

Summary
Lung nociceptors initiate cough and bronchoconstriction. To elucidate if these fibers also contribute to allergic airway inflammation we stimulated lung nociceptors with capsaicin and observed increased neuropeptide release and immune cell infiltration. In contrast, ablating Nav1.8+ sensory neurons or silencing them with QX-314, a charged sodium channel inhibitor that enters via large pore ion channels to specifically block nociceptors, substantially reduced ovalbumin or house dust mite-induced airway inflammation and bronchial hyperresponsiveness. We also discovered that IL-5, a cytokine produced by activated immune cells, acts directly on nociceptors to induce release of vasoactive intestinal peptide (VIP). VIP then stimulates CD4+ and resident innate lymphoid type 2 cells, creating an inflammatory signaling loop that promotes allergic inflammation. Our results indicate that nociceptors amplify pathological adaptive immune responses and that silencing these neurons with QX-314 interrupts this neuro-immune interplay, revealing a potential new therapeutic strategy for asthma.
Introduction
Asthma is an inflammatory airway disease caused by environmental (allergens, air pollution, cold, smoking) and genetic interactions (Martinez, 2007). The disease affects 7–10% of the world’s population, causes ~250,000 deaths annually (Akinbami, 2006), and its prevalence is increasing (Ramsey and Celedon, 2005). Asthma symptoms include wheezing, coughing, chest tightness and shortness of breath, caused by increased airway hyperresponsiveness, inflammation, mucus hypersecretion and structural remodeling (Locksley, 2010). Histopathology shows goblet cell metaplasia, thickened basement membranes, increased airway smooth muscle and inflammatory cell infiltration (Locksley, 2010). Immune cells, particularly innate lymphoid type 2 cells (ILC2), T helper 2 (TH2) cells, and eosinophils, are central to the pathological airway transformation. Inhaled allergens such as house dust mites, viruses or bacteria are sensed mainly by dendritic cells in the lung mucosa, which promote precursor TH cell differentiation into TH2 cells. These, along with ILC2 cells, initiate an inflammatory response including recruitment and activation by cytokines of immune effector cells, with eosinophils contributing to bronchoconstriction, microvascular permeability and airway remodeling (Kumar et al., 2005; Locksley, 2010).
The lung is densely innervated by sensory fibers, most of which express markers of nociceptors, including the TRP channels TRPV1 and TRPA1 (Ni et al., 2006). Airway nociceptors respond to chemical, mechanical or thermal stimuli to initiate essential protective airway reflexes such as cough (Canning et al., 2006). Asthmatic patients have a denser network of these fibers around small airways (Barnes, 1996; Myers et al., 2002) and a reduced activation threshold in response to airborne irritants (Canning and Spina, 2009). Patients also display elevated neuropeptide levels in bronchoalveolar lavage fluids (BALF) (Lilly et al., 1995). These features indicate changes in, and excess activity of peptidergic sensory fibers (Patterson et al., 2007). The large-pore cation channels TRPV1 and TRPA1 are activated by exogenous chemical irritants, such as cigarette smoke (Kanezaki et al., 2012) and also directly and indirectly via GPCR- and receptor tyrosine kinase-coupling by many endogenous ligands generated during inflammation, including protons, lipids, endogenous cannabinoids, bradykinin and NGF (Szallasi et al., 2007). Stimulation of nociceptor peripheral terminals results in calcium-mediated vesicular release of neuropeptides like substance P and calcitonin gene related peptide (CGRP), to generate neurogenic inflammation, which is characterized by increased vascular permeability and vasodilatation. This is amplified and spreads by the antidromic reflex, where the sensory input in one branch of a sensory neuron initiates an action potential back down a connecting branch to its peripheral terminal (Chiu et al., 2012). Nociceptors may contribute to airway disease both by their capacity to produce bronchoconstriction (Trankner et al., 2014) and local neurogenic inflammation (Caceres et al., 2009; Hox et al., 2013). Supporting involvement of sensory fibers in key aspects of type-2 inflammation, genetic knockout or pharmacological antagonism of the TRPA1 channel reduced inflammation in a mouse model of allergic airway disease (Caceres et al., 2009), while ablation of TRPV1 afferents blocked bronchial hyperresponsiveness (Trankner et al., 2014). However, exactly how sensory neurons and immune cells cooperate to amplify immunopathology and direct various types of inflammation is unknown (Chiu et al., 2013; Liu et al., 2014; Nussbaum et al., 2013; Riol-Blanco et al., 2014; Wilson et al., 2013).
We have investigated the contribution of lung nociceptor neurons to the generation and resolution of allergic airway inflammation by asking if global ablation of all nociceptor neurons (NaV1.8 expressing neurons) attenuates allergen-induced immune responses. We also temporarily silenced nociceptors using inhaled permanently charged quaternary derivatives of sodium channel blockers (Binshtok et al., 2007). The charge of these quaternary compounds prevents their diffusion through neuronal membranes, making them ineffective as local anesthetics. However, their small size enables them to permeate into cells through activated large-pore channels, including TRPV1 (Binshtok et al., 2007) and TRPA1 (Lennertz et al., 2012). A combination of a TRP channel opener and the charged sodium blocker QX-314 produces prolonged (>9 hours) peripheral nociceptor silencing and a pain- or itch- specific local blockade (Binshtok et al., 2007; Brenneis et al., 2013; Roberson et al., 2013). This strategy enabled us to determine the consequences of blocking lung nociceptor activity on established allergic inflammation. We find that sensory neurons, in part through the VIP-VPAC2 axis, drive CD4+ and ILC2 cell production of cytokines including IL-5, and reciprocally that IL-5 activates sensory neurons. Our results indicate that nociceptors play a major role in amplifying adaptive immune responses in the lung and local pharmacological silencing of nociceptors interrupts the pro-inflammatory signaling loop between neurons and immune cells to enhance allergic airway inflammation resolution.
Results
Nociceptor activation and allergic airway inflammation
Allergic inflammation and bronchial hyperresponsiveness can be induced in mice by an initial sensitization to an allergen, in this case ovalbumin (OVA) (i.p. day 0 and 7) followed by inhaled OVA challenges (days 14–17) (figure 1A). To investigate if sensory fibers play a role in allergic airway inflammation we first performed lung nociceptor gain-of-function experiments in vivo. Mice were treated with intranasal instilled capsaicin, a selective TRPV1 agonist (Szallasi et al., 2007), and 24h later bronchoalveolar lavage fluid (BALF) was immunophenotyped. In naïve mice (no OVA exposure) capsaicin treatment increased CD45+ cells (marker of all immune cells) (figure 1B). In OVA-treated mice, capsaicin administration on day 18 (the day after completing OVA challenges and time of maximal inflammation), further increased the already elevated levels of CD45+ cells (figure 1B). These included eosinophils (figure 1C), macrophages (figure 1D) and lymphocytes (figure 1E). The pro-inflammatory effect of capsaicin was also observed in lungs of lymphocyte reporter mice (LCK+/−eGFP+/−; figure 1F–G). We conclude that exogenous activation of lung nociceptors increases airway leukocytes. The larger effect in OVA-challenged mice than naïve mice implies that factors released by nociceptors in inflamed lung augment immune cell recruitment.
Figure 1. Nociceptor activation promotes airway inflammation.

(A) Inflammation induced in mice following i.p. allergen (ovalbumin (OVA)) sensitization (day 0 and 7, with AlOH) and inhaled challenge (days 14–17). Drugs/vehicle administered at peak of OVA-induced inflammation (day 18) and impact assessed on day 21. Capsaicin (B–E; 1 μmol, red bar) or vehicle (white bar) instilled (intranasally) on Day 17 after vehicle or OVA challenge (24h prior to BALF measurement). Capsaicin increased airway CD45+ (total immune) (B), eosinophil (C), macrophage (D) and lymphocyte (E) cell counts. T-cell influx (green, LCK-Cre+/−eGFP+/−) in control lungs following vehicle (F) or capsaicin (G; 1 μmol) instillation. Scale bar 10 μm. Mean ± S.E.M; Two-tailed unpaired Student’s t-test (n = 6–10 animals/group; 2 cohorts).
