parameters for the model are shown in D (mean ± SEM), obtained from fitting n = 8 cells. E, these parameters yield a Ca2+ sensitivity curve matching data from Fig.3E (grey circles). F and G, simulations of step increases and decreases of intracellular Ca2+ show that the maximal ‘on’ and ‘off’ responses have time constants of 2 ms and 31 ms.
Figure 7. Voltage insensitivity of Ca2+-driven conversion to facilitated channels.

A, diagram of channel model illustrating the possibility of having multiple rate constants (ka–kf) driving channels towards facilitated states. B, multiple G–V curves are obtained with tail protocols before and after uncaging Ca2+. As the intracellular Ca2+ concentration drops over time, the peak tail currents correspondingly decrease. C, the peak Li+ currents plotted against the intracellular Ca2+ charts out the steady-state CDF vs. Ca2+ curves for the voltages assessed. D, normalized CDF vs. Ca2+ for voltages from −10 to 50 mV show that CDF at different voltages have the same Ca2+ sensitivity. E and F, as above, population data (mean ± STD) shown for n = 7 cells. All fits are Hill equations with nHill = 1.8 and EC50 = 0.5 μm.
In Figs5, 6 and 8, [Ca2+] and transmembrane voltage were supplied as inputs into the model, with the channel currents calculated as above, where:
 |
Figure 6. Physiologically relevant action potential waveforms have significant CDF.

A, parameters of channel model illustrated in Fig.5A in the unfacilitated and facilitated states. B and C, CaV2.1 E3A permeating Li+ currents are triggered by action potential waveforms recorded from cerebellar Purkinje neurons. Stimulated currents are steady at baseline Ca2+ (C, left). Ca2+ uncaging in the midst of the ∼50 Hz train causes large enhancement of Li+ current (C, right). An expanded view of traces before and after Ca2+ elevation (black) are shown in B, with simulation results overlaid (red). D and E, population data (mean ± SEM) for peak Li+ current as a function of time with n = 5 cells.
The voltage-dependent unitary current (iGHK) consists of a superposition of two GHK equations, because CaV2.1 E3A channels permeate both inward Li+ and outward Cs+ currents with relative permeabilities P1 and P2. Furthermore, outward current declines relative to expected GHK current at more depolarized voltages due to voltage-dependent pore block, represented here by b, a voltage-dependent Boltzmann factor.
Physiological model
A spherical cell/synapse was divided into three symmetrical compartments, representing a thin shell near the membrane, the cytosol and a reservoir for Ca2+ efflux (Fig.1D). CaV2.1 channels provide Ca2+ influx into the shell compartment, and the resulting Ca2+ can then diffuse into the remaining compartments, informed by the following differential equations:
where  , where F is Faraday's constant, and
, where F is Faraday's constant, and  . Po is determined from the 18-state model described above, where V was provided as an input, and Cashell was used as the Ca2+ signal to facilitate CaV2.1 channels. This 20-state model was then solved numerically with MATLAB using the stiff ODE solver as before.
. Po is determined from the 18-state model described above, where V was provided as an input, and Cashell was used as the Ca2+ signal to facilitate CaV2.1 channels. This 20-state model was then solved numerically with MATLAB using the stiff ODE solver as before.
Results
Minimizing Ca2+ block of Li+ currents carried by CaV2.1 channels
Figure 2A summarizes our initial attempt to utilize Ca2+ photo-uncaging to investigate CaM regulation of CaV2.1 channels fluxing Li+. Recombinant channels were heterologously expressed in HEK 293 cells. The left subpanel displays a robust Li+ current evoked by step depolarization to −10 mV. Current decays minimally during the voltage step, as expected for channels coexpressed with β2a auxiliary subunits to minimize VDI (Patil et al. 1998). Though Ca2+-loaded DM-nitrophen was present in the internal solution, intracellular Ca2+ concentration remained low throughout (∼0.1 μm) in the absence of ultraviolet (UV) illumination. The right subpanel summarizes the result during a subsequent voltage pulse during which Ca2+ was uncaged by a brief UV pulse (at vertical violet bar). Prior to uncaging, the initial current waveform (black trace) precisely tracked the control response from the left subpanel (reproduced as grey trace), substantiating minimal rundown. By contrast, upon Ca2+ uncaging to nearly 10 μm, the current abruptly decreased to a nadir in less than a few milliseconds, followed by slow recovery over tens of milliseconds. The precipitous decrement of current might have initially appeared perplexing because it occurred far too quickly to be explained by a slow CDI process (DeMaria et al. 2001). The subsequent recovery of current could have arisen from CDF developing over this unspecified inhibitory process. By recalling classic studies of ion permeation through L-type Ca2+ channels (McCleskey & Almers, 1985; Hess et al. 1986), we soon suspected that the Li+ flux through CaV2.1 might be exquisitely susceptible to block by Ca2+ binding to the pore (Fig. 2A, right subpanel diagram). The quick decline of current upon Ca2+ uncaging would accord with the sub-millisecond equilibration of Ca2+ block of monovalent currents fluxing through Ca2+ channels.
Figure 2. Relieving Ca2+ blockade of Li+ current.

A, Li+ permeation through CaV2.1 is blocked by Ca2+. CaV2.1 EFa fluxes Li+ current in response to a step depolarization at baseline intracellular Ca2+ (left). A brief UV pulse (violet bar) induces a step-like elevation of intracellular Ca2+ in the midst of the voltage step, resulting in a rapid block of Li+ current, followed by CDF (right). B, characterization of Ca2+ block. Li+ currents from voltage ramps were obtained in the presence of varying amounts of Ca2+ in the bath (Cao). Normalized peak currents show the decrement in peak currents of CaV2.1 in response to Ca2+. Exemplar current–voltage (I–V) traces (right) from the last trace of each Ca2+ solution (coloured circles, left) show explicitly the reduction in current amplitude without shifting of the reversal potential. C, similar traces for CaV2.1 E3A show a reduced Ca2+ sensitivity to block (left), and a left-shifting of the reversal potential (right). D, diagram of CaV2.1 channel highlighting the four glutamates (E1–E4) forming the selectivity filter (top). Population (mean ± SEM) Ca2+ block data for CaV2.1 (bottom): wild-type (WT; IC50 = 0.8 μm; n = 7), E1A (IC50 = 7 μm; n = 5), E2A (IC50 = 8 μm; n = 5), E3A (IC50 = 35 μm; n = 5), E4A (IC50 = 10 μm; n = 5). Fits to binding curves all have Hill coefficient of 1.
To test this hypothesis, and to minimize the potential confounding effects on characterizing CaM regulation, we investigated Ca2+ block of CaV2.1 channels. The fractional decrement of peak Li+ currents (evoked by voltage ramps) was characterized as a function of Ca2+ added to the bath (Fig.2B). Consistent with previous studies on CaV1.2 channels (Yang et al. 1993), CaV2.1 exhibited a high affinity for Ca2+, with an IC50 of ∼0.8 μm. Aware that permeation is markedly influenced by four specific glutamate residues in the selectivity filter of L-type channels (Ellinor et al. 1995), we reasoned that substituting alanines for the corresponding glutamates in CaV2.1 (Fig.2D, top) might also attenuate Ca2+ block. Accordingly, E→A substitutions were introduced into each of four homologous domains, and Ca2+ block was then quantified. Figure 2C summarizes the outcome for E→A substitution in domain III (E3A). Indeed, Ca2+ block was markedly reduced compared to wild-type CaV2.1. Nearly 30% of the initial current amplitude was maintained despite 104 μm Ca2+ in the bath. As expected, the reversal potential for E3A was also left-shifted relative to wild-type, with Cs+ being the main intracellular cation. There was also asymmetry with respect to the effects of alanine substitutions in the various CaV2.1 domains (Fig.2D, bottom). Notably, E3A was least sensitive to Ca2+ block (IC50 of 35 μm), whereas mutations in other domains (E1A, E2A, E4A) yielded smaller IC50 values of 7, 8 and 10 μm, respectively. Indeed, this rank order was similar to that in CaV1.2 (Ellinor et al. 1995) and CaV1.3 (Tadross et al. 2013). That said, we chose the E3A variant for subsequent Ca2+-uncaging experiments, presuming that the confounding decrement of current in Fig.2A (right subpanel) was indeed due to Ca2+ block.
