

Abstract
Purpose
The human endogenous retrovirus (HERV-K) envelope (env) protein is a tumor- associated antigen expressed on melanoma, but not normal cells. This study was designed to engineer a chimeric antigen receptor (CAR) on T cell surface, such that they target tumors in advanced stages of melanoma.
Experimental Design
Expression of HERV-K protein was analyzed in 220 melanoma samples (with various stages of disease) and 139 normal organ donor tissues using immuno-histochemical (IHC) analysis. HERV-K env-specific CAR derived from mouse monoclonal antibody was introduced into T cells using the transposon-based Sleeping Beauty (SB) system. HERV-K env-specific CAR+ T cells were expanded ex vivo on activating and propagating cells (AaPC), and characterized for CAR expression and specificity. This includes evaluating the HERV-K-specific CAR+ T cells for their ability to kill A375-SM metastasized tumors in a mouse xenograft model.
Results
We detected HERV-K env protein on melanoma, but not in normal tissues. After electroporation of T cells and selection on HERV-K+ AaPC, over 95% of genetically-modified T cells expressed the CAR with an effector memory phenotype and lysed HERV-K env+ tumor targets in an antigen specific manner. Even though there is apparent shedding of this TAA from tumor cells which can be recognized by HERV-K env-specific CAR+ T cells, we observed a significant anti-tumor effect.
Conclusion
Adoptive cellular immunotherapy with HERV-K env-specific CAR+ T cells represents a clinically-appealing treatment strategy for advanced-stage melanoma and provides an approach for targeting this TAA on other solid tumors.
INTRODUCTION
DNA from integrated retrovirus is found interspersed in the human genome and represents about 4.2% of our total chromosomal DNA. Of this, 8% is composed of human endogenous retroviral elements (HERVs) (1) which integrated into the human genome 1–5 million years ago. Transcriptional activity of the envelope (env) and polymerase proteins appears to have remained intact (2, 3) although HERVs do not apparently produce infectious virions, are poorly expressed in somatic cells, and are not expressed in other species (4). Stressors such as exposure to UV light and hormones (e.g., estrogen and progesterone) can increase transcriptional activity of HERV-K in tumor cells (2, 5). Approximately half of HERV-K long terminal repeats (LTRs) found in the human genome initiate transcription of non-repetitive coding regions, of which 30% of their gene products bind p53, suggesting a role in oncogenesis (6). Expression of the HERV-K envelope (env) protein is upregulated on infected cells (e.g., HIV-infected T cells) (7) and tumor cells including breast cancer (8), ovarian cancer (9), lymphoma (10), teratocarcinoma (11) and melanoma (12, 13). During the early stages of melanoma transformation, HERV-K mRNA can be induced via the BRAF-MEK-ERK signaling pathway and epigenetic changes associated with p16INK4A-CDK4 (12). Expression of the HERV-K env positively correlates with progression of primary melanoma to those with metastatic potential (13).
Adoptive transfer of genetically modified T cells redirected to tumor-associated antigens (TAAs) has shown promise although most of the clinical trials report on the therapeutic benefit from targeting hematologic malignancies. These therapies include a portfolio of T cells genetically modified to express (a) T-cell receptor (TCR) that recognizes TAA in the context of HLA (14) or (b) a chimeric antigen receptor (CAR) specific for a TAA independent of HLA (15). Clinical trials infusing non-genetically modified T cells include melanoma-specific tumor-infiltrating lymphocytes (TIL) for the investigational treatment of advanced-stage melanoma and have yielded favorable objective responses (16). This approach requires autologous tumor cells that process and present TAAs on HLAs in sufficient numbers to be recognized by endogenous specific TCRs (17). The ex vivo genetic modification to enforce expression of melanoma-specific CAR on T cells derived from peripheral blood is one approach to bypass the need to harvest tumor cells and overcome immune tolerance.
We and others have designed CD19-specific CARs for the investigational treatment of B-cell malignancies which are currently being evaluated in clinical trials (18). We express our CD19-specific CARs on T cells using a non-viral approach to gene transfer based on the Sleeping Beauty (SB) transposon/transposase system (19). T cells stably expressing the introduced CAR are selectively propagated on γ-irradiated artificial activating and propagating cells (AaPC) derived from K-562 cells (20). The two platform technologies of SB system and AaPC have been successfully used to generate CAR+ T cells for ongoing clinical trials (15).
We now report the successful generation of HERV-K env-specific CAR+ T cells using the SB system and their numeric expansion on HERV-K+ AaPC. In vitro, HERV-K env-specific CAR+ T cells lysed tumors cells expressing HERV-K env on the cell surface. These CAR+ T cells were also able to detect HERV-K env shed from the tumor cell surface. In vivo, infusion of HERV-K env-specific CAR+ T cells decreased tumor burden in a mouse model of metastatic melanoma. These data suggest that adoptive transfer of HERV-K env-specific CAR+ T cells is a promising therapeutic strategy for melanoma.
Methods
Generation and expansion of HERV-K env-specific CAR+ T cells
Generation and expansion of HERV-K env-specific CAR+ T cells were performed using a protocol previously described (20). Briefly, the 2x107 PBMC cells were washed and rested in complete RPMI supplemented with 10% fetal bovine serum (Thermo Scientific) and 1% glutamax (Gibco Life technologies) for 2 hrs. On day 0, these cells were then re-suspended in 100 μL of Nucleofector solution (Human T-cell Kit) (Lonza, Allendale, NJ), with supercoiled DNA coding for HERV-K env-specific CAR transposon (15 μg CoOp6H5CARCD28/pSBSO) and supercoiled DNA coding for SB transposase (5 μg pCMV-SB11), and transferred to a single cuvette and electroporated using the U-14 program (Lonza). The electroporated T cells were rested for 4 hours at 37°C in complete phenol red-free RPMI (Thermo Scientific) after which a half-media change was performed. The K562-derived clone 4 expresses endogenous HERV-K env (Supplemental Fig. S2A) and thus serves as AaPC to propagate HERV-K env-specific CAR+ T cells. The electroporated T cells cultured in RPMI containing 10% FBS were supplemented with γ-irradiated (100 Gy) K562-AaPC at a 1:2 T cell/AaPC ratio on day 1. Irradiated AaPC were re-added at the end of every week for T-cell stimulation at the same ratio. Soluble recombinant IL-21 (eBioscience, San Diego, CA) and IL-2 (Invitrogen) cytokines were supplemented at a concentration of 30 ng/mL and 50 U/mL respectively to complete RPMI media on a Monday, Wednesday, Friday schedule. Mock transfected negative “no DNA” control T cells propagated in presence of OKT3-loaded AaPC, IL-2, and IL-21. CD19-specific CAR+ T cells electroporated with CoOpCD19CARCD28/pSBSO and SB11 transposase and propagated under the same culture conditions as for HERV-K env-specific CAR+ T cells and served as a negative control. The T-cell cultures were monitored weekly for the contaminating presence of CD3negCD56+ NK cells which were depleted if this population exceeded 10% of the total population. A depletion usually occurred between 10 and 14 days of initial co-culture with AaPC and was carried out using CD56 beads (Miltenyi Biotech Inc, Auburn, CA) on autoMACS (Miltenyi Biotech) using the positive selection “possel” program according to manufacturer’s instruction. T-cell viability was assessed by trypan blue exclusion using a Cellometer automated cell counter (Auto T4 Cell Counter, Nexcelom Bioscience, Lawrence, MA). The fold expansion was calculated (compared to day 1) of total, CD3+, CD4+, CD8+, and CAR+ cells at the end of 7, 14, 28, and 32 days of co-culture on AaPC with cytokines. The average CAR+ T-cell growth of 4 donors was compared between the CD19-specific CAR+ T cells and no DNA control cells using a Student’s t-test.