Nociceptor ablation and allergic airway inflammation
We next used targeted diphtheria toxin-based cell ablation (Palmiter et al., 1987) to explore if nociceptor neurons are required for lung inflammation in the OVA mouse model. NaV1.8 is a TTX-resistant voltage-gated sodium channel expressed by nociceptors, including those in the nodose ganglion (Muroi and Undem, 2014) and the majority of nodose ganglion lung afferents (Kwong et al., 2008). NaV1.8-Cre+/−DTA+/− mice are devoid of all NaV1.8 expressing nociceptors (Stirling et al., 2005) and have no response to noxious heat, mechanical stimuli or capsaicin (Chiu et al., 2013). In WT mice and NaV1.8-Cre+/−DTA−/− littermate control mice (with intact nociceptors), numbers of CD45+ cells, including eosinophils and lymphocytes increased substantially in BALFs obtained on protocol day 21 (four days after the last inhaled OVA challenge) (figure 2A–D). In contrast, OVA-exposed NaV1.8-Cre+/−DTA+/− mice (with ablated nociceptors) showed significantly dampened immune cell infiltration on day 21 with fewer eosinophils, macrophages and lymphocytes (figure 2A–D).
Figure 2. Lung sensory neuron ablation reduces airway allergic inflammation.
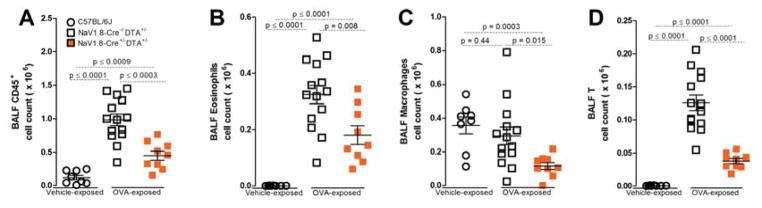
Four days after last aerosol challenge (day 21), OVA-exposed mice developed CD45+ cell count increases in BALF (A), including eosinophils (B) and lymphocytes (D), but not macrophages (C). Ablation of NaV1.8 (NaV1.8+/− DTA+/−, orange squares; A–D) expressing sensory neurons significantly decreased these levels. Mean ± S.E.M; Two-tailed unpaired Student’s t-test (n = 4–12 animals/group; 1–2 cohorts).
Silencing airway nociceptors in non-inflamed lungs
Since ablating nociceptors during development might conceivably cause compensatory changes in the lung and immune system, we shifted to a strategy to temporarily silence afferents in the adult lung using large pore ion channels as a drug entry port for charged sodium channel blockers to produce targeted action potential blockade (Binshtok et al., 2007; Brenneis et al., 2013). We first assessed the efficiency of sensory neuron silencing using QX-314, a permanently charged quaternary derivative of lidocaine, in reversing neuropeptide release from nociceptors and consequent vascular leakage in the lung. Activating lung nociceptors in naïve mice with capsaicin (1 μmol, 20 μl instilled intranasally) induced significant increases in calcitonin gene related peptide (CGRP) levels in BALFs collected 60 minutes later (figure 3A–D). QX-314 (100 μM). Pre-treatment with QX-314 by inhaled nebulizer 1h prior to the capsaicin (when no large pore channels are activated), failed to alter subsequent capsaicin-induced increases in the neuropeptide, showing that when administered by itself in the non-inflamed lung, QX-314 fails to block nociceptors, an expected result since there is no portal for entry into nociceptors and it is ineffective when administered extracellularly (Binshtok et al., 2007) (figure 3A); however, when administered just after capsaicin administration, which opens TRPV1 channels, QX-314 now blocked CGRP release into BALF (figure 3B). Co-administration of QX-314 with capsaicin also blocked development of plasma extravasation in the lung, a measure of capillary permeability due to neuropeptide release (figure 3C). These data show that when large pore channels are exogenously activated by TRP channel agonists, QX-314 can enter into lung nociceptors and inhibit them.
Figure 3. Airway sensory neuron silencing with QX-314 abolishes capsaicin-induced peptide release and vascular leak.
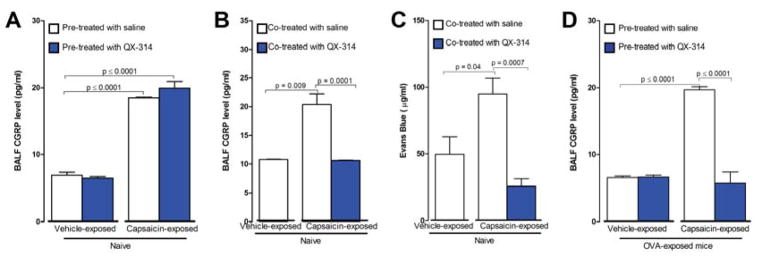
To assess nociceptor-mediated peptide release capsaicin (1 μmol, intranasal) was administered to naïve mice. BALF CGRP levels were increased one hour following the capsaicin-challenge (A–B). QX-314 (100 μM) treatment 1 hr prior to the capsaicin did not alter capsaicin-induced iCGRP levels (A) but QX-314 (100 μM) when administered immediately after the capsaicin, blocked the CGRP rise (B) (n = 4–15 animals/group; 2–3 cohorts). Reduction in vascular leak (C) following co-administration of capsaicin (1 μmol) with QX-314 (100 μM) (n = 4–5 animals/group; 1 cohort). BALF CGRP levels increased following capsaicin (1 μmol) instillation in OVA-challenged mice (D) and this was blocked by QX-314 pre-treatment one hour before (100 μM). Mean ± S.E.M; Two-tailed unpaired Student’s t-test (n = 5–12 animals/group; 2 cohorts).
Silencing airway nociceptors in inflamed lungs
Reduced OVA-induced allergic airway inflammation in TRPA1 knockout mice (Caceres et al., 2009) and following TRPV1-siRNA knockdown (Mabalirajan et al., 2013; Rehman et al., 2013) indicates activation of these channels by endogenous ligands during allergic inflammation, providing the opportunity to target QX-314 into nociceptors in inflamed lungs without need for administration of an exogenous TRP channel agonist. When QX-314 was administered 60 minutes prior to a capsaicin challenge to OVA-challenged mice on day 18, at the height of inflammation, capsaicin-evoked neuropeptide release was, unlike the situation in naive mice, markedly decreased (figure 3D), showing that QX-314 treatment by itself in OVA-exposed mice locally blocked nociceptors. To further asses afferent silencing, intranasal capsaicin (500 nmol) was instilled in OVA-challenged mice and nodose ganglion neuron activation measured by phospho-ERK (p-ERK) immuno-labelling. When OVA-challenged mice were pre-exposed to QX-314 (100 μM) 1 day prior to p-ERK measurements, fewer lung afferent neurons, identified by retrograde DiI-label (figure S1), were p-ERK positive than in vehicle treated mice; 18.5% versus 86.5% (figure S1A–D), demonstrating that inhaled QX-314 silences lung nociceptors in OVA-treated mice. Activation of lung nociceptor TRP channels either by capsaicin in naïve animals or by endogenous ligands during allergic inflammation permits sufficient QX-314 entry to block local nociceptor activity (figure S1E–G).
