

Abstract
Key points
A conserved proline in M1 causes a kink between α and π helical segments.
The kink is under greater tension in the resting versus active conformation.
The kink and the agonist do not interact directly.
The π-helix separates the gating functions of the extracellular and transmembrane domains.
Mutations of the conserved proline and propofol increase desensitization.
Abstract
Nicotinic acetylcholine receptors (AChRs) switch on/off to generate transient membrane currents (C↔O; closed-open ‘gating’) and enter/recover from long-lived, refractory states (O↔D; ‘desensitization’). The M1 transmembrane helix of the muscle endplate AChR is linked to a β-strand of the extracellular domain that extends to a neurotransmitter binding site. We used electrophysiology to measure the effects of mutations of amino acids that are located at a proline kink in M1 that separates π and α helices, in both α (N217, V218 and P221) and non-α subunits. In related receptors, the kink is straighter and more stable in O vs. C structures (gating is ‘spring-loaded’). None of the AChR kink mutations had a measureable effect on agonist affinity but many influenced the allosteric gating constant substantially. Side chains in the M1 α-helix experience extraordinarily large energy differences between C and O structures, probably because of a ∼2 Å displacement and tilt of M2 relative to M1. There is a discrete break in the character of the gating transition state between αN217 and αV218, indicating that the π-helix is a border between extracellular- and transmembrane-domain function. Mutations of the conserved M1 proline, and the anaesthetic propofol, increase a rate constant for desensitization. The results suggest that straightening of the M1 proline kink triggers AChR desensitization.
Introduction
The muscle AChR has an equatorial gate in the transmembrane domain (TMD) that regulates ion conductance and two sites in the extracellular domain (ECD) that can bind agonists to influence the gating equilibrium constant (Sine, 2012; Unwin, 2013; Auerbach, 2014; Changeux, 2014). The TMD of each subunit is a 4-helix bundle (M1–M4). M2 lines the pore and bends to open/close the gate (Sauguet et al. 2014). M1 has a proline kink and a π-helix (Hibbs & Gouaux, 2011) that are conserved in all pentameric ligand-gated ion channels (pLGICs). We measured changes in agonist binding, channel gating and receptor desensitization consequent to mutations of residues located at the M1 kink.
M1 is linked covalently to the β10-strand of the ECD that projects to an agonist site (Fig.1A). The kink is located just above the level of the TMD equator, close to a binding site for the anaesthetic propofol in a prokaryotic pLGIC (Fig.1B) (Nury et al. 2011). It has been proposed (Lee & Sine, 2005) and questioned (Purohit & Auerbach, 2013) that the principal pathway for the channel-opening transition starts with movements of ligands at agonist sites that perturb the β10 backbone and a salt bridge at its base to control the M2 gate. Below, we present results that show that the M1 π-helix and ECD have a common gating transition state but argue against an agonist–β10–M1 mechanical link.
Figure 1. M1 structure.
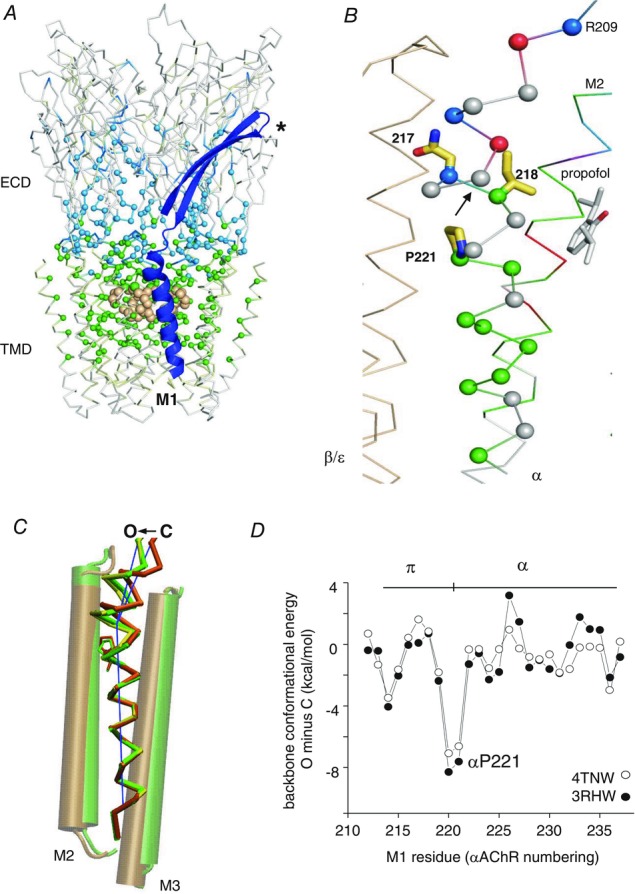
A, side view of the AChR homologue GLIC (PDB accession number 3P50; Nury et al. 2011). ECD, extracellular domain; TMD, transmembrane domain; blue, β9–10–M1; light brown spheres, equatorial M2 gate; *, neurotransmitter binding site (AChR α subunit). The αC atoms are coloured by gating φ value (AChR α subunit). The ECD is mostly ∼0.8 (cyan) and the TMD is mostly ∼0.6 (green). B, close-up of M1 (αAChR numbering). αN217–αV218 is a gating φ value border (arrow). The N-terminal residue of the π-helix, αF214, has a φ ∼0.3 (red). C, M1, O vs. C (GluCl) (Hibbs & Gouaux, 2011; Althoff et al. 2014). The π−α tilt angle is ∼7 deg shallower in O (green; 3RHW) vs. C (orange; 4TNV) and even shallower in a possibly desensitized GABA receptor structure (yellow; 4COF; Miller & Aricescu, 2014). D, the difference in M1 backbone conformational energy, O minus C (GluCl). The π−α boundary is more strained in C.
We used single-channel electrophysiology to assess the effects of mutations at three kink positions (αN217, αV218 and αP221) with regard to gating (Fig.2) and desensitization. The relative position of the peak free energy change experienced by a side chain within a reaction (for instance, C↔O or O↔D) is given by φ, the slope of a rate–equilibrium free energy relationship (REFER). In muscle AChRs residues in the αECD have a relatively early gating transition state (φ ∼0.8), most of those in the αTMD reach theirs approximately mid-reaction (φ ∼0.6) and those at the gate change energy (structure) near the end of the opening process (φ ∼0.3) (Purohit et al. 2013) (Fig.1A). This pattern led to the proposal that the channel-opening transition occurs as a longitudinal conformational ‘wave’ (Grosman et al. 2000), but recent results suggest that this process starts at the ECD–TMD interface rather than at the agonist sites.
Figure 2. Estimating gating energies from rate constants.