Cell lines and their propagation
A375, A624, and A888 were a gift from Dr. Lazlo Radvanyi (MDACC); EL4 parental were obtained from American Type Culture Collection (catalog # TIB-39, Rockville, MD); and A375-SM (super-metastatic melanoma cell line) was received from the Characterized Cell Line Core Facility (CCCF) at MDACC. All cell lines were obtained in the year 2010 during study initiation and were cultured in complete RPMI (Thermo Scientific, Rockford, IL) with 10% FBS (Thermo Scientific) and 5% glutamax (Gibco Life technologies, Grand Island, NY). All cell lines were authenticated by MDACC DNA Sequencing and Analysis Core using DNA profiling of short tandem repeat markers and further verified by morphology, and/or flow cytometry. They were routinely tested negative for Mycoplasma.
6H5 mAx
In order to measure the HERV-K env expression on tumor cell surface we developed a 6H5 mAx specific for the HERV-K env antigen. 6H5 mAx represents soluble CAR by swapping the CD28 and CD3ζ endodomain with a polyhistidine tag (Supplemental Fig. 3A). This construct was inserted into a DNA plasmid coding for lentiviral vector (Supplemental Fig. 3B) which was introduced into 293 METR cells (gift from Dr. Brian Rabinovich) using Lipofectamine 2000 (Life Technologies, Grand Island, NY), and conditioned supernatant was concentrated using 100 kDa cut-off centrifugal filter (catalog # UFC910024, EMD Millipore, Billerica, MA). The 6H5 mAx was then purified using a polyA column (Life Technologies) and analyzed on a SDS-PAGE gel to confirm purity.
In vivo CAR+ T cell and tumor cell activity measurement by photon quantification
All animal experiments were performed after the approval of Institutional Animal Care and Use Committee at MD Anderson Cancer Center (MDACC) in accordance to NIH guidelines for the Care and Use of Laboratory Animals. 5 week old female NOD.Cg-PrkdcscidIl2rgtm1wjl/SzJ (NSG, Jackson Laboratories, Bar Harbor, ME) mice were intravenously injected with 106 A375-SM-RmK cells on Day 0 (21). Mice in the treatment cohorts (n = 7) received 2×107 HERV-K env-specific CAR+ ffLuc+ T cells on Days 7, 14 and 21. 6×104 U IL-2 (eBioscience) was injected intraperitonealy (IP) on day of each T-cell infusion and twice on the day after. One cohort of mice (n = 6) bearing the tumor received no treatment while a control group of mice (n = 5) without tumor received a similar number of CAR+ T cells as in treatment group. Bio luminescence imaging (BLI) on mice in anterior-posterior position was performed weekly using a Xeno IVIS 100 series system (Caliper Life Sciences, Alameda, CA) to reveal the distribution and quantity of tumor and T cells as previously described (22). Mice were anesthetized and placed in for BLI To measure the HERV-K env-specific CAR+ffLuc+ T-cell activity 150 μL (200 μg/mouse) of D-Luciferin potassium salt (Caliper Life Sciences) was injected IP. Ten minutes after injection emitted photons were quantified using the Living Image 2.50.1 (Caliper Life Sciences) program. To image the tumor cell activity 100 μL (60 μM final concentration) of EnduRen (Promega, Fitchburg, WA) was injected IP. 20 minutes after injection the tumor activity was quantified similar to ffLuc. This experiment was performed twice and unpaired Student’s t-test was performed to establish statistical significance of the photon flux. Livers from tumor alone and treatment groups were isolated on day 25 and imaged directly for mKate expression using a Leica M205FA stereo microscope and quantified for number of metastatic foci on the liver.
RESULTS
Tumor-specific expression of HERV-K env on primary melanoma
The 6H5 HERV-K env-specific monoclonal antibody (mAb) (23) was used in immunohistochemistry (IHC) to investigate expression of the HERV-K env protein. Initially, we evaluated melanoma tumor-cell lines (A888, A624, A375, A375-super metastatic cells (SM) that are highly metastatic upon passage in mice (21), and K-562 parental cells) The 6H5 mAb revealed the presence of full length HERV-K env represented as a single band on western blot at 66 kDa under reducing conditions which was absent in the mouse parental EL4 T-cell line (Supplemental Fig. 1A) (23). This mAb was used to interrogate expression of HERV-K env on and in primary (stages I through IV) and metastatic melanoma and normal skin. Punctate cell surface expression (solid arrow) and diffuse cytoplasmic staining (dotted arrow) were observed in tumor cells in a mutually exclusive manner (Fig. 1A). IHC staining (scored from 0 to 3) on primary melanoma tissues were graded based on intensity of staining for HERV-K env (Fig. 1B). An H-index was calculated by multiplying the percentage of HERV-K env+ cells with the intensity of staining compared to the isotype control (24). The average H-index for melanoma was 9.2 (n = 220, standard error of mean (SEM) = 2.4) which was 206-fold greater (p <0.001) than benign skin tissue sampled from patients with melanoma (H-index = 0.045, n = 55, SEM = 0.03; Fig. 1C). The average H-index calculated for metastatic tissues (H-index = 10.65, n = 84, SEM = 3.9) was greater than that for primary tissues (H-index = 8.4, n = 136, SEM = 3.0) though this did not reach statistical significance (p = 0.07) (Supplemental Fig. 1B). 30% tumor cells positive for TAA in primary tissues and 15.5% metastatic tumor cells were positive for TAA (Supplemental Fig. 1C). To determine whether HERV-K env expression was restricted to tumor cells, tissues from 29 types of healthy organs from three different donors were also assessed by IHC. We observed apparent absence of HERV-K env protein expression from these samples including normal skin (Fig. 1D). In contrast, HERV-K env was observed on tumor cells originating from multiple anatomical sites (Supplemental Fig. 1D). These data support our contention that HERV-K env is a TAA and that as it is expressed on the cell surface it may be targeted by HERV-K env-specific CAR+ T cells.
Figure 1. TAA expression on primary tumor samples and normal tissues.
