

Abstract
Democratization of genomics technologies has enabled the rapid determination of genotypes. More recently the democratization of comprehensive proteomics technologies is enabling the determination of the cellular phenotype and the molecular events that define its dynamic state. Core proteomic technologies include mass spectrometry to define protein sequence, protein:protein interactions, and protein post-translational modifications. Key enabling technologies for proteomics are bioinformatic pipelines to identify, quantitate, and summarize these events. The Trans-Proteomics Pipeline (TPP) is a robust open-source standardized data processing pipeline for large-scale reproducible quantitative mass spectrometry proteomics. It supports all major operating systems and instrument vendors via open data formats. Here we provide a review of the overall proteomics workflow supported by the TPP, its major tools, and how it can be used in its various modes from desktop to cloud computing. We describe new features for the TPP, including data visualization functionality. We conclude by describing some common perils that affect the analysis of tandem mass spectrometry datasets, as well as some major upcoming features.
Keywords: bioinformatics, mass spectrometry
1. Introduction
Mass spectrometry-based proteomics has become the most widely used technique for the molecular characterization of large numbers of proteins in complex samples and the determination of their relative abundances. Several different workflows have been developed that encompass both data-dependent and data-independent analyses. Some, such as selected reaction monitoring (SRM), are tuned for high reproducibility and sensitivity on a relatively small number of targets per run. Other workflows, such as shotgun proteomics, are able to provide quantitative measurements on many thousands of proteins per run, although generally with lower sensitivity and specificity.
Informatics analysis of the output of mass spectrometers is a crucial element of the interpretation of the results. For SRM workflows, the individual transitions must be measured from the chromatogram peak traces and then grouped to yield accurate abundance and confident detection of each targeted peptide. Statistical methods can provide confidence metrics that the targeted peptide ion has truly been detected [1], as well as abundance uncertainty measures. Similarly for shotgun proteomics, the interpretation of thousands or millions of mass spectra is a multi-step process that usually includes statistical validation to provide identification and abundance confidence metrics.
Mass spectrometry-based proteomics technologies are dominant in both the academic research community and industrial research and development settings. However, it is only just gaining momentum for clinical use. SRM is routinely used for clinical verification and validation of biomarkers and more recently used in CLIA approved clinical assays for cancer [2–4]. Even shotgun proteomics is beginning to see clinical applications such as for the diagnosis of amyloidosis [5]. However, as instrumentation becomes faster and more sensitive, and analysis software becomes more powerful and easy to use, the application of MS proteomics in clinical settings will accelerate.
Although there are a tremendous number of individual tools for processing proteomics data [6] and some developed as pipelines [7, 8], the Trans-Proteomic Pipeline (TPP; [9, 10]) is the most widely used free and open source complete suite of tools for processing shotgun proteomics data with statistical validation from start to finish for typical workflows [11]. It encompasses initial conversion to open formats, sequence database- and spectral library-based searching, statistical post-validation of the search results, quantification information extraction, protein inference, and graphical exploration of the results. The TPP can be used on all major computing platforms and scales from the desktop to large computing clusters on the cloud for enterprise operation. It is embedded in many other applications and LIMS packages, which use the TPP as the core set of tools for data processing, such as Progenesis QI (Nonlinear Dynamics, Waters, USA). The TPP does not support analysis of data from SRM workflows, but the free and open source tool Skyline [12] is the leading software for such data.
In this review we will provide an overview of the many features of the TPP and discuss their implications. The features include tools that have been present the development of the TPP as well as new features that are now available in the latest 4.8 release of the TPP. Some of these advances are algorithmic, and others are visualization features. We will then discuss the clinical applications in biomarker discovery, identification of clinically relevant post-translational modifications by the TPP, and a few perils relevant to interpreting these kinds of data, and conclude with future directions for the software.