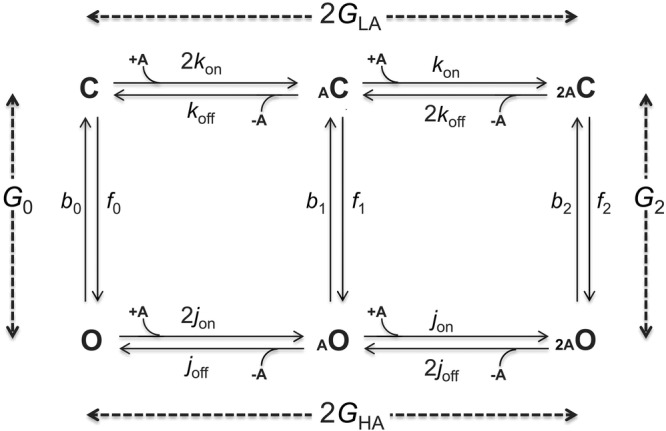
C, closed-channel structure; O, open-channel structure; a, agonist, shown in small font because it is a tiny structural perturbation (∼0.005% of the total mass). Vertical steps are ‘gating’ that occur by the same mechanism with or without bound agonist (Purohit & Auerbach, 2009). The free energy values (Gn, in kcal mol−1) are differences between the end states. Free energies were calculated from equilibrium constants (rate constant ratios) estimated from single-channel current interval durations. Horizontal steps are ‘binding’ (LA, lower affinity to C; HA, higher affinity to O). In mouse adult-type muscle AChRs the 2 binding steps are equivalent and independent. The total energy from the two affinity changes is ΔGB2 = 2(GHA – GLA). From microscopic reversibility, ΔGB2 = G2 – G0. G0 and ΔGB2 are proportional to the log of the allosteric and coupling constant, respectively.
To a first approximation AChRs have only two stable conformations, C (without agonists) and D. These have lifetimes of ∼minutes, whereas all others have sub-millisecond lifetimes. Hence, the O state is a transient intermediate within C↔D that is, however, the source of the physiological response. It is known that in AChRs, desensitization proceeds mainly from O states (Katz & Thesleff, 1957; Auerbach & Akk, 1998). Hence, an agonist molecule is a catalyst that reduces the activation energy of the C↔D transition state by increasing the stability of the O intermediate. Little is known about the structural changes that comprise AChR desensitization, but the structure of a possibly desensitized GABA receptor suggests that in addition to having conductance regulated by the equatorial gate, the pore can be occluded at its base by a constriction of the M2 helices (Miller & Aricescu, 2014).
Interesting observations regarding AChR gating function have been made previously regarding the M1 kink amino acids. The mutation αN217K causes a congenital myasthenic syndrome (CMS) (Wang et al. 1997). Many side chains throughout the protein change energy between C and O. The largest range of energy change measured so far is for αV218, where an alanine-to-tyrosine substitution (in both α subunits) increases the gating equilibrium constant more than a millionfold (Purohit et al. 2013). αP221 is conserved absolutely across the entire pentameric ligand-gated ion channel superfamily. Substitutions of natural side chains here that are expected to reduce kinking reduce or eliminate AChR currents, whereas ester backbone substitutions that allow kinking do not (England et al. 1999). We discovered that in our preparation natural mutations of αP221 are functional.
There are three main findings. (1) Mutations of the kink residues (only in the α subunit) change the unliganded (allosteric) gating equilibrium constant substantially but have no detectable effect on either low- or high-affinity agonist binding. (2) There is a sharp discontinuity in gating φ values in the π-helix. The αN217–αV218 backbone bond is a border that divides ECD and TMD gating function. (3) Mutations of αP221 are like others in αM1 with regard to gating but unusual because they increase desensitization, as does the anaesthetic propofol. The results suggest that the M1 π-helix is an important moving part in AChR gating and that straightening this kink initiates receptor desensitization.
Methods
Mutagenesis and expression
Human embryonic kidney (HEK293) cells were maintained at 37°C (95% air and 5% CO2) in Dulbecco's minimum essential medium supplemented with 10% (v/v) fetal bovine serum plus 1% (v/v) penicillin–streptomycin (pH 7.4). Mutations were created using Quik-Change site-directed mutagenesis kit (Stratagene, LA Jolla, CA, USA) and confirmed by dideoxy sequencing of the cDNA samples. HEK cells were transiently transfected with a mixture of cDNAs (αβδε, 2:1:1:1 ratio, ∼3.5 μg per 35 mm dish) encoding wild-type (WT) or mutant subunits by calcium phosphate precipitation. cDNA encoding green fluorescent protein (GFP) (0.1 μg μl−1) was added as a marker. The cells were washed after about 16 h and electrophysiological recording commenced within 24–48 h.
Electrophysiology
Single-channel currents were recorded in the cell-attached patch configuration at 23°C. The bath and pipette solution was phosphate-buffered saline (PBS) containing (mm): 137 NaCl, 0.9 CaCl2, 2.7 KCl, 1.5 KH2PO4, 0.5 MgCl2, and 8.1 Na2HPO4 (pH 7.4). In experiments with ligand, choline (Cho), carbamylcholine (CCh), acetylcholine (ACh) or propofol (100 μm) was added just to the pipette solution. Typically, the pipette potential was held at +70 mV (which corresponds to a membrane potential of approximately −100 mV). Unliganded AChR currents were measured under similar conditions except without any agonists added to the pipette solution. A separate pipette holder was used in these experiments to avoid any contamination. Patch pipettes were pulled from borosilicate capillaries to a resistance of ∼10 MΩ and coated with Sylgard (Dow Corning, Midland MI USA). Single-channel currents were recorded using a PC-505B amplifier (Warner Instrument Corp., Hamden, CT, USA) with external low-pass filtering (Warner Instruments; LPF-8) at 20 kHz, and were digitized at a sampling frequency of 50 kHz using an SCB-68 data acquisition board (National Instruments, Toronto, ON, Canada).
Kinetic analysis of gating
The single-channel currents were digitized, analysed and simulated using QuB software (Nicolai & Sachs, 2013). Clusters of single-channel openings that reflect mainly binding and gating events arising from individual AChRs were selected by eye. Intra-cluster currents were idealized into noise-free intervals (after digitally low-pass filtering at 10–15 kHz) by using the segmental k-means algorithm with a two-state, shut↔open model. Rate constants were estimated from the idealized interval durations by using a maximum log-likelihood algorithm after imposing a dead time correction of 50 μs.
Energy estimation
A cyclic reaction scheme was used to estimate the salient energies (Fig.2). In experiments with agonist, the diliganded opening (f2) and closing (b2) rate constants were measured using either Cho (20 mm), CCh (5 mm) or ACh (500 μm). These concentrations are ∼5 times larger than the corresponding resting-state equilibrium dissociation constant (Kd) and ensured that agonists were almost always present at both binding sites. The idealized intra-cluster interval durations were fitted by a shut↔open↔shut model. The first step of this scheme estimated f2 and b2 and the second step accommodated a state associated with brief desensitization (Elenes & Auerbach, 2002). From the rate constants for the first step we calculated the diliganded gating equilibrium constant (E2 = f2/b2) and gating free energy difference (G2 = −0.59lnE2; kcal mol−1).
The intrinsic (unliganded) C↔O equilibrium constant is called the allosteric constant and the corresponding energy difference, G0, is proportional to its logarithm. In order to estimate the effect of a mutation on the unliganded gating equilibrium constant (E0 = f0/b0), the frequency of constitutive openings was increased over the WT by using the triple-mutant background construct α(D97A + Y127F + S269I) (DYS). αD97 and αY127 are in the ECD (loop A and β-strand 6) and αS269 is in the TMD (αM2–3 linker). Together, these mutations increase E0 by ∼63,000-fold (−6.5 kcal mol−1) without affecting Kd or the low/high affinity ratio (Kd/Jd) (the ‘coupling’ constant) (Purohit & Auerbach, 2009). The mutated positions in M1 are not close to these background mutations. The fold-change in f0 and b0 over the DYS background caused by an M1 mutation was calculated from the idealized current interval durations to estimate a net unliganded gating equilibrium constant, E0mut+DYS. The free energy change caused by the mutations was ΔG0mut and was calculated as −0.59ln(E0mut+DYS/E0DYS).