Steady-state Ca2+ sensitivity of CaV2.1 CaM regulation revealed by Ca2+ uncaging
Before performing Ca2+ uncaging experiments with the E3A variant, we first verified that the pore mutation did not significantly alter CDF, a plausible expectation given that the molecular determinants of CDF reside within the channel carboxy terminus rather than the permeation pathway (DeMaria et al. 2001; Lee et al. 2003; Mori et al. 2008). For this purpose, we used a well-established prepulse protocol to coarsely estimate CDF as triggered by Ca2+ itself fluxing through channels (DeMaria et al. 2001; Chaudhuri et al. 2004). Corresponding wild-type CaV2.1 currents are displayed in Fig.3A. Upon presentation of an isolated test pulse to 5 mV (left subpanel), the activation of Ca2+ current follows a biphasic time course. The first phase comprises an initial rapid increase of current owing to the comparatively speedy voltage activation of channels, initially all in their ‘unfacilitated’ condition (Fig.3A, τfast). A subsequent second phase of current increase (Fig.3A, τslow) reflects the slower facilitation of channel opening by ongoing Ca2+ entering through channels. To characterize facilitation more fully, we examined facilitation produced during brief voltage prepulses, using a variation of this initial protocol. In the example shown (Fig.3A, right subpanel, black trace), the test-pulse current following a prepulse exhibits a much reduced slow-activation phase, because many channels have already undergone facilitation during the prepulse. For reference, the test-pulse current without prepulse has been reproduced in grey. The extent of prepulse facilitation (the metric CDF) turns out to be proportional to the area between black and grey traces ΔQ (right subpanel, shaded red area) divided by τslow (from left subpanel) (Chaudhuri et al. 2004). Plots of CDF as a function of prepulse voltage then yield the bell-shaped relation shown in Fig.3B, averaged over multiple E3A (red) and wild-type (black) CaV2.1 currents. This bell-shape accords with CDF reflecting a genuine Ca2+-triggered process, because CDF mirrors the amplitude of prepulse Ca2+ currents elicited at various prepulse voltages (Brehm & Eckert, 1978). When Ba2+ was substituted as charge carrier, the bell-shaped augmentation was largely eliminated (Fig.3B, open symbols) because Ba2+ binds poorly to CaM (Chao et al. 1984). Importantly, the CDF profiles for E3A and wild-type CaV2.1 channels were indistinguishable, permitting use of the E3A construct to gauge CDF in subsequent Ca2+-uncaging experiments.
Figure 3. Steady-state Ca2+ sensitivity of CaV2.1 CaM regulation revealed by Ca2+ uncaging.

A, characterization of CDF using the prepulse protocol. With Ca2+ as the charge carrier, a step depolarization to 5 mV induces an inward current with rapid and slow components due to a superposition of voltage activation and CDF (left). With a preceding voltage step to 20 mV, sufficient Ca2+ enters to partially facilitate the channels, such that the subsequent step to 5 mV has a markedly blunted slow component (right). The area between the two current traces (ΔQ), divided by τslow, gives an approximation of the CDF triggered by the prepulse. B, population data (mean ± SEM) for CDF vs. prepulse voltage of the pore mutant E3A (red, n = 9) and WT (black, n = 8). The open circles denote the use of Ba2+ as the charge carrier (incapable of triggering CDF), while the filled circles denote the use of Ca2+. C, E3A mutant permeating Li+ current shows negligible Ca2+ block. Ca2+ uncaging during a voltage step of a CDF-incapable splice variant of Cav2.1 (EFb −47) induces neither block nor CDF (middle, right). D, Ca2+ uncaging with the EFa variant (−47) showcases large and rapid induction of CDF (middle, right). E, population data for CaV2.1 EFa −47 (black circles, mean ± STD, n = 12 cells), quantifying CDF as a function of different uncaged Ca2+ concentrations during voltage steps to −10 mV yields a Ca2+ sensitivity curve, with EC50 = 0.6 μm and nHill = 1.8. Population data for CaV2.1 EFb −47 (grey circles, mean ± STD, n = 17 cells) show complete absence of CDF. F, similar experiments with CaV2.1 EFa −47 coexpressed with CaM1234, a mutant CaM incapable of binding Ca2+, resulted in the elimination of CDF (mean ± STD, n = 7 cells). G, similar experiments with CaV2.1 EFa –47 carrying an I(0)A mutation at the IQ domain show a knockout of CDF (mean ± STD, n = 8 cells).
Turning in earnest towards Ca2+ uncaging, we initially measured the effects of Ca2+ elevation on a splice variant of CaV2.1 thought to exhibit far weaker CDF (EFb −47) (Chaudhuri et al. 2004). Results from this construct explicitly test whether the E3A pore mutation suffices to minimize Ca2+ block effects in the precise configuration of uncaging protocols. Figure3C (left subpanel) displays exemplar Li+ currents through EFb −47 channels bearing the E3A pore mutation. Ca2+ remains low throughout (∼0.1 μm) in the absence of uncaging. Upon Ca2+ uncaging to just over 1 μm (middle subpanel, vertical violet bar), Li+ current appeared unchanged, demonstrating minimal Ca2+ block. Yet stronger Ca2+ elevation (right subpanel) also negligibly impacted Li+ current, substantiating insignificant Ca2+ block at levels approaching 10 μm. Likewise, Ca2+ elevations over a similar range produced imperceptible effects on a related EFb +47 splice variant of CaV2.1 (Chaudhuri et al. 2004) (data not shown). Having confirmed that Ca2+ block effects were adequately blunted by E3A mutation, we focused again on the prototypic CaV2.1 variant (EFa −47) that exhibits robust CDF, as shown earlier in Fig.3A and B. Figure3D summarizes the effects of Ca2+ uncaging, specifically on a version of these prototypic channels containing the E3A mutation. The left subpanel shows steady Li+ current evoked without Ca2+ uncaging. By contrast, upon Ca2+ uncaging (middle subpanel, grey-shaded region), a nearly two-fold augmentation of current was rapidly produced (red-shaded region). Stronger and more sustained Ca2+ elevation (right subpanel) induced a still larger and more sustained elevation of current. These effects directly substantiate a fully legitimate CDF process, one that appears considerably larger than previously estimated by indirect means such as in Fig.3B (Lee et al. 2000; DeMaria et al. 2001; Chaudhuri et al. 2005). Data obtained from multiple cells and uncaging experiments are summarized in Fig.3E. For EFa −47 channels, the steady-state relation between CDF and [Ca2+] defines a Hill function with a maximum of 2.22, midpoint EC50 of approximately 0.5 μm, and Hill coefficient nHill of 1.8 (red curve). CDF was here defined as the ratio of currents evoked before and after Ca2+ uncaging at steady state. For EFb −47 channels, CDF was negligibly elevated for [Ca2+] up to 10 μm (grey relation). Reassuringly, CDF was essentially eliminated in EFa −47 channels coexpressed with a dominant-negative CaM molecule (Fig.3F), confirming CaM as the sensor for CDF as found in previous studies (DeMaria et al. 2001; Chaudhuri et al. 2005, 2007). Moreover, no appreciable CDF was observed for EFa −47 channels bearing an isoleucine to alanine substitution within an IQ domain in the CaV2.1 carboxy terminus (Fig.3G), an outcome that fits with previous characterization of this mutant using more indirect measures (DeMaria et al. 2001).
Beyond the large magnitude of CDF, another unexpected observation was the absence of CDI (Lee et al. 1999; DeMaria et al. 2001) in all the records shown thus far, even though the E3A mutant was capable of undergoing CDI (Fig.4B and C). In particular, there was no hint of current decline in either EFb −47 or EFa −47 records after Ca2+ uncaging to several micromolar (Fig. 3C and D, rightmost subpanels). It turns out that Ca2+ elevations greater than 10 μm are required to trigger CDI (Fig.4D and E). Owing to technical constraints, it was difficult to uncage Ca2+ to such large concentrations, while reliably achieving low baseline [Ca2+] prior to uncaging. For this reason, we focused the remainder of experiments on the CDF process, which could be studied in isolation because of the low Ca2+ sensitivity of the CDI mechanism.
Figure 4. High Ca2+ requirements for CDI.
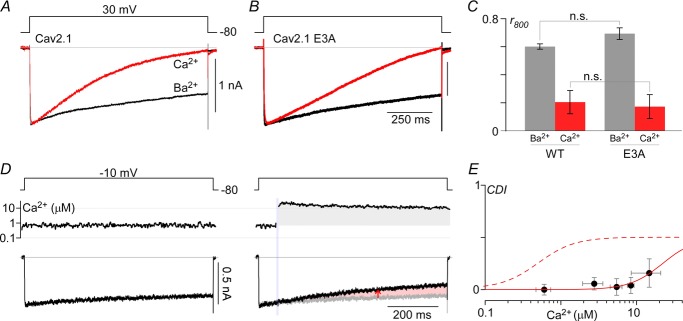
A, exemplar currents from CaV2.1 permeating either Ca2+ (red) or Ba2+ (black) during a 1 s depolarization to 30 mV. B, similar protocol for CaV2.1 E3A. In both cases, 40 Ca2+/40 Ba2+ (mm) were used to increase current density. C, population data for WT and E3A channels, plotting the ratio of currents at 800 ms to peak current for Ba2+ and Ca2+ (mean ± SEM). Both WT and E3A have n = 5 cells each, with comparison between E3A and WT done using unpaired Student's t test (P > 0.05). D, Li+ currents through CaV2.1 EFb E3A channels are stable at ∼0.7 μm (left). Uncaging Ca2+ to ∼14 μm elicits a small amount of CDI (right). E, compiling traces from n = 19 cells where Ca2+ was uncaged to various levels yields a Ca2+ dose–response curve for CDI (red continuous line), defined here as CDI = r400,before – r400,after (red arrow in D), where r400 = I(tuncaging)/I(tuncaging + 400ms). E, data points shown are mean ± STD, where fit is a Hill function with nHill = 2, IC50 = 22 μm. Fit for CDF's Ca2+ sensitivity is reproduced as red dashed line for comparison.