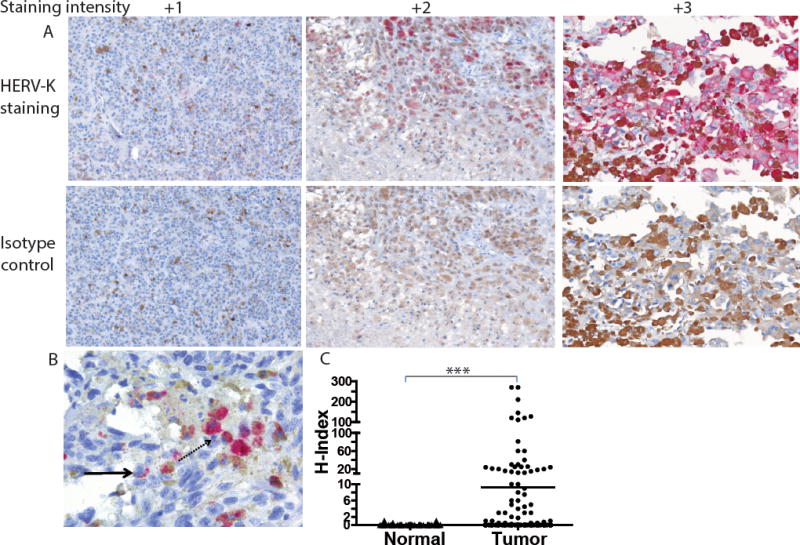
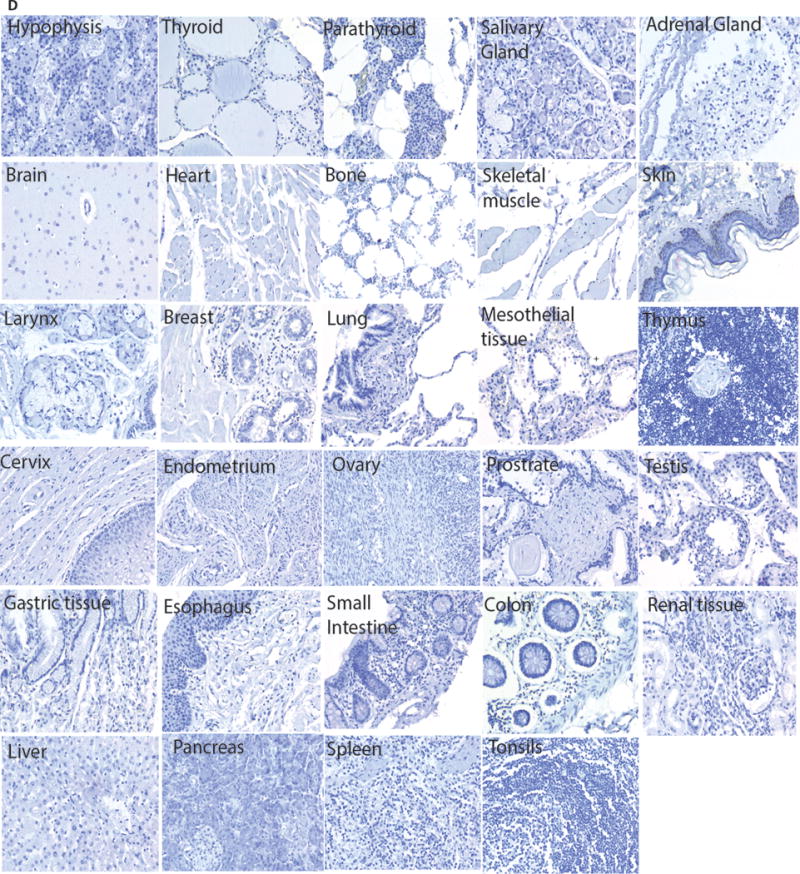
(A) Representative images of primary melanoma tumor cells (200×) with varying staining intensity (scored 0 to 3) of HERV-K env expression (top panel) when compared to isotype IgG2a control staining (bottom panel). (B) Tumor cells (400×) showing HERV-K env expression on cell membrane (punctate pattern; solid arrow) or cytoplasmic staining (diffuse pattern; dotted arrow). (C) Dot plot representing H-index (measured as product of intensity of staining by percent HERV-K env+ tumor cells) of benign tissue obtained from patient with melanoma (n = 55) and melanoma primary tumor samples (n = 220). Each dot in the dot plot represents a biopsy of an individual patient; unpaired Student’s t-test with Mann-Whitney post-test; ***p < 0.001. (D) Representative pictures (200×) of lack of HERV-K env antigen expression on tissues sections from 29 normal organs. The H-index was calculated as zero since no staining was observed in or on these tissues.
Generation and characterization of HERV-K env-specific CAR derived from 6H5 mAb
We engineered a CAR with specificity for HERV-K env based upon the VH (at amino terminus) and VL domains derived from the 6H5 mAb which were joined via the Whitlow linker to form a single chain variable fragment (scFv). This was fused in frame to a modified human IgG4 hinge/Fc (25) stalk, CD28 transmembrane domain, and a combination of CD28 and CD3ζ intracellular domains. This design is similar to our 2nd generation CD19-specific CAR (designated CD19RCD28) currently employed in clinical trials (20). We stably expressed this CAR on T cells derived from peripheral blood mononuclear cells (PBMC) using the SB transposon/transposase system (Fig. 2A). HERV-K env-specific CAR+ T cells were then selectively propagated on AaPC (26) (designated clone 4) derived from K-562 cells which expresses endogenous HERV-K env and were previously genetically modified by lentivirus transduction to co-express CD64, CD86, CD137L, and a first-generation membrane-bound IL-15 (Supplemental Fig. 2A) (20). Control T cells from PBMC were (i) mock electroporated without DNA (“no DNA control”) and expanded on AaPC clone 4 pre-loaded via transgenic CD64 with a CD3-specific mAb (OKT3) and (ii) CD19-specific CAR+ T cells generated per published method for our clinical trials (20). 95% of the electroporated/propagated T cells stably expressed the HERV-K env-specific CAR on their cell surface after 28 days of co-culture with AaPC in the presence of soluble recombinant IL-2 and IL-21 (Fig. 2B). Compared with CD19-specific CAR+ T cells, no appreciable difference was seen in (a) growth kinetics of HERV-K env-specific CAR+ T cells (Fig. 2C), (b) percentage outgrowth of CD3+CAR+ T cells (Fig. 2D), (c) ratio of CD4+ to CD8+ T cells (Supplemental Fig. 2B), or ratio of expression of CAR to CD3 T cells (Supplemental Fig. 2C). The average copy number of integrated transposon was 1.6 (n = 3, standard deviation (SD) = 0.03) per T-cell genome based on a Jurkat cell clone with a single integration of CAR (27) (Fig. 2E). This appears less than that reported after virus-mediated transduction of T cells to express a CAR (28). We evaluated the therapeutic potential of the T cells (n = 4) using multi-parameter flow cytometry which revealed the percentage of CAR+ T cells with naive (CD45ROnegCD45RA+CD28+; mean 7.79 ± SD 2.3), central memory (CD45RO+CD45RAnegCD28+; 12.02 ± 10.4), effector memory (CD45RO+CD45RAnegCD28+; 87.9 ± 10.9) and effector memory RA phenotypes (CD45ROnegCD45RA+CD28+; 91.04 ± 3.4) (Supplemental Fig. 2D). These effector memory T cells were further characterized as CD27+CCRneg (33.22% ± 24) similar to our previous reported data for CD19-specific CAR+ T cells (not shown) (29). We observed the outgrowth of granzyme-B+ HERV-K env-specific CAR+ T cells (98.17% ± 2.3, n = 4) suggesting that these cells should be cytotoxic (Fig. 2F). In addition to flow cytometry, we employed a set of 500 bar-coded probes to digitally count mRNA molecules to reveal selected gene expression. Compared to no DNA control T cells (n = 3), the HERV-K env-specific CAR+ T cells (n = 3) had significantly (p <0.5) increased levels of mRNA species coding for (a) chemokines and receptors (CCR1, CCR5, CCL3 and CCL4) (30), (b) transcriptional regulators (LRP5, PAX5, TCF7) (c) T-cell activators (CD80, RORA, KIR3DL2) and (d) genes participating in lysis (perforin1 and granzyme H) suggesting that the HERV-K env-specific CAR+ T cells are capable of producing an effective response against the target TAA (Supplemental Fig. 2E). In aggregate, these data demonstrate that T cells could be modified with the SB system to express a CAR from 6H5 and propagated on HERV-K env+ AaPC generating a biologic product with therapeutic potential for melanoma.
Figure 2. Generation of HERV-K env-specific CAR+ T cells.