Within the forward A2C→A2O isomerization there is a low→high affinity change at each of the two neurotransmitter binding sites (Fig.1C). The low/high equilibrium dissociation constant ratio at each site is the coupling constant, and ΔGB1 is proportional to its logarithm. From the cycle (and assuming detailed balance), the difference in binding free energy for two sites combined (ΔGB2) is equal to the difference between the free energies for diliganded vs. unliganded gating. For each mutation, this energy was calculated as: ΔGB2mut = (G2mut − G0mut).
Correcting for additional background perturbations
Part of our basic strategy for measuring the energies was to use background mutations to engineer the allosteric constant so that the emergent single-channel current interval durations were in an optimal range for detection and analysis (∼0.1–10 ms) (Jadey et al. 2011). When high [agonist] was used, fast channel block by the ligand was reduced by depolarization, a perturbation that also changes G0. The measured rate constants were then corrected for the effects of the background, to estimate their values under a standard condition (adult WT AChRs, −100 mV).
The f2ACh values for the mutations αV218F and Y were too fast to be reliably measured using the WT construct. We therefore added background perturbations to slow the opening rate constant (make G0 more positive) in order to obtain estimates that were more accurate. The background perturbations were +100 mV depolarization, αT422V and εI257A. These reduce E0WT by ∼13-, 23- and 59-fold (increase G0WT by +1.5, +1.8 and +2.4 kcal mol−1), respectively, without affecting ΔGB2. The energy changes caused by these three background perturbations were assumed to be independent and, therefore, energetically additive. In combination, the net ΔG0 values were: αT422V + depolarization (+3.3 kcal mol−1), εI257A + depolarization (+3.9 kcal mol−1) and αT422V + εI257A + depolarization (+5.7 kcal mol−1). These background energies add to that of the adult WT (+8.3 kcal; Nayak et al. 2012) to set the net G0 for the background construct.
Kd estimation
The low affinity agonist association (kon) and dissociation (koff) rate constants were estimated by fitting globally idealized intra-cluster open and shut interval durations obtained at three different agonist concentrations (see Fig.4). As shown below, the αN217K mutation increased E0 by ∼42-fold (ΔG0 = −2.2 kcal mol−1) and made f2 with the potent agonists acetylcholine and carbamylcholine too fast to be measured reliably. Therefore, we expressed αN217K using a distant, loss-of-function background mutation (αY127C) that reduced E0 by ∼180-fold (ΔG0 = +3.1 kcal mol−1) but did not affect KdACh. The background was WT when choline was the agonist. The kon and koff estimates were optimized assuming equivalent binding sites (Jha & Auerbach, 2010; Nayak et al. 2014) using a two binding-step, linear activation scheme: A + C↔AC + A↔A2C↔A2O (where A is the agonist; Fig.2). Kd was calculated as koff/kon.
Figure 4. αN217K binding.

A–C, interval duration histograms and example clusters from the CMS mutation αN217K, activated by three different agonists. The dissociation/association rate constants (s−1 and μm−1 s−1) for ACh, carbamylcholine (CCh) and choline (Cho) were 18,200/100, 8930/26 and 9639/9.2, respectively. D, the mutation did not change the resting equilibrium dissociation constant (Kd) significantly. The background construct was WT in C and αY127C in A and B. This mutation has no effect on binding but reduces the allosteric constant by ∼200-fold (+3.1 kcal mol−1) (Purohit & Auerbach, 2007b).
Desensitization modelling
The connectivity between the four or five desensitized states apparent in diliganded WT AChRs has not been established (Elenes & Auerbach, 2002). We used a linear C↔O↔D1↔D2↔D3 scheme to estimate rate and equilibrium constants for gating and desensitization of unliganded AChRs. Other, less-coupled schemes produced similar results for the O↔D1 equilibrium constant, but these could not be distinguished on the basis of log-likelihood value.
We measured desensitization parameters for αP221 (and other subunit) mutations, or after the addition of propofol. In these experiments the background construct was DYS and no agonist molecules were present. The error limits on the desensitization rate constants were large, in part because there were only a small number of long-lived desensitized states in the current recordings. Also, the D3→D2 rate constant depends on the number of channels in the patch, which was variable.
REFER analyses
The rate and equilibrium constants for a series of mutations of one position were plotted in log–log format to generate a rate–equilibrium free energy relationship (REFER). The slope of the linear fit to the REFER is called φ and gives the extent to which a change in the equilibrium constant was caused by a change in the forward vs. backward rate constant, on a scale from 1 to 0. φ estimates the reaction progress at transition state for the perturbed position (1 is early and 0 is late). REFERs for other αM1 residues have been presented elsewhere (Purohit et al. 2013).
Structure analyses
π-helix identification
We used the following criteria for identifying a π-helix (Cooley et al. 2010). (i) At least two (n–n+5) backbone H-bonds (in AChR α subunits, between positions 214–219 and 215–220). (ii) Dihedral angles: (ψn + φn+1) ≈ −125 deg in the π-helix region (≈ −105 deg in the α-helix region). In GluCl, this sum for the first residue of the π-helix is ∼−95 deg, as expected. (iii) Seven amino acids, with those in the αAChR π-helix (FIINVII) being characteristic (Fodje & Al-Karadaghi, 2002). We cross-checked our π-helix identification using the pi-HUNT code (Cooley et al. 2010).
The π–α tilt angle was that between the central axes of the helices. The central axis was defined by the least squares linear regression fit of the coordinates of the backbone atoms. The regression fit was calculated by a Singular Value Decomposition technique. Residues 214–220 and 223–236 (AChR α subunit numbers) were used for the π and α axes.
Dihedral angles
The dihedral angles φ and ψ were obtained from the Ramachandran plot using VMD version V1.9 (Humphrey et al. 1996). All M1 residues (including the proline) are in the trans conformation in all pLGIC structures. Therefore, the rotation angle Ω per residue in the helix could be calculated by using:
Backbone bond energy
The conformational energy of the backbone of the M1 helix was calculated using CHARMM (Brooks et al. 2009). The bonded energy terms (the bond, angle, Urey-Bradley, dihedral and improper energy) were calculated using the CHARMM27 force field (MacKerell et al. 1998) with CMAP corrections.
Results
M1 structures
The M1 kink proline (αP221 in AChRs) separates a π-helix (α214–α220) and an α-helix (α221–α233). π-helices are common secondary structures that are often located at active sites (Cooley et al. 2010). The M1 backbone of GLIC (Sauguet et al. 2013), GluCl (Hibbs & Gouaux, 2011), ELIC (Hilf & Dutzler, 2008) and a GABAA receptor (Miller & Aricescu, 2014) fits the definition: seven residues, two n + 5 backbone H-bonds, (ψ n + φn+1) ≈ 125 deg, characteristic side chains and a following proline (Fodje & Al-Karadaghi, 2002). The M1 π-helix is present in all pLGIC X-ray structures reported so far, with the possible exception of 5-HT3 (Hassaine et al. 2014). Our search did not identify any other π-helices in GluCl, GLIC, ELIC or GABA structures, but one is present in the intracellular domain of the 5-HT3 receptor, just before M4 (positions 321–329).