Contrasts between fully facilitated and non-facilitated channels at various voltages
Thus far, our data nicely revealed features of CDF, but only as viewed during voltage steps to −10 mV. We therefore sought next to examine the contrasts in opening at different voltages, as exhibited by unfacilitated CaV2.1 channels compared to those fully facilitated by Ca2+. In particular, Li+ currents evoked before Ca2+ uncaging (Fig.5A, left subpanel, black trace) reflect the opening and closing of channels governed by transitions among closed states (C) and a predominant open configuration (O) that comprise an unfacilitated mode of gating (middle subpanel, top) (Chaudhuri et al. 2007). Most of the transitions are voltage dependent, while the final transition into O is largely voltage insensitive (Chaudhuri et al. 2007). By contrast, after strong uncaging to fully facilitate channels with respect to Ca2+, Li+ currents evoked by an identical voltage pulse protocol (right subpanel, black trace) would largely reflect transitions within a ‘facilitated’ gating mode (middle subpanel, bottom), where opening is favoured over that in the unfacilitated mode (middle subpanel, top). That said, a critical feature of CDF concerns the relative advantage in opening between these two gating modes over a range of voltages.
Figure 5B displays the results of a protocol that specifically reveals this advantage. Tail Li+ currents through CaV2.1 EFa E3A −47 channels were evoked immediately following a series of brief activating voltage pulses ranging from −30 to 50 mV, initially in the absence of Ca2+ uncaging (blue circles). The response for the activating pulse to −10 mV (highlighted in grey zone) is displayed on an expanded scale in Fig.5A (left subpanel, black trace). Plotting the peak amplitude of these tail currents as a function of voltage in corresponding activating pulses (Fig.5C, blue symbols and relation) yields the steady-state voltage activation profile of unfacilitated channels. For convenient comparison across cells, the data have been normalized to the largest tail current amplitude. Immediately following acquisition of this initial set of tail currents, current was evoked by a sustained voltage pulse to −10 mV, and Ca2+ uncaged to 3.4 μm to produce the usual facilitation of current (note events in Fig.5B near vertical violet bar) as illustrated previously in Fig.3D. As this level of Ca2+ saturated facilitation with respect to Ca2+ (Fig.3E), subsequent acquisition of tail currents (Fig.5B, red circles) sufficed to approximate the steady-state voltage activation profile for fully facilitated channels (Fig.5C, red symbols and relation). These data have been normalized to the maximal tail current before Ca2+ uncaging. Viewed in this way, one can readily deduce that CDF produces both an absolute upward scaling of the voltage activation profile (Fig.5C, effect a) and a hyperpolarizing shift of the relation (effect b) (Chaudhuri et al. 2007). Dividing the relations for facilitated and unfacilitated channels yields the fractional increase in open probability produced by CDF as a function of voltage (Fig.5D). These results were fully corroborated over multiple cells (Fig. 5E and F). Intriguingly, the net result of effects a and b is to produce a Boltzmann-like CDF profile (Fig.5D and F). To confirm that all effects under this protocol were strictly related to CDF, we replicated experiments using CaV2.1 EFb E3A −47 channels (which lack facilitation, Fig.3C), and observed a complete absence of CDF behaviour (Fig.5G and H).
Two notable features merit attention. First, the magnitudes of CDF at voltages less than zero are remarkably larger than expected from traditional protocols (Fig.3A), reaching values exceeding two. CDF values of this order are consistent with the results of Ca2+ uncaging experiments performed earlier during step depolarizations to −10 mV (Fig.3D, right subpanel), but now we can appreciate that such large facilitation would extend over a range of voltages less than zero. Second, physiological activation of CaV2.1 channels, such as during neuronal action potentials, would draw heavily from this hyperpolarized voltage range, so one might expect the effects of CDF to be especially pronounced during physiological spike activation.
To explore further this inference about action potential behaviour, we constructed channel kinetic mechanisms corresponding to the gating modes in Fig.5A, using the parameters in Fig.6A. Quantitative simulations reproduced several important features: the whole-cell current waveforms evoked in Fig.5A (red curves) and B; all the voltage activation and CDF profiles in Fig.5E and F; and published single-channel open times (Chaudhuri et al. 2007). As previously suggested (Chaudhuri et al. 2007), differences between unfacilitated and facilitated gating modes were mainly restricted to the rightmost opening and closing steps according to fixed scaling factors f and g (Figs 5A and 6A). With our gating mechanisms thus established, we turned to predictions of the maximal impact of CDF on currents evoked by physiological action potential waveforms, taken from cerebellar Purkinje neurons (Chaudhuri et al. 2005). For channels resident within the unfacilitated mode, simulated activation by a typical neuronal action potential (Fig.6B, left, red curve) predicts well the actual CaV2.1 current (black trace). Upon maximal Ca2+-induced conversion towards the facilitated mode, the corresponding simulated current increases by nearly twofold (Fig.6B, right, red curve). This outcome fits with the intuition that CDF could strongly enhance currents during physiological activation. That said, Fig.6C tests experimentally for CDF during trains of action potential waveforms. For an exemplar cell, with Li+ currents fluxing through recombinant CaV2.1 EFa E3A −47 channels, evoked currents remained steady over multiple action potential waveforms in the absence of Ca2+ uncaging (left subpanel), substantiating stability of the preparation. By contrast, upon uncaging to a saturating level of Ca2+, corresponding currents increased by nearly twofold (Fig.6C, right subpanel). The impact of CDF on current is further emphasized by the expanded time base plots of exemplar currents evoked just before and after Ca2+ uncaging (Fig.6B, black traces, corresponding to Fig.6C responses shaded in green). Additionally, data averaged over many cells confirmed that full induction of CDF can strongly amplify spike current (Fig. 6D and E).
Voltage insensitivity of Ca2+-driven conversion to facilitated channels
Still uncertain, though, is whether strong interconversion towards the facilitated mode would actually be produced by physiological Ca2+ elevations. In particular, only at a fixed potential of −10 mV has the Ca2+ dependence of CDF been determined (Fig.3E). This Ca2+ sensitivity reflects the steady-state distribution of channels between upper and lower modes (Fig.7A), as governed by vertical transitions aligned with closed and/or open states most heavily populated at −10 mV (say, for example, kd transitions). At different potentials, the most frequented closed and/or open states may differ considerably, emphasizing different vertical transitions (for example, ka, kb, kc, ke or kf) that may yield alternative Ca2+ sensitivities, knowledge of which would be fundamental to appreciating the physiological relevance of CDF. Accordingly, we took advantage of the fact that it takes 1–3 min for Ca2+ to return to baseline after large Ca2+ uncaging events, owing to the relatively slow diffusion of fresh DM-nitrophen from the pipette into the cell interior. Figure7B displays results for an exemplar cell undergoing such a protocol. As Ca2+ gradually recedes, we could continue periodic assessment (every 5–30 s) of voltage activation curves using a tail-current protocol (format as in Fig.5B). In this manner, we could assess the status of near steady-state channel partitioning between unfacilitated and facilitated configurations, over a broad range of smoothly waning Ca2+ levels. Most of this partitioning would occur at the holding potential of −80 mV (5–30 s sojourns), punctuated by significant visitations to a wide range of other potentials during the tail-current protocols that last ∼0.5 s. If the vertical transitions in Fig.7A were appreciably different, we would anticipate CDF–Ca2+ relations that deviate considerably from those obtained at −10 mV throughout (Fig.3E). Figure7C displays the result for this exemplar cell, where fold increase in tail-current amplitude is plotted versus Ca2+ concentration. For example, for responses following brief activation to 0 mV, tail-current amplitudes denoted by the cyan symbols in Fig.7B were normalized to that seen before Ca2+ uncaging, and these normalized amplitudes were then plotted against corresponding Ca2+ concentrations to yield the cyan CDF–Ca2+ relation in Fig.7C. Analogous analysis yielded CDF–Ca2+ curves relating to different activation potentials (other coloured relations in Fig.7C). Normalizing all of these relations yields the single Ca2+ response profile in Fig.7E, which is indistinguishable from the steady-state function obtained at −10 mV in Fig.3E (EC50 = 0.5 μm and nHill = 1.8). Note that currents obtained after Ca2+ returned to baseline (Fig.7B, far right) were indistinguishable from those obtained before uncaging (Fig.7B, far left), excluding appreciable run-down phenomena and permitting fold changes in tail current amplitude to be directly attributed to CDF. Averaged data from seven cells fully corroborated this finding (Fig.7D and F). Thus, Ca2+-driven conversions between unfacilitated and facilitated conformations (Fig.7A) are insensitive to membrane potential.