Nociceptor silencing and allergic airway inflammation
Single 100 μM treatment with QX-314 on day 18, after OVA inhalation challenges on days 14, 15, 16 and 17 in OVA sensitized mice, substantially reduced the immune cell infiltration normally seen on day 21, with decreased BALF CD45+ cells (total immune cells) and decreased eosinophils, macrophages and lymphocytes (figure 4A–D). Similar results were seen in Hematoxilin and Eosin stained lung tissue (figure 4E–G). The QX-314 effects were independent of sex, age (8 vs 16 weeks) and strain (C57Bl/6 vs BALB/c) (data not shown). In addition, this treatment decreased by 1.75 fold airway basement membrane thickening (figure 4H). We also tested if sensory neuron silencing reversed type 2 inflammation in the clinically-relevant house dust mite (HDM) model (Johnson et al., 2004). Briefly, 8 week male BALB/c mice were sensitized with HDM (day 1–5) and challenged on day 8–10. On day 15, HDM challenged mice showed increased BALF CD45+ cells (figure 3I) and lymphocytes (figure 3J), both of which were reduced by QX-314 (100 μM, day 12) treatment. To establish if this was a general to all types of allergic inflammation, we analyzed the impact of silencing in a type 1-skewed allergic airway inflammation model. Here, instead of sensitizing mice with aluminum hydroxide and OVA, as in all experiments above, which produces a TH2-skewed inflammation (Bogaert et al., 2011), mice were sensitized with a mixture of complete Freund adjuvant (CFA) and OVA (Bogaert et al., 2011) which models non-eosinophilic TH1-skewed asthma. These mice had a ~2 fold increase in the TH1/TH2 ratio, (defined as CD4+IFNγ+ cells/CD4+IL4+ cells), compared to AlOH/OVA sensitized mice (figure 4K). QX-314 failed to impact levels of CD45+ cells (figure 4L), eosinophils, macrophages and lymphocytes on day 21 in this model (data not shown). Sensory neuron effects on inflammation are context-dependent and contribute to type 2 models of allergic airway inflammation but not a type 1 model.
Figure 4. Airway sensory neuron silencing reduces lung inflammation and hyperresponsiveness.

OVA-exposed mice develop increased CD45+ immune cell counts in BALF (A), that include eosinophils (B) and lymphocytes (D), but not macrophages (C). Silencing lung sensory neurons with QX-314 (100 μM, 72h prior to measurement, blue squares) decreased these immune cell responses (A–H). Representative Hematoxylin and Eosin (H&E)-stained sections of OVA exposed lungs treated with saline (E) or QX-314 (100 μM; F). Scale 100 μm. Immune cell infiltration severity (G) and basement membrane thickening (H) measured by tissue morphometry in HE- sections. House dust mite (HDM) challenge increased BALF CD45+ (I) and lymphocyte counts (J), an effect reversed by QX-314 (100 μM). CFA/OVA sensitization produced a greater increase in Th1/Th2 cell ratio than AlOH/OVA sensitization (K). QX-314 (100 μM) reduced BALF CD45+ cell counts in the AlOH/OVA but not CFA/OVA sensitized model (L). Mean ± S.E.M; Two-tailed unpaired Student’s t-test (n = 4–25 animals/group; 4 cohorts). Change in airway reactivity measured as ED200RL (M), Resistance (N) Elastance (O) in OVA-exposed mice treated with QX-314 (D–F; 100 μM, day 18) or saline. Difference vehicle- (***) and OVA-exposed (+++) groups P <0.001. OVA-exposed mice performed less voluntary wheel running (P) (1h assessment) than control mice, an effect reversed by QX-314 (100 μM, day 18) assessed 24h after silencing. Two-tailed unpaired Student’s t-test (n = 4–40 animals/group; 2–3 cohorts).
Inhaled lidocaine, a short-acting sodium channel blocker, has anti-inflammatory effects on murine asthma models (Serra et al., 2012) and in patients (Weiss and Patwardhan, 1977), but directly suppresses immune cells (Serra et al., 2012). We therefore investigated if QX-314 has direct effects on immune cells. QX-314 had no impact on in vitro survival, chemotaxis and activation of lung macrophages and eosinophils (data not shown), and exposure to QX-314 (up to 1 mM) had no effect on TH0, TH1 and TH2 cell proliferation or cytokine production in vitro (data not shown). We conclude that the immune suppressing actions of QX-314 are due to nociceptor silencing. The “anti-inflammatory” effect of nociceptor silencing with QX-314 was replicated with a chemically distinct sodium channel blocker, a permanently charged derivative of mexiletine, N-methyl-mexiletine (NMM). When NMM was administered by nebulizer (on day 18) to OVA-treated mice, BALF-harvested immune cell counts were significantly reduced (figure S2) albeit with less potency than QX-314. The same dose of uncharged mexiletine failed to resolve the OVA-mediated allergic airway inflammation (figure S2). Thus, the ability of charged compounds to penetrate into nociceptors through activated large pore ion channels enables blockade of sodium channels for longer periods and this is critical to how silencing sensory neurons reduces inflammation.
We next examined the effect of sensory neuron silencing on bronchial hyperresponsiveness. Both genetic ablation of lung afferent neurons (data not shown) and QX-314 silencing on day 18 reversed OVA-mediated bronchial hyperresponsiveness, as measured on day 21 by ED200RL (figure 4M), lung elastance (figure 4N) and resistance (figure 4O). Specifically, the dose of methacholine (MCh) required to produce a 200% increase in RL (i.e., log ED200RL) decreased significantly (P<0.001) from 6.38 ± 0.74 in control mice to 0.79 ± 0.06 after OVA challenge, while QX-314 increased by ~3 fold the log ED200RL values in OVA-challenged mice (figure 4M). N-methyl-mexiletine also reduced airway hyperresponsiveness (data not shown).
To functionally explore the consequences of airway hyperresponsiveness in mice, we measured daily voluntary wheel running. Individual animals were followed over the course of the allergen protocol (figure 1A) and their daily level of voluntary exercise (1h, dark, 21h00-1h00) was analyzed. At the peak of inflammation (day 18) mice showed a significant decrease in travel distance ~25% compared to their pre-challenge period (day 13) (figure 4p) and this was matched by an equivalent decrease in exercise duration. Voluntary wheel running distance was rescued by QX-314 treatment, measured 24h later (figure 4P). Local silencing of lung nociceptors by a single exposure to a charged sodium channel blocker at the peak of allergen-induced inflammation reduced two cardinal features of allergic asthma; immune infiltration and bronchial hyperresponsiveness.
IL-5 activates lung nociceptors
Our data suggest that nociceptors are activated during exposure to allergens in sensitized animals but the mechanisms responsible are unknown. Skin nociceptors are activated by the cytokine IL-1β (Binshtok et al., 2008) and we hypothesized that similar signaling could occur during allergic airway inflammation. To look for candidates, we asked if silencing lung nociceptors with QX-314 reduced BALF levels of cytokines, and discovered decreased IL-5 (figure 5A), eotaxin-2, IL-4, IL-9, IL-10, IL-13, IP-10, MCP-1, TNF-α and TARC, but not IL-17, IL-23, IL-27, IL-28β, INF-γ and RANTES (table S1). Transcript profiling of the naïve nodose ganglion revealed that lung NaV1.8+ nociceptors express receptors for some of these cytokines, including IL-33R, INFg-R, IL-4R, IL-9R and IL-5R (figure 5B). We therefore explored if eotaxin-2, IL-13 and IL-5 could activate cultured nodose neurons, using calcium imaging of neurons from control, OVA sensitized (day 13) and OVA sensitized and challenged (day 21) mice, and observed that the latter condition produced maximal responses (data not shown), and chose to perform subsequent experiments on neurons from OVA challenged mice. Exposing neurons to eotaxin-2 or IL-13 at concentrations up to 10 μg/ml produced small concentration-dependent calcium increases in a few neurons; for IL-13 (3/469 KCL+ cells, 0.6 %) and eotaxin-2 (26/403 KCL+ cells, 6.45%). However, larger responses in more neurons were observed for IL-5 (150/788 KCL+ cells, 19.04%) (figure 5C).
Figure 5. IL-5 activates NaV1.8+ nodose ganglion neurons.