The proline causes M1 to kink at boundary between the π and α helices (Fig.1C). In GluCl C→O (Hibbs & Gouaux, 2011; Althoff et al. 2014), the kink angle becomes shallower by ∼7 deg. Figure1D shows that the conformational energy of the backbone is lower (more stable) in O, in particular at the π–α boundary (α220–α221; AChR α subunit numbers). The AChR channel-opening conformational change appears to be ‘spring-loaded’ in so far as the M1 kink relaxes, C→O.
Mutations of M1 α-helix residues αV218, αC222, αF225, αS226 and αL228 have large effects on the diliganded gating equilibrium constant (Purohit et al. 2013). This indicates that the gating rearrangement of the αTMD helix bundle alters the local environments of these M1 side chains, with significant energy consequences. Figure1C shows the position of the M1 α-helix relative to M2 in GluCl. We speculate that the ∼2 Å upward relative displacement of M2, O vs. C, is the basis for the large gating energy changes in the αM1 side chains.
αN217
Figure 3A shows low time-resolution views of single-channel activity from αN217K AChRs with and without agonist molecules at the two transmitter binding sites. In both conditions the clusters of openings are mainly C↔O gating events and the silent periods between clusters are sojourns in desensitized states. Cluster durations reflect the time required to enter a long-lived desensitized state and the durations of the gaps between clusters reflect the time required to recover. Our analyses of this π-helix residue were restricted to intra-cluster, gating events. Qualitatively, there was no apparent effect of αN217 mutations on entry into (cluster duration) or recovery from (long-gap duration) desensitization.
Figure 3. αN217 gating.

A, low time-resolution view of single-channel currents. The clusters of openings (down) are gating events and the long gaps between clusters are desensitization. Top, with 20 mm choline (WT background); bottom, constitutively-active background (DYS). B, higher-resolution views of clusters for αN217 mutants, with and without agonist. In both conditions, K and T mutations increase cluster PO. C, for all mutations, the change in the C vs. O free energy difference (ΔGn) was the same regardless of whether or not agonists were present (n = 2 or 0). D, REFERs. In the α subunit the linear slope (φ, given below) is the same either with (filled circles) or without (open circles) agonists. For mutations in non-α subunits the change in gating equilibrium constant was too small to allow φ estimation.
Relative to the WT, the CMS mutation K increased the cluster open probability (PO) substantially, either when choline was present or in the absence of agonists (Fig.3B). An increase in PO indicates an increased gating equilibrium constant and a relatively more stable (more negative) O-state free energy. An F substitution also increased cluster PO but Q, E and A substitutions had little or the opposite effect. The effect of each mutation on the O vs. C free energy difference was approximately the same regardless of whether the binding sites were occupied by choline or just water (Fig. 3C). Hence, all of the tested mutations of αN217 only changed the allosteric constant (the unliganded gating equilibrium constant) but did not alter significantly the energy generated by affinity changes for the two choline molecules (the coupling constant).
The coupling constant of each binding site is proportional to the log of the ratio of equilibrium dissociation constants, C vs. O (Fig.2). It was possible, therefore, that the αN217K mutation decreased each constant to the same extent so that their ratio remained unchanged. Also, the effect of the αN217K mutation could be different for choline than for the neurotransmitter, ACh. We therefore measured Kd in this mutant by fitting single-channel currents intervals across different concentrations, using three different agonists (Fig.4). The estimated Kd values for choline (1.0 mm), CCh (0.34 mm) and ACh (0.18 mm) were approximately the same as in the WT. None of the αN217 substitutions, including the CMS mutant K, had a measurable effect on agonist binding.
Figure 3D (left) shows REFERs for αN217, with and without agonists. The slopes for both conditions were similar, with φ ∼0.8. This indicates that this π-helix side chain reaches its gating transition state relatively early, at about the same position as many other residues in the αECD.
We also examined the effects of several mutations of the corresponding M1 position in the non-α subunits. E, K and A substitutions at βN228, δN231 and εN226 had little or no effect on the diliganded gating equilibrium constant so no φ value could be estimated.
αV218
Figure 5A shows example currents of Y and S mutations of another π-helix residue, αV218, with and without agonists. As was the case for αN217, the changes in di- vs. unliganded gating energies were approximately equivalent (Fig.5B). These mutations altered the allosteric constant but did not influence the coupling constant.
Figure 5. αV218 gating.

A, example currents with and without 20 mm choline (current amplitude is smaller with agonist because of fast channel block). In both conditions the Y mutation increases and the S mutation decreases PO. B, the effect of the mutations on the gating equilibrium constant was the same with or without agonists. C, REFERs. f, opening rate constant; E, gating equilibrium constant. In the α subunit most mutations cause a slightly greater fold-change in the forward vs. backward gating rate constant (φ = 0.6). Inset, the 3 kinetic modes for the W mutation have a higher φ value than for other mutations. The φ value for the β subunit is smaller than for α. Mutations do not change gating significantly in δ and ε.
Figure 5C shows REFERs for this position in α and non-α subunits. Mutations had the largest effects on gating in α, but those in the β subunit, too, were substantial. The αV218 φ value was ∼0.6, a value that is characteristic of many other residues in the αTMD. The β229 φ value was lower, indicating a somewhat later transition state. Mutations of δV232 and εN227 had little or no effect on the diliganded gating equilibrium constant so a φ value could not be estimated.
The αV218W mutant was unusual because three different cluster populations could be identified based on their PO values. We do not know the mechanism that generates this heterogeneity, but because we did not observe mode-switching within clusters this process must be slow relative to desensitization. We analysed each population separately to generate a REFER for just αV218W (Fig.5C, left inset). The resulting φ value was ∼0.8, which is higher than those for the other αV218 mutants and the same as for αN217.
There are two α subunits in each AChR. To ascertain whether or not the energetic consequences of the two αV218 mutations were symmetric both WT and mutant α subunits were co-transfected along with WT β, δ and ε subunits (Fig.6). Accordingly, some receptors have two WT α subunits, some have two mutant α subunits and some would be ‘hybrids’ having one WT and one mutant α subunit, in two different ways. For αV218Y, 2 hybrid populations were apparent. The change in gating energy for the double mutant was equal to the sum of these energies calculated for the individual hybrid populations. αV218Y mutations have energetically distinct but independent effects in the two α subunits, but the experiments do not reveal whether the ∼−1.3 kcal mol−1 more favourable energy arises from the α–ε or α–δ subunit. The REFER shows that the hybrids have a φ value that is about the same as the double-mutant series. For αV218F, only one hybrid population was apparent. The gating energy change for the double-mutant was exactly twice that of the hybrid. The αV218F mutation is energetically symmetrical in the two α subunits, so that the two hybrids appear as a single population. Again, the φ value for the hybrid was the same as for the double mutant series.
Figure 6. Subunit symmetry of αV218 mutants.