Dynamic Ca2+ responsiveness of CDF
The key remaining biophysical unknown concerned the temporal response of CDF to Ca2+ signals. Uncaging Ca2+ during a sustained voltage step allows direct millisecond resolution of CDF induction. In this regard, a first important set of observations emerged when we overlaid multiple current traces of CaV2.1 EFa E3A −47 channels (Fig.8A, grey waveforms) corresponding to different-sized Ca2+ steps exceeding 3 μm (Fig.8A). The black waveform shows the average of these. These data indicate that CDF can evolve quickly with a monoexponential time constant τ ∼2 ms (Fig.8A), and that τ does not further decrease with larger Ca2+ steps. The latter property implicates the existence of a Ca2+-independent and rate-limiting step leading to the facilitated mode of gating. Thus, a minimal mechanism would in reality require three modes of gating (Fig.8B), as follows. The transition from unfacilitated (top) to prefacilitated modes of gating (middle) would be Ca2+ dependent. Notably, the propensity for opening in these two gating modes would be largely equivalent, so as to allow for the rate limitation of a subsequent transition (Ca2+ independent) to an outright facilitated mode with enhanced opening (bottom). These three gating modes correspond to previously described conformations underlying CaM regulation of Ca2+ channels (Tadross et al. 2008) (see legend, Fig.8B). Elucidating this simple three mode mechanism would permit prediction of how CDF evolves in response to the complex Ca2+ waveforms present physiologically in neurons.
Towards this end, Ca2+ photo-uncaging was used to deliver Ca2+ waveforms with variable magnitudes and time courses as shown in Fig.8C (top), while simultaneously recording currents (below). The different Ca2+ waveforms were produced by varying UV pulse intensity. Weak pulses liberated comparatively few Ca2+ ions that could be rapidly buffered via free cytoplasmic DM-nitrophen, yielding weaker Ca2+ transients with rapid decays. Strong pulses would uncage so many Ca2+ ions that free cytoplasmic DM-nitrophen would be consumed, such that minutes might be required to rebind released Ca2+ ions via fresh DM-nitrophen diffusion from the pipette. Moderate UV pulses would produce Ca2+ transients with intermediate properties.
Given this rich data set, we proceeded to constrain parameters by fitting Ca2+ waveforms with double exponential functions (Fig.8C, top, smooth curves), and then inputting these functions to the kinetic scheme in Fig.8B. The parameters (Fig.8D) were then adjusted until simulated outputs matched both experimental kinetic data (Fig.8C, bottom, smooth curves) and steady-state Ca2+ responsiveness (Fig.8E, smooth curve) determined earlier. By repeating this procedure for additional cells, we obtained the final mean parameters for intermodal exchange shown in Fig.8D (from n = 8 cells). Our model also incorporates a Ca2+ concentration exponent nHill of 1.8. Rate constants within the various gating modes were identical to those established in Fig.6A. With these parameters, the simulated time course of CDF in response to Ca2+ step increases and decreases can be shown in Fig. 8F and G. The maximal predicted ‘on’ and ‘off’ responses exhibit time constants of 2 ms and 31 ms, respectively, seemingly fast enough for physiological relevance.
Devising strategies for resolving native CaV2.1 properties
Use of the CaV2.1 E3A construct permitted Li+ currents to be directly studied in Ca2+ uncaging experiments, allowing unprecedented access to the Ca2+ dependence of CDF. However, the strategies employed thus far could not be used to study native CaV2.1 channels because of the problem of Ca2+ block (i.e. Fig.2A, right subpanel). It was nonetheless crucial to substantiate our biophysically rigorous understanding of recombinant CaV2.1 channels in the native setting.
We thus devised a ‘sparse-tail-current’ variant of our Ca2+ uncaging protocol. If Ca2+ (rather than Li+) could be used as the charge carrier, then little Ca2+ block would occur upon uncaging of intracellular Ca2+ (Yang et al. 1993), even in native channels with wild-type permeation properties. However, Ca2+ influxing through channels would constitute local Ca2+ sources with attendant elevations of nanodomain Ca2+ that would be difficult to measure with bulk-distributed Ca2+ fluorescent dyes (Tay et al. 2012). Indeed, this complexity, as outlined earlier in Fig.1A, motivated our use of Li+ currents in the first place. If, however, we applied only brief voltage pulses separated by comparatively long periods of quiescence (Fig.9A, left subpanel), we might be able to assess the propensity for opening (during brief voltage pulses) while the spatially uniform global Ca2+ component specifies the extent of CDF during brief opening (owing to the predominance of global Ca2+ during long interpulse segments). Importantly, bulk-distributed Ca2+ fluorescent dye would reliably measure this global Ca2+ concentration (Fig.9A, right subpanel). Figure 9B schematizes further the hoped-for outcome. Brief (1 ms) activating pulses would be alternately presented to 0 and 60 mV, with long (>50 ms) quiescent periods separating pulses. Also obtained would be corresponding tail currents, proportional to the open probability at the end of 1 ms activating voltage pulses. These tail currents (cyan circle after 0 mV pulse; rose circle after 60 mV pulse) would thus monitor the extent of CDF at two key points along the overall voltage-dependent profile of CDF (i.e. Fig.5F). A critical point is that although Ca2+ influx during channel openings would sharply increase nanodomain Ca2+ via a local component (Fig. 9B, yellow shaded area), this local perturbation would rapidly dissipate such that nanodomain Ca2+ during most of the period preceding voltage pulses would be equivalent to the global Ca2+ component measured by bulk Ca2+ fluorescent dye (grey shaded area). Moreover, it is precisely this preceding global Ca2+ concentration that should specify the extent of CDF reflected in tail currents, because the change in CDF that occurs during a 1 ms voltage pulse should be small, as follows. First, the Ca2+ entry during 1 ms activating voltage pulses would be limited, because there would be little Ca2+ influx during 60 mV steps (near the reversal potential), or voltage activation would be comparatively slow at 0 mV. Second, even under saturating Ca2+, CDF can increase no faster than specified by a ∼2 ms time constant (Fig.8A), and the voltage pulses in question only span 1 ms. All told, if all these scenarios were to hold true, then the CDF versus Ca2+ relations deduced via this variant protocol should accord with those obtained more directly using Li+ currents fluxing through CaV2.1 E3A channels (Fig.7E).
Figure 9. Towards resolving native CaV2.1 CDF.

A and B, schema outlining the strategy to resolve Ca2+ dependence of native CaV2.1 channels. A modified tail current protocol with very brief steps to 0 or 60 mV for 1 ms (brief gating) before eliciting tail currents at −50 mV minimizes the impact of local Ca2+ on the CaM regulatory module whilst still allowing assessment of the channel's state at two voltages. Uniform Ca2+ uncaging then provides an independent and measurable input to the regulatory module, pushing channels into the facilitated mode. C, exemplar traces from a single cell, illustrating Ca2+ tail currents evolving with time and global Ca2+. Voltage pulse protocol is displayed in the top row, and global Ca2+ in the second row. Tail currents triggered by preceding step to 0 mV are scaled up for clarity and shown in black in the third row (tails triggered by preceding 60 mV steps in grey), with cyan circles marking the peaks. The last row shows the tail currents triggered by preceding 60 mV steps in black, with peaks marked by red circles. D, expanded time frame views of tail currents before and after Ca2+ uncaging, normalized to peak levels at baseline Ca2+. E, steady-state CDF vs. Ca2+ curves at the two voltages assessed are obtained by plotting normalized peak tail currents against instantaneous Ca2+ levels. F, CDF vs. Ca2+ curves of the 2 voltages are normalized, showing the same Ca2+ sensitivity. G and H, population data (mean ± STD) show similar results, with n = 9 cells. Fitted curves have nHill = 1.5, EC50 = 0.3 μm.
Figure 9C displays the experimental outcome for an exemplar cell expressing CaV2.1 EFa −47 channels fluxing Ca2+. The voltage pulse protocol is displayed in the top row, with the global Ca2+ concentration readout shown below in the second row. The same resulting tail-current traces are displayed at two gains in the remaining rows. In the third row, the higher-gain trace shows well the smaller tail currents following pulses to 0 mV (black segments of trace, cyan circles) and the segments containing the larger tail currents after 60 mV pulses are plotted in grey and clipped to the frame. The fourth row plots the lower-gain trace optimized for display of larger tail currents after 60 mV pulses (black segments of trace, rose circles), and the segments containing tails after 0 mV pulses are shown in grey. With this setup in mind, the manifestation of CDF can be readily appreciated. Prior to Ca2+ uncaging, tail currents remain steady between pulses and immediately following Ca2+ uncaging (vertical violet bar) there is strong CDF as demonstrated by the substantial enhancement of tail currents following pulses to both 0 and 60 mV. This observation is corroborated by expanded time base displays of current immediately before and after the uncaging event (Fig.9D), which support nearly twofold enhancement of tails following 0 mV pulses. Following Ca2+ uncaging, global Ca2+ levels gradually relaxed over the course of minutes, as shown in the remainder of Fig.9C via a display of four time segments punctuated by time-axis breaks. Likewise, tail current amplitudes diminished in parallel with the decline of Ca2+, where the horizontal cyan and rose lines reference amplitudes before Ca2+ elevation. Importantly, upon the return of Ca2+ to baseline (rightmost segment), tail currents were closely similar to those prior to uncaging, allowing interpretation of intervening currents in terms of CDF apart from run-down. We thus plotted fold increase in tail current amplitude as a function of instantaneous Ca2+ to obtain the quasi-steady-state CDF–Ca2+ correlations in Fig.9E. Reassuringly, the functions for 0 and 60 mV closely resembled the actual steady-state relations obtained using the original protocol with Li+ currents (Fig.7C). As expected, the larger CDF observed for 0 versus 60 mV pulses reflects the voltage dependence of unfacilitated versus facilitated modes of gating (Fig.5A and F). Indeed, amplitude scaling of both plots (Fig.9F) furnished a normalized relation tightly resembling that obtained at steady state (Fig.7E), with nearly the same half-Ca2+ level (EC50 = 0.3 μm) and slightly diminished Hill coefficient of 1.5 (compared with EC50 = 0.5 μm and nHill = 1.8 in Fig.7E). The small differences here may reflect imperfect exclusion of the local Ca2+ component in this sparse-tail-current protocol. Similar analysis in multiple cells furnished equivalent results (Fig.9G and H). Thus, we established that this sparse-tail-current protocol could assess CDF–Ca2+ relations with near quantitative precision while employing Ca2+ as the charge carrier through channels.