IL-5 levels in BALFs increased in OVA challenged mice, an effect reversed by QX-314 (100 μM) (A). Transcript profiling of naïve nodose ganglia reveals basal expression of cytokine receptors including IL-5R (B). Cultured nodose ganglion neurons from OVA-exposed mice show a dose-dependent calcium increase (C) in response to IL-5 (0.3–10 μg/ml). Venn diagram shows overlapping populations of KCL+IL-5+ and AITC or capsaicin responsive neurons (D). IL-5 responsive cells are mainly small and medium size (E) and nearly null in OVA-exposed Nav1.8-Cre +/− DTA+/− mice (F). IL5 mediated calcium increase absent in 0 external Ca2+ (G). Current-clamp recordings of small cells (capacitance <45 pF) showing that IL-5 (1 μg/ml) depolarizing resting membrane potential (RMP) by ~6 mV (H, I; n = 12 cells). The current needed to trigger an action potential (rheobase) is smaller when IL-5 is present (J, K; n = 7 cells). Mean ± S.E.M; Two-tailed unpaired (A–G) and paired (H–K) Student’s t-test (n = 3 biological replicate per group).
IL-5, along with IL-13, is a major effector cytokine in asthma, implicated in airway inflammation, hyperresponsiveness and remodeling as well as mucus metaplasia (Ortega et al., 2014). To characterize nodose neurons that responded to IL-5 we exposed them to different TRP channel ligands and found that 55 % (42/78) of IL-5 responsive cells also responded to capsaicin and 41% (32/78) to AITC (figure 5D), while 55 % of capsaicin and 100% of AITC responsive cells were IL-5 responsive (data not shown). IL-5 responsive nodose ganglion neurons were predominantly of small and medium size (figure 5E). Only ablated mice responded to IL-5 in comparison to ~25% in 1.3% of cultured nodose ganglion from NaV1.8 littermate controls (figure 5F). Removing extracellular calcium eliminated the IL-5 signal (figure 5G) suggesting flux through membrane bound channels. Whole-cell patch clamp recordings in current-clamp mode from small cells (capacitance <45 pF) that had responded to IL-5 by calcium imaging, showed that IL-5 perfusion (1 μg/ml) depolarized the cells by ~6 mV (figure 5H, 5I; n = 12 cells), an effect that was reversible after extensive washout (more than 20 min). Before IL-5 perfusion, cells had a stable RMP with no spontaneous action potential firing, however occasional spontaneous spiking was observed following IL-5 treatment (2/12 patched cells). The current needed to trigger an action potential (rheobase) was smaller when IL-5 was present (figure 5J, 5K; n = 7 cells), but no difference in spiking frequency during a ramp current was observed (not shown). We conclude that nociceptors can be directly activated by IL-5.
Lung nociceptors activate ILC2 and lymphocytes
Lung epithelial and dendritic cells are activated upon an allergen challenge to stimulate resident lung ILC2 cells (Licona-Limon et al., 2013). ILC2 cells are implicated in the onset of asthma, and are directly activated by IL-25 and IL-33 released from goblet and dendritic cells, to act upstream of TH2 cells, B cells, eosinophils, smooth muscle cells and macrophages. ILC2 cells produce IL-5, IL-9 and IL-13, and are resident in the airways, where they orchestrate type-2 immunity (Barnig et al., 2013; Licona-Limon et al., 2013). Activation of ILC2 cells typically precedes that of TH2 cells, although transactivation and cooperation occur (Halim et al., 2014; Licona-Limon et al., 2013). Typically activated ILC2 cells produce IL-13 and have higher levels of the IL-33R, ST2, while a subset of TH2 cells produces IL-5 and IL-13, the predominant effector cytokines in asthma. We therefore set out to assess if nociceptor ablation had any effect on ILC2 cell or CD4+ cell activation, as measured by surface markers and cytokine production. To do this, we examined early immune changes after two OVA challenges on day 16. Relative to littermate controls (NaV1.8-Cre−/−DTA+/−), OVA-exposed mice with ablated nociceptors (NaV1.8-Cre+/−DTA+/−) showed decreased BALF levels of CD45+, as seen previously (figure 2A, 6A). In addition, there were fewer CD4+ (figure 6B) and ILC2 (figure 6C) cells. Moreover, nociceptor ablated mice demonstrated decreased numbers of IL-13+ ILC2 (figure 6D), and lower expression of CD25 (figure 6E) and ST2 (figure 6F), ILC2 activation markers (Wilhelm et al., 2011). Similarly, QX-314 administration at the peak of inflammation (100 μM, day 18) reduced the rise in total CD4+, CD4+IL-5+, and CD4+IL-13+ cells (BALF, on day 21). Thus, both genetic ablation and and pharmacologic silencing of nociceptors result in decreased activation of ILC2s and ILC2 and Th2 effector cytokine production.
Figure 6. Sensory neurons control ILC2 and CD4+ cell recruitment and activation in the lung.

On day 16 (A–F), OVA-exposed mice (pink bars) with intact nociceptors (littermate; NaV1.8−/−DTA+/−) had higher whole lung CD45+ (A), CD4+ (B), ILC2 (C), ILC2 IL-13+ cell counts (D), as well as ILC2 CD25 (E) and ST2 mean fluorescence intensity MFI) (F) than mice with ablated nociceptors (NaV1.8+/−DTA+/−). QX-314 treatment (100 μM, day 18; blue squares) decreased OVA-induced CD4+ cells on day 21 (G), including CD4+IL5+ (H) and CD4+IL13+ (I) cells, relative to control mice (white squares). Mean ± S.E.M; Two-tailed unpaired student’s t-test (n = 4–9 animals/group; 1–2 cohorts).
Lung nociceptors release VIP
The effect of sensory neurons on ILC2 and CD4+ cells likely involves multiple pathways and several candidate neuropeptides could be involved. Genomic analysis of lung resident ILC2 cells has revealed expression of receptors for several neuropeptides released by sensory neurons, including SP, CGRP and VIP (Saenz et al., 2013), providing an opportunity for nociceptors to directly communicate with these cells. We chose to examine VIP, both because it is present at high levels in nodose ganglion neurons (Figure 7A) and because its two G protein coupled receptors VPAC1 and VPAC2 are expressed by and exert a powerful regulatory effect on T cells, including differentiation, migration and cytokine production (Delgado et al., 2000). We found that NaV1.8+ nodose ganglion neurons express VIP (Figure 7B), including lung afferents in OVA-exposed mice (DiI-retrogradely labeled) (figure S7A). Cultured nodose ganglion neurons stimulated with capsaicin (figure S3B) or IL-5 (figure 7C) released VIP while BALF from OVA-exposed mice contained elevated VIP compared to vehicle-challenged mice and this was reduced by inhaled QX-314 (figure 7D). Nodose ganglions from OVA-exposed mice express more VIP transcripts than niave mice (data not shown). These data indicate that VIP is released in the inflamed lung and this can be blocked by silencing neurons with a charged sodium channel blocker.
Figure 7. IL-5 provokes sensory neuron release of VIP to activate ILC2 and recruit CD4+ cells.

Gene profiling of naïve NaV1.8+ nodose ganglion neurons reveals expression of nociceptor markers and VIP (A). A VIP reporter mouse (B, VIP-Cre+/−EGFP/td-tomato+/− green) reveals that ~80% of NaV1.8+ (magenta) nodose ganglion neurons express VIP (green) (scale bar = 50 μm) (B). IL-5 (1h, 3 ug/ml) induced VIP release from nodose ganglion neurons from OVA-exposed mice (C). These mice have elevated BALF VIP levels on day 21 compared to vehicle-exposed mice (white bar), which is decreased by QX-314 treatment (100 μM, day 18; blue bar) (D). In OVA-exposed lungs, the VPAC2 antagonist PG 99465 (100 nM, every 12h for 48h starting on day 16; pink squares) did not impact resident ILC2 numbers (E) but reduced ILC2activation; IL-13+ cells (F). The VPAC2 agonist BAY 55-9837 (10nM, every 12h for 96h; red squares) increased, and the VPAC2 antagonist PG 99465 (100 nM, every 12h for 96h; pink squares) decreased CD4+ cell numbers(G) and IL-5 mRNA expression in FACS-sorted CD4+ cells (H). Mean ± S.E.M; Two-tailed unpaired Student’s t-test (n = 3–14 animals/group; 1–2 cohorts).