2. Basic Workflow
The overall basic steps of the workflow of shotgun proteomics have not changed significantly since we described them previously [11]. The general process of data analysis follows the steps of raw data conversion, identification of the spectra, validation and modeling of the identification step, abundance measurements, protein inference, storage of the data in local database systems, and then final deposition into public data repositories. However, the maturity of the tools has improved markedly in the past few years, enabling a more thorough analysis of data than was previously possible. In the following sections we describe each of these steps in the workflow, and how recent advances have made each step more robust and effective. Figure 1 displays the main set of TPP tools underpinned by standard formats.
Figure 1.
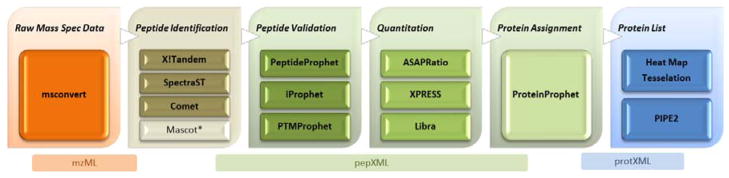
The TPP provides tools for format conversion, spectrum identification, search result validation, protein assignment and quantification. It allows for the use of multiple algorithms at any step where they exist (e.g., Comet, X!Tandem, Mascot or SpectraST for database search). Currently each of these steps is run directly by the user using either the TPP Petunia graphical user interface or via command-line interface scripts.
Each of the instrument vendors has their own format for storing the raw data generated by the mass spectrometers. These formats have evolved in subtle ways as new technologies have emerged, but remain basically similar. The msconvert tool within the free and open source ProteoWizard [13, 14] toolkit remains the standard for converting each of the vendor formats into the Proteomics Standards Initiative (PSI; [15]) mzML format [16]. This is accomplished via the use of DLLs (dynamic link libraries) obtained directly from the vendors. These DLLs, which contain the software necessary to read the vendor formats, are not open source, but may be used by any other software to access data in those files. Thus, msconvert has no code to read the individual vendor formats directly, but rather uses the vendor-supplied DLLs for accessing the data indirectly. This has the advantage that ProteoWizard does not have to keep up with vendor format changes, but rather just ensure that the vendors continue to supply up-to-date DLLs. Beyond the msconvert tool, any software that uses the ProteoWizard library is able to read the vendor files directly. However, the significant disadvantage is that the vendor DLLs are only available under the Microsoft Windows OS. Therefore, under most workflows, conversion with msconvert must happen on a Windows computer, but then all subsequent analysis may happen on any platform. Although not a TPP-developed tool per se, msconvert comes bundled with the TPP and is accessible via the GUI, and it is therefore easy for TPP users to convert their raw data files to mzML, on which the rest of the TPP tools depend. The full landscape of formats commonly used in MS-based proteomics has been recently reviewed [17].
The next step in data processing is the interpretation or “searching” of all mass spectra in a dataset. There are 3 broad approaches to searching mass spectra. Sequence searching is the most common approach, wherein each spectrum is normalized and scored against a set of theoretically generated spectra selected from a set of candidate peptide sequences extracted from a FASTA-formatted protein list. The spectral library searching approach matches each new spectrum against spectra selected from a reference file of previously observed and identified spectra. The third approach is de novo searching, in which one attempts to derive the peptide sequence by measuring the m/z values of individual peaks and intervals between peaks to infer the peptide sequence directly, without the use of a reference; this is typically only possible with spectra of extraordinary quality. Some software tools combine some of the approaches as well. The TPP is now packaged with two open-source sequence search engines, X!Tandem [18] with the k-score plugin [19], and Comet [20].
There are many other sequence search engines [21], and most of the popular ones are supported by the TPP tools in downstream validation and processing, but are not bundled with the TPP itself. The TPP tool SpectraST [22] is a highly advanced spectral library searching tool, which is also capable of building spectral libraries [23]. There is currently no support for de novo searching in the TPP, but since modern mass spectrometers coming into common use are now capable of generating spectra of sufficient quality for de novo sequence, support for this approach will soon follow.
