

Abstract
Key points
Although learning can arise from few or even a single trial, synaptic plasticity is commonly assessed under prolonged activation. Here, we explored the existence of rapid responsiveness of synaptic plasticity at corticostriatal synapses in a major synaptic learning rule, spike-timing-dependent plasticity (STDP).
We found that spike-timing-dependent depression (tLTD) progressively disappears when the number of paired stimulations (below 50 pairings) is decreased whereas spike-timing-dependent potentiation (tLTP) displays a biphasic profile: tLTP is observed for 75–100 pairings, is absent for 25–50 pairings and re-emerges for 5–10 pairings.
This tLTP induced by low numbers of pairings (5–10) depends on activation of the endocannabinoid system, type-1 cannabinoid receptor and the transient receptor potential vanilloid type-1.
Endocannabinoid-tLTP may represent a physiological mechanism operating during the rapid learning of new associative memories and behavioural rules characterizing the flexible behaviour of mammals or during the initial stages of habit learning.
Abstract
Synaptic plasticity, a main substrate for learning and memory, is commonly assessed with prolonged stimulations. Since learning can arise from few or even a single trial, synaptic strength is expected to adapt rapidly. However, whether synaptic plasticity occurs in response to limited event occurrences remains elusive. To answer this question, we investigated whether a low number of paired stimulations can induce plasticity in a major synaptic learning rule, spike-timing-dependent plasticity (STDP). It is known that 100 pairings induce bidirectional STDP, i.e. spike-timing-dependent potentiation (tLTP) and depression (tLTD) at most central synapses. In rodent striatum, we found that tLTD progressively disappears when the number of paired stimulations is decreased (below 50 pairings) whereas tLTP displays a biphasic profile: tLTP is observed for 75–100 pairings, absent for 25–50 pairings and re-emerges for 5–10 pairings. This tLTP, induced by very few pairings (∼5–10) depends on the endocannabinoid (eCB) system. This eCB-dependent tLTP (eCB-tLTP) involves postsynaptic endocannabinoid synthesis, requires paired activity (post- and presynaptic) and the activation of type-1 cannabinoid receptor (CB1R) and transient receptor potential vanilloid type-1 (TRPV1). eCB-tLTP occurs in both striatopallidal and striatonigral medium-sized spiny neurons (MSNs) and is dopamine dependent. Lastly, we show that eCB-LTP and eCB-LTD can be induced sequentially in the same neuron, depending on the cellular conditioning protocol. Thus, while endocannabinoids are usually thought simply to depress synaptic function, they also constitute a versatile system underlying bidirectional plasticity. Our results reveal a novel form of synaptic plasticity, eCB-tLTP, which may underlie rapid learning capabilities characterizing behavioural flexibility.
Introduction
Cardinal cognitive abilities can display rapid learning dynamics. Forming new associative memories and behavioural rules can be learned within a few (5–10) or even a single trial (Schultz et al. 2003; Pasupathy & Miller, 2005; Tse et al. 2007; Quilodran et al. 2008; Ito & Doya, 2009). In cortex and striatum, neurons that respond to behaviourally relevant events (cues, actions or rewards) fire very few spikes (one to a dozen) upon each trial (i.e. they typically discharge at a frequency of 5–25 Hz during a 0.1–0.5 s period; Schultz et al. 2003; Pasupathy & Miller, 2005; Quilodran et al. 2008). This evidence suggests that the discharge of a small number (2–50) of spikes should be sufficient to induce synaptic plasticity, a substrate for learning and memory (Martin & Morris, 2002). However, typical cell conditioning protocols for initiating long-term plasticity, such as high- or low-frequency stimulations, rely on the repetition of hundreds of pre- or postsynaptic spikes. Noticeable exceptions are reports showing the existence of single-shock LTD in visual cortex or single-burst LTP in hippocampus (Holthoff et al. 2004; Remy & Spruston, 2007). Besides these reports introducing the possibility of bidirectional plasticity induced by limited stimulation, the possible existence of rapid responsiveness of synaptic plasticity still needs to be extended to other synapses and cell conditioning protocols.
Here, we tested the hypothesis that a low number of spikes could lead to long-term synaptic plasticity. For this purpose, we chose spike-timing-dependent plasticity (STDP) as a synaptic Hebbian learning paradigm. Indeed, STDP (tLTP and tLTD) depends on the relative timing between pre- and postsynaptic spikes, and relies on much fewer events (around 100 paired stimulations) than the high- or low-frequency stimulation protocols (hundreds of stimulations) (Sjöström et al. 2008; Feldman, 2012). We first investigated if limited occurrences of paired stimulations, from 2 to100 pre–post or post–pre pairings, could induce STDP at corticostriatal synapses. In the striatum, bidirectional STDP with NMDA receptor (NMDAR)-mediated tLTP and endocannabinoid (eCB)-mediated tLTD has previously been reported with 100 paired stimulations (Fino et al. 2005, 2010; Shen et al. 2008; Pawlak & Kerr, 2008; Fino & Venance, 2010; Paillé et al. 2013). In the present study we report that these forms of plasticity disappear when the number of paired stimulations decreases. However, a reliable and robust tLTP re-emerges for a low number of paired stimulations (∼5–10). We show that this tLTP is not dependent on NMDAR but is eCB-mediated. This eCB-tLTP depends on the activation of type-1 cannabinoid receptor (CB1R) and transient receptor potential vanilloid type-1 (TRPV1). eCB-tLTP can be induced in both striatopallidal and striatonigral MSNs and is dopamine dependent. Finally, we observe that eCB-tLTP and eCB-tLTD can be sequentially induced at the same synapse, thus demonstrating that eCBs serve as a generic signalling system able to encode for bidirectional plasticity. eCBs have emerged as a major signalling system in learning and memory because of their powerful influence on synaptic plasticity which has been associated with the depression of neuronal communication on short or long timescales (Kano et al. 2009; Katona & Freund, 2012; Castillo et al. 2012; Mathur & Lovinger, 2012; Melis et al. 2014). The eCB-tLTP reported here shows that eCBs in fact support bidirectional plasticity. This new form of plasticity may underlie the quick reactivity necessary for synaptic weight adaptation during rapid learning.
Methods
Animals
All experiments were performed in accordance with local animal welfare committee (Centre for Interdisciplinary Research in Biology and EU guidelines, directive 2010/63/EU). Every precaution was taken to minimize stress and the number of animals used in each series of experiments. Sprague–Dawley rats (Charles River, L'Arbresle, France) and C57BL/6 mice, CB1R−/− and CB1R+/+ littermates (Ledent et al. 1999), and D1-eGFP mice were used for brain slice electrophysiology. Animals were housed in standard 12 h light–dark cycles and food and water were available ad libitum.
Brain slice preparation
Horizontal brain slices containing the somatosensory cortex and the corresponding corticostriatal projection field were prepared according to methods previously published (Fino et al. 2005; Paillé et al. 2013). Corticostriatal connections (between somatosensory cortex layer 5 and dorsal striatum) are preserved in a horizontal plane. Briefly, horizontal brain slices with a thickness of 330 or 300 μm were prepared, respectively, from rats (males and females, postnatal days 17–25) or mice (males and females, postnatal days 17–25 and 60–90) using a vibrating blade microtome (VT1200S, Leica Micosystems, Nussloch, Germany). Brains were sliced in a 95% CO2–5% O2-bubbled, ice-cold cutting solution containing (in mm) 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 1 pyruvic acid, and then transferred into the same solution at 34°C for 1 h and then moved to room temperature.
Electrophysiology recordings
Patch-clamp recordings were performed as previously described (Fino et al. 2010; Paillé et al. 2013). Briefly, for whole-cell recordings borosilicate glass pipettes of 4–6 MΩ resistance contained (in mm): 105 potassium gluconate, 30 KCl, 10 Hepes, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na, 0.3 EGTA (adjusted to pH 7.35 with KOH). The composition of the extracellular solution was (mm): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, with 10 μm pyruvic acid, bubbled with 95% O2 and 5% CO2. Signals were amplified using EPC10-2 amplifier (HEKA Elektronik, Lambrecht, Germany). All recordings were performed at 34°C using a temperature control system (Bath-controller V, Luigs & Neumann, Ratingen, Germany) and slices were continuously superfused at 2–3 ml min−1 with the extracellular solution. Slices were visualized on an Olympus BX51WI microscope (Olympus, Rungis, France) using a ×4/0.13 objective for the placement of the stimulating electrode and a ×40/0.80 water-immersion objective for localizing cells for whole-cell recordings. Medium-sized spiny neurons (MSNs), the striatal output neurons, were distinguished from interneurons based on passive and active membrane properties (Fino et al. 2008). Series resistance was not compensated. Current-clamp recordings were filtered at 2.5 kHz and sampled at 5 kHz and voltage-clamp recordings were filtered at 5 kHz and sampled at 10 kHz using the Patchmaster v2×32 program (HEKA Elektronik).