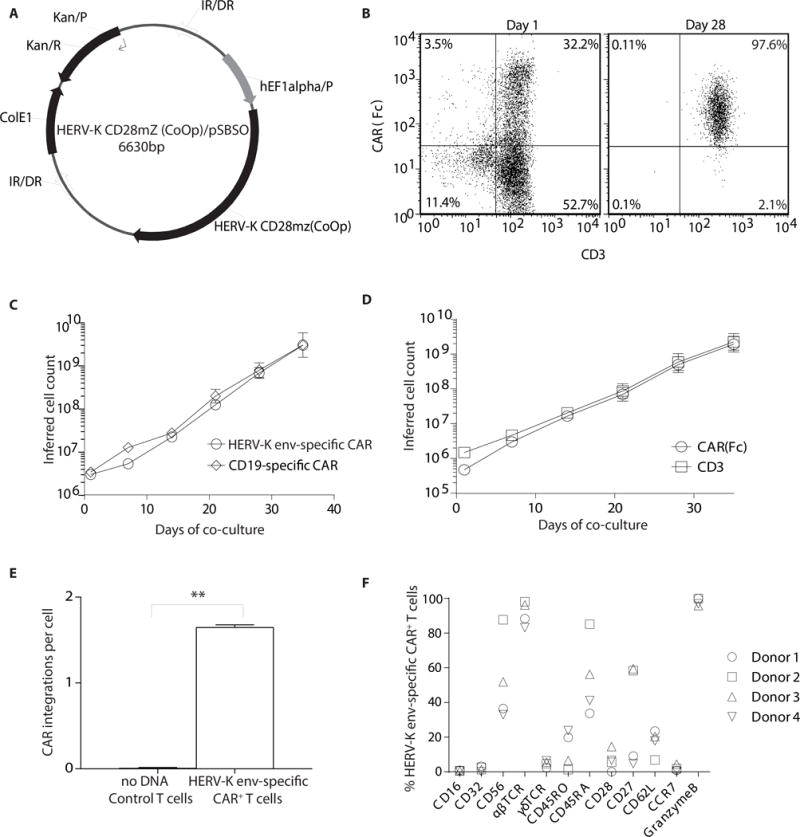
(A) SB-derived DNA transposon (HERV-K CD28mZ (CoOp)/pSBSO) designed to express HERV-K env-specific CAR. Abbreviations: IR/DR: inverted repeat/direct repeat, hEF-1alpha/p: human elongation factor-1α hybrid promoter, HERV-K CD28mZ (CoOp): codon-optimzed HERV-K envelope scFv, ColE1: E. coli origin of replication, Kan/R: kanamycin resistance, Kan/p: kanamycin resistance promoter. PBMCs (n = 4) were electroporated with SB plasmids, HERV-K-specific CAR and SB11 transposase and co-cultured with AaPCs with cytokines. (B) Representative flow plots of transient (day 1) and stable (day 28) CAR (binding of Fc-specific mAb) and CD3 expression on HERV-K env-specific CAR+ T-cell cultures. (C) Rate of expansion of total HERV-K env-specific CAR+ T cells and CD19-specific CAR+ T cells propagated on aAPC and cytokines. There was no difference in the kinetics of expansion. (D) Rate of expansion of CD3+ and CAR+ HERV-K env-specific T cells propagated on aAPC and cytokines. (E) Measurement of the copy number of CAR (transposon) in T-cell genome after electroporation and propagation. (F) Multiparameter flow cytometry to evaluate the phenotype of HERV-K env-specific CAR+ T cells. Each symbol represents individual donor. Data represented as mean or mean ± SD (n = 4); unpaired Student’s t-test; **p < 0.01.
Generation and characterization of HERV-K env-specific maxibody derived from 6H5 mAb
To help define the specificity of the CAR derived from 6H5, we generated a soluble version of this immunoreceptor as a maxibody (mAx) (31). The mAx is a soluble immunoreceptor that approximates the membrane-bound immunoreceptor as it is based CAR design, but lacks sequence anchoring it to the cell membrane. Indeed, the HERV-K env-specific 6H5 mAx consisted of the 6H5 scFv (used to generate the CAR) fused to full-length human IgG4 using the same modified hinge and Fc region employed in the CAR construct (Supplemental Fig. 3, A and B). We evaluated cell-surface expression of HERV-K env on EL4, AaPC clone 4, A888, A375-SM and A624 cells using 6H5 mAb (Fig. 3A) and 6H5 mAx (Fig. 3B). There was a positive trend between the H-indices calculated using 6H5-derived mAb and mAx at three different time points (days 1, 2, and 3 after plating the cells on day 0; spearman correlation coefficient, ρ = 0.5, p = 0.06) (Fig. 3C). Despite the variation in binding efficiency of 6H5 mAx and 6H5 mAb due to structural differences, the correlation plot indicates that these two soluble species apparently recognize the same TAA. We also performed an ELISA and observed a comparable level of binding of 6H5-derived mAb and mAx to plate-bound purified recombinant HERV-K env antigen, designated K10 (23) (Fig. 3D). K10 was expressed as a fusion protein with glutathione S-transferase and purified as previously described (8). CD19-specific mAb (Fig. 3D) and plate-bound CD19 antigen were used as a negative control (Supplemental Fig. 3C). As both soluble reagents inhibit the binding of each other, the mAb and mAx thus appear to bind to a shared epitope on K10 (Supplemental Fig. 3D). Taken together, the 6H5-derived mAb and mAx, appear to recognize HERV-K env lending support to the concept that the CAR expressed on T cells will also recognize this TAA in an antigen specific manner.
Figure 3. Comparison of 6H5 mAb versus 6H5 mAx.
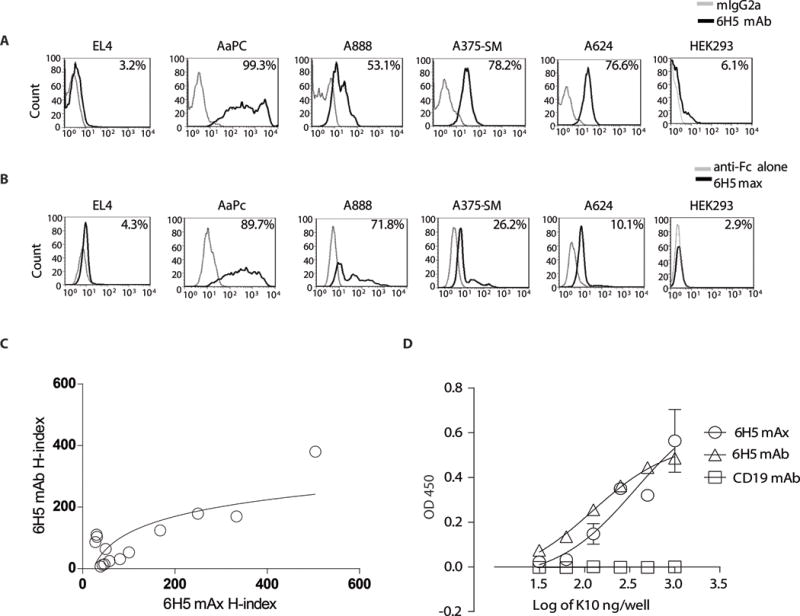
Histograms representing surface antigen expression of HERV-K env using (A) 6H5 mAb (black) and (B) 6H5 mAx (black). Isotype (grey) and secondary antibody (grey) were used as controls. (C) Correlation plot between tumor cell surface (A888, A624, A375, A375-SM, EL4) H-indices of 6H5 mAb or 6H5 mAx staining of tumor cells grown at three different time point with varying cell density (spearman correlation coefficient ρ = 0.5033, p = 0.067). (D) Binding of 6H5 mAx and 6H5 mAb to purified HERV-K env (K10) protein by ELISA. CD19 mAb was used as negative control. H-index was calculated as product of intensity of HERV-K env staining (MFI) on tumor cell surface and percent tumor cells positive for antigen expression. Data represents mean ± SD from triplicate measurements that were pooled from two independent experiments for ELISA experiments.