A crucial set of components of the TPP beyond the software tools themselves are the common data formats that allow the TPP tools to interoperate efficiently. The pepXML and protXML formats [9] were developed 10 years ago to allow efficient exchange of data among TPP tools. They have never become official standards, but have become de facto standards supported by many tools. Some of the search engines supported by the TPP write their results in pepXML directly. However, for others there is a software utility in the TPP that can convert the native output of the search engine into pepXML, so that it may be fed into the rest of the TPP tools.
A hallmark of these search tools is that they will produce a best-match result for each spectrum with a corresponding score, but many of these best matches are incorrect. The key aspect then of the TPP that sets it apart from many other solutions is the tools that can develop mixture models to discriminate between correct and incorrect identifications, and importantly, assign probabilities of being correct to each result. The primary tool is PeptideProphet [24], which works directly with the search engine output. It models the output scores of each peptide-spectrum match (PSM) along with other metrics such as m/z difference to assign each PSM a probability that it belongs to the population of correct identifications.
We have recently developed some additional modeling tools that refine the models and probabilities derived from PeptideProphet. The iProphet tool [25] takes one or more pepXML files from PeptideProphet and refines the probabilities based on many lines of corroborating evidence. For example, in cases where multiple search engines have identified the same PSM, where a peptide has been identified in multiple charge states, or where a peptide has been identified with different mass modification configurations, the confidence is higher that each sibling PSM is correct. Each dataset is modeled independently and therefore each of these aspects will have a different effect on improving or degrading each probability.
Another new tool in the TPP suite is PTMProphet [26], which is designed to model the confidence with which mass modifications are correctly localized for each peptide. All of the popular search engines can identify that mass modifications are present for a peptide, but it is difficult to know the confidence with which the assignments are made. PTMProphet considers all of the possible configurations, and applies a statistical model to predict which modification sites are most probable based on the spectrum evidence.
For most experiments it is very important to be able to quantify the relative peptide and protein abundances among the different conditions. This can be accomplished either via labeling of the different conditions or quantifying the number of ions observed without the aid of a label. The TPP includes several software tools to aid in quantitation of peptide ions and proteins. The Libra [27] tool is used for data from samples that have been labeled with an isobaric label such as TMT or iTRAQ [28]. For isotopically labeled samples, when using such labels as ICAT [29], SILAC [30], and N14/N15, the ASAPRatio [31] tool or XPRESS tool can be used. The XPRESS tool can also be used to measure ion current abundances in label-free data, and ProteinProphet output is used for spectral counting quantification.
These tools first derive a quantitative measure of ion intensity for each PSM along with an uncertainty estimate. ASAPRatio is able to derive intensity measures for other sibling ions of different charge. These PSM intensities are then aggregated to yield a peptide-level relative abundance measures along with combined uncertainties. The tools then use the weights apportioned to the peptides by ProteinProphet to contribute to the final protein relative abundance and corresponding uncertainty or p-value. Only peptides with a weight greater than 0.5 contribute to a protein abundance. The measured peptide abundances could potentially be used to infer from which protein they are derived in cases of ambiguity, but such logic is planned for future versions of these quantitation tools.
Once peptides have been identified with statistical metrics of confidence, the next step is to apply a protein inference algorithm. Since many peptides can map to multiple proteins, it can be difficult to know from which protein the peptides have been detected. Although some researchers report the maximal list of proteins to which the peptides map, it is becoming accepted to report a parsimonious list of proteins, i.e. the minimum number of proteins that are required to explain the observed peptides. The ProteinProphet [32] software tool pioneered this approach and with periodic improvements, continues to be the state-of-the-art in protein inference. Each protein with mapping peptides is assigned a probability that it is present in the sample, based on the combination of individual peptide probabilities derived from previous analysis by PeptideProphet and iProphet. The proteins are grouped together based on the peptides they share. With the developments in mass spectrometry over the recent years, the use of high-resolution and high mass accuracy assignment of MS/MS spectra provides increased confidence on the results obtained and aids in the determination of correct spectral assignment of peptides.