VIP acts on ILC2 and TH2 cells
We next asked if VIP can act on ILC2 and T cells. We isolated CD4+ T cells from spleens and lymph nodes of naïve mice, and differentiated them in vitro under TH1, TH2 and TH17 skewing conditions, followed by exposure to VIP. When CD4+ T cells cultured under TH2 skewing conditions were exposed to recombinant mouse VIP (500 nM), their transcript levels of IL-13 (data not shown) and IL-5 (figure S3C) increased, suggesting that VIP contributes to the competence of TH2 cells to transcribe these type II regulatory cytokines. VIP (0–1.0 μM) did not affect transcription of IL-5 or IL-13 in T cells cultured under TH0, TH1 and TH17 skewing conditions (data not shown). The VPAC2 antagonist PG 99465 administered early in the OVA challenge (100 nM in 50 μl, intra-nasally, every 12h for 48h starting on day 14) significantly decreased CD4+ cells in the whole lung on day 16, particularly those producing IL-13 (figure S3D, E). Resident lung ILC2 numbers were not affected by VPAC2 blockade (figure 7E), but their activation was significantly reduced, as indicated by decreased IL-13 intracellular staining (figure 7F) and decreased surface expression of ST2 (figure S3G). Impaired ILC2 activation may be important given the key role for this population in the initiation of allergic airway inflammation (Halim et al., 2014). When the VPAC2 antagonist was given for a longer period, over the full duration of OVA challenge (twice daily on days 14–18), it reduced BALF total immune (CD45+) (figure S3F) and CD4+ cell numbers (figure 7G) as well as IL-5 mRNA expression in CD4+ T cells FACS sorted from BALF on day 19 (figure 7H). In contrast, a VPAC1 antagonist did not alter immune cell number or activation (data not shown). Activation of VPAC2 in OVA-exposed mice with a selective agonist further increased CD4+ cell numbers (figure 7G; S3H, I) and IL-5 mRNA levels (figure 7H). VIP signaling through VPAC2 may play, therefore, a significant role in the activation of ILC2 cells and cytokine expression by CD4+ T cells, and may account, at least partially, for the ability of nociceptors to amplify type II inflammation in the lung.
Discussion
The nervous and immune systems work in concert to protect against environmental dangers and can also fail together in disease states, including asthma. Nociceptor-immune interactions appear to contribute to arthritis-induced inflammation (Levine et al., 1984), colitis (Engel et al., 2011), psoriasis (Riol-Blanco et al., 2014) and acute lung injury (Su et al., 2005); however, the mechanisms by which this happens remain uncertain. Here, we demonstrate a complex interplay between nociceptors and immune cells. Through pharmacological silencing and genetic ablation of nociceptors in the ovalbumin and house dust mite-induced mouse models of airway inflammation, we have uncovered a major upstream contribution of these fibers to the generation and persistence of type 2 allergic airway inflammation and bronchial hyperresponsiveness. Whereas direct stimulation of lung nociceptors with capsaicin promotes neuropeptide release and immune cell infiltration, silencing sensory neurons or ablation of NaV1.8+ nociceptors substantially reduces immune cell infiltration and bronchial hyperresponsiveness. We have also identified a means of communication between sensory neurons and those select set of immune cells pivotal for the initiation and amplification of type 2 allergic inflammation. We find that IL-5, a type II effector cytokine produced by multiple immune cells, directly activates sensory neurons leading to secretion of the neuropeptide VIP, and that VIP acts on ILC2 and CD4+ cells to induce cytokine production. In addition, we discover that charged sodium channel blockers which specifically and locally silence TRP-activated sensory neurons, interrupt this signaling loop and reduce well-established inflammation.
Several studies have suggested that nociceptors play a role in adaptive immune responses. A reduction in allergic airway inflammation was shown in TRPA1-KO mice (Caceres et al., 2009) and upon TRPV1-siRNA knockdown (Mabalirajan et al., 2013; Rehman et al., 2013) while C-fiber denervation in rats (produced by neonatal high dose capsaicin exposure) decreased OX-6+ dendritic cell numbers in the lung, pulmonary lymphoid infiltration after allergen challenge (hen egg lysozyme) (Kradin et al., 1997) and PAR2-mediated airway constriction, pulmonary inflammation and edema (Su et al., 2005). Collectively, these findings together with studies on atopic dermatitis and psoriasis (Riol-Blanco et al., 2014; Wilson et al., 2013), indicate that nociceptors can potentiate adaptive immune responses; however, the mechanisms responsible were not identified in these studies. Here, we show that nociceptors are crucial to the generation and persistence of type 2 airway inflammation, identify a pharmacological approach for using this insight therapeutically, and identify some specific mechanisms for these neuro-immune interactions.
Allergic airway hyperresponsiveness may result both from local signaling by immune cells directly to airway smooth muscle cells and through nociceptor-mediated stimulation of sub-epithelial parasympathetic efferents through central and local reflexes. A recent study found that ablation of a subset of TRPV1+ sensory neurons reduced bronchial hyperresponsiveness (Trankner et al., 2014). We replicate this finding using NaV1.8 ablation, and extend it by showing similar effects with the pharmacological silencing of pain fibers, including on daily voluntary exercise, a new functional readout of airway hyperresponsiveness. Trankner et al., reported that ablation of TRPV1 sensory neurons did not reverse airway inflammation, in contrast to our pharmacological and genetic ablation data which reveal such an effect. Explanations for this discrepancy include different allergy models (intranasal allergen vs nebulisation), timing of measurements (24h vs 96h) and assessment methods (Wright-Giemsa stain vs FACS immunophenotyping) but most importantly, different targeted neuron populations (TRPV1 vs NaV1.8). NaV1.8 is expressed by ≥80% of nociceptors (Muroi and Undem, 2014) and the majority of nodose ganglion lung afferents (Kwong et al., 2008) while TRPV1 is expressed by only ~50% of Nav1.8 nociceptors. This suggests that a broad population of nociceptors needs to be silenced or ablated to interrupt their pro-inflammatory actions.
TH2 and ILC2 cells are both critical to the pathogenesis of allergic airways disease. We found that after an allergen challenge in TH2 skewed sensitized mice, sensory neurons stimulate cytokine production in CD4+ and ILC2 cells. ILC2 are implicated in the onset of asthma and are directly activated by IL-25 and IL-33 released from goblet and dendritic cells, and act upstream of TH2 cells, B cells, eosinophils, smooth muscle cells and macrophages (Walker et al., 2013). Therefore, activation of sensory neurons on allergen exposure may be a very early event in igniting inflammation in a sensitized animal. We asked how sensory neurons may communicate with TH2 and ILC2 cells to potentiate/initiate inflammation. Activation of nociceptors, through calcium influx, induces release of neuropeptide transmitters from vesicles in the peripheral terminals. In the lung, release of these neuropeptides occurs in response to chemical agents (capsaicin, citric acid, nicotine, cigarette smoke, histamine and bradykinin) and physical factors (mechanical probing, dry gas and hypertonic aerosols) (Bessac and Jordt, 2008). Classically, such neuropeptide release (with a focus on substance P and CGRP), was thought to trigger only local increased capillary permeability and vasodilation but there have been suggestions that nociceptor-mediated peptide release may contribute to inflammation in arthritis (Levine et al., 1984), colitis (Engel et al., 2011), psoriasis (Riol-Blanco et al., 2014) and acute lung injury (Su et al., 2005). Lung resident ILC2 cells express receptors for the ensory neuron neuropeptide SP, CGRP and VIP (Saenz et al., 2013). Established neuropeptide-mediated immune cell regulation include, the CGRP and VIP biasing of dendritic cells toward the TH2/TH1 response, and enhanced dendritic cell migration to the lymph node (Ding et al., 2008), as well as activation of mast (Ansel et al., 1993), Langerhans (Hosoi et al., 1993), TH2 cells (Delgado et al., 2000; Ding et al., 2008) and ILC2 cells (Nussbaum et al., 2013).