Specificity of CAR+ T cells for HERV-K env
The redirected effector function of CAR+ T cells was evaluated to establish specificity for HERV-K env. We modified HERV-K envneg EL4 cells to express HERV-K env (Supplemental Fig. 4A) to serve as targets (Fig. 4A). HERV-K env-specific CAR+ T cells specifically killed HERV-K env+ EL4, but not parental EL4 cells (Fig. 4B). HERV-K env-specific CAR+ T cells also mediated lysis of melanoma cells expressing endogenous HERV-K env. Compared to control no DNA control T cells, killing of melanoma cell lines A888, A624, A375, A375-SM, which are recognized by 6H5, was significantly greater using HERV-K env-specific CAR+ T cells (p <0.001) (Fig. 4C). CD19-specific CAR+ T cells served as an additional negative control and as expected failed to lyse A888, A375-SM, A624 and CD19neg EL4 tumor cells above background. However, the CD19-specific CAR+ T cells exhibited specific lysis of melanoma and EL4 cells genetically modified to express CD19 antigen (22) (Supplemental Fig. 4B and C). These control data reveal that the apparent absence of an allo-mediated killing effect by CAR+ T cells targeting melanoma tumor cells. Although the cell surface expression of HERV-K env varied at different time points during cell culture (days 1, 2, and 3 after plating the tumor cells on day 0) as assessed using 6H5mAx by flow cytometry (Fig. 4D), we observed a positive correlation between lysis of melanoma cell lines by HERV-K env-specific CAR+ T cells and the density of HERV-K env expression on the tumor cell surfaces as calculated using H-index on (ρ = 0.68, p <0.001) (Supplemental Fig. 4D). To further assess the specificity of the HERV-K-specific CAR, a lentivirus encoding HERV-K env-specific shRNA was transduced into A888 cells to knockdown (KD) the TAA (designated A888 KD). Immunoblot analysis showed a decrease of approximately 50% HERV-K env expression compared to a scrambled shRNA control (Supplemental Fig. 4E). We observed a 3-fold decrease in killing of A888 KD compared to parental A888 cells (Supplemental Fig. 4F).We also examined the redirected lysis by HERV-K env-specific CAR+ T cells by video time-lapse microscopy (VTLM) (32). The genetically modified T cells were co-cultured with HERV-K env+ A888 or A375 tumor cells or HERV-K envneg HEK293 at an effector to target (E:T) ratio of 1:5. Twenty-five target cells were individually monitored from each cell line for 15 hours and the increase in fluorescence (associated with SYTOX in the media entering damaged tumor cells and intercalating with DNA) was calculated for A888 (mean fluorescent intensity (MFI) = 445, SD = 67.8), A375 (MFI = 434, SD = 20.6) which was significantly greater than HEK293 cells (MFI = 394, SD = 19.1) (p = 0.016) (Fig. 4E, Supplemental Fig. 4G, Supplementary Video A, B &C). In addition to killing, we evaluated whether CAR+ T cells could be activated for IFN-γ production upon co-culture with melanoma cells. These tumor targets were co-cultured with HERV-K env-specific CAR+ T cells or CARneg no DNA control T cells at a 10:1 E:T ratio. Compared to control T cells, the percentage of IFN-γ+ HERV-K env-specific CAR+ T cells was 2.9, 5.1, 5.9, and 2.6 fold greater when incubated with A888, A375, A375-SM and A624, respectively (Fig. 4F). These data reveal that CAR+ T cells are specifically activated for killing and cytokine effector functions by HERV-K env.
Figure 4. Redirected specificity of HERV-K env-specific CAR+ T cells.
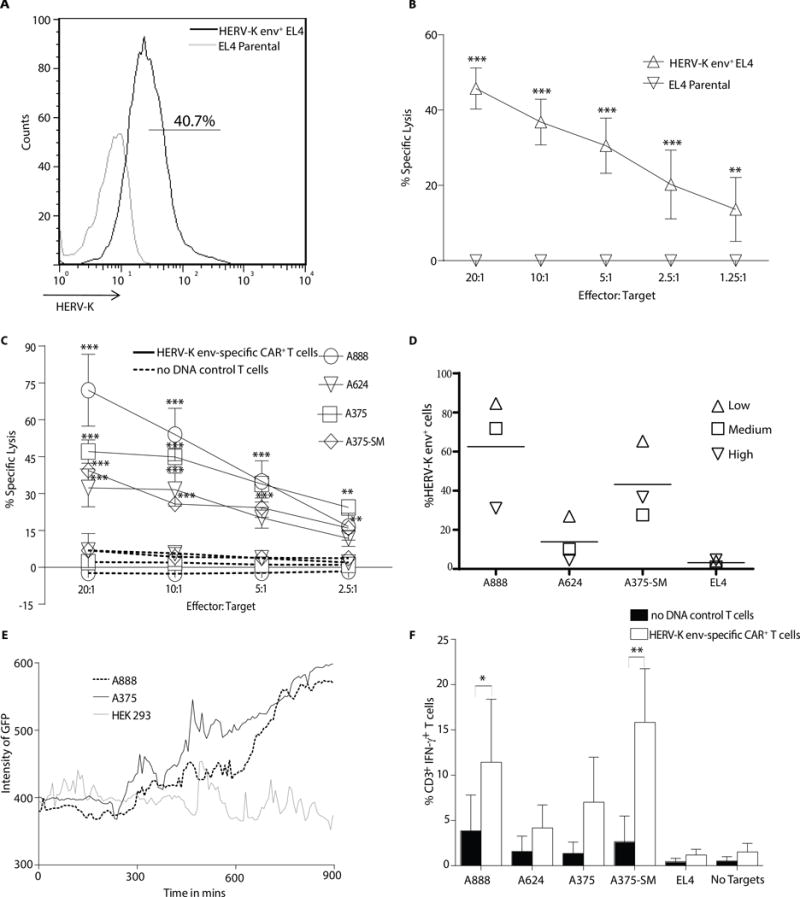
(A) Representative flow plots (n = 3) of HERV-K env expression on EL4 cells transduced with HERV-K env antigen compared with HERV-K envneg EL4 parental. (B) A four hour chromium release assay (CRA) using HERV-K env-specific CAR+ T cells as effectors and either EL4 parental or HERV-K env+ EL4 cells as targets. (C) A 4 hour CRA of HERV-K env-specific CAR+ T cells (solid line) compared to no DNA control T cells (dotted lines) using melanoma tumor cells as targets. (D) Percentage expression of HERV-K env protein on tumor cell surface using 6H5 mAx. Each symbol represents a low (20%), medium (50%) and high (70%) confluence in the tissue culture flask (n = 3) and horizontal line the mean. (E) Video time-lapse microscopy (VTLM) to assess killing by HERV-K env-specific CAR+ T cells over a 15 hour period co-cultured at 5:1 ratio with A888, A375, or HEK293 target cells in media with SYTOX. Green cells in the FITC channel were recorded as dead cells and the intensity of the fluorescence was measured. Data represented as mean ± SD from 25 tumor cells pooled from two independent experiments. (F) IFN-γ production by CAR+ and CARneg T cells upon incubation with targets. Data represent mean ± SD from three healthy donors (average of triplicate measurements for each donor). Two way ANOVA with Bonferroni post-test was performed on CRA and IFN-γ production assay between the HERV-K env-specific CAR+ T cells and no DNA control cells *p < 0.05, **p < 0.01 and ***p < 0.001.
HERV-K env is shed and can be detected by HERV-K env-specific CAR+ T cells
Since the percentage of HERV-K env expression appeared to vary at different time points when cultured tumor cells were analyzed by flow cytometry (Figure 4D), we assessed whether cell-surface expression of this TAA was modulated as a result of shedding and re-attachment over time. Western blot analysis (under reducing condition) of the concentrated supernatant from culture of tumor cells (HERV-K env+ A375-SM, A375, A888, clone 4 and HERV-K envneg EL4 probed with 6H5 mAb revealed a single band at 55 kDa consistent with cleaved HERV-K env lacking the transmembrane domain (in contrast to the 66 kDa band observed from the whole tumor cell lystes which includes the surface and the transmembrane domains) (10) (Fig. 5A). Pre-incubation of the 6H5 mAb with soluble K10 for one hour at 37°C reduced the intensity of staining on the blot, supporting our contention that the band detected in the cultured supernatant was HERK-K env (Fig. 5B). A similar band was not was observed in the flow through after concentrating conditioned supernatant through a 100 kDa filter suggesting that shed non-denatured HERV-K env is more than 100 kDa in size (Fig. 5C). We investigated whether concentrated media from A375-SM could block the ability of HERV-K env-specific CAR+ T cells to target A375-SM tumor cells. This conditioned supernatant, compared with media collected without exposure to tumor cells, resulted in approximately 80% reduction in tumor lysis (p <0.05) (Fig. 5D). We also analyzed whether the shed TAA could trigger a response by genetically modified T cells. HERV-K env-specific CAR+ T cells produced 13-fold increase in IFN-γ when co-cultured with parental EL4 cells upon exposure to concentrated supernatant collected from AaPC clone 4. This cytokine production was evaluated compared with concentrated supernatant from cultured EL4 cells or when control CD19-specific CAR+ T cells were used as effectors (p <0.001) (Fig. 5E). As the shed TAA used in these experiments was concentrated it is likely above physiological levels for IHC staining of tumor samples did not reveal shed TAA or presence of HERV-K env expression in benign tissues surrounding tumor cells. These data indicate that HERV-K env protein is apparently present in supernatant concentrated from cultured tumor cells, however, the physiologic relevance of this observation and impact on ability of CAR+ T cells to recycle effector functions remains to be determined.