The TPP itself does not provide data management functionality. It leaves data management and organization up to the user. On account of this, several groups have created proteomics data management systems that embed the TPP as their data processing infrastructure, on top of which they provide users with data organization and annotation functionality that suits their needs. Some examples of such management systems are SBEAMS [33], CPFP [34], CPAS [35], and YPED [36].
A desirable endpoint for most datasets acquired for research purposes is deposition in public data repositories. When this is performed, the data can be reused by other researchers and reanalyzed as part of concerted efforts to define MS-observable proteomes, such as PeptideAtlas [37, 38], PRIDE Cluster [39] and GPMdb [40]. When datasets are deposited in public data repositories, they can continue to contribute to the field by being incorporated into public resources. The ProteomeXchange [41] consortium of repositories is now in place so that datasets deposited to one repository can be propagated to the others or other interested projects when they are announced.
3. Significant Advantages and New Features
The TPP offers several significant advantages over the huge variety of software tools available for the processing of proteomics data. These features include modularity, support for common XML formats, robust modeling and statistic metrics, data visualization features, and platform independence. In the subsections below we describe these features in detail.
Rather than being a single monolithic tool, the TPP is composed of several tools that each performs one function well. This gives great flexibility to the user for processing data by stringing together several tools as is appropriate for the data that was generated. This makes the TPP amenable for use within workflow engines where the TPP tools can be combined with other tools within a workflow. The TPP has been adapted [26] to the Taverna [42] workflow platform, as well as others.
A key component to being modular and working well with workflow engines is the ability to easily pass data from one tool to another. In addition to the co-development and support for mzML described above, the pepXML and protXML formats [9] were specifically developed for TPP tools to interoperate efficiently. Based on XML, these formats allow complex data and metadata to be read as input for each tool and stored as output. The formats have continued to be updated to support the new features of the TPP since the original publication, and have become de facto standard formats for many tools beyond the TPP. The PSI, with participation from ISB, has developed a trio of new standard formats, mzIdentML [43], mzQuantML [44], and mzTab [45]. Since pepXML and protXML are specifically tailored to the TPP tools, and maintaining flexibility is important, it is unlikely that TPP tools will move away from pepXML and protXML for internal representation and exchange. However, TPP output can already be converted to mzIdentML, and efforts to export final TPP results to the other formats are underway. This enables easy deposition into public repositories and transfer of results to other tools.
One of the hallmarks of the TPP has been its statistical modeling such that global and individual confidence metrics can be assigned at many levels in the data. PeptideProphet derives a mixture model for the PSMs from a search engine and assigns probabilities to each PSM. The iProphet tool further refines this model using corroborating information from related PSMs to build a model at the distinct peptide sequence level, as well as refine the PSM-level results. ProteinProphet uses these individual probabilities as part of the protein inference described above to derive probabilities that each of the proteins have been detected by their constituent peptides. At each of these levels, decoy search results may be used to inform the models, or merely to verify the correctness of models derived without knowledge of the decoys. The ASAPRatio tool for isotopic labeling quantification and the Libra tool for isobaric labeling both calculate uncertainties for each of their abundance ratios so that users may easily determine which ratios are significant. The new PTMProphet tool calculates the probability that a detected mass modification is localized to a specific site, in cases where multiple sites are possible. The end result of application of these tools is a statistically sound set of models that enables users to report their results at confidence thresholds of their choice, along with confidence metrics for each datum.
Ensuring that results derived from the TPP remain accurate as development of the tools continues is crucial, and therefore test-driven software development is playing an increasing role for the TPP tools. For example the new TPP components that enable cloud computing for proteomics datasets incorporate standard unit testing mechanisms in the source code. We have also set up a suite of test datasets that are run through the TPP in an automated fashion after builds, and the results compared with a reference result. Deviations from the reference result are reported and examined by developers. If the new result is deemed an acceptable improvement, it becomes the new reference result. The infrastructure to allow a new installation to be verified by processing and comparing one or more of the test datasets is an upcoming feature.