Chemicals
Chemicals were bath applied or injected only in the recorded postsynaptic neuron through the patch-clamp pipette. dl-2-amino-5-phosphono-pentanoic acid (d-AP5, 50 μm; Tocris, Ellisville, MO, USA), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP hydrochloride, 10 μm; Tocris), 5,11-dihydro-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one dihydrochloride (pirenzepine dihydrochloride, 1 μm; Sigma) were dissolved directly in the extracellular solution and bath applied. N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251, 3 μm; Tocris), picrotoxin (50 μm; Sigma), 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 2-methyloxyethyl 1-methylethyl ester (nimodipine, 1 μm; Tocris) and (2E)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide (AMG9810, 1 μm; Tocris), R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH-23390, 4 μm; Sigma) and (S–)-5-aminosulfonyl-N-[(1-ethyl-2-pyrrolidinyl)methyl]-2-methoxybenzamide (sulpiride, 10 μm; Tocris) were dissolved in ethanol and then added in the external solution at a final concentration of ethanol of 0.01–0.1%. BAPTA (1 mm; Sigma) and GDP-ß-S (2 mm) were dissolved directly into the intracellular solution and applied via the patch-clamp pipette. U73122 (5 μm; Sigma) was dissolved in ethanol and then added to the intracellular solution at a final concentration of ethanol of 0.033%. Tetrahydrolipstatin (THL, 10 μm; Sigma) was dissolved in DMSO and applied internally via the patch-clamp pipette at a final DMSO concentration of 0.1%. (S)-α-methyl-4-carboxyphenylglycine (MCPG, 500 μm; Tocris) was dissolved in 1.1 equiv NaOH, and then added in the external solution. N-[2-(4-chlorophenyl)ethyl]-1,3,4,5-tetrahydro-7,8-dihydroxy-2H-2-benzazepine-2-carbothioamide (capsazepine, 10 μm; Tocris) and 2-arachidonoylglycerol (2-AG, 100 μm; Tocris) were dissolved in DMSO and then added to the external solution at final DMSO concentrations of 0.0025 and 0.1%, respectively. We verified that DMSO (0.1%) or ethanol (0.033%) when applied intracellularly did not alter the tLTP observed in control conditions (165 ± 11%, P < 0.0001, n = 27). DMSO (0.1%), the vehicle used to dilute iTHL, did not preclude the tLTP induced with 10 post–pre pairings (213 ± 22%, P = 0.0153, n = 4; P = 06564 when compared with control). Likewise, ethanol (0.033%), the vehicle used to dilute iU73122, did not prevent tLTP (176 ± 20%, P = 0.0139, n = 6; P = 01220 when compared with control).
Note that none of the bath-applied drugs had a significant effect on basal EPSC amplitudes (Table1).
Table 1.
Effects of bath-applied drugs on corticostriatal EPSC baseline amplitude
| Normalized EPSC (baseline | ||
|---|---|---|
| Bath-applied drugs | drugs/baseline control) amplitude | n, Pa |
| d-AP5 (50 μm) | 98 ± 4% | n = 6, P = 0.6076 |
| AM251 (3 μm) | 99 ± 5% | n = 8, P = 0.7988 |
| MCPG (500 μm) | 94 ± 6% | n = 5, P = 0.4029 |
| MPEP (10 μm) | 100 ± 4% | n = 4, P = 0.9243 |
| Pirenzepine (1 μm) | 92 ± 7% | n = 6, P = 0.3739 |
| Nimodipine (1 μm) | 96 ± 4% | n = 4, P = 0.3790 |
| Capsazepine (10 μm) | 100 ± 4% | n = 5, P = 0.9778 |
| AMG9810 (1 μm) | 97 ± 8% | n = 5, P = 0.6800 |
| Picrotoxin (50 μm) | 96 ± 3% | n = 6, P = 0.1493 |
| SCH23390 (4 μm) + sulpiride (10 μm) | 102 ± 2% | n = 7, P = 0.4468 |
None of the bath-applied drugs had a significant effect on EPSC baseline amplitude. In all cases, only the baselines with the bath-applied drugs were compared to EPSC baselines measured after 60 min recording.
Spike-timing-dependent plasticity induction protocols
Electrical stimulations were performed with a bipolar electrode (Phymep, Paris, France) placed in layer 5 of the somatosensory cortex (Fino et al. 2005, 2010). Electrical stimulations were monophasic at constant current (ISO-Flex stimulator, AMPI, Jerusalem, Israel). Currents were adjusted to evoke 50–200 pA EPSCs. Repetitive control stimuli were applied at 0.1 Hz. STDP protocols consisted in pairings of pre- and postsynaptic stimulations (at 1 Hz) with the two events separated by a specific temporal interval (ΔtSTDP). Presynaptic stimulations corresponded to cortical stimulations and the postsynaptic stimulation of an action potential evoked by a depolarizing current step (30 ms duration) in MSNs. Suprathreshold depolarization for 30 ms was chosen to mimic corticostriatal summation of EPSPs induced by cortical activity as observed in in vivo studies (Mahon et al. 2006). MSNs were maintained throughout the STDP experiments at a constant holding membrane potential which corresponds to their initial resting membrane potential (–75 ± 1 mV, n = 103). Thus, EPSCs during baseline recording or after the STDP protocol were mesured at the same membrane potential (in voltage-clamp mode); note that the STDP pairings (performed in current-clamp mode) were also conducted at the same holding membrane potential. Neurons were recorded for 10 min during baseline amplitude and for at least 60 min after the STDP protocol; long-term synaptic efficacy changes were measured after 50 min. Thirty successive EPSCs (at 0.1 Hz) were individually measured and then averaged. Variation of series resistance beyond 20% led to the rejection of the experiment. After recording of 10 min control baseline amplitude, drugs were applied in the bath. A new baseline with drugs was recorded after a time lapse of 10 min (to allow the drug to be fully perfused) for 10 min before the STDP protocol (see effects of the bath-applied drugs on baseline amplitude in Table1). Drugs were present until the end of the recording (except when specified). In a subset of experiments (for U73122, THL, BAPTA and GDP-ß-S) drugs were applied intracellularly through the patch-clamp pipette. Once the cell patched, drugs were allowed to diffuse into the cell for at least 15 min before starting recording of the baseline amplitude. Local applications of 2-AG were performed through a patch-clamp pipette placed in the vicinity (50 μm) of the recorded neuron and linked to a Picospritzer II system (Parker, USA), which supplies repeatable pressure pulses.
It should be noted that STDP protocols consisting of 5–10 post–pre pairings (with a single postsynaptic spike) were sufficient to induce potent tLTP in rats while in C57BL/6 mice 15 pairings (with 2–3 postsynaptic spikes per posynaptic discharge) were necessary to trigger tLTP.
Electrophysiological data analysis
Off-line analysis was performed using Fitmaster (Heka Elektronik) and Igor-Pro 6.0.3 (Wavemetrics, Lake Oswego, OR, USA). Statistical analysis was performed using Prism 5.0 software (San Diego, CA, USA). In all cases n refers to the number of repetitions of an experiment (each experiment being performed on different brain slices) from single slice. Experimenters were blind to the genotype of CB1R−/− and CB1R+/+ littermate mice during electrophysiological recordings and analysis. All results were expressed as means ± SEM in the text and, for visualization purposes, as means ± SD in the figures, and statistical significance was assessed using two-sided Student's t test or the one sample t test when appropriate at the significance level (P) indicated.
Results
We investigated whether a low number of post- and presynaptic paired stimulations induced plasticity in a major synaptic learning rule such as STDP. As previously described (Fino et al. 2005; Shen et al. 2008; Pawlak & Kerr, 2008; reviewed in Kreitzer & Malenka, 2008; Fino & Venance, 2010; Feldman, 2012), 100 pairings induced bidirectional STDP in MSNs: post–pre pairings (–30<ΔtSTDP<0 ms) induced tLTP (mean value of the EPSC amplitude recorded 50 min after STDP protocol: 142 ± 16%, P = 0.0262, n = 10), while pre–post pairings (0<ΔtSTDP<+30 ms) induced tLTD (66 ± 10%, P = 0.0124, n = 7) (Fig.1A–C and I). We observed a similar bidirectional STDP with 75 pairings: post–pre and pre–post pairings induced tLTP (167 ± 26%, P = 0.0378, n = 8) and tLTD (64 ± 5%, P = 0.0010, n = 6), respectively (Fig.1D and I). We then decreased the number of pairings to 50 and observed contrasting effects on synaptic plasticity: while a potent tLTD persisted with pre–post pairings (66 ± 6%, P = 0.0013, n = 7), the tLTP usually associated with post–pre pairings disappeared (103 ± 12%, P = 0.7902, n = 7) (Fig.1E and I). In turn tLTD disappeared for 25 pre–post pairings (94 ± 3%, P = 0.0801, n = 8) (Fig.1F and I). On the post–pre side there was still no significant plasticity (132 ± 24%, P = 0.1985, n = 11) following 25 pairings, although half of the cells displayed tLTP and the other half no plasticity (Fig. 1F and I). Unexpectedly, decreasing the number of paired stimulations further unveiled another trend: whereas 10 pre–post pairings failed to induce significant plasticity (99 ± 10%, P = 0.9267, n = 9), 10 post–pre pairings were sufficient to induce a potent tLTP (165 ± 11%, P < 0.0001, n = 27) (Fig.1G and I). A similar picture was obtained even with 5 pairings: post–pre pairings induced tLTP (139 ± 13%, P = 0.0087, n = 16) while pre–post pairings induced no significant plasticity (93 ± 6%, P = 0.2417, n = 6) (Fig.1H and I). Finally, no significant plasticity was detected with 2 post–pre pairings (108 ± 9%, P = 0.4013, n = 6) (Fig.1I). Note that a single postsynaptic burst (composed of 3–4 spikes) paired with a single presynaptic spike in a post–pre manner also failed to induce significant plasticity (data not shown). In conclusion, tLTD disappears with decreasing numbers of paired stimulations whereas tLTP displays a biphasic profile since tLTP is observed for 75–100 pairings and 5–10 pairings (with similar amplitudes) and is absent for 25–50 pairings.