Prominent CDF in native CaV2.1 in cerebellar Purkinje neurons
Thus armed, we turned to characterization of CDF in native CaV2.1 currents in cerebellar Purkinje neurons, renowned for the predominance of these Ca2+ channels (Mintz et al. 1995). Acutely dissociated neurons from P14–P18 mice were used (Fig.10A, inset), so as to maximize the presence of EFa versus EFb variants (∼85% EFa transcripts; Chang et al. 2007). Although the presence of CDF in these currents had been detected previously (Chaudhuri et al. 2005; Adams et al. 2010), the means of quantification and characterization were indirect, the extents of CDF seemingly modest, and the quantitative dependence on Ca2+ concentration unspecified.
Figure 10. CDF in native CaV2.1 channels within cerebellar Purkinje cells.

A, acutely dissociated Purkinje cells were identified by their large size and tear-drop shape (picture inset). CaV2.1 Ca2+ currents stimulated by ∼50 Hz action potential waveforms are stable and show little facilitation near baseline Ca2+. B, Ca2+ uncaging in the midst of the action potential train triggers large CDF. C–E, Ca2+ sensitivities of CDF at 2 voltages were determined with a protocol similar to that in Fig.9C. Steady currents are seen in the absence of uncaging (C), while uncaging to different Ca2+ levels triggers large increases in current (D and E). F, tail currents around the time of Ca2+ uncaging are shown with expanded time base, normalized to the peak levels prior to uncaging. G, relative increase in peak tail currents plotted against Ca2+ for the above exemplar cell yields steady-state CDF vs. Ca2+ curves for −10 and 40 mV. H, data in G, normalized, demonstrate the same Ca2+ sensitivity for the two voltages. I and J, population data (mean ± STD) show similar results, with n = 5 cells. Fitted curves have nHill = 1.3, EC50 = 0.5 μm.
To corroborate directly the existence of strong CDF in native CaV2.1 channels, we evoked native Ca2+ currents with trains of action potential waveforms (50 Hz), and simultaneously introduced Ca2+ uncaging events. During baseline conditions without uncaging (Fig.10A), currents appeared steady (horizontal cyan line) from the beginning to end of a train, and Ca2+ readouts remained constant near 0.1 μm. Thus, it appeared that the local Ca2+ component (Fig.9B) was insufficient to trigger appreciable CDF under these conditions. In the next train of action potentials (Fig.10B), currents were initially indistinguishable from those in the previous train (horizontal cyan line). However, upon strong uncaging to several micromolar Ca2+ (vertical violet bar), pronounced ∼1.6× amplification of currents was produced, indicating the potential for large native CDF matching that observed in previous figures for recombinant channels.
To quantify the Ca2+ sensitivity of this CDF, we applied the sparse-tail-current protocol devised in Fig.9B. Results from a single exemplar Purkinje neuron are reported in Fig.10C–E, using the same display format as in Fig.9C. Absent Ca2+ uncaging (Fig.10C) currents remained steady and bulk Ca2+ persisted near 0.1 μm, all indicative of a stable baseline. Upon moderate Ca2+ uncaging (Fig.10D), currents exhibited modest amplification which decayed as Ca2+ levels relaxed. Upon intense Ca2+ uncaging (Fig.10E), currents manifested marked and sustained enhancement of nearly twofold for −10 mV pulses. This modulation can be further appreciated in the expanded time base plots of currents immediately before and after uncaging (Fig.10F). Because of slow drift of currents following large Ca2+ elevations in these neurons, we could not in general compile traces over the course of minutes (as in Fig.9C), but were instead limited to analysing data from several trace segments, each containing a different-sized uncaging event. Using this strategy in this exemplar neuron, plots of fold enhancement of tail currents as a function of Ca2+ yield the CDF–Ca2+ relations in Fig.10G. Reassuringly, these bear a strong resemblance to those obtained for recombinant CaV2.1 channels (Fig.9E) and amplitude scaling yields an aggregate normalized relationship (Fig.10H, EC50 = 0.5 μm and nHill = 1.3) closely similar to that of recombinant channels undergoing the same protocol (Fig.9F). Population data from n = 5 neurons replicate these findings (Fig.10I and J) and firmly establish strong native CDF, whose parameters largely accord with those of recombinant channels. Compared to recombinant channels, the slightly diminished maximal CDF in native currents (Fig.10I) fits with a modest fraction of CaV2.1 EFb variants that lack CDF (∼15% transcripts; Chang et al. 2007).
Discussion
This study reveals the long-sought speed and extent of Ca2+-dependent facilitation (CDF) of CaV2.1 channels. Using Ca2+ photo-uncaging as a signal generator of intracellular Ca2+, we demonstrate that CDF is considerably larger, faster, and more Ca2+ sensitive than previously imagined. This performance profile positions CaV2.1 channels for extensive CDF under neurophysiological activity, offering the potential for strong activity-dependent adjustment to downstream signalling pathways. Additionally, the new and extensive Ca2+ and electrophysiological constraints furnished by our approach significantly advance mechanistic understanding of this regulatory system, which is formalized by a realistic biophysical model of CaV2.1 gating and CDF. Overall, these findings hold key implications for CDF in relation to structural determinants, functional mechanisms and neurophysiological roles, as follows.
Structural determinants of CDF
During baseline characterization via Ca2+ uncaging, important aspects of the structural basis of CDF were refined. From qualitative measures of CDF, the carboxy tail of CaV2.1 has previously been suggested to be essential for CDF (Mori et al. 2008), just as for CaM regulation of CaV1 channels (Ben-Johny et al. 2013). In this regard, a first advance in the present study was to revise previous suggestions that alternative splicing at exon 37 (encoding EFa or EFb modules within the carboxy tail) enables or disables CDF, but in a manner modulated by inclusion of exon 47 on the extreme channel carboxy terminus (Chaudhuri et al. 2004). Using the higher-resolution Ca2+ uncaging assay used here, we now demonstrate that the EFa/EFb module fully switches CDF on and off (Fig.3C and E), and that inclusion or exclusion of exon 47 does not affect CaV2.1 CDF. This simplifying feature strengthens the view that CDF may be exquisitely tuned in the human brain, since the EFa:EFb variant ratio is regulated in an age-specific, region-specific and sex-specific manner (Chaudhuri et al. 2004, 2005; Chang et al. 2007). Furthermore, the complete switching of CDF at the EF locus offers a simple strategy to create transgenic animal models that lack or possess CDF, thereby aiding clarification of its physiological role. Similarly, one wonders whether diseases of aberrant splicing (Tazi et al. 2009) might act at this EF locus. Second, beyond the EF element, substituting alanine for the central isoleucine within the IQ domain also eliminates CDF under the Ca2+ uncaging regime (Fig.3G), affirming further the critical role of the IQ element for CDF (DeMaria et al. 2001; Lee et al. 2003; Mori et al. 2008). Third, the elimination of CDF by the Ca2+-insensitive mutant CaM1234 (Fig.3F) consolidates the role of Ca2+-free CaM (apoCaM) as a resident CDF sensor bound to the channel carboxy terminus (DeMaria et al. 2001). Finally, our finding of a Hill coefficient near 2 for CDF–[Ca2+] relations (Figs 3E and 6F) affirms previous conclusions that CDF is predominantly triggered by Ca2+ binding to the C-terminal lobe of CaM (DeMaria et al. 2001), which contains two highly cooperative EF-hand Ca2+ binding sites.
Advances in functional mechanisms
Our experiments confirm an ‘enhanced-opening’ mechanism of CDF whereby the maximal open probability of channels at saturating depolarization is enhanced (Chaudhuri et al. 2007). That said, with the improved resolution in the present study, we now also detect a 5 mV left shift of the voltage activation relation (Fig.5C and E) (Borst & Sakmann, 1998), whereas our previous study lacked the sensitivity to detect this effect (Chaudhuri et al. 2007). Though modest, it is this shift in voltage dependence that amplifies CDF significantly during physiological patterns of activation (Figs 5D and F, and 6B and E). These effects could be almost entirely explained by enhancing the last transition to the open state (Fig.5, kf) while reducing the closing rate from the open state (kr), leaving all other transitions largely unchanged.