4. Platforms
The TPP was originally conceived as a set of Linux-based command-line tools to be used for high throughput data analysis of proteomics data on the ISB Linux cluster. However, it was soon realized that there was great interest in a version of the TPP that could be used on Microsoft Windows-based computers as well and OS X platforms. Not only is Windows a very common platform, it became clear that for students in our proteomics courses to run tutorials, it would be best if Windows were supported. Therefore, the TPP has been written so that it can be compiled and run under Windows and OS X as well as Linux.
To enable easier access to the command-line tools, a web-based graphical user interface (GUI) was developed as front end to the command-line tools. Although a native desktop interface can provide a smoother interface than a web interface, developing desktop interfaces that are also platform neutral is quite difficult. The added benefit of a web interface is that it can be used locally on a single machine as well as remotely on any computer with a network connection. This platform neutrality has made the TPP and its tools usable both on a laptop as well as a large high throughput compute cluster. The single exception to complete neutrality is the conversion from proprietary vendor formats to mzML, as described above in section 2, since this relies on vendor-supplied Windows-only DLLs.
This design choice has also made the port to cloud computing relatively easy. The TPP has been ported to the Amazon.com Amazon Web Services (AWS). Two modes have been developed [46]. The first is one where a user with an AWS account can trivially launch an instance of a single Linux computer running the TPP on the cloud, connect to it via the web interface, and interact with it in the same manner as on a local computer. The second mode is to use a set of scripts to launch one or more AWS instances from the command line and automatically distribute larger computing jobs onto a cluster of AWS instances that expand and contract dynamically (and eventually shutdown completely) based on the computing demand.
5. Visualization
Although a web interface has disadvantages when trying to design a highly responsive GUI, recent advances in JavaScript libraries have opened many opportunities for GUI improvements and dynamic visualizations of the analysis results. Considerable recent effort has therefore gone into updating the TPP GUI, called Petunia, to make navigation and interpretation of the output of the tools easier.
A new “dashboard” view has been written to allow users to interactively explore the models that were derived by the various tools, replacing previous static images. The graphical plots depict ROC curves, mixture models, as well as numbers of identifications within various categories. This view allows the user to quickly assess the quality of the PeptideProphet and iProphet results, as well as the overall quality of their data.
The protXML Viewer has been completely rewritten to move from text-based presentation of results to a view that displays confidence metrics and quantification information in a graphical form. Individual protein group entries are collapsed by default so that high-level information is visible at first, but then may be expanded to reveal additional details. The interface has been tuned to perform quickly even with very large experiments, which were sometimes impossible to view with the previous interface.
In order to allow users to compare the relative abundances of proteins among different conditions, a heatmap viewer has been implemented. The heatmap Y-axis represents proteins, and the X-axis represents an unlimited number of different conditions. The abundance of each protein is displayed as a pixel of variable color. The user may zoom and pan through the visualization, with more information provided on mouse hover, and full information in a new window available with a mouse click. Figure 2 depicts an example of the heatmap view using a set of 13 conditions.
Figure 2.

A partial screenshot showing a self-organizing map shown as a heatmap. On the X-axis are the 13 experiments ordered by clustering results. Plotted across the Y-axis is the union of all proteins in these samples, also ordered by clustering results. The view is zoomed such that many more proteins are out of view on this screen shot. The colors of the boxes range from blue for low abundance to red for high abundance, in this case based on label-free spectral counting quantification. Gray boxes represent missing data.
To visualize the classifications of proteins, the TPP now implements a tessellation visualization interface, within which proteins are grouped according to a set of arbitrary categories and displayed with a shape size proportional to the abundance. If the proteins are given UniProtKB [47] identifiers, then Gene Ontology [48] classifications within the function, process, and location categories are shown. Several options allow customizations of the display, including tessellation shape and colors. Figure 3 depicts an example screenshot of a tessellation visualization of Gene Ontology function categories of the proteins in a sample dataset.
Figure 3.