Figure 5. HERV-K env-specific CAR+ T cells recognize shed TAA.
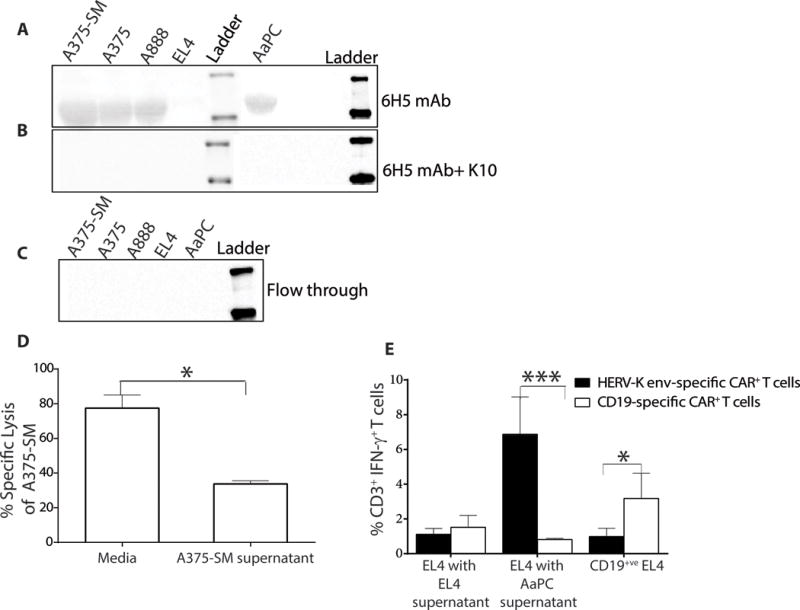
(A) Immunoblot (n = 3) revealing presence of HERV-K env in conditioned culture supernatant harvested from tumor cells. (B) Immunoblot assay (n = 3) shows specificity of 6H5 mAb by using recombinant protein (K10) to block binding to shed TAA. Conditioned supernatant from tumor cells containing shed HERV-K env protein was incubated with 6H5 and K10. (C) Immunoblot showing the absence of HERV-K env particles in the flow through after conditioned culture media was concentrated through a 100 kDa membrane. (D) A 4 hour CRA (n = 3; effector: target is 1: 10) using HERV-K env-specific CAR+ T cells as effectors against A375-SM tumor targets incubated with concentrated A375-SM tumor supernatant. Concentrated RPMI media treated were used as control. (E) IFN-γ production by HERV-K env-specific and CD19-specific CAR+ T cells upon incubation with EL4 targets in the presence of concentrated supernatant harvested from EL4 cells or AaPC(clone 4). Data represent mean ± SD from three healthy donors (average of triplicate measurements for each donor). One-way ANOVA for CRA and IFN-γ production assay with Tukey’s multiple comparison test was used *p < 0.05.
Reduced primary tumor and metastatic burden after infusion of HERV-K env-specific CAR+ T cells
We investigated whether the anti-melanoma activity observed in vitro could be extended to an in vivo mouse model. T cells co-expressing Firefly luciferase (ffLuc) and HERV-K env-specific CAR were generated by “double transposition” using the SB system (Supplemental Fig. 5A) (33). The growth kinetics, cytotoxicity, and specificity of HERV-K env-specific CAR+ffLuc+ T cells and HERV-K env-specific CAR+ T cells were not significantly different (p >0.05) (Supplemental Fig. 5, B and C). Next, we developed a xenogenic NSG mouse model of metastatic melanoma in which A375-SM tumor cells (21) were genetically modified to co-express Renilla luciferase 8.6535 (rLuc) and mKate S158A fluorescent protein (34) from a bi-cistronic vector (A375-SM-RmK) and intravenously (IV) injected (defined as day 0) (Supplemental Fig. 5D). Following tumor cell engraftment in lung and metastasis to liver, 2×107 HERV-K env-specific CAR+ffLuc+ T cells were IV administered with IL-2 (Supplemental Fig. 5E). Bioluminescent imaging (BLI) was employed to serially measure rLuc-derived tumor burden on days 3, 10, 17 and 25 (Fig. 6A). Localization of HERV-K env-specific CAR+ffLuc+ T cells was assessed the day after each T-cell injection. We monitored HERV-K env-specific CAR+ffLuc+ T cell activity on days 7, 13, and 20 (Supplemental Fig. 5F). Twenty-five days after injection of tumor, we observed 4.7-fold increased rLuc activity in untreated tumor bearing mice compared to mice with tumor receiving HERV-K env-specific CAR+ffLuc+ T cells (p <0.001) (Fig. 6B). By this time point, the untreated mice had a hunch-back posture and appeared moribund. In contrast, mice treated with HERV-K env-specific CAR+ffLuc+ T cells were active and apparently healthy (Supplemental Fig. 5G and Supplementary video E). The morphology of the liver in untreated mice appeared shriveled and necrotic compared to treated mice (Supplemental Fig. 5H) and we used optical imaging at necropsy to observe a 75% reduction in fluorescent tumor colonies in the livers of the T-cell treatment group versus the untreated group (p <0.05) (Fig. 6, C and D). These data indicate that genetically modified CAR+ T cells can be used to treat mice with disseminated HERV-K env+ tumor.
Figure 6. In vivo killing of melanoma by HERV-K env-specific CAR+ffLuc+ T cells.

NSG mice were intravenously injected with 106 A375-SM-RmK cells on Day 0. The treatment group received 2x107 HERV-K env-specific CAR+ffLuc+ T cells on days 7, 14, and 21 along with three doses of 6×104 U IL-2 (IP) (A) BLI images of rLuc-derived tumor cell activity in tumor group (n = 6) and treatment group (n = 7) mice from days 3 to 25. (B) Plot of average rLuc flux (mean ± SD) in treated (n = 7) and untreated (n = 6) mouse groups over time. (C) Postmortem imaging of mKate-derived fluorescence representing melanoma metastatic foci in livers of treated and untreated mouse groups. (D) Bar graph of average number of tumor metastatic foci on treated and untreated mouse groups. All data are representation of two independent mouse experiments with similar sample size for each group. Statistics performed with two-way ANOVA with Bonferroni’s post-tests to calculate rLuc activity and unpaired Student’s t-test to calculate liver metastatic foci between treated and untreated mice. *p < 0.05, **p < 0.01 and ***p < 0.001.
Discussion
The env protein from an endogenous retrovirus may represent an attractive TAA for CAR-mediated T-cell therapy. This is based on our demonstration that melanoma, but not normal tissues, express HERV-K env protein and that a HERV-K env-specific CAR can recognize this TAA in vitro and in vivo.