Figure 1. A low number of paired stimulations induces spike-timing-dependent potentiation.
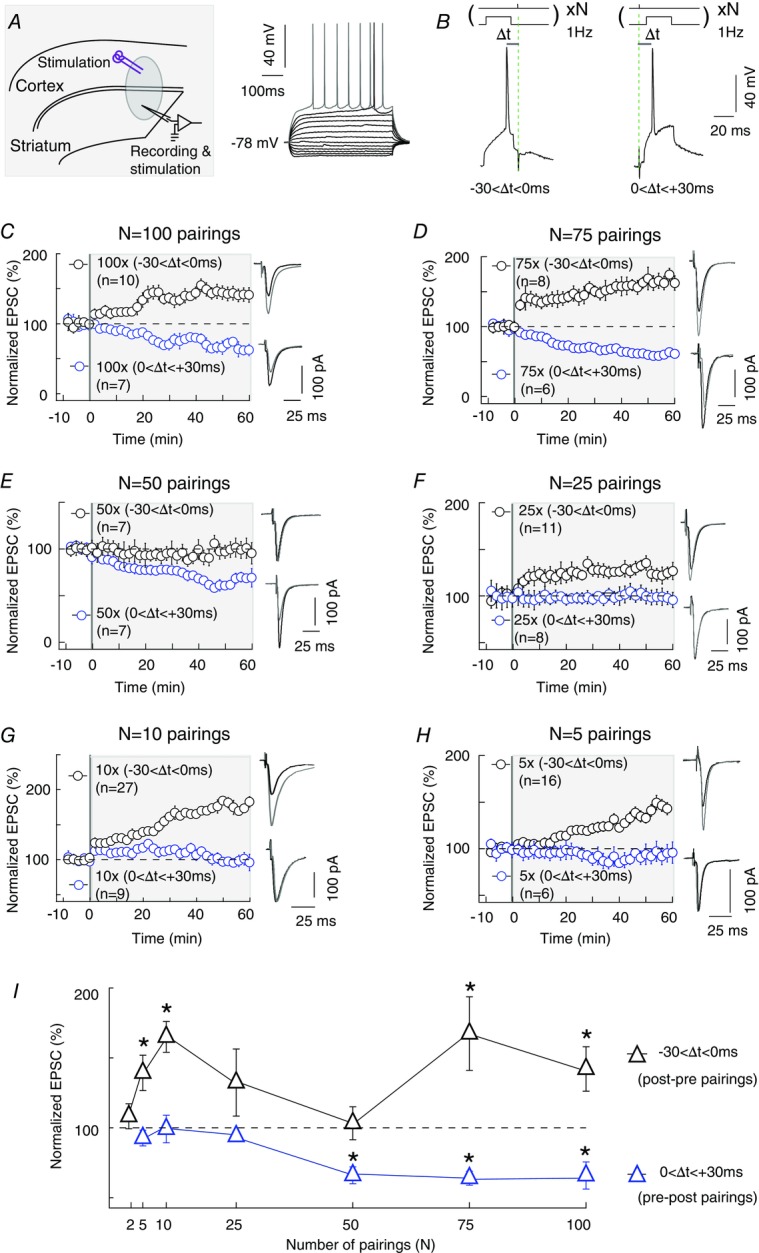
A, left: scheme of the recording and stimulating sites in corticostriatal slices. A, right: characteristic voltage responses of a MSN to a series of 500 ms current pulses from –150 to +180 pA with current steps increasing by 30 pA (black traces) and to +60 pA above spike threshold (grey trace). B, STDP protocol: a spike evoked in striatal neuron was paired with cortical stimulation N times at 1 Hz. Δt indicates the time delay between pre- and postsynaptic stimulations. –30<Δt<0 ms and 0<Δt<+30 ms refers to post–pre and pre–post pairings, respectively. C, 100 post–pre and pre–post pairings (n = 10 and 7) induced bidirectional plasticity, i.e. tLTP and tLTD, respectively. D, 75 post–pre and pre–post pairings (n = 8 and 6) induced tLTP and tLTD, respectively. E, 50 post–pre and pre–post pairings (n = 7 and 7) induced unidirectional plasticity, i.e. no plasticity and tLTD, respectively. F, 25 post–pre and pre–post pairings (n = 11 and 8) did not induce significant plasticity. G, 10 post–pre and pre–post pairings (n = 27 and 9) induced unidirectional plasticity, i.e. tLTP and no plasticity, respectively. H, 5 post–pre and pre–post pairings (n = 16 and 6) induced tLTP and no plasticity, respectively. I, summary graph showing the effect of different numbers of pairings (from 100 to 2) on long-term plasticity induction. There is an absence of corticostriatal tLTP with 50, 25 or 2 post–pre pairings while 75–100 or 5–10 post–pre pairings induced significant tLTP. Bidirectional (tLTD and tLTP) STDP is observed for 75–100 pairings, unidirectional (tLTD) STDP for 50 pairings and unidirectional (tLTP) STDP for 5–10 pairings. Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 50 min after STDP protocol (grey traces). Error bars represent SD. *P < 0.05.
tLTP induced by 10 pairings is NMDAR independent
We then questioned the mechanism of tLTD and tLTP induced by these different numbers of pairings. We observed that the corticostriatal 100 pairings-induced tLTD was CB1R dependent, as previously demonstrated (Shen et al. 2008; Pawlak & Kerr, 2008; Fino et al. 2010; Paillé et al. 2013). Indeed, pharmacological inhibition of CB1R with AM251 (3 μm) prevented the expression of 100 pairings-induced tLTD (106 ± 6%, P = 0.4153, n = 5; Fig.2A). Note that AM251 alone (without electrical stimulation) had no effect on basal synaptic transmission (99 ± 5%, P = 0.7988, n = 8; Table1), indicating that CB1R had no constitutive activity at corticostriatal synapses. Similarly to the 100 pairings-induced tLTD, the 50 pre–post pairings- induced tLTD was prevented with AM251 (3 μm) (114 ± 17%, P = 0.4541, n = 7; Fig.2B). Thus, the pre–post corticostriatal tLTD was CB1R mediated.
Figure 2. tLTP induced by 10 pairings is NMDAR independent.
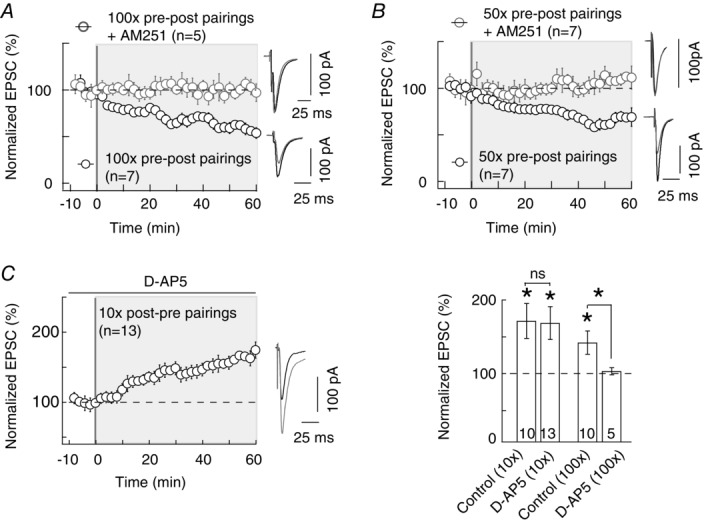
A and B, tLTD induced with 100 (A) or 50 (B) pre–post pairings is CB1R mediated. 100 and 50 pre–post pairings induced a potent tLTD (n = 7 and 7, respectively), which was prevented by AM251 treatment (3 μm, n = 5 and 7, respectively). C, tLTP induced with 10 post–pre pairings was not prevented by d-AP5 (50 μm, n = 13), indicating that this tLTP was not NMDAR mediated. Summary bar graphs illustrating that tLTP induced by 100 post–pre pairing is NMDAR mediated since it is prevented by d-AP5 treatment, while tLTP induced by 10 post–pre pairings is NMDAR independent. Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 50 min after STDP protocol (grey traces). Error bars represent SD. *P < 0.05; ns, not significant.