A partial screenshot depicting proteins organized by Gene Ontology (GO) category with an example dataset. The size of each rectangle is relative to the abundance (spectral counts in this case) of each protein (inner rectangles) and GO annotation (outer rectangles). The colors are autogenerated. GO categories that have many proteins in them are shown as rectangles with many other rectangles inside. Rectangles with one or few inner rectangles represent GO categories with few associated proteins. The rectangles are ordered in order of relative abundance associated with each GO category. More information for each rectangle can be viewed via mouse over, with hyperlinks out to UniProtKB. The word “stress” is typed in the search term filter box, thereby shading yellow all GO terms that have the substring “stress” in them.
6. Clinical relevance
Discovery based proteomics is a driving component for biomarker discovery, functional protein modifications and cellular response to perturbations. Key to these applications in clinical proteomics is the both the depth of proteome coverage and the confidence in proteomic identification of each of the data sets involved. The use of robust tools such as the TPP provides the groundwork for the development of these datasets that enables clinically relevant biological proteomics to be collected, interpreted, consolidate and ultimately shared [49, 50].
Proteomic analysis of cellular response is a key aim of human disease research. However, proteomics also plays just as significant a role in plant, animal and environmental research as each of these also impact human health and well-being, Therefore, comprehensive discovery proteomics offers the means to measure biochemical impacts of genomic abnormalities, including expression of variant proteins encoded by gene mutations, protein copy number differences, gene amplification, deletion, silencing or changes in microRNA expression across many human studies as well as agricultural and environmental studies. Using the TPP in these studies provides a readily available system to link technically challenging mass spectrometry data collection to consolidated, statistically valid results for the biologist to understand.
With the full realization that proteins are the drivers of cellular response and ultimately define phenotypic characteristics, it is implicit that the tools required for proteomics readout must be sufficiently developed to perform basic data processing but also provide a high level of confidence in the results. In addition, quantitative analysis has now become the standard for proteomics, and although there are many technological approaches, the processed results must provide a degree of confidence in the data with respect to quantitative differences and consistency in identification from the data extraction. The current development of the TPP has focused directly on these aims for seamless and consistent data extraction, quantitative identification and statistical analysis of these results. In future releases of the TPP as detailed below, the integration of proteomics data with genomics data through the same simple pipeline will provide operator ease and increased single point data sharing greatly expand the relevance of proteomics data analysis in a multi-omics approach.
7. Data analysis perils
There are some limitations and potential perils when using the TPP tools that are worth highlighting. One is specific to the TPP, while others are generic to analysis of shotgun proteomics data. Avoiding them can avert situations where the processing results are misinterpreted and erroneous conclusions are made.
One fundamental assumption of the primary parametric model of PeptideProphet is there is both a population of correct identifications (positive distribution) and incorrect identifications (negative distribution) of sufficient size. If the population of incorrect results is removed, for example by filtering our all low scoring PSMs before processing with PeptideProphet, then the modeling results can be unpredictable, with the positive distribution modeled as the negative. Similarly, in cases where there is no positive distribution because all identifications are incorrect, the modeling may fail completely, or in some cases a positive model may be fit to a small population of the highest scoring incorrect identifications. It is therefore important to use the dashboard tool to view the modeling results to ensure that the models are reasonable. In cases where there are fewer than a few hundred PSMs with a high modeled probability, the results should be manually spot checked to ensure that the spectrum identifications are convincing.
A generic peril of shotgun proteomics is using an inappropriate database. The appropriate database for any sequence search should contain as many of the protein sequences that may well be present in the sample as is reasonable. Limiting the sequence database to not contain sequences that are known not to be present is a reasonable strategy to decrease search time and increase sensitivity. However, if sequences that correspond to some spectra in the sample are excluded, there is a significant risk that those spectra may match with the incorrect peptide sequence with a high score, or even with a correct peptide sequence that will be associated with the wrong species. Known common contaminants should always be included in the search database. A suitable database for these contaminants that can be customized to the user’s workflow is the cRAP database (http://www.thegpm.org/crap/). In cases where a sample contains multiple organisms of different species, none should be excluded simply because they are not of interest. Rather, all sequences should be presented to the search engine, modeling performed with the TPP tools, and only at the end should the identifications of no interest be discarded. A discussion of this issue along with past cases where this has led to incorrect conclusions has been previously presented in detail [51].