As for the Ca2+-induced transitions among unfacilitated and prefacilitated modes of gating (vertical arrows in Fig.8B), we find that these transitions can be approximated as the same, regardless of position along the activation pathway. Physically speaking, this feature suggests that Ca2+ binding to calmodulin is largely insensitive to the status of voltage activation. We further suggest that the transitions between prefacilitated and facilitated gating modes are also insensitive to voltage activation, except when the actual open states are reached (Fig.8B; also, see ‘Simulations: CaV2.1 channel models’ in Methods). This latter attribute hints that the binding of Ca2+–CaM to a channel effector site is mainly sensitive to the final concerted opening step, rather than movements of voltage-sensing paddles.
Together, all these features can be incorporated within an explicit biophysical gating mechanism (Fig.8B), arguably the most extensive for calmodulin regulation of any channel. Indeed, the CDI of CaV1.2/3 channels, as triggered by the C-lobe of CaM (Peterson et al. 1999; Yang et al. 2006), may turn out to be described by an analogous mechanism (Fig.8B), except that kf/kr in the bottom gating mode would be reduced, and the modes would be described as non-inactivated, pre-inactivated, and inactivated.
A final novel functional feature concerns the stark contrast in the Ca2+ sensitivity of CDF versus CDI (Fig. 4E), far more than imagined previously. Whereas the EC50 for CDF is ∼0.5 μm Ca2+, the corresponding value for CDI would approximate ∼22 μm. Recalling the earlier finding that CaV2.1 CDI is triggered by Ca2+ binding to the N-lobe of CaM (DeMaria et al. 2001), as well as the mechanistic framework developed previously (Tadross et al. 2008), we suggest that this relative Ca2+ insensitivity of CDI may arise from a far greater channel affinity for the N-terminal lobe of apoCaM compared with Ca2+–CaM. The wide divergence of EC50 values for CDF and CDI would allow facilitation to develop in isolation over sustained periods of Ca2+ elevation of less than ∼10 μm (Tanaka et al. 2007). The induction of CDI might be reserved for clustered channels positioned at local subcompartments where Ca2+ may achieve concentrations exceeding that in bulk cytoplasm. The previous observation that CDI is only apparent in a fraction of cerebellar Purkinje neurons (Chaudhuri et al. 2005) may coincide with this large EC50 for CDI, and the sporadic attainment of these Ca2+ levels. Interestingly, previous estimates of Ca2+ sensitivity for CDF and CDI in the calyx of Held (EC50 values of 2.5 μm and 6 μm, respectively; Lin et al. 2012) are lower than the estimates provided here. This apparent discrepancy is most likely the result of differences in channel isoform expression. For example, calyces from young, P8–P10 rats used in the previous report express a significant percentage of CaV2.2 and CaV2.3 channels (Iwasaki & Takahashi, 1998; Wu et al. 1998, 1999; Chaudhuri et al. 2005), which do not have appreciable CDF (Liang et al. 2003) and may have lower Ca2+ requirements for CDI than CaV2.1 (Nakamura et al. 2008). In contrast, the Ca2+ currents recorded from Purkinje neurons isolated from older animals (P14–P18) in our present study are predominantly from the facilitating EFa subtype of CaV2.1 (Regan, 1991; Chang et al. 2007), allowing a more specific assessment of the Ca2+ responsiveness of CaV2.1 channels. Furthermore, Lin et al. used the ratio of cumulative Ca2+ current during the first 3 ms of a 20 ms test pulse with or without Ca2+ uncaging (Q3ms,Ca/Q3ms,ctrl) as their metric for CDF. However, our measure of CDF kinetics (τon ∼2 ms, Fig.8) indicates that a significant fraction of channels shift into the facilitated state within the first 3 ms of a test pulse. Because their baseline measure (Q3ms,ctrl) includes prefacilitated channels, their assay likely underestimates the magnitude and sensitivity of CDF.
Neurophysiological implications
Our experiments in cerebellar Purkinje neurons (Fig.10) not only substantiate the existence of CDF within these neurons (Chaudhuri et al. 2005), but reveal just how large CDF can be in this setting (∼1.8× in Fig.10G and I). The similarity of Ca2+ sensitivity for CDF in these neurons (EC50 ∼0.5 μm in Fig.10J), compared to that for recombinant channels (∼0.5 μm in Fig.7F), further supports CaM as the Ca2+ sensor for CDF (Chaudhuri et al. 2005). Given the prominence of Ca2+ signalling in Purkinje neurons (Gruol et al. 2012), along with the primacy of CaV2.1 as a Ca2+ entry portal in this locus (Usowicz et al. 1992; Hartmann & Konnerth, 2005), the CDF of CaV2.1 channels seems particularly well poised for physiological influence in this context.
A first potential role concerns recent exciting findings that coordinated motor control may require cerebellar Purkinje neurons to spontaneously fire simple spikes with regular and uniform rate (Hoebeek et al. 2005; Otis & Jen, 2006; Walter et al. 2006). Empirically speaking, this requirement can be verified by correlating altered coordination with pharmacological and/or genetic modulation of spike regularity (Walter et al. 2006). From the theoretical standpoint, the need for constant spiking may relate to promising synchronization encoding mechanisms wherein motor information is conveyed through selected deep cerebellar nuclei only at appropriate times, in particular when inhibitory inputs from multiple convergent Purkinje neurons become synchronized (Medina & Khodakhah, 2012; Person & Raman, 2012a). Achieving such synchrony, for example following a shared complex-spike phase reset among convergent Purkinje neurons, would require these neurons to manifest a regular and uniform spontaneous simple spike rate (Person & Raman, 2012b). We can connect this fundamental substrate of motor coordination to CaV2.1 CDF by noting that phasic activation of small-conductance Ca2+-activated K+ channels (SK channels), which drives after-hyperpolarization following spikes, is a prime determinant of spontaneous simple spike rate (Swensen & Bean, 2003). Moreover, the dominant trigger of SK channels involves preferential Ca2+ signalling from colocalized CaV2.1 channels fluxing peak Ca2+ current during the repolarizing phase of spikes (e.g. Fig.5B) (Swensen & Bean, 2003; Womack et al. 2004). Accordingly, attenuation of either SK or CaV2.1 channels, such as in heritable forms of ataxia, begets rate variability and motor dyscoordination (Otis & Jen, 2006). We suggest that a potential role of CaV2.1 CDF could be to enhance the regularity of simple spike rate in an elegant servo control mechanism, as follows: perturbations increasing the spiking rate would heighten global Ca2+, boost CaV2.1 CDF and SK channel activation, and thereby downwardly restore rate towards an intrinsic setpoint (Fig.1A). Conversely, transient rate decreases would lower global Ca2+, weaken CDF and SK channel activity, and consequently upwardly restore rate (Fig.1B). Thus, we hypothesize that CaV2.1 CDF may play a role in optimizing motor coordination. Indeed, the postnatal developmental increase of CDF (due to switching from CaV2.1 EFb to EFa variants; Chaudhuri et al. 2005) closely parallels the time course of motor coordinative improvements (Arata & Ito, 2005)11.
Figure 11. Neurophysiological implications of CaV2.1 CDF.

A, diagram illustrating the role of CDF in homeostatic rate control of Purkinje cells. Increased rate of Purkinje cell spiking elevates global Ca2+, facilitating CaV2.1 opening via CDF. Increased CaV2.1 activity in turn activates colocalized SK channels, resulting in stronger membrane hyperpolarization that then reduces spiking rate. B, similarly, decreased spiking rate leads to lower global Ca2+, reduced CDF, and less SK activation, finally resulting in weaker hyperpolarization and restoration of spiking rate. C, Ca2+ dose–response curves for CDF (continuous black line) and neurotransmitter secretion (red line). Additionally, Ca2+ sensitivity of paired-pulse facilitation due to residual Ca2+ (Regehr et al. 1994) (dashed line) shows an additional mechanism specifying the extent of transmitter release. D, generic nerve terminal is modelled as a sphere with CaV2.1 channels uniformly expressed on the membrane. A thin shell compartment adjacent to the membrane forms the nanodomain within which the CDF Ca2+ sensor CaM, which acts as a channel subunit, actually senses Ca2+. Ca2+ within the shell compartment can diffuse to and from the much larger cytosolic compartment. The cytosolic compartment is in turn connected to a third and much larger compartment representing the axon branch itself. The Ca2+ concentration within this axonal compartment is set at a fixed resting level, and thus serves to returns Ca2+ within the terminal back to its resting levels after periods of activity. The parameters for this system are: rsphere = 8 μm, rcyto = 7.9 μm, Nchan = 60,000, ksc = kcs = 6 × 10−13 dm3 ms−1, and kefflux = 6 × 10−14 dm3 ms−1, with the parameters adjusted such that simulations in E match data from Cuttle et al. (1998). Note that the simulated volume is larger than physical terminals because Ca2+ buffers are not explicitly modelled here. E, simulations of the system in D in response to 100 Hz step depolarizations to −20 mV. CaV2.1 currents are elicited (bottom row) by the voltage stimulation (top row), which results in a build-up of Ca2+ in both the shell (second row) and cytosolic (third row) compartments. The increased Ca2+ induces CDF of CaV2.1 channels, resulting in gradual amplification of ICa, Cashell and Cacyto. F, CDF at steady state is inversely related to stimulation frequency (blue circles and continuous line), largely due to frequency-dependent accumulation of residual Ca2+ (100 Hz example in red).