Concerning the post–pre pairing-induced tLTP, we confirmed that the 100 post–pre pairings-induced tLTP was NMDAR mediated since it was prevented with the selective NMDAR blocker d-AP5 (50 μm) (104 ± 5%, P = 0.4310, n = 5; Fig.2C), as previously reported (Shen et al. 2008; Pawlak & Kerr, 2008; Fino et al. 2010; Paillé et al. 2013). We then explored the mechanism of the tLTP induced by 10 post–pre pairings and found that it does not rely on the same signalling pathway (i.e. NMDAR). Indeed, the tLTP induced by 10 pairings was not significantly affected by d-AP5 (169 ± 22%, P = 0.0098, n = 13; Fig.2C), questioning the identity of the signalling pathways underlying this new form of tLTP.
tLTP induced by 10 pairings involves postsynaptic 2-AG signalling
The corticostriatal synapse is glutamatergic and we first tested whether glutamatergic G-protein-coupled receptors were required for the expression of tLTP induced by 10 pairings. MSNs express group-I metabotropic glutamate receptors (mGluRs) (Testa et al. 1994) belonging to the class of Gq/11-coupled receptors. tLTP induced by 10 pairings was prevented by the inhibition of group-I mGluR with MCPG (500 μm) (100 ± 8%, P = 0.9636, n = 5; Fig.3A and B). More specifically, among group-I mGluRs, MSNs express prominently the mGluR5 isoform (Uchigashima et al. 2007). MPEP (10 μm), a blocker of mGluR5, prevented the induction of 10 pairings-induced tLTP and a slight depression was observed (68 ± 8%, P = 0.0108, n = 6; Fig.3B). Besides glutamate, acetylcholine is also released following cortical activation of the corticostriatal synapses, since striatal cholinergic interneurons are tonically active and directly contacted by cortical pyramidal cells. Interestingly, MSNs express the M1 muscarinic receptors which are also another class of Gq/11-coupled receptors (Hersch et al. 1994; Yamasaki et al. 2010). We thus tested whether these receptors could be involved in the 10 pairings-induced tLTP. We found that the inhibition of M1 muscarinic receptors with pirenzepine (1 μm) prevented tLTP (95 ± 20%, P = 0.8037, n = 6; Fig.3B). Altogether, these results show that the tLTP triggered by 10 pairings requires the concomitant activation of mGluR5 and M1 muscarinic receptors. We investigated the localization of these receptors with the application in the recorded postsynaptic neuron of a non-hydrolysable nucleotide GDPβS which prevents G-protein activation (2 mm i-GDPβS applied intracellularly through the patch-clamp pipette). i-GDPβS precluded tLTP (90 ± 10%, P = 0.3249, n = 10) indicating that mGluR5 and/or M1 receptors were postsynaptically located (Fig.3B). Group-I mGluRs and M1 receptors are Gq/11-coupled receptors and thus activate phospholipase Cβ (PLCβ; Rebecchi & Pentyala, 2000). We then tested if PLCβ activation was involved in the 10 pairings-induced tLTP. In the presence of a PLCβ inhibitor applied intracellularly through the patch-clamp pipette (i-U73122, 5 μm) 10 post–pre pairings failed to induce any significant plasticity (94 ± 7%, P = 0.4255, n = 5; Fig.3B), confirming the implication of postsynaptic PLCβ.
Figure 3. Induction of tLTP by 10 pairings involves postsynaptic 2-AG signalling.

A and B, summary graphs of pharmacological experiments delineating the intracellular signalling pathways involved in 10 pairings-induced tLTP. A, tLTP was prevented by inhibition of group-I mGluR with MCPG (500 μm, n = 5) or of DAGLα by i-THL (10 μm, n = 5; the prefix ‘i’ indicates that the drug was applied in the recorded postsynaptic neuron through the patch-clamp pipette). B, summary bar graphs showing that LTP was mediated by mGluR5 and M1R. Indeed, tLTP was prevented by inhibition of group-I mGluR with MCPG (500 μm, n = 5) and more specifically by inhibition of mGluR5 with MPEP (10 μm, n = 6); inhibition of M1R also prevented tLTP (1 μm pirenzepine, n = 6). Downstream of these receptors, inhibition of postsynaptic G-protein-coupled receptors (with 2 mm i-GDP-β-S, n = 10), PLCβ (with 5 mm i-U73122, n = 5), DAGLα (with 10 μm i-THL, n = 5) shows the involvement of PLCβ and 2-AG synthesis. In addition, bar graphs show the involvement of postsynaptic intracellular calcium (inhibited by 10 mm i-BAPTA, n = 5) and VSCCs (inhibited by 1 μm nimodipine, n = 5), since their blockade prevented the expression of tLTP. C, repeated brief application of 2-AG induces LTD. A serie of 100 2-AG puffs (100 μm, 300 ms duration each) delivered at 1 Hz at the vicinity (50–100μm) of the recorded striatal neuron, induced LTD in the absence of any STDP protocol (n = 8). This 2-AG-mediated LTD was prevented by AM251 (3 μm, n = 6). D, limited brief application of 2-AG induces LTP. Application of 10 2-AG puffs (100 μm, 300 ms duration each) delivered at 1 Hz was able to induce LTP in the absence of any STDP paired stimulation (n = 8). Inhibition of CB1R with bath-applied AM251 (3 μm, n = 5) prevented the induction of LTP by 2-AG puffs. Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 50 min after STDP protocol (grey traces). Error bars represent SD. *P < 0.05.
After activation by Gq/11-coupled receptors, PLCβ increases the levels of calcium ions (Rebecchi & Pentyala, 2000). We therefore tested the involvement of calcium in the expression of 10 pairings-induced tLTP. We first showed that postsynaptic calcium elevation, in the recorded striatal neuron, was mandatory for tLTP induction. Indeed, specific loading of the recorded postsynaptic neuron with the fast calcium buffer BAPTA (10 mm i-BAPTA applied intracellularly) prevented tLTP induction (108 ± 10%, P = 0.4774, n = 5; Fig.3B). We next demonstrated that calcium entry via L-type voltage-sensitive calcium channels (VSCCs), the main type of activated VSCCs in MSNs (Carter & Sabatini, 2004), is responsible since their blockade (1 μm nimodipine, 95 ± 9%, P = 0.6092, n = 5) precluded tLTP (Fig.3B). Further downstream in the signalling pathway, these concomitant activations are expected to promote diacylglycerol lipase-α (DAGLα) activity and therefore 2-arachidonoylglycerol (2-AG) synthesis (Hashimotodani et al. 2005; Piomelli et al. 2007; Di Marzo, 2008; Kano et al. 2009; Tanimura et al. 2010; Alger & Kim, 2011). 2-AG is produced from the PLCβ product diacylglycerol by calcium-activated DAGLα and is the principal eCB involved in modulating synaptic strength by selectively activating CB1R (Piomelli et al. 2007). We found that a DAGLα inhibitor, tetrahydrolipstatin (10 μm i-THL applied intracellularly), prevented tLTP (91 ± 7%, P = 0.2607, n = 5; Fig.3A and B). Importantly, since i-THL application was restricted to the recorded neuron, this result indicates that the production of 2-AG needed to activate CB1R arises from the postsynaptic striatal neuron engaged in the paired stimulations. In summary, tLTP induced with 10 pairings involves the 2-AG synthesis pathway.
To further demonstrate the key role of 2-AG in bidirectional STDP (tLTD and tLTP with 100 pre–post pairings and 10 post–pre pairings, respectively), we applied local puffs of 2-AG of different duration in the vicinity (50–100 μm) of the recorded striatal neuron. We first applied 100 brief (300 ms) puffs of 2-AG (100 μm) at 1 Hz, i.e the same total duration as a 100 pairings STDP protocol at 1 Hz. In these conditions (i.e. in the absence of STDP protocols), we observed that local application of 2-AG was able to induce a significant LTD (68 ± 10%, P = 0.0156, n = 8; Fig.3C) with a magnitude similar to the tLTD induced by 100 pre–post electrical pairings (P = 0.8624). This form of LTD involves CB1R activation since 2-AG puffs did not induce any more plasticity when AM251 (3 μm) was bath applied (96 ± 5%, P = 0.5000, n = 6; Fig.3C).
We then aimed at mimicking the 10 pairing-induced LTP, by applying brief puffs (300 ms) of 2-AG (100 μm) 10 times at 1 Hz, thus with the same total duration as a 10 pairings STDP protocol at 1 Hz. We observed that even in the absence of STDP protocol, local application of 2-AG was able to induce a significant LTP (139 ± 24%, P = 0.0391, n = 8; Fig.3D) with a magnitude similar to the tLTP induced by 10 post–pre pairings (P = 0.3705). This LTP involved CB1R activation since 2-AG puffs did not induce any more plasticity in the presence of AM251 (3 μm) (92 ± 4%, P = 0.1542, n = 5; Fig.3D).
tLTP induced by 10 pairings is CB1R activation mediated and presynaptic
Since 2-AG is a specific ligand of CB1Rs (Piomelli et al. 2007; Di Marzo et al. 2008; Alger & Kim, 2011; Katona & Freund, 2012), we then asked whether tLTP induced by 10 pairings was indeed CB1R mediated. Pharmacological inhibition of CB1R with AM251 (3 μm) prevented the expression of 10 pairings-induced tLTP (80 ± 11%, P = 0.1424, n = 6; Fig.4A).
Figure 4. tLTP induced by 10 pairings is CB1R mediated.

A, tLTP induced by 10 pairings is prevented by a specific CB1R inhibitor, AM251 (3 μm, n = 6). B, 10 post–pre pairings induced tLTP in wild-type CB1R+/+ mice (n = 10) while no plasticity was observed in CB1R−/− mice (n = 16). C, summary bar graphs with CB1R−/− and CB1R+/+ mice illustrate that eCB-tLTP is CB1R mediated both in juvenile (postnatal days 18–25) and adult (postnatal days 60–90) animals. D, representative traces and summary bar graphs (n = 9) of paired-pulse cortical stimulation with 50 ms interstimulus interval illustrate a decrease of facilitation after STDP. This indicates a presynaptic locus of the eCB-tLTP. E, mean variance analysis (CV−2, n = 20) indicates a presynaptic locus of eCB-tLTP maintenance. Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 50 min after STDP protocol (grey traces). Error bars represent SD. *P < 0.05.