Another peril comes when combining multiple datasets after processing. For a single dataset, by using the models and probabilities calculated by the TPP tools, a user can set a threshold of their choosing, and the resulting FDR estimated by the model is usually quite accurate. However, if two or more datasets are subsequently merged after this processing, the FDR will be different than it was for the individual datasets, and likely higher in cases where the samples are similar. The reason for this is that the correct identifications tend to be similar among datasets that are merged, while the incorrect identifications scatter randomly across the proteome. Consider an example of two datasets that have been each applied a separate threshold to a 1% protein-level FDR and claim 1000 proteins, therefore yielding 990 correct proteins and 10 incorrect ones. If the two datasets are replicates and identify the same 990 correct proteins, but different incorrect ones (since they are random), then the merged FDR will be ~2% (20/1010). At the other extreme, if none of the proteins overlap at all, only then will the FDR still be 1% (20/2000). In most real world cases, the situation is much closer to the former than the latter. For further discussion of this and related topics, see Reiter et al. (2009) [52].
A final peril presented here is the problem of choosing insufficiently stringent thresholds for large datasets. The TPP tools are capable of calculating FDRs at the PSM level, distinct peptide sequence level, and the protein level. And in typical cases for large modern datasets, the protein-level FDR will be much larger than the peptide-level FDR, which in turn will be much larger than the PSM-level FDR. While a PSM-level FDR of 1% may sound reasonably small, and can be appropriate for small datasets, for a dataset that has 1 million PSMs above threshold, there will be 10,000 incorrect PSMs. Since these tend to scatter randomly across the proteome, it becomes easy to falsely cover a large fraction of the entire proteome. The remedy for this is to be far more stringent at the PSM threshold or otherwise insist on suitably stringent thresholds at the protein level. See Reiter et al. [52] and Farrah et al. [53] and references therein for further exploration of this topic.
8. Future work
The TPP is currently primarily designed for the analysis of shotgun proteomics datasets, but several new workflows seem promising to extract greater information from protein samples. The first development is the concept of using RNA-seq data to achieve an improved proteomics analysis. RNA-seq can potentially improve the proteomics workflow described in the sections above in four ways: 1) reduce the size of the sequence search database based on an assumption that proteins will only be detected where transcripts are also detected; 2) allow an improved sequence database that is customized to the sequence variants specific to the individual from which the sample was extracted; 3) aid in protein inference under the assumption that the presence of a particular protein will correlate with transcript presence; and 4) enable transcript abundance and protein abundance comparisons to gain better insight into the biological system. The TPP will soon support the full analysis of paired RNA-seq and proteomics datasets, both locally and using the AWS framework described above.
The second new technology that will soon be supported by the TPP is SWATH-MS [54] data analysis. In the SWATH-MS workflow, MS2 fragmentation spectra are collected using wide ~25 m/z isolation windows rather than the typical ~2 m/z isolation windows; further, the instrument steps across nearly all precursor masses within a cycle lasting a few seconds. This has the advantage that fragmentation spectra are collected for nearly all precursor ions every few seconds to create a complete record of all peptide ions in each sample. The disadvantage is that all MS2 spectra are quite highly multiplexed, rendering traditional search strategies unusable, and therefore requiring new processing techniques. However, these new search techniques are being developed, the results of which will be compatible with downstream TPP tools and therefore many of the benefits described above.
9. Conclusion
The Trans-Proteomic Pipeline release 4.8 is a mature and robust system that is ready for deployment in clinical settings. It can be deployed on any of the major operating systems, is fully usable in single desktop configurations as well as with compute clusters. The TPP now fully supports cloud computing using the AWS infrastructure. It has an advanced web-based graphical user interface and many client-side, interactive visualization features for exploration of data. Yet, since it primarily consists of command-line tools, it is completely amenable to scripting, automation, and embedding within custom data management systems. And perhaps best, it is fully open source with a broad user base, and can thus be built upon or modified to suit a variety of needs. Development continues on this system and several advanced new features are being finished for upcoming releases. Additional information, documentation, and downloads are available at http://tools.proteomecenter.org/TPP.