Our data suggest that release of VIP by sensory fibers in the lung activates ILC2 and CD4+ T cells, in addition to their canonical activation by cytokines, and that this contribution is sufficiently large that silencing sensory neurons substantially reduces and increases resolution of allergic inflammation (figure 8). The action of VIP on immune cells is context and cell type dependent. Unstimulated TH cells have high basal expression of VPAC1 (Harmar et al., 1998). T cell antigen receptor stimulation however, upregulates VPAC2 and diminishes VPAC1 expression, and in consequence, VPAC2 is the dominant transducer of VIP’s effects on TH2 cells (Lara-Marquez et al., 2001). This shift from VPAC1 to VPAC2 in activated T cells shifts the balance from the delayed-type cellular protective hypersensitivity responses mediated by TH1 cells to the immediate-type allergic immune responses mediated by TH2 cells. Supporting a role for VPAC2 signaling in type II immune responses, transgenic mice with high constitutive VPAC2 expression in T cells show blood eosinophilia, elevated serum IgE levels, greater IgE antibody responses and cutaneous immediate-type reactions to allergy-producing antigens (Voice et al., 2001), while VPAC2-KO mice show TH1-associated responses (Goetzl et al., 2001). Nociceptor activation and consequent VIP release may elicit therefore, different types of immune activation, depending on immune cell state. Activation of ILC2 cells by VIP results in IL-5 induction (Nussbaum et al., 2013), something we reproduce here. We further show that ILC2 activation in the OVA model of airway allergic inflammation, is reduced by a VPAC2 antagonist. However, because of the bronchodilator actions of VIP on smooth muscle (Linden et al., 2003), it is unlikely that VPAC2 antagonists could be used therapeutically for asthma.
Figure 8. Model of nociceptor involvement in type-2 inflammation.

During allergen exposure, lung afferents are activated by dendritic and epithelial cells. Dendritic cells also polarize precursor TH cells into TH2 cells. Activated nociceptors release VIP, which stimulates lung resident ILC2 and newly differentiated TH2 cells via the VPAC2 receptor. Type 2 cytokines, including IL-5 and IL-13, are released by ILC2 and TH2 cells and initiate the chemotaxis and activation of eosinophils and macrophages, IgE secretion by B cells, mucus production by goblet cells, and smooth muscle contraction, culminating in allergic inflammation and bronchial hyperresponsiveness. IL-5 activates nociceptors to trigger VIP and other neuropeptide release leading to additional IL-5 production. Scheme inspired by Licona-Limon et al., 2013; Vercelli, 2008.
What may lead to nociceptor activation after an allergen challenge? Because nodose ganglion neurons express a wide variety of receptors for many cytokines and chemokines upregulated in asthma, we explored the possibility that select mediators could drive nociceptor activation, and found that IL-5 directly activates nociceptors, leading to calcium influx and release of VIP, which act on immune cells to trigger more IL-5 production. IL-5 is known to be central to asthma-associated pathologies where promotes eosinophil recruitment, triggers release of eosinophil cationic protein (ECP) and transforming growth factor-β (TGFβ), to induce airway smooth muscle inflammation and hyperreactivity (Shi et al., 1998).
We have uncovered a positive feed-forward loop, involving IL-5, nociceptors, VIP, ILC2 and TH2 cells, that amplifies type 2 inflammation (figure 8). There may well be additional mechanisms to activate sensory neurons during such inflammation, for example thymic stromal lymphopoeitin, which is implicated in asthma (Gauvreau et al., 2014). TSLP is released from keratinocytes in a mouse model of atopic dermatitis to directly activate a subset of TRPA1+ sensory neurons (as well as immune cells) to trigger itch (Wilson et al., 2013). Furthermore, in psoriasis-like conditions, skin allergen-sensitized dendritic cells make direct contact with afferent sensory neurons and activate them (Riol-Blanco et al., 2014) although the mediator responsible is not yet identified.
Currently there is no treatment that accelerates resolution of inflammation in asthma (Levy et al., 2012). Several drugs control disease symptoms and help abort attacks (Fanta, 2009). Bronchodilators, β2-agonists or anti-cholinergics, give immediate relief through relaxation of smooth airway muscle (Green et al., 2003). Immune modulators inhibit inflammation, with corticosteroids the primary treatment modality (Green et al., 2003); however, chronic steroid use leads to adverse effects (Locksley, 2010). Despite the effectiveness of current therapy for mild asthma, patients with severe asthma have frequent exacerbations and excess morbidity. Asthma is a heterogeneous condition but a therapeutic approach that targeted nociceptors upstream of type 2 inflammation, might provide a broadly effective treatment for many patients (Barnes, 2004). Nevertheless, sensory neuron interaction with immune cells appears to be highly cell and context dependent; for example, during pathogen invasion release of neuropeptides (CGRP, galanin and somatostatin) supresses innate immune responses (Chiu et al., 2014), in contrast to the amplifying role we report here for type 2 allergic airway inflammation, and treatment would need to evaluated and individualized accordingly.
In conclusion, the silencing and genetic ablation of nociceptors has uncovered a major upstream contribution of these fibers in the generation and persistence of type-2 allergic airway inflammation and bronchial hyperresponsiveness. Sensory neurons on activation by allergen exposure in sensitized animals, control through local neuropeptide release at their peripheral terminals, type-2 inflammation, acting on ILC2 and CD4+ cells. These cells secrete cytokines such as IL-5 that recruit effector immune cells integral to the disease state, and also further activate sensory neurons. Silencing sensory neurons provides, therefore, a potential strategy worth exploring for the treatment of type-2 allergies, potentially including conjunctivitis, rhinitis and asthma. This strategy is highly specific, only targeting neurons whose TRP channels are activated by inflammation, with expected low systemic side effects (positively charged drugs have limited capability of spreading) and a long duration of action (positively charged compounds should remain trapped inside neurons for prolonged periods), constituting a novel therapeutic approach to allergic airway inflammation centered on interrupting sensory neuron immune signaling.
Experimental procedures
All procedures were approved by the Institutional Animal Care and Use Committees of Boston Children’s Hospital. Allergic airway inflammation was studied in an ovalbumin (OVA) based model (Haworth et al., 2008). On day 0 and 7, mice were sensitized by a 200 μl i.p. injections of a solution containing 1 mg/ml ovalbumin (Sigma-Aldrich) and 5 mg/ml aluminum hydroxide (Sigma-Aldrich, Boston, Ma). On day 14–17 (10:00 am) mice were exposed to 6% OVA aerosol for 25min. QX-314 (100 μM) was nebulized on day 18 and mice sacrificed on day 21, BALF harvested; cells isolated, counted and immunophenotyped using FACS. We also investigated impact of sensory neuron silencing in a TH1-skewed model of allergic airway inflammation (Bogaert et al., 2011) following s.c. sensitization with OVA (1 mg/ml) in a 200 μl emulsion of sterile PBS and 50% Complete Freund Adjuvant (CFA) on day 0 and with 50% Incomplete Freund Adjuvant (IFA) on day 7. Another type 2 inflammation model was induced by house dust mite (Johnson et al., 2004). Lightly anesthetized (isoflurane) mice were sensitized (day 1–5) and challenged (day 8–10) with house dust mite (HDM; 25 μg/10μl, intranasal) and exposed to QX-314 (100 μM, day 12). Mice were sacrificed on day 15, BALF harvested; cells isolated, counted and immunophenotyped using FACS
Statistics
Data expressed as mean ± S.E.M. from 4–36 mice. Statistical significance determined by two-tail (unpaired or paired (patch clamp)) Student’s t-test. P values less than 0.05 were considered significant. Numbers of animals are defined in figure legends.