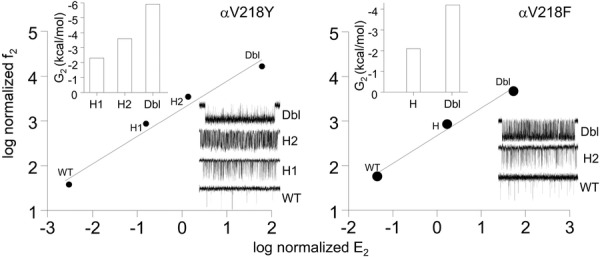
Left, 2 hybrid populations were apparent in αV218Y. The change in gating energy for the double mutant (Dbl) was equal to the sum of these energies calculated for the individual hybrid populations (H1 and H2). Right, one hybrid population was apparent in αV218F. The gating energy change for the double-mutant was exactly twice that of the hybrid. The αV218F mutation is energetically symmetric in the two α subunits, so that the two hybrids appear as a single population. For both F and Y substitutions, the φ values for the hybrids were the same as for the double mutant.
αP221: gating
In muscle AChRs there are three prolines in the extended αM1 region. One is in the β10–π linker (αP210), another is at the kink (αP221) and the third is near the cytoplasmic limit of the α-helix (αP236). Previously, it was found that mutations of αP210 have little or no effect on the gating equilibrium constant (Purohit & Auerbach, 2007a). Apparently, this side chain experiences little energy change between C and O. We tested five side chain substitutions at αP236 (A, C, G, F and S) but in all cases no single-channel openings were apparent (5–10 patches of each mutant; ∼30 min recording time per patch). These AChRs may have failed to fold and express, or they could have prevented opening because of a tiny gating or an enormous desensitization equilibrium constant.
We examined 10 different substitutions of αP221 (G, A, S, V, C, L, T, R, F and Y) (Fig.7). All of the mutations gave rise to functional AChRs, as evidenced by typical single-channel current amplitudes and a concentration-dependent increase in gating activity between 30 and 500 μm ACh. AChRs having an S, L or R mutation did not produce clusters of openings at 500 μm ACh and were not analysed further. The other mutations produced clusters that, however, had complicated kinetics (see below).
Figure 7. αP221 gating.

A, low time-resolution views of αP221G activity, with and without agonists. In both conditions the clusters are interrupted by long gaps that reflect sojourns in desensitized states. B, higher-resolution views of clusters for mutants activated by 30 μm (left) or 500 μm (right) ACh. C, the effect of αP221 mutations on the gating equilibrium constant was the same with or without agonists, indicating no effect on the coupling constant. D, REFERs. The gating φ value for the α subunit proline is 0.6 and similar to that for αV218; φ is smaller in β.
We assumed that at high [ACh] the predominant shut and open interval components within clusters represented A2C↔A2O gating. Accordingly, we could estimate diliganded gating rate and equilibrium constants and construct a REFER. For A and G mutations the change in gating was approximately the same with or without agonists (Fig.7C). For the P, G, A, V and Y side chains the φ value for the α subunit proline was ∼0.6, with the largest energy change being for a Y to G substitution (−4.3 kcal mol−1). Overall, αP221 mutations resemble those of αV218 in so far as they produce large energy differences between C and O only by changing the allosteric constant and reach their transition state just before the midpoint of the gating isomerization.
Figure 7D also shows gating REFERs for A and G mutations of the corresponding proline in the non-α subunits. Mutations of βP232 had small but measureable effects, with a φ value that was lower than that for αP221. Mutations of δP235 and εP230 had little or no effect on the diliganded gating equilibrium constant so a φ value could not be estimated.
αP221: desensitization
In whole cell currents from muscle AChRs, desensitization is manifest as a decline in the response in the continued presence of agonist. Many side chain substitutions slow the time constant of the decline (τD), but often this happens only because of a reduction in the gating equilibrium constant and, hence, the equilibrium occupancy of the intermediate O state (the state from which desensitization occurs) rather than by altering the microscopic rate constants of the desensitization process itself. Mutations at the gate region of M2, too, can slow τD, but this happens simply because the A2O state has been made more stable so that all exit rate constants are slowed, including those for channel closing (O→C) and desensitization (O→D). Hence, both of these classes of mutation reveal little about the actual desensitization mechanism; a weak agonist and a channel blocker will have similar effects on τD.
The overall architecture of clusters was visibly different with αP221 mutations, indicating that they changed the desensitization process itself (Fig.8). To quantify this difference we examined the kinetics of the long, desensitization gaps in G and A mutants of αP221 and its non-α homologues. In order to avoid interference from open-channel noise from fast, unresolved channel block by the agonist, we studied unliganded activity using a constitutively active background.
Figure 8. αP221 desensitization.
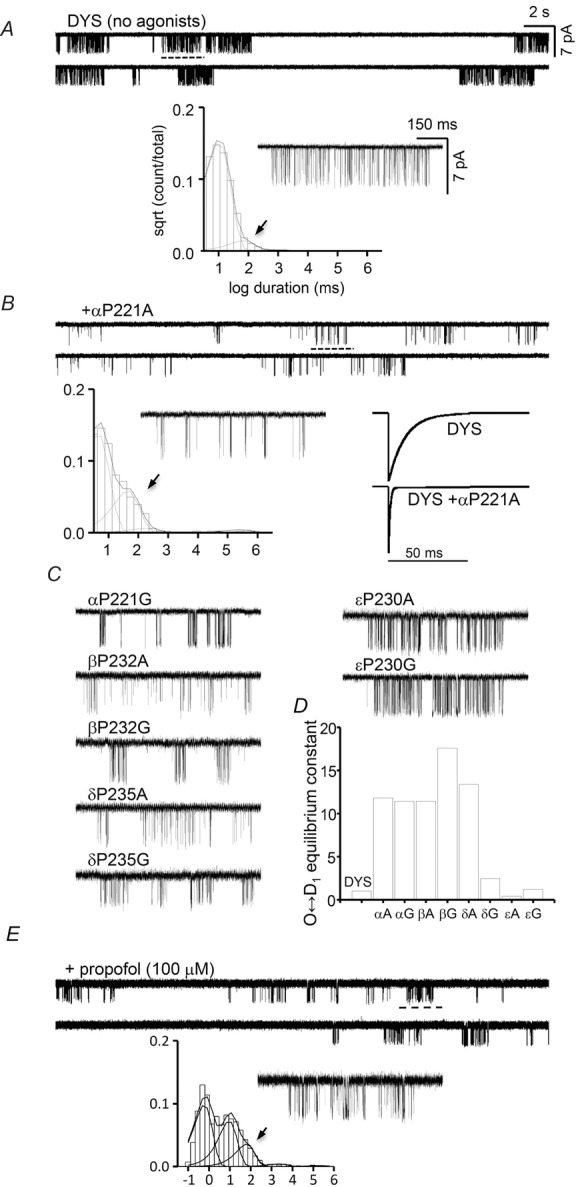
A, unliganded activity of the constitutively active background. Below, an example cluster and the global shut-interval duration histogram. The briefest component is gating and that with a time constant of τ ≈ 0.1 s (arrow) reflects sojourns in a brief desensitized state; longer desensitized components were infrequent and not visible in the histogram. B, unliganded activity of αP221A expressed on the same background. This mutation causes a substantial increase in the prevalence of the τ ≈ 0.1 s shut component. Bottom right, simulated whole-cell currents for a step increase to saturating [ACh] based on the rate constants shown in Table1. C, example clusters for other proline mutants. D, O↔D1 desensitization equilibrium constants (see Table1). A and G substitutions cause a ∼10-fold increase in this constant in the α and β subunits, as did A in δ. E, adding 100 μm propofol (same background, no proline mutation) causes a similar increase in the prevalence of the τ ≈ 0.1 s shut component.