This pharmacological result was further confirmed by experiments with CB1R-knockout (CB1R−/−) mice (Ledent et al. 1999), where no significant plasticity was observed following 15 pairings (93 ± 4%, P = 0.0882, n = 16) whereas tLTP could be induced in the littermate wild-type CB1R+/+ mice (135 ± 5%, P = 0.0001, n = 10; Fig.4B and C). Note that in C57BL/6 mice 15 pairings with 2–3 postsynaptic APs were required to efficiently induced tLTP while 5–10 pairings with a single postsynaptic AP per pairing were sufficient to trigger tLTP in rats. This CB1R-mediated tLTP was expressed up to adulthood since we observed a reliable tLTP in postnatal days 60–90 CB1R+/+ mice (137 ± 8%, P = 0.0109, n = 5), which was absent in postnatal days 60–90 CB1R−/− mice (93 ± 6%, P = 0.3048, n = 5; Fig.4C). Pharmacological and genetic evidence demonstrated that tLTP induced by 10 pairings is eCB mediated. We thus refer to this new form of LTP as eCB-tLTP.
The results described above show that synthesis and release of eCBs in eCB-tLTP relies on signalling pathways that are located in the postsynaptic neuron. We then searched for the locus of eCB-tLTP maintenance. As shown above, eCB-tLTP is CB1R activation dependent and since CB1Rs are expected to be located on presynaptic corticostriatal terminals (Katona & Freund, 2012), the locus of eCB-tLTP maintenance is likely to be presynaptic. We used paired-pulse stimulations to test this hypothesis. Paired-pulse intervals of 50 ms induced a significant EPSC paired-pulse facilitation (PPF) in striatal output neurons (Goubard et al. 2011). Thus, we applied paired pulses with 50 ms interpulse interval before and after STDP protocol. We observed a significantly decreased PPF (PPFplasticity/baseline = 0.912 ± 0.037, P = 0.4366, n = 9), indicating a presynaptic origin of the plasticity (Fig.2D). This was further confirmed by the mean variance analysis of EPSCs: we obtained a CV–2 value of 3.1 ± 0.6 (P = 0.0014, n = 20), which also indicated a presynaptic maintenance of eCB-tLTP (Fig.2E). In conclusion, our results indicate that eCB-tLTP is induced postsynaptically through the synthesis and release of eCBs, and maintained presynaptically downstream of CB1Rs.
eCB-LTP induction involves TRPV1
Besides 2-AG, the production of another eCB, anandamide, can also be increased upon cellular activity (Piomelli et al. 2007; Alger & Kim, 2011). Whereas 2-AG is a specific ligand of CB1R, anandamide activates both CB1R (albeit less potently than 2-AG) and TRPV1. TRPV1 is a non-selective cationic channel (Ross, 2003; Starowicz et al. 2007; Di Marzo, 2008) involved in eCB-mediated short- and long-term depression (Gibson et al. 2008; Maione et al. 2009; Chávez et al. 2010; Grueter et al. 2010; Puente et al. 2011). We therefore tested whether TRPV1 was implicated in eCB-tLTP. Note that in the absence of paired stimulation, application of capsazepine (10 μm), a TRPV1 antagonist, had no significant effect on basal EPSC (100 ± 4%, P = 0.9778, n = 5; Table1), indicating that TRPV1 has no constitutive activity at corticostriatal synapses. We then found that the application of capsazepine (10 μm) during the STDP stimulation protocol (10 post–pre pairings) efficiently blocked eCB-tLTP (83 ± 11%, n = 6, P = 0.1133; Fig.5). To confirm this result, we used AMG9810, another competitive TRPV1 antagonist, structurally distinct from capsazepine, and observed that AMG9810 (1 μm) also blocked eCB-tLTP (93 ± 6%, P = 0.3046, n = 5; Fig.5B).
Figure 5. eCB-LTP is also TRPV1 mediated.
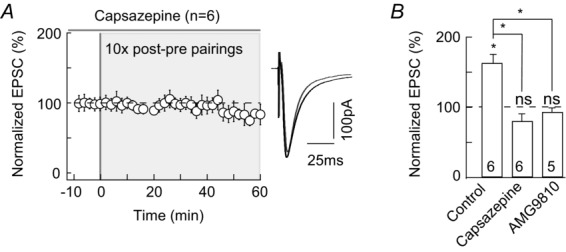
A, tLTP induced by 10 pairings was prevented when TRPV1 was inhibited by capsazepine (10 μm, n = 6). B, summary bar graphs show that capsazepine (10 μm, n = 6) or AMG9810 (1 μm, n = 5) prevented the 10 pairings-induced tLTP. Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 50 min after STDP protocol (grey traces). Error bars represent SD. *P < 0.05; ns, non-significant.
Altogether, our results demonstrate that 10 pairings-induced tLTP is mediated by eCB (2-AG and anandamide), acting on both CB1R and TRPV1.
eCB-tLTP occurs in both striatopallidal and striatonigral MSNs and is dopamine dependent
Pyramidal cells from cortex layer 5 contact two MSN subpopulations belonging to the direct (striatonigral) or indirect (striato-pallido-subthalamo-nigral) trans-striatal pathways (Gerfen & Surmeier, 2011; Calabresi et al. 2014). We investigated whether eCB-tLTP is induced in both striatopallidal and striatonigral MSNs. The two MN subtypes express different dopaminergic receptors (D1R-like and D2R-like for the direct and indirect pathways, respectively) allowing us to identify them with transgenic D1-eGFP mice and to investigate eCB-tLTP occurrence in D1+ and non-D1+ MSNs (Fig.6A and B). We observed that 15 post–pre pairings (see Methods) induced tLTP in both D1+ (165 ± 18%, P = 0.0166, n = 7) and non-D1+ (156 ± 20%, P = 0.0215, n = 9) MSNs (Fig.6A and B). This indicates that tLTP can be induced with few pairings in both striatopallidal and striatonigral MSNs.
Figure 6. eCB-tLTP is induced in both striatopallidal and striatonigral MSNs and is dopamine dependent.
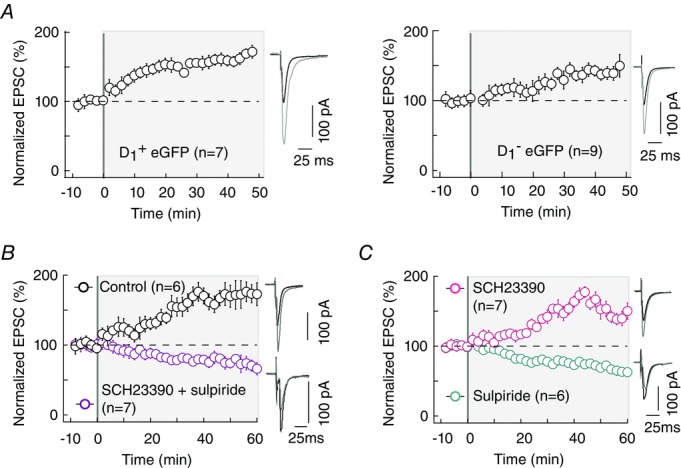
A, tLTP induced by 15 pairings is observed in both striato-pallidal (D1-eGFP positive neurons, D1+, n = 7) and striato-nigral (D1-eGFP negative neurons, D1–, n = 9) MSNs. B, co-application of antagonists of D1R and D2R, SCH23390 (4 μm) and sulpiride (10 μm), prevents eCB-tLTP (n = 7). C, tLTP was induced in presence of the D1R antagonist SCH23390 (4 μm, n = 7), while no plasticity was observed with the D2R antagonist sulpiride (10 μm, n = 6). Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 40 (A) and 50 (B and C) min after STDP protocol (grey traces). Error bars represent SD.
Striatum receives excitatory afferents from the cortex as well as a dense innervation from midbrain dopaminergic neurons. Dopamine, a key regulator of action selection and associative learning (Yin & Knowlton, 2006; Redgrave & Gurney, 2006; Schultz, 2007), efficiently modulates corticostriatal synaptic plasticity and particularly the ‘classical’ eCB-LTD (Kreitzer & Malenka, 2008; Di Filippo et al. 2009; Gerfen & Surmeier, 2011). We then asked whether dopamine was involved in eCB-tLTP induction. We examined which dopaminergic receptor subtype is involved in eCB-tLTP. For this purpose we first bath applied a mixture of D1R and D2R antagonists, SCH23390 (4 μm) and sulpiride (10 μm), respectively. We found that this cocktail prevented the induction of eCB-tLTP and a depression was observed (75 ± 2%, P < 0.0001, n = 7; Fig.6B). Thus, eCB-tLTP is dopamine dependent. We then selectively inhibited either D1R or D2R. When we applied the D1R antagonist SCH23390 (4 μm) alone, we observed a potent tLTP (149 ± 16%, P = 0.0211, n = 7) while tLTD could be elicited when the D2R antagonist sulpiride (10 μm) was bath applied alone (68 ± 10%, P = 0.0209, n = 6; Fig. 6C). This indicates that eCB-tLTP is D2R mediated and not dependent on D1R.