Supplementary Material
Highlights.
Nociceptor sensory neuron ablation reduces allergic airway inflammation.
QX-314 enters through large pore ion channels to silence airway sensory neurons.
IL-5 triggers sensory neuron release of peptides which drive immune cell responses.
Silencing nociceptors is a new strategy to treat type-2 inflammation and allergies.
Acknowledgments
This work was supported by the Hood foundation (CJW), National Institute of Health (NIH) grants PO1NS072040 (CJW and BPB), R37NS039518 (CJW), RO1-HL122531; AI068084; P01-GM095467 (BDL). ST, REA, PRB, NG, IMC, respectively hold postdoctoral fellowships from the Canadian Institute of Health and Research (CIHR), NIH (5T32HL007633), CIHR and NRSA F32 fellowship (NS076297-01). NIH-P30-HD18655 supported the cores used. We thank Stella J. Lee, Seungkyu Lee, Priscilla Riva, Catherine Ward, Olusegun Babaniyi and Kush Gupta for their assistance, respectively with MATLAB, ELISA, tail-vein injections, mice genotyping and the Flexivent apparatus.
Footnotes
Author Contributions
ST, REA, PRB, SL, SC, MP, CL, SLF, JT, NL, IMC, MD, BA conducted the experiments. CvH, OH and HS initiated the study. ST, REA, PRB, CL, MP, SLF, NG, DR, IMC, VKK, BPB, BDL and CJW designed the study. ST, JMP, VKK, BPB, BDL and CJW wrote manuscript.
The authors have declared that no conflicts of interest exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akinbami L. The state of childhood asthma, United States, 1980–2005. Adv Data. 2006:1–24. [PubMed] [Google Scholar]
- Ansel JC, Brown JR, Payan DG, Brown MA. Substance P selectively activates TNF-alpha gene expression in murine mast cells. J Immunol. 1993;150:4478–4485. [PubMed] [Google Scholar]
- Barnes PJ. Neuroeffector mechanisms: the interface between inflammation and neuronal responses. J Allergy Clin Immunol. 1996;98:S73–81. discussion S81–73. [PubMed] [Google Scholar]
- Barnes PJ. New drugs for asthma. Nat Rev Drug Discov. 2004;3:831–844. doi: 10.1038/nrd1524. [DOI] [PubMed] [Google Scholar]
- Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, Wechsler ME, Israel E, Levy BD. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5:174ra126. doi: 10.1126/scitranslmed.3004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessac BF, Jordt SE. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology. 2008;23:360–370. doi: 10.1152/physiol.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449:607–610. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1beta sensors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert P, Naessens T, De Koker S, Hennuy B, Hacha J, Smet M, Cataldo D, Di Valentin E, Piette J, Tournoy KG, Grooten J. Inflammatory signatures for eosinophilic vs. neutrophilic allergic pulmonary inflammation reveal critical regulatory checkpoints. American journal of physiology Lung cellular and molecular physiology. 2011;300:L679–690. doi: 10.1152/ajplung.00202.2010. [DOI] [PubMed] [Google Scholar]
- Brenneis C, Kistner K, Puopolo M, Segal D, Roberson D, Sisignano M, Labocha S, Ferreiros N, Strominger A, Cobos EJ, et al. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:315–326. doi: 10.1523/JNEUROSCI.2804-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres AI, Brackmann M, Elia MD, Bessac BF, del Camino D, D’Amours M, Witek JS, Fanger CM, Chong JA, Hayward NJ, et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9099–9104. doi: 10.1073/pnas.0900591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning BJ, Mori N, Mazzone SB. Vagal afferent nerves regulating the cough reflex. Respir Physiol Neurobiol. 2006;152:223–242. doi: 10.1016/j.resp.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Canning BJ, Spina D. Sensory nerves and airway irritability. Handb Exp Pharmacol. 2009:139–183. doi: 10.1007/978-3-540-79090-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature. 2013;501:52–57. doi: 10.1038/nature12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. 2012;15:1063–1067. doi: 10.1038/nn.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M, Leceta J, Sun W, Gomariz RP, Ganea D. VIP and PACAP induce shift to a Th2 response by upregulating B7.2 expression. Ann N Y Acad Sci. 2000;921:68–78. doi: 10.1111/j.1749-6632.2000.tb06952.x. [DOI] [PubMed] [Google Scholar]
- Ding W, Stohl LL, Wagner JA, Granstein RD. Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J Immunol. 2008;181:6020–6026. doi: 10.4049/jimmunol.181.9.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel MA, Leffler A, Niedermirtl F, Babes A, Zimmermann K, Filipovic MR, Izydorczyk I, Eberhardt M, Kichko TI, Mueller-Tribbensee SM, et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology. 2011;141:1346–1358. doi: 10.1053/j.gastro.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Fanta CH. Asthma. N Engl J Med. 2009;360:1002–1014. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- Gauvreau GM, O’Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, FitzGerald JM, Boedigheimer M, Davis BE, Dias C, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. The New England journal of medicine. 2014;370:2102–2110. doi: 10.1056/NEJMoa1402895. [DOI] [PubMed] [Google Scholar]
- Goetzl EJ, Voice JK, Shen S, Dorsam G, Kong Y, West KM, Morrison CF, Harmar AJ. Enhanced delayed-type hypersensitivity and diminished immediate-type hypersensitivity in mice lacking the inducible VPAC(2) receptor for vasoactive intestinal peptide. Proc Natl Acad Sci U S A. 2001;98:13854–13859. doi: 10.1073/pnas.241503798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RH, Brightling CE, Pavord ID, Wardlaw AJ. Management of asthma in adults: current therapy and future directions. Postgrad Med J. 2003;79:259–267. doi: 10.1136/pmj.79.931.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, Takei F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40:425–435. doi: 10.1016/j.immuni.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Arimura A, Gozes I, Journot L, Laburthe M, Pisegna JR, Rawlings SR, Robberecht P, Said SI, Sreedharan SP, et al. International Union of Pharmacology. XVIII. Nomenclature of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Pharmacol Rev. 1998;50:265–270. [PMC free article] [PubMed] [Google Scholar]
- Hosoi J, Murphy GF, Egan CL, Lerner EA, Grabbe S, Asahina A, Granstein RD. Regulation of Langerhans cell function by nerves containing calcitonin gene-related peptide. Nature. 1993;363:159–163. doi: 10.1038/363159a0. [DOI] [PubMed] [Google Scholar]
- Hox V, Vanoirbeek JA, Alpizar YA, Voedisch S, Callebaut I, Bobic S, Sharify A, De Vooght V, Van Gerven L, Devos F, et al. Crucial role of transient receptor potential ankyrin 1 and mast cells in induction of nonallergic airway hyperreactivity in mice. Am J Respir Crit Care Med. 2013;187:486–493. doi: 10.1164/rccm.201208-1358OC. [DOI] [PubMed] [Google Scholar]
- Johnson JR, Wiley RE, Fattouh R, Swirski FK, Gajewska BU, Coyle AJ, Gutierrez-Ramos JC, Ellis R, Inman MD, Jordana M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. American journal of respiratory and critical care medicine. 2004;169:378–385. doi: 10.1164/rccm.200308-1094OC. [DOI] [PubMed] [Google Scholar]
- Kanezaki M, Ebihara S, Gui P, Ebihara T, Kohzuki M. Effect of cigarette smoking on cough reflex induced by TRPV1 and TRPA1 stimulations. Respir Med. 2012;106:406–412. doi: 10.1016/j.rmed.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Kradin R, MacLean J, Duckett S, Schneeberger EE, Waeber C, Pinto C. Pulmonary response to inhaled antigen: neuroimmune interactions promote the recruitment of dendritic cells to the lung and the cellular immune response to inhaled antigen. Am J Pathol. 1997;150:1735–1743. [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS. Robbins and Cotran pathologic basis of disease. 7. Philadelphia: Elsevier Saunders; 2005. [Google Scholar]