In WT AChRs exposed to high [ACh] there are five shut interval components (≥10 μs) in the single-channel record, with the briefest corresponding to gating and the other four reflecting sojourns in desensitized states of various durations (Elenes & Auerbach, 2002). Desensitization kinetics of unliganded AChRs have not yet been reported so we first measured the shut components for the background construct alone (no agonists and no αP221 mutation) (Fig.8A). We observed one C state but only three D states. This difference (and the desensitized time constants) compared to the diliganded WT pattern (Elenes & Auerbach, 2002) are interesting, but because our goal was to measure the effects of proline mutations we did not pursue this further. Using a linear (chain) scheme, in the background the three desensitization equilibrium constants were all ∼5, but with >10-fold slower rate constants with progression through the chain (Table1). The briefest desensitized component had a lifetime of τ ≈ 100 ms that, unlike the slowest component, is not affected by the number of channels in the patch.
Table 1.
Desensitization rate and equilibrium constants for the DYS background and proline mutants (all rate constants, s−1; n is the number of patches
| Construct | O→D1 | ±SEM | D1→O | ±SEM | E(O↔D1) | ±SEM | n |
|---|---|---|---|---|---|---|---|
| DYS | 101.6 | 35.0 | 18.8 | 5.0 | 6.3 | 2.0 | 4 |
| αP221A | 1101.0 | 126.0 | 17.8 | 5.0 | 74.8 | 26.0 | 3 |
| αP221G | 268.7 | 40.0 | 3.8 | 0.2 | 71.9 | 13.0 | 3 |
| βP232A | 1794.0 | — | 25.0 | — | 71.8 | — | 1 |
| βP232G | 435.7 | 207.0 | 7.4 | 3.0 | 70.6 | 24.0 | 3 |
| δP235A | 303.5 | 94.0 | 3.9 | 2.0 | 84.8 | 15.0 | 2 |
| δP235G | 382.0 | — | 25.0 | — | 15.3 | — | 1 |
| εP230A | 74.7 | 6.0 | 39.0 | 15.0 | 2.4 | 0.7 | 3 |
| εP230G | 213.8 | 63.0 | 14.4 | 4.0 | 16.7 | 4.5 | 3 |
| Construct | D1→D2 | ±SEM | D2→D1 | ±SEM | E(D1↔D2) | ±SEM | n |
| DYS | 3.3 | 1.0 | 0.8 | 0.2 | 5.9 | 3.4 | 4 |
| αP221A | 1.8 | 0.8 | 1.7 | 1.0 | 1.8 | 0.9 | 3 |
| αP221G | 0.9 | 0.0 | 0.6 | 0.3 | 3.9 | 2.7 | 3 |
| βP232A | 1.0 | — | 1.3 | — | 0.8 | — | 1 |
| βP232G | 0.8 | 0.3 | 2.3 | 1.0 | 0.7 | 0.4 | 3 |
| δP235A | 0.7 | 0.1 | 0.3 | 0.2 | 2.2 | 0.2 | 2 |
| δP235G | 6.7 | — | 1.6 | — | 4.2 | — | 1 |
| εP230A | 7.4 | 2.4 | 4.7 | 2.6 | 2.4 | 0.8 | 3 |
| εP230G | 4.1 | 1.3 | 1.0 | 0.4 | 6.5 | 3.3 | 3 |
| Construct | D2→D3 | ±SEM | D3→D2 | ±SEM | E(D2↔D3) | ±SEM | n |
| DYS | 0.33 | 0.26 | 0.110 | 0.090 | 6.7 | 2.7 | 4 |
| αP221A | 0.26 | 0.05 | 0.022 | 0.002 | 11.9 | 3.1 | 3 |
| αP221G | 0.24 | 0.12 | 0.030 | 0.010 | 6.9 | 3.5 | 3 |
| βP232A | 0.13 | — | 0.009 | — | 14.4 | — | 1 |
| βP232G | 0.04 | — | 0.004 | — | 10.0 | — | 1 |
| δP235A | 0.12 | 0.10 | 0.006 | 0.001 | 20.2 | 18.0 | 2 |
| δP235G | 1.10 | — | 0.050 | — | 22.0 | — | 1 |
| εP230A | 1.40 | 0.70 | 0.230 | 0.100 | 6.6 | 1.7 | 3 |
| εP230G | 0.92 | 0.20 | 0.060 | 0.010 | 16.2 | 5.8 | 3 |
We observed a similar number of shut components in all of the eight constructs we examined using the same background but with different mutations of the kink proline (Fig.8B and C). In five of the mutants, αP221 (A and G), βP232 (A and G) and δP235A, the main effect of the mutation was to increase the prevalence of the τ ≈ 100 ms shut component. Kinetic analyses showed that in these mutants the O↔D1 equilibrium constant increased on average by ∼12-fold, usually by an increase in the forward, entry rate constant (Fig.8D). In these constructs the subsequent desensitization steps were similar to those in the background and changed only by ≤3-fold. The G mutation in δ and both A and G mutations in ε had little or no effect on any of the desensitization equilibrium constants. Figure8B shows simulated whole-cell currents based on the rate constants shown in Table1.
Figure 8E shows the effects of the anaesthetic propofol on unliganded activity in the same, constitutively active background (no proline mutation). The cluster architecture resembled those of the αP211 mutants. With propofol, two shut components (τ ∼10 ms and ∼100 ms) were more prevalent than in the background.
Discussion
Allosteric constant
Mutations of many αM1 residues have substantial effects on the diliganded gating equilibrium constant, which indicates this region of the protein changes its energy between C and O. However, the kink mutations influence only the unliganded gating equilibrium constant (the allosteric constant) and have little or no effect on agonist binding (the resting affinity or the coupling constant). Despite being linked covalently to a β-strand that reaches up to an agonist site, substituting side chains at αN217, αV218 and αP221 have no detectable, long-range interactions with agonist.
This pattern of locality for side chain gating energy change is echoed throughout the AChR. So far, mutations of only a few residues in the immediate vicinity of the agonist sites have been found to alter substantially the coupling constant, and mutant-cycle analyses show that residue pairs separated by <∼10 Å show only a small amount of interaction energy in gating (Purohit et al. 2013). In the AChR, most side chain gating rearrangements, including those in αM1, have mainly local energetic consequences that sum to set the overall O vs. C energy difference and, hence, the allosteric constant (Purohit et al. 2013). When a mutation away from an agonist site shifts the midpoint of a concentration–response profile it is probable that the effect is mediated through the global, intrinsic gating conformational change.
We draw two conclusions from this pattern. First, to a good approximation natural selection of AChR side chains appears to be an independent, stepwise search for a combination that places the allosteric constant into an optimal range for physiology and survival. Second, most long-distance interactions in gating probably occur through the backbone rather than the side chains. In AChRs, the side chains appear to comprise a viscous liquid that coats the backbone and does not propagate energy over long distances.