Corticostriatal eCB-tLTP is paired-activity dependent
To confirm that eCB-tLTP requires both pre- and postsynaptic activity, we performed paired recordings of two neighbouring MSNs (perisomatic distance < 50μm) in which one neuron was subject to 10 post–pre pairings (STDP protocol) while the second received only 10 presynaptic stimulations (n = 6 pairs; Fig.7A). We observed a potent tLTP (150 ± 13%, P = 0.0143, n = 6) only in the neuron subject to post–pre pairings, while the neighbouring neuron, which received only the presynaptic stimulation, did not show any significant plasticity (106 ± 3%, P = 0.0833, n = 6), indicating that corticostriatal eCB-tLTP is paired-activity dependent.
Figure 7. Corticostriatal eCB-tLTP is paired activity dependent.
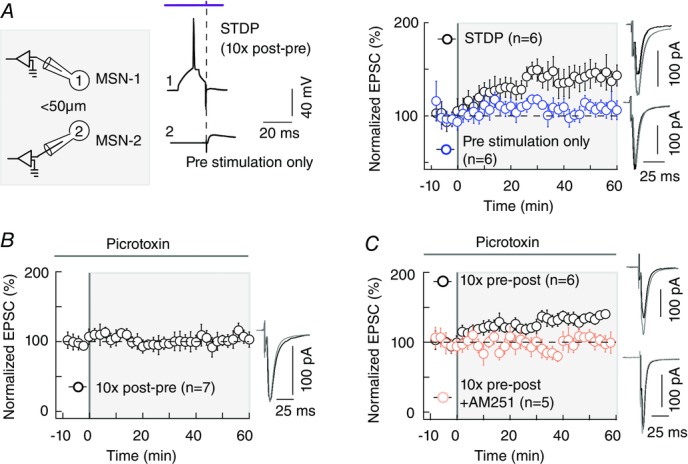
A, scheme and raw traces illustrating paired recordings of MSNs (perisomatic distance < 50μm): one neuron received a 10 pairings STDP protocol (pre- and postsynaptic stimulations) while the other one only received 10 presynaptic stimulations. tLTP was induced exclusively in the neuron with the STDP protocol (10 post–pre pairings, black circles, n = 6) while no significant plasticity was observed in the neighbouring neuron (presynaptic stimulations only, n = 6). B, inhibition of the GABAA transmission with picrotoxin did not affect eCB-tLTP magnitude but controled the time dependence of eCB-tLTP. With bath-applied picrotoxin (50 μm), a potent tLTP was induced by 10 pre–post pairings (n = 6; blue symbols) while no significant plasticity was observed with 10 post-pairings (n = 7; black). The occurrence and magnitude of tLTP were not affected by a blockade of GABAA transmission since induced plasticities were not significantly different from the ones observed in control conditions. This tLTP induced with 10 pre–post pairings under GABAA receptor blockade is mediated by eCB since it was prevented with bath application of AM251 (3 μm). GABAergic microcircuits are not involved in the synaptic efficacy changes themselves but control the time dependence of the eCB-tLTP. Representative traces are the average of 15 EPSCs during baseline recording (black traces) and 50 min after STDP protocol (grey traces). Error bars represent SD.
In the hippocampus, facilitation of LTP via eCB-induced presynaptic depression of GABAergic transmission has been reported (Carlson et al. 2002; Chevaleyre & Castillo, 2004; Zhu & Lovinger, 2007). Here, the observed eCB-tLTP could arguably arise from a decrease of GABA release, through an activation of CB1Rs located on GABA terminals, thus decreasing the inhibitory tonus during the pairing protocol. To test this hypothesis, we blocked the GABAA receptors with picrotoxin (50 μm). A significant tLTP was still observed for 10 pairings (135 ± 12, n = 6; P = 0.0300; Fig.7B and C). The magnitude of 10 pairings-induced tLTP was not affected by the blockade of GABAA transmission since there was no significant difference compared to control conditions (P = 0.1200). In agreement with our recent report of STDP induced by 100 pairings (Paillé et al. 2013), GABA controls the polarity of the timing dependance of STDP: with picrotoxin, tLTP was observed with 10 pre–post pairings (n = 6; Fig.7C) while 10 post–pre pairings did not induce plasticity (96 ± 7, P = 0.5865, n = 7; Fig.7B). We confirmed that the tLTP observed in the presence of picrotoxin was still eCB -mediated: the co-application of picrotoxin (50 μm) and AM251 (3 μm) prevented the induction of tLTP (103 ± 7%, P = 0.7454, n = 5; Fig.7C). In conclusion, GABAergic microcircuits are not involved in the synaptic efficacy changes induced by 10 pairings but control the polarity of the timing dependence of the eCB-tLTP.
Bidirectional eCB-STDP in the same neuron
eCB-tLTP and eCB-tLTD could represent functional inverses of each other. This could be demonstrated if both phenomena could be sequentially triggered in the same neuron to modify the synaptic weight and then bring it back to its baseline. We tested this hypothesis by applying successively two protocols leading to unidirectional plasticity, which exclusively imply eCBs: 10 post–pre pairings (eCB-tLTP; Figs1 and 4) and 50 pre–post pairings (eCB-tLTD; Fig.2B). We found that eCB-tLTP and eCB-tLTD can indeed be induced sequentially in the very same neuron independently of the order of induction protocols (P < 0.0001 for both forms of plasticity, comparing the 30 EPSCs preceding the STDP protocols to the last 30 EPSCs 25–30 min after the STDP protocols, n = 5; Fig.8). Following eCB-LTP induction by 10 post–pre pairings, the potentiated synaptic weight can be decreased back to its basal level by applying 50 pre–post pairings (Fig.8A). Conversely, the synaptic weight depressed by an eCB-tLTD could be increased again by eCB-tLTP induction in the same neuron (Fig.8B). These results demonstrate that eCB-tLTP and eCB-tLTD can be induced sequentially in the same neuron.
Figure 8. Bidirectional eCB synaptic plasticity in a single neuron.
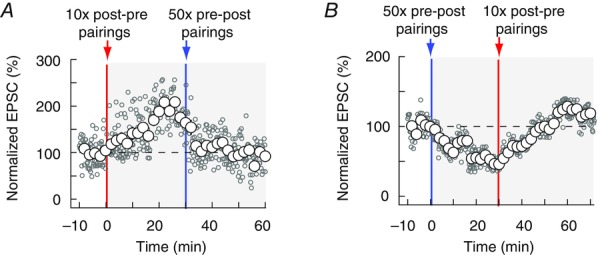
A and B, eCB-tLTP and eCB-tLTD can be induced sequentially in the same neuron. Representative experiments illustrate in A an eCB-tLTP event (induced by 10 post–pre pairings, red vertical line) followed by an eCB-tLTD occurrence (induced by 50 pre–post pairings, blue vertical line) and in B the reversed sequence (eCB-tLTD followed by eCB-tLTP). In both cases, the EPSC of the neurons returned to baseline level after the full sequence. Single EPSC amplitudes (empty grey circles) and averaged data (empty white circles) are represented.
Discussion
Corticostriatal long-term plasticity provides a fundamental mechanism for the function of the basal ganglia in action selection and in procedural learning (Yin & Knowlton, 2006; Yin et al. 2009; Koralek et al. 2012). Thus, characterizing the striatal plasticity repertoire in physiological conditions is crucial. The striatum receives a wide range of patterns of cortical activities from isolated trains of few spikes to prolonged bursting events. While corticostriatal plasticity under prolonged activation is well elucidated, its occurrence in response to few spikes has remained unexplored. Here, we report the existence of an eCB-tLTP induced by a low number of pairings in the striatum of both juvenile and adult rodents. Indeed, a few coincident pre- and postsynaptic spikes (5–10) were found to increase synaptic efficacy through a signalling pathway that relies on the eCB system. eCB-tLTP induction relies on activation of CB1R and TRPV1 and on 2-AG elevations triggered by coupled rises of calcium and DAGLα activity (mediated by mGluR5, muscarinic M1 receptors and VSCCs) in MSNs. Both activation of glutamatergic afferents from cerebral cortex and striatal cholinergic interneurons (which are monosynaptically contacted by cortical pyramidal cells (Fino et al. 2008)) promote the induction of eCB-tLTP.
We also found that eCB-tLTP is dopamine dependent. More precisely, eCB-tLTP is D2R mediated and not dependent on D1R. We then investigated the localization (pre- or postsynaptic) of the D2R involved in eCB-tLTP. The postsynaptic localization at MSNs was a priori less likely. Indeed, due to the segregation of expression of D1R and D2R among MSNs (Kreitzer & Malenka, 2008; Gerfen & Surmeier, 2011; Calabresi et al. 2014), roughly half of MSNs are expected to be D2R-expressing neurons. If eCB-tLTP was supported by the postsynaptic D2R MSNs, one would expect to induce eCB-tLTP in ∼50% of the (randomly chosen) MSNs. In our experiments, eCB-tLTP was successfully induced in 83% of the (randomly chosen) MSNs in rats, thus suggesting a presynaptic localization of the D2R. This was confirmed with experiments performed in D1R-eGFP mice, which show that eCB-tLTP can be induced in both striatopallidal and striatonigral MSNs. This suggests that the D2Rs involved in eCB-tLTP are not postsynaptically located. Presynaptic D2Rs are expressed at three different locations: the nigrostriatal dopaminergic afferents (De Mei et al. 2009), the cholinergic interneurons (Hersch et al. 1995) and the corticostriatal glutamatergic afferents (Bamford et al. 2004). The precise locus of presynaptic D2R involved in eCB-tLTP remains to be determined.