Secondly, given the predominance of CaV2.1 channels in triggering transmitter release across the CNS (Uchitel et al. 1992; Wheeler et al. 1994; Dunlap et al. 1995), CDF might contribute presynaptically to short-term synaptic plasticity, either to boost outright facilitation or to buffer the extent of depression. Indeed, trains of spike-like depolarizations induce large 1.8× facilitation of rat (P9–P17) CaV2.1 currents in the calyx of Held (Cuttle et al. 1998). Given that neurotransmitter release probability is proportional to Ca2+ concentration to the third or fourth power (Schneggenburger & Neher, 2000; Xu et al. 2007), this CDF could orchestrate order-of-magnitude (Fig.10G–J, 1.84 ∼10) modulation of synaptic strength. Of note, reports of weaker CDF in calyceal CaV2.1 currents restricted experiments to conditions minimizing vesicle depletion (Muller et al. 2008), with the result that Ca2+ levels (252 nm) may have been insufficient to recruit much of CDF by our Ca2+ sensitivity measures (arrow in Fig.1C). Additionally, it is worth noting that whereas CaM is well accepted as the trigger for CaV2.1 CDI in the calyx (Xu et al. 2007; Nakamura et al. 2008), neuronal calcium sensor-1 (NCS-1) has been proposed to mediate CDF (Tsujimoto et al. 2002). However, biophysical studies in other cell types suggest NCS-1 alters CaV2.1 current, with little to no effect on CDF (Weiss et al. 2010; Yan et al. 2014). As such, we propose that CaM is the predominant molecular sensor for CaV2.1 CDF in the calyx, although NCS-1 may modulate CaV2.1 currents indirectly through other pathways (Weiss et al. 2010). More broadly, we anticipate a strong contribution of CDF to synaptic plasticity at other synapses. When we embedded our in silico channel mechanism (Fig.8B) within a generic three-compartment spherical terminal (Fig.1D), we reproduced (Fig.1E) both the kinetics and magnitude of experimentally observed CDF in the calyx (Cuttle et al. 1998). In this scheme (Fig.1D), Ca2+ enters initially through CaV2.1 channels into a submembranous shell compartment, followed by rapid diffusion into a larger cytosolic compartment. This cytosolic space is in turn connected with an efflux pathway into a large axonal compartment set at resting Ca2+. When simulated CaV2.1 channels are opened with a 1 ms depolarization to −20 mV (Fig.1E, top), peak Ca2+ currents (bottom row) caused Ca2+ elevation in the shell and cytoplasmic compartments (Cashell and Cacyto in rows 3–4). The shell compartment experiences large Ca2+ spikes coincident with channel opening, each followed by rapid relaxation to the cytosolic (global) pedestal. The global cytoplasmic Ca2+ rises with each stimulus during a 100 Hz train, eventually reaching a quasi plateau where Ca2+ influx matches its efflux. Interestingly, even though CaM physically senses local Ca2+ within the shell, it is the global Ca2+ component in this compartment that drives much of CDF when brief spikes are sufficiently spread out. Thus, the time course of CDF tracks the rise and fall of global Ca2+, much as in our sparse-tail protocol (Fig.9A). Altering stimulus frequency reveals that CDF quickly decays with slower pacing (Fig.1F), projecting substantial contributions to short-term synaptic plasticity.
In this light, it is worth quantifying how CDF fits with other facilitatory mechanisms underlying rapid synaptic upregulation (Fig.1C). The red curve plots the Ca2+ sensitivity of outright neurotransmitter release, presumably as specified by Ca2+ binding properties at C2 sites on synaptotagmin (Schneggenburger & Neher, 2000). High Ca2+ concentrations (>10 μm) may be required for this process, such as produced by clusters of CaV2.1 channels near neurotransmitter vesicle complexes (Sheng et al. 2012). However, accumulation of ‘residual’ presynaptic Ca2+ from prior activity (Zucker & Regehr, 2002), insufficient to trigger release, may nonetheless preload unspecified high-affinity Ca2+ binding sites (Regehr et al. 1994; Awatramani et al. 2005) to boost ensuing release according to a residual Ca2+ (RC) mechanism (Fig.1C, dashed RC curve). Here, our Ca2+ uncaging experiments project CaV2.1 CDF as another relevant system, in this case with intermediate Ca2+ sensitivity (Fig.1C, black curve). Of note, synapses in the calyx of Held of transgenic mice lacking CaV2.1 (compensated by CaV2.2) exhibit little to no short-term synaptic facilitation (Inchauspe et al. 2007). That said, we do not believe that RC and CDF need be one and the same process, as synaptic paired-pulse facilitation in the calyx of Held can be observed in the absence of CDF (Muller et al. 2008). Overall, it seems likely that CDF and RC processes will differ in their relative contributions at various synapses, and the RC category may encompass several as yet loosely defined processes. That said, our extensive biophysical clarification of CaV2.1 CDF, in both recombinant and native currents, now leaves little doubt that this Ca2+ regulation will figure prominently in the CNS.
Acknowledgments
We thank Dr Terry Snutch for providing parental CaV2.1 clones, Dr Ye Chen-Izu for providing flash lamp equipment, Dr Indira Raman for providing Purkinje cell action potential waveforms, Manu Ben-Johny and Ivy Dick for guidance with Ca2+ uncaging experiments, Wanjun Yang for technical support, and the Calcium Signals Lab for valuable comments.
Glossary
- CaM
calmodulin
- CDF
calcium-dependent facilitation
- CDI
calcium-dependent inactivation
- RC
residual calcium mechanism of paired-pulse facilitation
- VDI
voltage-dependent inactivation
Additional information
Competing interests
The authors have no conflicts of interest to declare.
Author contributions
All experiments were performed in the Calcium Signals Laboratory at the Johns Hopkins University School of Medicine. S.-R.L. created pore mutants, performed electrophysiological experiments, analysed data and wrote the code for the simulations; P.J.A. isolated cerebellar Purkinje neurons, and advised on their electrophysiological characterization. S.-R.L. and D.T.Y. conceived and designed the project, created figures, and wrote the initial manuscript. S.-R.L. and P.J.A. critically reviewed and revised the manuscript and gave final approval of the version to be published.
Funding
This study was supported by grants from NIMH (D.T.Y.), Parkinson Society Canada (P.J.A.) and NIH MSTP grant (S.-R.L.).