Subunit symmetry
The extent of change in the allosteric constant caused by mutations has been measured for many α and non-α positions throughout the pentamer. Mutations of many TMD residues produce larger mutational changes in the α subunits compared to β, δ and ε, for example in the pre-M1 (Bruhova & Auerbach, 2010), the M2–3 linker (Jha et al. 2009), M3 and M4 (Purohit et al. 2013). A comparison of the sensitivities of M1 side chains in different subunits suggests that mutations here, too, have larger effects in α. It appears that most positions in M1, M3 and M4 of the non-α subunits show little or no energy change between C and O, which suggests that they do not undergo a substantial structural change in gating. This pattern, however, is inverted in M2 at the gate region, where larger energy changes prevail in δ, β and ε compared to α (Purohit et al. 2013). This suggests that the M2 gating rearrangements in the non-α subunits are triggered by movements of the α subunits rather than by intra-subunit movements.
At the M1 kink, the β subunit showed the next-largest sensitivity after α. Mutations of the M1 proline increased desensitization to similar extents in the α, β and ε subunits, so this process may be more subunit-symmetric than for gating. With regard to the α-subunit symmetry at αV218, F mutations had indistinguishable effects but for Y these were different. This suggests that at this level of the αTMD the two V218 side chains (that face the bundle core rather than an adjacent subunit) are not acting identically in gating.
φ boundary
The Cα atoms of αM1 residues are coloured by φ value in Fig.1B. Between positions αV218 and αL231, all residues that are sufficiently sensitive to allow a gating φ measurement have a value of 0.60 ± 0.03 (mean ± SD; n = 9). This indicates that the C↔O transition state for all of these side chains is reached about midway through the process and suggests that this segment (that includes αP221) moves as a rigid body between C and O. Many α subunit side chains in M2, M3 and M4 that face the helix bundle core also have φ values of ∼0.6, so in AChRs the TMD helices appear to unpack as a concerted event near the midpoint of the gating reaction.
In our measurements, the gating behaviours of αP221 mutants were typical of those in the M1 α-helix. They only change the allosteric constant, with a φ value of ∼0.6 and with an energy range that is similar to those for other positions. Previous results showing that natural mutations of the kink proline reduce activity probably derive from enhanced desensitization rather than a loss of binding or gating function.
The φ value for αN217 is larger than for αV218 and the α-helix residues. This indicates that the π-helix reaches its gating transition state earlier than the distal portion of αM1. Putting the low-φ residues in M1 aside for the moment (see below), measurable positions between αR209 and αN217 have an average φ of 0.79 ± 0.06 (n = 3), which is typical for the αECD. Residues between αV218 and αL231 have an average φ of 0.6, which is typical for the αTMD. Hence, in the α subunit the αN217–αV218 bond is a discrete border that separates the gating actions of the ECD and the TMD. The αM1 π-helix separates the gating functions of the sensor and effector domains of the AChR. In the C→O transition, the β10-strand, salt bridge arginine and part of the π-helix (α198–α217) comprise a contiguous structural and functional element that projects from an agonist site to near the hydrophobic gate.
The break in gating function at the kink is correlated with the M1 hydrogen bond pattern in GLIC. Because of αP221, the αN217 backbone carbonyl is free. However, in GLIC the (partly conserved) side chain here bonds to the backbone of the proximal, n-4 residue (αY213 in AChRs; not conserved). One residue down, the backbone bond of αV218 is part of the hydrogen bond network of the α-helix. The αV218W substitution has an ECD-like φ value of ∼0.8. We speculate that the indole nitrogen connects this side chain with the proximal, rather than distal, hydrogen bond network, to the effect of increasing its φ value. There are other discrete φ boundaries in AChR gating; it is possible that these, too, derive from transitions between hydrogen bond networks.
There are two low-φ positions in or near the π-helix, αF214 and αL210 (φ = 0.32 and 0.35; coloured red in Fig.1B). Both of these non-polar side chains reach the external surface of the protein, close to the level of the extracellular surface of the membrane. As noted previously (Purohit et al. 2013), all of the low-φ positions in the TMD appear to be in contact with either water or lipid, including at the M2 gate region and at a patch in αM3 that corresponds to an ivermectin/lipid binding site of GluCl (Hibbs & Gouaux, 2011). We hypothesize that the low φ values in M1 reflect a late gating energy change arising from interactions with lipid molecules. It is possible that the entire annulus of boundary lipids is perturbed as a single, structural unit at the end of the gating process, to lower φ values of separated, membrane-facing residues.
Conformational change
The backbone and gating φ values are contiguous, from loop C at the top of β10, the αR029 salt bridge and the M1 π-helix (αY198–αN217). However, there is little evidence to support the idea that the agonist generates a force that is transmitted mechanically to the TMD equator through this track in the gating isomerization. (i) Deletion of loop C (in the α subunits) eliminates agonist binding but has almost no effect on constitutive gating (Purohit & Auerbach, 2013). (ii) The gating equilibrium constant (with agonists) is normal in AChRs that lack the αR209 salt bridge (Purohit & Auerbach, 2007a). (iii) The effects of αM1 mutations (including αP221) are approximately the same with or without agonists. (iv) Conversely, αM1 mutations had no detectable effect on agonist binding, and the energy from the agonist affinity change has no apparent effect on αM1 (Purohit & Auerbach, 2013). Despite the physical connection between the agonist site and αM1, the results so far suggest that energy changes associated with gating (but perhaps not desensitization) along this pathway do not occur by a rigid-body linkage.
Lacking time-resolved signals of AChR internal gating motions, we use φ values to place the αM1 energy changes into a sequence, regardless of whether these quantify relative transition state positions or time. Without agonists the highest φ values (earliest transition states) are in the αM2–3 linkers (∼0.95), followed by residues at the agonist sites (Purohit & Auerbach, 2010) and the rest of the αECD (∼0.8), most of the αTMD (∼0.6) and the M2 equator (∼0.3). This map suggests that AChR opening starts with a spontaneous fluctuation in the αM2–3 backbone (Lummis et al. 2005; Bafna et al. 2008), followed by a tilt/twist of the αECD and the π-helix, a bend in M2 and then expansion of the gate. The high φ value for αN217 suggests that the decrease in the π–α tilt angle precedes M2 bending and, hence, that it is a cause rather than a consequence.
αP221 mutations alter the desensitization process. With αP221A and βP232A, the rate constant for entry into a desensitized state is >10 times faster than without the mutation. This translates to a desensitization φ value ∼1, which implies that these positions reach their transition state near the onset of this reaction. Propofol produced effects that were similar, but not identical, to those caused by the αP221 mutations, so we hypothesize that in muscle AChRs this anaesthetic reduces activity by increasing the occupancy of desensitized states. We did not study in detail the effects of propofol on gating, so other binding sites (Ghosh et al. 2013) and actions of this drug are possible.
The structural events that undergird AChR desensitization are not well understood. Regarding β10–M1, in α7 nicotinic receptors mutations of a proline near the middle of the companion β9-strand slows desensitization (McCormack et al. 2010). The mutation of the M1 proline is expected to modify both the π–α angle and the α-helix twist. In AChRs, these changes appear to be more important energetically in the α and β subunits.
Because the O conformation is a necessary intermediate for desensitization, we hypothesize that the straightening of M1 in C→O apparent in GluCl continues in O→D (and, perhaps, propofol-inhibition). The high desensitization φ value for some αP221 mutants suggests that changes in energy (structure) occur at the M1 π–α border early in the desensitization process, and, hence, may be a trigger. It is possible that greater M1 straightening allows M2 to occlude the bottom of the pore, as suggested by a possibly desensitized GABA receptor structure (Miller & Aricescu, 2014).