We describe here a paired-activity-dependent tLTP in mammals, wherein eCB signalling directly underlies both the induction and the long-term maintenance of synaptic weight increase. eCB signalling exhibits bidirectional plasticity with noth eCB-tLTP and eCB-tLTD. Bidirectionality is of paramount functional importance since it allows LTP and LTD to reverse each another at a single synapse.
eCBs have emerged as a major actor in learning and memory because of their powerful influence on synaptic plasticity (Katona & Freund, 2012; Castillo et al. 2012; Melis et al. 2014). The eCB system is mainly composed of biolipids synthesized and released on demand, acting as retrograde neurotransmitters on presynaptic CB1R (one of the most abundant G protein-coupled receptors in the brain) and postsynaptic TRPV1. eCBs have been reported to depress synaptic weight, i.e. short- or long-term depression, through the activation of CB1R (Kano et al. 2009; Katona & Freund, 2012; Castillo et al. 2012; Melis et al. 2014) or TRPV1 (Gibson et al. 2008; Maione et al. 2009; Chávez et al. 2010; Grueter et al. 2010; Puente et al. 2011). Noticeable exceptions are reports of an indirect role of eCBs in promoting LTP at mixed (chemical and electrical) synapses of the goldfish Mauthner cell via intermediary dopaminergic neurons (Cachope et al. 2007) or at hippocampal CA1 synapses via a GABAA receptor-mediated mechanism (Lin et al. 2011; Xu et al. 2012). Likewise, facilitation of LTP in the hippocampus via eCB-induced presynaptic depression of GABAergic transmission (Carlson et al. 2002; Chevaleyre & Castillo, 2004; Zhu & Lovinger, 2007), and mediation of heterosynaptic short-term potentiation via intermediary astrocytes (Navarrete & Araque, 2010) have been reported. However, to our knowledge, the present study is the first report of a paired-activity eCB-dependent LTP in mammals, with direct implication of eCBs in the induction and long-term maintenance of spike-timing-dependent potentiation of the stimulated synapse itself.
mGluR5 and M1R need to be simultaneously activated to elicit eCB-tLTP. Both receptors (mGluR5 and M1) are Gq/11-coupled receptors and positively coupled to PLCβ, thus leading to DAG production, and favouring the synthesis of 2-AG. Whereas cholinergic activation is not sufficient to trigger eCB-LTP, it remains necessary for eCB-LTP induction. Assuming that mGluR5 and M1 are localized postsynatpically (which remains to be demonstrated), our hypothesis is that eCB-LTP is induced only when large levels of 2-AG are produced. Our results from M1, mGluR5, VSCC and TRPV1 blocking experiments indicate that it is mandatory to activate all possible cumulative contributions to 2-AG production (PLCβ activation for DAG production, VSCC and TRPV1 to increase calcium surge, thus activating DAGLα) in order to reach high levels of 2-AG, which would promote eCB-tLTP.
Just as in hippocampal pyramidal cells (Shouval et al. 2002; Graupner & Brunel, 2012), postsynaptic calcium levels (or time course) could be crucial in the induction of eCB-STDP in the striatum. Since many of the steps along the eCB pathways are Ca2+ dependent (including 2-AG and anandamide synthesis), the ‘Ca2+ hypothesis’ would translate to the CB1R pathway. This would lead to a scenario where low to moderate peak levels of eCB would lead to LTD whereas high eCB levels would yield LTP. According to this scenario, our results could be reconciled if the first 5–20 post–pre pairings produce very large peak levels of 2-AG, and thus tLTP. If the amplitude of the 2-AG peaks decreases for subsequent post–pre pairings, this initial tLTP would be de-potentiated by the subsequent pairings, thus restricting the expression of eCB-tLTP to the first 5–20 pairings. On the other hand, the LTP observed with 100 post–pre pairings entirely results from an increase of the postsynaptic weight through activation of the CaMKII pathway by NMDARs, with no additional contribution of the eCB-LTP; indeed, when CB1R was inhibited, the NMDAR-tLTP induced with 100 post–pre pairings was not significantly affected (Fino et al. 2010). Thus, according to this scenario, eCB-LTP would start to be expressed after 5–20 post–pre pairings and subsequent pairings would then erase this potentiation while, independently, triggering the expression of NMDA-LTP.
There is a large diversity of the STDP rules at play in the brain and even within the same structure, variety seems to be the rule (Feldman, 2012). Indeed, in striatum the main neuronal population, the MSNs, express NMDAR-tLTP and eCB-tLTD (Shen et al. 2008; Pawlak & Kerr, 2008; Fino et al. 2010; Paillé et al. 2013) and eCB-tLTP (the present report). Whereas neighbouring striatal fast-spiking GABAergic interneurons express solely NMDAR-dependent STDP, both for LTP and LTD, for 100 pairings (Fino et al. 2008) but lack plasticity at low numbers of pairings (data not shown). In addition, our results provide evidence that eCB-tLTP is anti-Hebbian at corticostriatal synapses and tightly controlled by GABAergic interneurons, similar to the bidirectional corticostriatal STDP (i.e. NMDAR-tLTP and eCB-tLTD; Paillé et al. 2013). It has been reported that the endocannabinoid-mediated LTP at hippocampal CA1 synapses induced with high-frequency (Lin et al. 2011), low-frequency (Zhu & Lovinger, 2007) or paired stimulations (Xu et al. 2012) were prevented not only by inhibition of CB1R but also by inhibition of GABAA receptors. Here, we show that GABA is not involved in eCB-tLTP induction or magnitude at corticostriatal synapses but controls the polarity of eCB-tLTP.
Due to their on-demand intercellular signalling modus operandi (Alger & Kim, 2011), eCB biosynthesis and release are evoked by precisely timed and positioned physiological stimuli (Katona & Freund, 2008). However, evidence for TRPV1 activation by physiological neuronal activity patterns was lacking. As previously described, our study confirms that STDP indeed efficiently triggers eCB signalling and we demonstrate that STDP is able to engage the TRPV1 signalling pathway. Being a cationic channel highly permeable to calcium (Ross, 2003; Starowicz et al. 2007; Di Marzo, 2008), TRPV1 may contribute to eCB-tLTP induction by boosting the calcium transients in the postsynaptic element. As recently described for short- and long-term depression (Puente et al. 2011), our results illustrate the versatility of eCB signalling as a system displaying polymodal activation through CB1R and TRPV1, to trigger LTP.
eCB-LTP is promoted by very low numbers of pairings (∼5–10), therefore providing a mechanism whereby synapses react to the very first occurrences of incoming activity. This ability contrasts strongly with NMDAR-dependent LTP which requires the iteration of at least 75–100 paired stimulations to be expressed in the classical (1 Hz) STDP context. In mammals, associative memories and behavioural rules can be learned within a few trials (5–10) or even sometimes within a single trial (Schultz et al. 2003; Pasupathy & Miller, 2005; Tse et al. 2007; Quilodran et al. 2008; Ito & Doya, 2009). In the cortex or striatum, neurons with behaviour-related activities fire a few spikes upon behaviourally relevant events during each trial (i.e. at a frequency of 5–25 Hz and during 0.1-0.5 s, typically < 10 spikes; Schultz et al. 2003; Pasupathy & Miller, 2005; Quilodran et al. 2008), suggesting that a few trials should be sufficient to induce synaptic plasticity. eCB-tLTP may be used for learning and memorizing salient events from a few spikes. Hence, eCB-LTP may represent a molecular substrate operating during rapid learning of new arbitrary associative memories and behavioural rules characterizing the flexible behaviour of mammals or during the initial stages of slower habit learning (Barnes et al. 2011). Considering a specific context, the impairment of working memory by marijuana intoxication can be re-evaluated in light of our results. This impairment was hitherto interpreted solely as the effect of cannabinoids on the promotion of synaptic depression. Our results, together with recent reports (Lin et al. 2011; Xu et al. 2012), open new perspectives since they suggest that synaptic potentiation may as well be implied in the effects of marijuana.
Acknowledgments
We thank O. Manzoni, S. Hormuzdi, and P. Faure for helpful suggestions and critical comments; We thank C. Ledent (ULB, Belgium) and Denis Hervé (Institut du fer a Moulin, France) for kindly providing, respectively, CB1R−/− and D1R-eGFP mice.