The 6H5 mAb was used to evaluate the cellular expression of HERV-K env. We observed that only tumor cells (cell lines and primary biopsy material) express HERV-K env in contrast to benign skin from the same patients. Furthermore, the transition from primary to metastatic melanoma was associated with increased expression of this TAA. This supports results by others that high HERV-K env levels correlates with increased metastatic capacity (35). Two distinct patterns of expression of this TAA were observed. Punctate and diffuse distribution of HERV-K env was found in the cell membrane and cytosol, respectively. Punctate staining may be associated with HERV-K env protein involved in (a) recycling of the HERV-K env between endosomes and the cell membrane (b) budding of exosomes and/or (c) packaging of virions. The latter two phenomena may aid in detachment and metastasis of tumor cells to distant anatomical sites (36–38).
In addition to detecting HERV-K env on and in tumor cells, we also found this TAA in conditioned and concentrated supernatant harvested from tumor cells. Although HERV-K env could be concentrated using a 100 kDa filter, we assume the shed HERV-K is either oligomerized or associated with other proteins which are dissociated when denatured to its 55-kDa form. However, immunoprecipitation of this protein for mass spectrometry analysis with 6H5 mAb or 6H5 mAx did not yield data indicating other proteins complexed with HERV-K env (data not shown). Endogenous retroviral particles are capable of extruding or budding from the tumor cell membrane as immature provirus (39, 40). Indeed, shed HERV-K env is present in human plasma of patients with lymphoma and breast cancer (10). The shed HERV-K env is likely incapable of binding or infecting normal cells surrounding the tumor due to presence of multiple stop codons (39, 40). In vitro we could show that HERV-K env was recognized by CAR+ T cells and this interfered with the ability to target this TAA on tumor cells. However, this was under conditions when the shed TAA was concentrated. This is in contrast to our observations in vivo when the adoptive transfer of CAR+ T cells resulted in an anti-tumor effect.
HERV-K env has been described on and in other tumor cells including breast cancer, ovarian cancer, prostate cancer, lymphoma, renal cell carcinoma, teratocarcinoma and infected T cells during AIDS and thus has been studied as a target antigen for developing new treatment strategies (7–11, 41–43). For example, 6H5 mAb against HERV-K env expressed on breast cancer cells produced a 10-fold reduction in tumor volume in a breast cancer mouse model (23). Prophylactic vaccination against HERV-K env precluded the formation of renal cell carcinoma tumor and subsequent metastasis in mice (43). HERV-K env-specific CD8+ T cells obtained from patients with HIV were found in vitro to cross-react and eliminate HIV-infected cells in an antigen specific manner (7). Furthermore, anti retroviral drugs such as efavirenze and RNA interference (RNAi) of HERV-K retroviral element also reduced the growth and tumorigenic potential of melanoma in a mouse model (44). A four-fold increase in HERV-K+ melanoma tumor cell lysis was observed using HERV-K env-specific TCR expressed on T cells compared to HERV-Kneg melanoma cell line in vitro (45).
Our work using the SB system is a non-viral approach that utilizes a transposon (encoding CAR) and transposase (responsible for genome integration) that is suitable for clinical translation. We were able to selectively propagate HERV-K env-specific CAR+ T cells on AaPC along with IL-2 and IL-21 to numbers similar to CD19-specific CAR+ T cells used in our trials for the treatment of B-lineage malignancies (15). 50% of propagated HERV-K env-specific CAR+ T cells were of effector memory phenotype, previously shown to be more effective in vivo than other T-cell phenotypes, such as short lived effector T cells (46). These HERV-K env-specific CAR+ T cells lysed tumor cells in an antigen-specific manner in vitro and in vivo implying the specificity of the CAR+ T cells to target HERV-K env protein. Thus, targeting the envelope protein HERV-K by CAR+ T cells may constitute a broad based immunotherapeutic option for multiple cancers including solid and hematological tumors and infectious diseases such as AIDS.
Supplementary Material
Statement of Translational Relevance.
Patients with metastatic melanoma have a poor prognosis due to resistance to conventional therapies such as chemotherapy, radiation, and surgery. We use a non-viral gene therapy method whereby T cells are engineered to express HERV-K envelope (env)-specific CAR. Our pre-clinical data suggests that we can specifically target and kill tumor cells expressing HERV-K env antigen while not attacking the normal cells. This therapy has implications for the treatment of human diseases beyond melanoma. HERV-K env is expressed on tumor cells derived from hematologic malignancies and solid tumors as well as after infection such as CD4 T cells bearing HIV. This provides an attractive opportunity that one CAR design targeting HERV-K env to be used to treat a variety of disease states.
Acknowledgments
We thank the flow cytometry, cytogenetic (for fingerprinting tumor cells) and histology cores (MDACC). We thank Dr. Carl June and colleagues (University of Pennsylvania) for help generating K-562-derived AaPC, Dr. Perry Hackett (University of Minnesota) for assistance with SB, Dr. Jae Chen (MDACC) for scoring the human melanoma tissues, Dr. Bipulendu Jena (MDACC) for providing CD19 mAb and CD19 recombinant protein, Dr. Jason Roszik (MDACC) and Dr. Alvaro Laureano (Universidad Federal do Rio Grande do Sul) for assistance with VTLM, Dr. Eric Davis (MDACC) for assistance with analysis of digital mRNA data, Dr. Harry Zhou (Fabion) for his assistance with 6H5 mAx purification, and Dr. Jailiang Dai (MDACC) for his assistance with biostatistics.
Funding
Janani Krishnamurthy is a Joanne M foundation Fellow in Cancer Research (UT-GSBS-Houston), and a Teal Pre-doctoral Scholar (Department of Defense grant BCRP W81XWH-11-1-0002). This work was supported by funding from: Cancer Center Core Grant (CA16672); RO1 (CA124782, CA120956, CA141303; CA141303); R33 (CA116127); P01 (CA148600); Albert J. Ward Foundation; Burroughs Wellcome Fund; Cancer Prevention and Research Institute of Texas; CLL Global Research Foundation; Department of Defense; Estate of Noelan L. Bibler; Gillson Longenbaugh Foundation; Harry T. Mangurian, Jr., Fund for Leukemia Immunotherapy; Fund for Leukemia Immunotherapy; Institute of Personalized Cancer Therapy; Leukemia and Lymphoma Society SCOR; Lymphoma Research Foundation; Miller Foundation; Mr. Herb Simons; Mr. and Mrs. Joe H. Scales; Mr. Thomas Scott; MDACC Moon Shot; National Foundation for Cancer Research; Pediatric Cancer Research Foundation; Production Assistance for Cellular Therapies (PACT); TeamConnor; Thomas Scott; William Lawrence and Blanche Hughes Children’s Foundation; Department of Defense W81XWH-12-1-0223 for Dr. Wang-Johanning.
Footnotes
Author contribution
JK designed, performed, the experiments, and wrote the manuscript. BR designed the 6H5 maxibody experiments and provided advice. TM and KS assisted with in vivo experiments. SO assisted with designing the CAR and bidirectional SB vectors. SM assisted with NanoString nCounter data. JBP provided purified K10 fusion protein, as well as purified and conjugated 6H5 mAb. HS, PK, HH, and BR assisted in designing experiments and interpreting data. FWJ helped establish the research concept. LJNC generated the idea, conceptualized the experiments, and edited the manuscript.
Conflict of interest statement
LJNC has patents with Sangamo BioSciences regarding artificial nucleases. He consults with Targazyme, Inc. (formerly American Stem cells, Inc.), GE Healthcare, Ferring Pharmaceuticals, Fate Therapeutics, Janssen Pharmaceuticals, and Bristol-Myers Squibb. He receives honoraria from Miltenyi Biotec. LJNC has recently licensed his technology to Intrexon and Ziopharm. Patent application based on this manuscript filed February 13, 2014.