Acknowledgments
We thank M. Merritt, M. Shero and M. Teeling for technical assistance and David Cadugan for early screening of some mutants.
Glossary
- AChR
acetylcholine receptor
- CMS
congenital myasthenic syndrome
- DYS
triple-mutant background construct α(D97A + Y127F + S269I)
- ECD
extracellular domain
- pLGIC
pentameric ligand-gated ion channel
- REFER
rate–equilibrium free energy relationship
- TMD
transmembrane domain
- WT
wild-type
Additional information
Competing interests
None declared.
Author contributions
P.P. designed, implemented and analysed the electrophysiology experiments. S.C. designed, implemented and analysed the structural computations. A.A. participated in the design of all experiments and wrote the paper. All authors approved the final version of the manuscript.
Funding
This work was funded by grants from NIH (NS064969 and NS023513).
References
- Althoff T, Hibbs RE, Banerjee S. Gouaux E. X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature. 2014;512:333–337. doi: 10.1038/nature13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. Agonist activation of a nicotinic acetylcholine receptor. Neuropharmacology. 2014 doi: 10.1016/j.neuropharm.2014.10.004. (in press; DOI: 10.1016/j.neuropharm.2014.10.004 ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach A. Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J Gen Physiol. 1998;112:181–197. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafna PA, Purohit PG. Auerbach A. Gating at the mouth of the acetylcholine receptor channel: energetic consequences of mutations in the αM2-cap. PLoS One. 2008;3:e2515. doi: 10.1371/journal.pone.0002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM. Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhova I. Auerbach A. Subunit symmetry at the extracellular domain-transmembrane domain interface in acetylcholine receptor channel gating. J Biol Chem. 2010;285:38898–38904. doi: 10.1074/jbc.M110.169110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux J-P. Protein dynamics and the allosteric transitions of pentameric receptor channels. Biophys Rev. 2014;6:311–321. doi: 10.1007/s12551-014-0149-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley RB, Arp DJ. Karplus PA. Evolutionary origin of a secondary structure: π-helices as cryptic but widespread insertional variations of α-helices that enhance protein functionality. J Mol Biol. 2010;404:232–246. doi: 10.1016/j.jmb.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenes S. Auerbach A. Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. J Physiol. 2002;541:367–383. doi: 10.1113/jphysiol.2001.016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England PM, Zhang Y, Dougherty DA. Lester HA. Backbone mutations in transmembrane domains of a ligand-gated ion channel: implications for the mechanism of gating. Cell. 1999;96:89–98. doi: 10.1016/s0092-8674(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Fodje MN. Al-Karadaghi S. Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng. 2002;15:353–358. doi: 10.1093/protein/15.5.353. [DOI] [PubMed] [Google Scholar]
- Ghosh B, Satyshur KA. Czajkowski C. Propofol binding to the resting state of the Gloeobacter violaceus ligand-gated ion channel (GLIC) induces structural changes in the inter- and intrasubunit transmembrane domain (TMD) cavities. J Biol Chem. 2013;288:17420–17431. doi: 10.1074/jbc.M113.464040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Zhou M. Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, Graff A, Stahlberg H, Tomizaki T, Desmyter A, Moreau C, Li XD, Poitevin F, Vogel H. Nury H. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature. 2014;512:276–281. doi: 10.1038/nature13552. [DOI] [PubMed] [Google Scholar]
- Hibbs RE. Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJC. Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A. Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jadey SV, Purohit P, Bruhova I, Gregg TM. Auerbach A. Design and control of acetylcholine receptor conformational change. Proc Natl Acad Sci U S A. 2011;108:4328–4333. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A. Auerbach A. Acetylcholine receptor channels activated by a single agonist molecule. Biophys J. 2010;98:1840–1846. doi: 10.1016/j.bpj.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Purohit P. Auerbach A. Energy and structure of the M2 helix in acetylcholine receptor-channel gating. Biophys J. 2009;96:4075–4084. doi: 10.1016/j.bpj.2009.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY. Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- Lummis SC, Beene DL, Lee LW, Lester HA, Broadhurst RW. Dougherty DA. Cis–trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 2005;438:248–252. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- McCormack TJ, Melis C, Colón J, Gay EA, Mike A, Karoly R, Lamb PW, Molteni C. Yakel JL. Rapid desensitization of the rat α7 nAChR is facilitated by the presence of a proline residue in the outer β-sheet. J Physiol. 2010;588:4415–4429. doi: 10.1113/jphysiol.2010.195495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D. Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Miller PS. Aricescu AR. Crystal structure of a human GABAA receptor. Nature. 2014;512:270–275. doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak TK, Bruhova I, Chakraborty S, Gupta S, Zheng W. Auerbach A. Functional differences between neurotransmitter binding sites of muscle acetylcholine receptors. Proc Natl Acad Sci USA. 2014;111:17660–17665. doi: 10.1073/pnas.1414378111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak TK, Purohit PG. Auerbach A. The intrinsic energy of the gating isomerization of a neuromuscular acetylcholine receptor channel. J Gen Physiol. 2012;139:349–358. doi: 10.1085/jgp.201110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai C. Sachs F. Solving ion channel kinetics with the QuB software. Biophys Rev Lett. 2013;8:191–211. [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M. Corringer PJ. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Purohit P. Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the ‘pre-M1’ linker. J Gen Physiol. 2007a;130:559–568. doi: 10.1085/jgp.200709857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P. Auerbach A. Acetylcholine receptor gating: movement in the α-subunit extracellular domain. J Gen Physiol. 2007b;130:569–579. doi: 10.1085/jgp.200709858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P. Auerbach A. Unliganded gating of acetylcholine receptor channels. Proc Natl Acad Sci U S A. 2009;106:115–120. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P. Auerbach A. Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J Gen Physiol. 2010;135:321–331. doi: 10.1085/jgp.200910384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P. Auerbach A. Loop C and the mechanism of acetylcholine receptor-channel gating. J Gen Physiol. 2013;141:467–478. doi: 10.1085/jgp.201210946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Gupta S, Jadey S. Auerbach A. Functional anatomy of an allosteric protein. Nat Commun. 2013;4:2984. doi: 10.1038/ncomms3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L, Poitevin F, Murail S, Van Renterghem C, Moraga-Cid G, Malherbe L, Thompson AW, Koehl P, Corringer PJ, Baaden M. Delarue M. Structural basis for ion permeation mechanism in pentameric ligand-gated ion channels. EMBO J. 2013;32:728–741. doi: 10.1038/emboj.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L, Shahsavar A, Poitevin F, Huon C, Menny A, Nemecz A, Haouz A, Changeux JP, Corringer PJ. Delarue M. Crystal structures of a pentameric ligand-gated ion channel provide a mechanism for activation. Proc Natl Acad Sci U S A. 2014;111:966–971. doi: 10.1073/pnas.1314997111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM. End-plate acetylcholine receptor: structure, mechanism, pharmacology, and disease. Physiol Rev. 2012;92:1189–1234. doi: 10.1152/physrev.00015.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N. Nicotinic acetylcholine receptor and the structural basis of neuromuscular transmission: insights from Torpedo postsynaptic membranes. Q Rev Biophys. 2013;46:283–322. doi: 10.1017/S0033583513000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Auerbach A, Bren N, Ohno K, Engel AG. Sine SM. Mutation in the M1 domain of the acetylcholine receptor αsubunit decreases the rate of agonist dissociation. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]