Glossary
- 2-AG
2-arachidonoylglycerol
- AP
action potential
- AM251
N-(piperidin-1-yl)- 5-(4- iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AMG9810
(2E)-N-(2,3-dihydro- 1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide
- CB1R
type-1 cannabinoid receptor
- D1R
type-1 dopaminergic receptor
- D2R
type-2 dopaminergic receptor
- DAGLα
diacylglycerol lipase-α
- d-AP5
dl-2-amino-5-phosphono-pentanoic acid
- eCB
endocannabinoid
- EPSC
excitatory postsynaptic current
- LTD
long-term depression
- LTP
long-term potentiation
- MCPG
(S)-α-methyl-4-carboxyphenylglycine
- mGluR
metabotropic glutamate receptors
- MPEP
2-methyl-6-(phenylethynyl)pyridine hydrochloride
- MSN
medium-sized spiny neuron
- PPF
paired-pulse facilitation
- PLCβ
phospholipase Cβ
- STDP
spike-timing-dependent plasticity
- THL
tetrahydrolipstatin
- tLTD
spike-timing-dependent long-term depression
- tLTP
spike-timing-dependent long-term potentiation
- TRPV1
transient receptor potential vanilloid-type-1
- VSCC
voltage-sensitive calcium channels
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Y.C., H.B. and L.V. conceived and designed of the experiments; Y.C., V.P., H. X. and E.F. performed electrophysiological experiments and analysis; H.B., B.D. and S.G. contributed to analytical tools; Y.C., H.B. and L.V. wrote the manuscript; all authors have edited and corrected the manuscript.
Funding
This work has been supported by grants from INSERM, INRIA, Collège de France, the Agence Nationale pour la Recherche and the Ecole des Neurosciences de Paris.
References
- Alger BE. Kim J. Supply and demand for endocannabinoids. Trends Neurosci. 2011;34:304–315. doi: 10.1016/j.tins.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C. Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes TD, Mao JB, Hu D, Kubota Y, Dreyer AA, Stamoulis C, Brown EN. Graybiel AM. Advance cueing produces enhanced action-boundary patterns of spike activity in the sensorimotor striatum. J Neurophysiol. 2011;105:1861–1878. doi: 10.1152/jn.00871.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mackie K, Triller A, O'Brien J. Pereda AE. Potentiation of electrical and chemical synaptic transmission mediated by endocannabinoids. Neuron. 2007;56:1034–1047. doi: 10.1016/j.neuron.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Ghiglieri V. Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat Neurosci. 2014;17:1022–1030. doi: 10.1038/nn.3743. [DOI] [PubMed] [Google Scholar]
- Carlson G, Wang Y. Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Carter AG. Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chavez AE. Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AE, Chiu CQ. Castillo PE. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci. 2010;13:1511–1518. doi: 10.1038/nn.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V. Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- De Mei C, Ramos M, Iitaka C. Borrelli E. Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol. 2009;9:53–58. doi: 10.1016/j.coph.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Filippo M, Picconi B, Tantucci M, Ghiglieri V, Bagetta V, Sgobio C, Tozzi A, Parnetti L. Calabresi P. Short-term and long-term plasticity at corticostriatal synapses: Implications for learning and memory. Behav Brain Res. 2009;199:108–118. doi: 10.1016/j.bbr.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;60:1–24. doi: 10.1007/112_0505. [DOI] [PubMed] [Google Scholar]
- Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–571. doi: 10.1016/j.neuron.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Deniau JM. Venance L. Cell-specific spike-timing-dependent plasticity in GABAergic and cholinergic interneurons in corticostriatal rat brain slices. J Physiol. 2008;586:265–282. doi: 10.1113/jphysiol.2007.144501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J. Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM. Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E. Venance L. Spike-timing dependent plasticity in the striatum. Front Synaptic Neurosci. 2010;2:6. doi: 10.3389/fnsyn.2010.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ. Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubard V, Fino E. Venance L. Contribution of astrocytic glutamate and GABA uptake to corticostriatal information processing. J Physiol. 2011;589:2301–2319. doi: 10.1113/jphysiol.2010.203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M. Brunel N. Calcium-based plasticity model explains sensitivity of synaptic changes to spike pattern, rate, and dendritic location. Proc Natl Acad Sci U S A. 2012;109:3991–3996. doi: 10.1073/pnas.1109359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G. Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi Shin H-S. Kano M. Phospholipase Cβ serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H. Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15:5222–5237. doi: 10.1523/JNEUROSCI.15-07-05222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst C, Rees HD, Heilman CJ. Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;17:3334–3342. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff K, Kovalchuk Y, Yuste R. Konnerth A. Single-shock LTD by local dendritic spikes in pyramidal neurons of mouse visual cortex. J Physiol. 2004;560:27–36. doi: 10.1113/jphysiol.2004.072678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Doya K. Validation of decision-making models and analysis of decision variables in the rat basal ganglia. J Neurosci. 2009;29:9861–9874. doi: 10.1523/JNEUROSCI.6157-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosahu T, Hashimotodani Y, Uchigashima M. Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Koralek AC, Jin X, Long JD, Costa RM. Carmena JM. Corticostriatal plasticity is necessary for learning intentional neuroprosthetic skills. Nature. 2012;483:331–335. doi: 10.1038/nature10845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I. Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Katona I. Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Böhme GA, Imperato A, Pedrazzini T, Roques BP, et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- Lin QS, Yang Q, Liu DD, Sun Z, Dang H, Liang J, Wang YX, Chen J. Li ST. Hippocampal endocannabinoids play an important role in induction of long-term potentiation and regulation of contextual fear memory formation. Brain Res Bull. 2011;86:139–145. doi: 10.1016/j.brainresbull.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Mahon S, Vautrelle N, Pezard L, Slaght SJ, Deniau JM, Chouvet G. Charpier S. Distinct patterns of striatal medium spiny neuron activity during the natural sleep-wake cycle. J Neurosci. 2006;26:12587–12595. doi: 10.1523/JNEUROSCI.3987-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Cristino L, Migliozzi AL, Georgiou AL, Starowicz K, Salt TE. Di Marzo V. TRPV1 channels control synaptic plasticity in the developing superior colliculus. J Physiol. 2009;587:2521–2535. doi: 10.1113/jphysiol.2009.171900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ. Morris RG. New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus. 2002;12:609–636. doi: 10.1002/hipo.10107. [DOI] [PubMed] [Google Scholar]
- Mathur BN. Lovinger DM. Endocannabinoid-dopamine interactions in striatal synaptic plasticity. Frontiers Pharmacol. 2012;3:1–11. doi: 10.3389/fphar.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Greco B. Tonini R. Interplay between synaptic endocannabinoid signaling and metaplasticity in neuronal circuit function and dysfunction. Eur J Neurosci. 2014;39:1189–1201. doi: 10.1111/ejn.12501. [DOI] [PubMed] [Google Scholar]
- Navarrete M. Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron. 2010;68:113–126. doi: 10.1016/j.neuron.2010.08.043. [DOI] [PubMed] [Google Scholar]
- Paillé V, Fino E, Du K, Morera Herreras T, Perez S, Hellgren Kotaleski J. Venance L. GABAergic circuits control spike-timing-dependent plasticity. J Neurosci. 2013;33:9353–9363. doi: 10.1523/JNEUROSCI.5796-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasupathy A. Miller EK. Different time courses of learning-related activity in the prefrontal cortex and striatum. Nature. 2005;433:873–876. doi: 10.1038/nature03287. [DOI] [PubMed] [Google Scholar]
- Pawlak V. Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Astarita G. Rapaka R. A neuroscientist's guide to lipidomics. Nat Rev Neurosci. 2007;8:743–754. doi: 10.1038/nrn2233. [DOI] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P. Manzoni OJ. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci. 2011;14:1542–1547. doi: 10.1038/nn.2974. [DOI] [PubMed] [Google Scholar]
- Quilodran R, Rothé M. Procyk E. Behavioral shifts and action valuation in the anterior cingulate cortex. Neuron. 2008;57:314–325. doi: 10.1016/j.neuron.2007.11.031. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ. Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Redgrave P. Gurney K. The short-latency dopamine signal: a role in discovering novel actions? Nat Rev Neurosci. 2006;7:967–975. doi: 10.1038/nrn2022. [DOI] [PubMed] [Google Scholar]
- Remy S. Spruston N. Dendritic spikes induce single-burst long-term potentiation. Proc Natl Acad Sci U S A. 2007;104:17192–17197. doi: 10.1073/pnas.0707919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- Schultz W, Tremblay L. Hollerman JR. Changes in behavior-related neuronal activity in the striatum during learning. Trends Neurosci. 2003;26:321–328. doi: 10.1016/S0166-2236(03)00122-X. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P. Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF. Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A. Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Starowicz K, Nigam S. Di Marzo V. Biochemistry and pharmacology of endovanilloids. Pharmacol Ther. 2007;114:13–33. doi: 10.1016/j.pharmthera.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazakim M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, et al. The Endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB. Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse D, Langston RF, Bethus I, Wood ER, Witter MP. Morris RG. Schemas and memory consolidation. Science. 2007;316:76–82. doi: 10.1126/science.1135935. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M. Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JY, Zhang J. Chen C. Long-lasting potentiation of hippocampal synaptic transmission by direct cortical input is mediated via endocannabinoids. J Physiol. 2012;590:2305–2315. doi: 10.1113/jphysiol.2011.223511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Matsui M. Watanabe M. Preferential localization of muscarinic M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J Neurosci. 2010;30:4408–4418. doi: 10.1523/JNEUROSCI.5719-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH. Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilário MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM. Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ. Lovinger D. Persistent synaptic activity produces long-lasting enhancement of endocannabinoid modulation and alters long-term synaptic plasticity. J Neurophysiol. 2007;97:4386–4389. doi: 10.1152/jn.01228.2006. [DOI] [PubMed] [Google Scholar]