References
- 1.Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011;8:90. doi: 10.1186/1742-4690-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hohenadl C, Germaier H, Walchner M, Hagenhofer M, Herrmann M, Sturzl M, et al. Transcriptional activation of endogenous retroviral sequences in human epidermal keratinocytes by UVB irradiation. J Invest Dermatol. 1999;113:587–94. doi: 10.1046/j.1523-1747.1999.00728.x. [DOI] [PubMed] [Google Scholar]
- 3.Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14572–9. doi: 10.1073/pnas.0404838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hughes JF, Coffin JM. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc Natl Acad Sci U S A. 2004;101:1668–72. doi: 10.1073/pnas.0307885100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan AS, Muller J, Sears JF. Early detection of endogenous retroviruses in chemically induced mouse cells. Virus Res. 2001;79:39–45. doi: 10.1016/s0168-1702(01)00280-5. [DOI] [PubMed] [Google Scholar]
- 6.Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, et al. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci U S A. 2007;104:18613–8. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones RB, Garrison KE, Mujib S, Mihajlovic V, Aidarus N, Hunter DV, et al. HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates. J Clin Invest. 2012;122:4473–89. doi: 10.1172/JCI64560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, et al. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008;68:5869–77. doi: 10.1158/0008-5472.CAN-07-6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang-Johanning F, Liu J, Rycaj K, Huang M, Tsai K, Rosen DG, et al. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int J Cancer. 2007;120:81–90. doi: 10.1002/ijc.22256. [DOI] [PubMed] [Google Scholar]
- 10.Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008;82:9329–36. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lower R, Lower J, Tondera-Koch C, Kurth R. A general method for the identification of transcribed retrovirus sequences (R-U5 PCR) reveals the expression of the human endogenous retrovirus loci HERV-H and HERV-K in teratocarcinoma cells. Virology. 1993;192:501–11. doi: 10.1006/viro.1993.1066. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Sheng T, Wan X, Liu T, Wu H, Dong J. Expression of HERV-K correlates with status of MEK-ERK and p16INK4A-CDK4 pathways in melanoma cells. Cancer Invest. 2010;28:1031–7. doi: 10.3109/07357907.2010.512604. [DOI] [PubMed] [Google Scholar]
- 13.Reiche J, Pauli G, Ellerbrok H. Differential expression of human endogenous retrovirus K transcripts in primary human melanocytes and melanoma cell lines after UV irradiation. Melanoma Res. 2010;20:435–40. doi: 10.1097/CMR.0b013e32833c1b5d. [DOI] [PubMed] [Google Scholar]
- 14.Eshhar Z. Tumor-specific T-bodies: towards clinical application. Cancer Immunol Immunother. 1997;45:131–6. doi: 10.1007/s002620050415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jena B, Maiti S, Huls H, Singh H, Lee DA, Champlin RE, et al. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS One. 2013;8:e57838. doi: 10.1371/journal.pone.0057838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, Lopez-Botet M, Duggan-Keen M, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today. 1997;18:89–95. doi: 10.1016/s0167-5699(96)10075-x. [DOI] [PubMed] [Google Scholar]
- 18.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper LJ, Al-Kadhimi Z, DiGiusto D, Kalos M, Colcher D, Raubitschek A, et al. Development and application of CD19-specific T cells for adoptive immunotherapy of B cell malignancies. Blood Cells Mol Dis. 2004;33:83–9. doi: 10.1016/j.bcmd.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–71. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Price JE, Fan D, Zhang RD, Bucana CD, Fidler IJ. Correlation of growth capacity of human tumor cells in hard agarose with their in vivo proliferative capacity at specific metastatic sites. J Natl Cancer Inst. 1989;81:1406–12. doi: 10.1093/jnci/81.18.1406. [DOI] [PubMed] [Google Scholar]
- 22.Singh H, Serrano LM, Pfeiffer T, Olivares S, McNamara G, Smith DD, et al. Combining adoptive cellular and immunocytokine therapies to improve treatment of B-lineage malignancy. Cancer Res. 2007;67:2872–80. doi: 10.1158/0008-5472.CAN-06-2283. [DOI] [PubMed] [Google Scholar]
- 23.Wang-Johanning F, Rycaj K, Plummer JB, Li M, Yin B, Frerich K, et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst. 2012;104:189–210. doi: 10.1093/jnci/djr540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald JW, Pilgram TK. Nuclear expression of p53, p21 and cyclin D1 is increased in bronchioloalveolar carcinoma. Histopathology. 1999;34:439–46. doi: 10.1046/j.1365-2559.1999.00632.x. [DOI] [PubMed] [Google Scholar]
- 25.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 26.Manuri PV, Wilson MH, Maiti SN, Mi T, Singh H, Olivares S, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 2010;21:427–37. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiti SN, Huls H, Singh H, Dawson M, Figliola M, Olivares S, et al. Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother. 2013;36:112–23. doi: 10.1097/CJI.0b013e3182811ce9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583–93. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 30.Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nat Immunol. 2001;2:102–7. doi: 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 31.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–36. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 32.Serrano LM, Pfeiffer T, Olivares S, Numbenjapon T, Bennitt J, Kim D, et al. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. 2006;107:2643–52. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci U S A. 2014;111:10660–5. doi: 10.1073/pnas.1312789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–6. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 35.Hahn S, Ugurel S, Hanschmann KM, Strobel H, Tondera C, Schadendorf D, et al. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res Hum Retroviruses. 2008;24:717–23. doi: 10.1089/aid.2007.0286. [DOI] [PubMed] [Google Scholar]
- 36.Tonjes RR, Boller K, Limbach C, Lugert R, Kurth R. Characterization of human endogenous retrovirus type K virus-like particles generated from recombinant baculoviruses. Virology. 1997;233:280–91. doi: 10.1006/viro.1997.8614. [DOI] [PubMed] [Google Scholar]
- 37.Boller K, Konig H, Sauter M, Mueller-Lantzsch N, Lower R, Lower J, et al. Evidence that HERV-K is the endogenous retrovirus sequence that codes for the human teratocarcinoma-derived retrovirus HTDV. Virology. 1993;196:349–53. doi: 10.1006/viro.1993.1487. [DOI] [PubMed] [Google Scholar]
- 38.Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, et al. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boller K, Schonfeld K, Lischer S, Fischer N, Hoffmann A, Kurth R, et al. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J Gen Virol. 2008;89:567–72. doi: 10.1099/vir.0.83534-0. [DOI] [PubMed] [Google Scholar]
- 40.Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007;3:e10. doi: 10.1371/journal.ppat.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang-Johanning F, Frost AR, Jian B, Epp L, Lu DW, Johanning GL. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene. 2003;22:1528–35. doi: 10.1038/sj.onc.1206241. [DOI] [PubMed] [Google Scholar]
- 42.Bieda K, Hoffmann A, Boller K. Phenotypic heterogeneity of human endogenous retrovirus particles produced by teratocarcinoma cell lines. J Gen Virol. 2001;82:591–6. doi: 10.1099/0022-1317-82-3-591. [DOI] [PubMed] [Google Scholar]
- 43.Kraus B, Fischer K, Buchner SM, Wels WS, Lower R, Sliva K, et al. Vaccination directed against the human endogenous retrovirus-K envelope protein inhibits tumor growth in a murine model system. PLoS One. 2013;8:e72756. doi: 10.1371/journal.pone.0072756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oricchio E, Sciamanna I, Beraldi R, Tolstonog GV, Schumann GG, Spadafora C. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene. 2007;26:4226–33. doi: 10.1038/sj.onc.1210214. [DOI] [PubMed] [Google Scholar]
- 45.Schiavetti F, Thonnard J, Colau D, Boon T, Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 2002;62:5510–6. [PubMed] [Google Scholar]
- 46.Chapuis AG, Thompson JA, Margolin KA, Rodmyre R, Lai IP, Dowdy K, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci U S A. 2012;109:4592–7. doi: 10.1073/pnas.1113748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.