- Kwong K, Carr MJ, Gibbard A, Savage TJ, Singh K, Jing J, Meeker S, Undem BJ. Voltage-gated sodium channels in nociceptive versus non-nociceptive nodose vagal sensory neurons innervating guinea pig lungs. J Physiol. 2008;586:1321–1336. doi: 10.1113/jphysiol.2007.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Marquez M, O’Dorisio M, O’Dorisio T, Shah M, Karacay B. Selective gene expression and activation-dependent regulation of vasoactive intestinal peptide receptor type 1 and type 2 in human T cells. J Immunol. 2001;166:2522–2530. doi: 10.4049/jimmunol.166.4.2522. [DOI] [PubMed] [Google Scholar]
- Lennertz RC, Kossyreva EA, Smith AK, Stucky CL. TRPA1 mediates mechanical sensitization in nociceptors during inflammation. PLoS One. 2012;7:e43597. doi: 10.1371/journal.pone.0043597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Clark R, Devor M, Helms C, Moskowitz MA, Basbaum AI. Intraneuronal substance P contributes to the severity of experimental arthritis. Science. 1984;226:547–549. doi: 10.1126/science.6208609. [DOI] [PubMed] [Google Scholar]
- Levy BD, Vachier I, Serhan CN. Resolution of inflammation in asthma. Clin Chest Med. 2012;33:559–570. doi: 10.1016/j.ccm.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nature immunology. 2013;14:536–542. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- Lilly CM, Bai TR, Shore SA, Hall AE, Drazen JM. Neuropeptide content of lungs from asthmatic and nonasthmatic patients. Am J Respir Crit Care Med. 1995;151:548–553. doi: 10.1164/ajrccm.151.2.7531100. [DOI] [PubMed] [Google Scholar]
- Linden A, Hansson L, Andersson A, Palmqvist M, Arvidsson P, Lofdahl CG, Larsson P, Lotvall J. Bronchodilation by an inhaled VPAC(2) receptor agonist in patients with stable asthma. Thorax. 2003;58:217–221. doi: 10.1136/thorax.58.3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XJ, Zhang Y, Liu T, Xu ZZ, Park CK, Berta T, Jiang D, Ji RR. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014 doi: 10.1038/cr.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabalirajan U, Rehman R, Ahmad T, Kumar S, Singh S, Leishangthem GD, Aich J, Kumar M, Khanna K, Singh VP, et al. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Scientific reports. 2013;3:1349. doi: 10.1038/srep01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FD. Genes, environments, development and asthma: a reappraisal. Eur Respir J. 2007;29:179–184. doi: 10.1183/09031936.00087906. [DOI] [PubMed] [Google Scholar]
- Muroi Y, Undem BJ. Targeting voltage gated sodium channels NaV1.7, Na V1.8, and Na V1.9 for treatment of pathological cough. Lung. 2014;192:15–20. doi: 10.1007/s00408-013-9533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AC, Kajekar R, Undem BJ. Allergic inflammation-induced neuropeptide production in rapidly adapting afferent nerves in guinea pig airways. Am J Physiol Lung Cell Mol Physiol. 2002;282:L775–781. doi: 10.1152/ajplung.00353.2001. [DOI] [PubMed] [Google Scholar]
- Ni D, Gu Q, Hu HZ, Gao N, Zhu MX, Lee LY. Thermal sensitivity of isolated vagal pulmonary sensory neurons: role of transient receptor potential vanilloid receptors. Am J Physiol Regul Integr Comp Physiol. 2006;291:R541–550. doi: 10.1152/ajpregu.00016.2006. [DOI] [PubMed] [Google Scholar]
- Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, Locksley RM. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502:245–248. doi: 10.1038/nature12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, Humbert M, Katz LE, Keene ON, Yancey SW, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. The New England journal of medicine. 2014;371:1198–1207. doi: 10.1056/NEJMoa1403290. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Behringer RR, Quaife CJ, Maxwell F, Maxwell IH, Brinster RL. Cell lineage ablation in transgenic mice by cell-specific expression of a toxin gene. Cell. 1987;50:435–443. doi: 10.1016/0092-8674(87)90497-1. [DOI] [PubMed] [Google Scholar]
- Patterson RN, Johnston BT, Ardill JE, Heaney LG, McGarvey LP. Increased tachykinin levels in induced sputum from asthmatic and cough patients with acid reflux. Thorax. 2007;62:491–495. doi: 10.1136/thx.2006.063982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey CD, Celedon JC. The hygiene hypothesis and asthma. Curr Opin Pulm Med. 2005;11:14–20. doi: 10.1097/01.mcp.0000145791.13714.ae. [DOI] [PubMed] [Google Scholar]
- Rehman R, Bhat YA, Panda L, Mabalirajan U. TRPV1 inhibition attenuates IL-13 mediated asthma features in mice by reducing airway epithelial injury. International immunopharmacology. 2013;15:597–605. doi: 10.1016/j.intimp.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, Paust S, Wood JN, von Andrian UH. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature. 2014;510:157–161. doi: 10.1038/nature13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson DP, Gudes S, Sprague JM, Patoski HA, Robson VK, Blasl F, Duan B, Oh SB, Bean BP, Ma Q, et al. Activity-dependent silencing reveals functionally distinct itch-generating sensory neurons. Nature neuroscience. 2013;16:910–918. doi: 10.1038/nn.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, Peterson LW, Wherry EJ, Goldrath AW, Bhandoola A, Artis D. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. The Journal of experimental medicine. 2013;210:1823–1837. doi: 10.1084/jem.20122332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra MF, Anjos-Valotta EA, Olsen PC, Couto GC, Jurgilas PB, Cotias AC, Pao CR, Ferreira TP, Arantes AC, Pires AL, et al. Nebulized lidocaine prevents airway inflammation, peribronchial fibrosis, and mucus production in a murine model of asthma. Anesthesiology. 2012;117:580–591. doi: 10.1097/ALN.0b013e31826687d5. [DOI] [PubMed] [Google Scholar]
- Shi HZ, Xiao CQ, Zhong D, Qin SM, Liu Y, Liang GR, Xu H, Chen YQ, Long XM, Xie ZF. Effect of inhaled interleukin-5 on airway hyperreactivity and eosinophilia in asthmatics. American journal of respiratory and critical care medicine. 1998;157:204–209. doi: 10.1164/ajrccm.157.1.9703027. [DOI] [PubMed] [Google Scholar]
- Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH, Nassar MA. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain. 2005;113:27–36. doi: 10.1016/j.pain.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Su X, Camerer E, Hamilton JR, Coughlin SR, Matthay MA. Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol. 2005;175:2598–2605. doi: 10.4049/jimmunol.175.4.2598. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357–372. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- Trankner D, Hahne N, Sugino K, Hoon MA, Zuker C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11515–11520. doi: 10.1073/pnas.1411032111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercelli D. Discovering susceptibility genes for asthma and allergy. Nature reviews Immunology. 2008;8:169–182. doi: 10.1038/nri2257. [DOI] [PubMed] [Google Scholar]
- Voice JK, Dorsam G, Lee H, Kong Y, Goetzl EJ. Allergic diathesis in transgenic mice with constitutive T cell expression of inducible vasoactive intestinal peptide receptor. FASEB J. 2001;15:2489–2496. doi: 10.1096/fj.01-0671com. [DOI] [PubMed] [Google Scholar]
- Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol. 2013;13:75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- Weiss EB, Patwardhan AV. The response to lidocaine in bronchial asthma. Chest. 1977;72:429–438. doi: 10.1378/chest.72.4.429. [DOI] [PubMed] [Google Scholar]
- Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, Sparwasser T, Helmby H, Stockinger B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nature immunology. 2011;12:1071–1077. doi: 10.1038/ni.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, Pellegrino M, Estandian DM, Bautista DM. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. 2013;155:285–295. doi: 10.1016/j.cell.2013.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.