References
- Abbott LF. Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Adams PJ, Rungta RL, Garcia E, van den Maagdenberg AM, MacVicar BA. Snutch TP. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc Natl Acad Sci U S A. 2010;107:18694–18699. doi: 10.1073/pnas.1009500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arata A. Ito M. Synaptic transmission and long-term depression in Purkinje cells in an in vitro block preparation of the cerebellum isolated from neonatal rats. Prog Brain Res. 2005;148:111–123. doi: 10.1016/S0079-6123(04)48010-8. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD. Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Ben-Johny M, Yang PS, Bazzazi H. Yue DT. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat Commun. 2013;4:1717. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M, Yang PS, Niu J, Yang W, Joshi-Mukherjee R. Yue DT. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell. 2014;157:1657–1670. doi: 10.1016/j.cell.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG. Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm P. Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- Brody DL, Patil PG, Mulle JG, Snutch TP. Yue DT. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J Physiol. 1997;499:637–644. doi: 10.1113/jphysiol.1997.sp021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Leal K. Nanou E. Calcium channels and short-term synaptic plasticity. J Biol Chem. 2013;288:10742–10749. doi: 10.1074/jbc.R112.411645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SY, Yong TF, Yu CY, Liang MC, Pletnikova O, Troncoso J, Burgunder JM. Soong TW. Age and gender-dependent alternative splicing of P/Q-type calcium channel EF-hand. Neuroscience. 2007;145:1026–1036. doi: 10.1016/j.neuroscience.2006.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, Suzuki Y, Zysk JR. Cheung WY. Activation of calmodulin by various metal cations as a function of ionic radius. Mol Pharmacol. 1984;26:75–82. [PubMed] [Google Scholar]
- Charlton MP, Smith SJ. Zucker RS. Role of presynaptic calcium ions and channels in synaptic facilitation and depression at the squid giant synapse. J Physiol. 1982;323:173–193. doi: 10.1113/jphysiol.1982.sp014067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Alseikhan BA, Chang SY, Soong TW. Yue DT. Developmental activation of calmodulin-dependent facilitation of cerebellar P-type Ca2+ current. J Neurosci. 2005;25:8282–8294. doi: 10.1523/JNEUROSCI.2253-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Chang SY, DeMaria CD, Alvania RS, Soong TW. Yue DT. Alternative splicing as a molecular switch for Ca2+/calmodulin-dependent facilitation of P/Q-type Ca2+ channels. J Neurosci. 2004;24:6334–6342. doi: 10.1523/JNEUROSCI.1712-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Issa JB. Yue DT. Elementary mechanisms producing facilitation of Cav2.1 (P/Q-type) channels. J Gen Physiol. 2007;129:385–401. doi: 10.1085/jgp.200709749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig PJ, McAinsh AD, McCormack AL, Smith W, Beattie RE, Priestley JV, Yip JL, Averill S, Longbottom ER. Volsen SG. Distribution of the voltage-dependent calcium channel α1A subunit throughout the mature rat brain and its relationship to neurotransmitter pathways. J Comp Neurol. 1998;397:251–267. [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID. Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS. Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- De Waard M. Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. J Physiol. 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI. Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- Ellinor PT, Yang J, Sather WA, Zhang JF. Tsien RW. Ca2+ channel selectivity at a single locus for high-affinity Ca2+ interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ. Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- Finch EA, Tanaka K. Augustine GJ. Calcium as a trigger for cerebellar long-term synaptic depression. Cerebellum. 2012;11:706–717. doi: 10.1007/s12311-011-0314-x. [DOI] [PubMed] [Google Scholar]
- Gruol D, Manto M. Haines D. Ca2+ signaling in cerebellar Purkinje neurons–editorial. Cerebellum. 2012;11:605–608. doi: 10.1007/s12311-012-0404-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J. Konnerth A. Determinants of postsynaptic Ca2+ signaling in Purkinje neurons. Cell Calcium. 2005;37:459–466. doi: 10.1016/j.ceca.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB. Tsien RW. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J Gen Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman D, Chen S, Aung TT, Cherksey B, Sugimori M. Llinás RR. Localization of P-type calcium channels in the central nervous system. Proc Natl Acad Sci U S A. 1991;88:7076–7080. doi: 10.1073/pnas.88.16.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK. Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hoebeek FE, Stahl JS, van Alphen AM, Schonewille M, Luo C, Rutteman M, van den Maagdenberg AM, Molenaar PC, Goossens HH, Frens MA. De Zeeuw CI. Increased noise level of Purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45:953–965. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Inchauspe CG, Forsythe ID. Uchitel OD. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J Physiol. 2007;584:835–851. doi: 10.1113/jphysiol.2007.139683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indriati DW, Kamasawa N, Matsui K, Meredith AL, Watanabe M. Shigemoto R. Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J Neurosci. 2013;33:3668–3678. doi: 10.1523/JNEUROSCI.2921-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S. Takahashi T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J Physiol. 1998;509:419–423. doi: 10.1111/j.1469-7793.1998.419bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F. Minor DL., Jr Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure. 2008;16:1455–1467. doi: 10.1016/j.str.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont MG. Weber JT. The role of calcium in synaptic plasticity and motor learning in the cerebellar cortex. Neurosci Biobehav Rev. 2012;36:1153–1162. doi: 10.1016/j.neubiorev.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Lee A, Scheuer T. Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T. Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Lee A, Zhou H, Scheuer T. Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of Cav2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA. Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lin KH, Erazo-Fischer E. Taschenberger H. Similar intracellular Ca2+ requirements for inactivation and facilitation of voltage-gated Ca2+ channels in a glutamatergic mammalian nerve terminal. J Neurosci. 2012;32:1261–1272. doi: 10.1523/JNEUROSCI.3838-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleskey EW. Almers W. The Ca channel in skeletal muscle is a large pore. Proc Natl Acad Sci U S A. 1985;82:7149–7153. doi: 10.1073/pnas.82.20.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina JF. Khodakhah K. Neuroscience: Spikes timed through inhibition. Nature. 2012;481:446–447. doi: 10.1038/481446a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL. Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Yamasaki M, Hashimoto K, Yamazaki M, Abe M, Usui H, Kano M, Sakimura K. Watanabe M. Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J Neurosci. 2012;32:1311–1328. doi: 10.1523/JNEUROSCI.2755-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Few AP, Scheuer T. Catterall WA. Regulation of presynaptic CaV2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–216. doi: 10.1016/j.neuron.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Mori MX, Vander Kooi CW, Leahy DJ. Yue DT. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+ Structure. 2008;16:607–620. doi: 10.1016/j.str.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T. Numa S. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- Muller M, Felmy F. Schneggenburger R. A limited contribution of Ca2+ current facilitation to paired-pulse facilitation of transmitter release at the rat calyx of Held. J Physiol. 2008;586:5503–5520. doi: 10.1113/jphysiol.2008.155838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yamashita T, Saitoh N. Takahashi T. Developmental changes in calcium/calmodulin-dependent inactivation of calcium currents at the rat calyx of Held. J Physiol. 2008;586:2253–2261. doi: 10.1113/jphysiol.2007.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS. Jen JC. Blessed are the pacemakers. Nat Neurosci. 2006;9:297–298. doi: 10.1038/nn0306-297. [DOI] [PubMed] [Google Scholar]
- Patil PG, Brody DL. Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Person AL. Raman IM. Purkinje neuron synchrony elicits time-locked spiking in the cerebellar nuclei. Nature. 2012a;481:502–505. doi: 10.1038/nature10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AL. Raman IM. Synchrony and neural coding in cerebellar circuits. Front Neural Circuits. 2012b;6:97. doi: 10.3389/fncir.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP. Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Regan LJ. Voltage-dependent calcium currents in Purkinje cells from rat cerebellar vermis. J Neurosci. 1991;11:2259–2269. doi: 10.1523/JNEUROSCI.11-07-02259.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG. Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol. 2012;4:a005702. doi: 10.1101/cshperspect.a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR. Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R. Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Sheng J, He L, Zheng H, Xue L, Luo F, Shin W, Sun T, Kuner T, Yue DT. Wu LG. Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat Neurosci. 2012;15:998–1006. doi: 10.1038/nn.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong TW, DeMaria CD, Alvania RS, Zweifel LS, Liang MC, Mittman S, Agnew WS. Yue DT. Systematic identification of splice variants in human P/Q-type channel α12.1 subunits: implications for current density and Ca2+-dependent inactivation. J Neurosci. 2002;22:10142–10152. doi: 10.1523/JNEUROSCI.22-23-10142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM. Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Dick IE. Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Tsien RW. Yue DT. Ca2+ channel nanodomains boost local Ca2+ amplitude. Proc Natl Acad Sci U S A. 2013;110:15794–15799. doi: 10.1073/pnas.1313898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Khiroug L, Santamaria F, Doi T, Ogasawara H, Ellis-Davies GC, Kawato M. Augustine GJ. Ca2+ requirements for cerebellar long-term synaptic depression: role for a postsynaptic leaky integrator. Neuron. 2007;54:787–800. doi: 10.1016/j.neuron.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Tay LH, Dick IE, Yang W, Mank M, Griesbeck O. Yue DT. Nanodomain Ca2+ of Ca2+ channels detected by a tethered genetically encoded Ca2+ sensor. Nat Commun. 2012;3:778. doi: 10.1038/ncomms1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazi J, Bakkour N. Stamm S. Alternative splicing and disease. Biochim Biophys Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Jeromin A, Saitoh N, Roder JC. Takahashi T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science. 2002;295:2276–2279. doi: 10.1126/science.1068278. [DOI] [PubMed] [Google Scholar]
- Uchitel OD, Protti DA, Sanchez V, Cherksey BD, Sugimori M. Llinás R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc Natl Acad Sci U S A. 1992;89:3330–3333. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usowicz MM, Sugimori M, Cherksey B. Llinás R. P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron. 1992;9:1185–1199. doi: 10.1016/0896-6273(92)90076-p. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C. Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Hui H. Burgoyne RD. Neuronal calcium sensor-1 regulation of calcium channels, secretion, and neuronal outgrowth. Cell Mol Neurobiol. 2010;30:1283–1292. doi: 10.1007/s10571-010-9588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP. Catterall WA. Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DB, Randall A. Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P. Tsien RW. CaV1 and CaV2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack M. Khodakhah K. Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar Purkinje neurons. J Neurosci. 2002;22:10603–10612. doi: 10.1523/JNEUROSCI.22-24-10603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C. Khodakhah K. Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci. 2004;24:8818–8822. doi: 10.1523/JNEUROSCI.2915-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD. Khodakhah K. Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J Neurosci. 2004;24:3511–3521. doi: 10.1523/JNEUROSCI.0290-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Borst JG. Sakmann B. R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci U S A. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA. Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, He L. Wu LG. Role of Ca2+ channels in short-term synaptic plasticity. Curr Opin Neurobiol. 2007;17:352–359. doi: 10.1016/j.conb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Yan J, Leal K, Magupalli VG, Nanou E, Martinez GQ, Scheuer T. Catterall WA. Modulation of CaV2.1 channels by neuronal calcium sensor-1 induces short-term synaptic facilitation. Mol Cell Neurosci. 2014;63:124–131. doi: 10.1016/j.mcn.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ellinor PT, Sather WA, Zhang JF. Tsien RW. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 1993;366:158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]
- Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA. Yue DT. Switching of Ca2+-dependent inactivation of Cav1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]