Acknowledgments
This work was funded in part by the American Recovery and Reinvestment Act (ARRA) funds through National Institutes of Health from the NHGRI grant No. RC2 HG005805; the NIGMS, under grant No’s. R01 GM087221 and 2P50 GM076547 Center for Systems Biology, the National Institute of Biomedical Imaging and Bioengineering grant No. U54EB020406, the National Science Foundation MRI Grant No. 0923536, and from EU FP7 grant ‘ProteomeXchange’ grant number 260558.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Reiter L, Rinner O, Picotti P, Huttenhain R, et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods. 2011;8:430–435. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- 2.Li XJ, Hayward C, Fong PY, Dominguez M, et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Science translational medicine. 2013;5:207ra142. doi: 10.1126/scitranslmed.3007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boja ES, Rodriguez H. Regulatory considerations for clinical mass spectrometry: multiple reaction monitoring. Clinics in laboratory medicine. 2011;31:443–453. doi: 10.1016/j.cll.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Boja ES, Fehniger TE, Baker MS, Marko-Varga G, Rodriguez H. Analytical Validation Considerations of Multiplex Mass Spectrometry-based Proteomic Platforms for Measuring Protein Biomarkers. J Proteome Res. 2014 doi: 10.1021/pr500753r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theis JD, Dasari S, Vrana JA, Kurtin PJ, Dogan A. Shotgun-proteomics-based clinical testing for diagnosis and classification of amyloidosis. Journal of mass spectrometry : JMS. 2013;48:1067–1077. doi: 10.1002/jms.3264. [DOI] [PubMed] [Google Scholar]
- 6.Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73:2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 8.Kohlbacher O, Reinert K, Gropl C, Lange E, et al. TOPP--the OpenMS proteomics pipeline. Bioinformatics. 2007;23:e191–197. doi: 10.1093/bioinformatics/btl299. [DOI] [PubMed] [Google Scholar]
- 9.Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005.0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deutsch EW, Lam H, Aebersold R. Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics. Physiol Genomics. 2008;33:18–25. doi: 10.1152/physiolgenomics.00298.2007. [DOI] [PubMed] [Google Scholar]
- 12.MacLean B, Tomazela DM, Shulman N, Chambers M, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chambers MC, Maclean B, Burke R, Amodei D, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deutsch EW, Albar JP, Binz P-A, Eisenacher M, et al. Development of Data Representation Standards by the Human Proteome Organization Proteomics Standards Initiative. JAMIA. 2014 doi: 10.1093/jamia/ocv001. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martens L, Chambers M, Sturm M, Kessner D, et al. mzML--a community standard for mass spectrometry data. Mol Cell Proteomics. 2011;10:R110 000133. doi: 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deutsch EW. File formats commonly used in mass spectrometry proteomics. Mol Cell Proteomics. 2012;11:1612–1621. doi: 10.1074/mcp.R112.019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 19.MacLean B, Eng JK, Beavis RC, McIntosh M. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics. 2006;22:2830–2832. doi: 10.1093/bioinformatics/btl379. [DOI] [PubMed] [Google Scholar]
- 20.Eng JK, Jahan TA, Hoopmann MR. Comet: an open source tandem mass spectrometry sequence database search tool. Proteomics. 2012 doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 21.Eng JK, Searle BC, Clauser KR, Tabb DL. A face in the crowd: recognizing peptides through database search. Mol Cell Proteomics. 2011;10:R111 009522. doi: 10.1074/mcp.R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam H, Deutsch EW, Eddes JS, Eng JK, et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics. 2007;7:655–667. doi: 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- 23.Lam H, Deutsch EW, Eddes JS, Eng JK, et al. Building consensus spectral libraries for peptide identification in proteomics. Nat Methods. 2008;5:873–875. doi: 10.1038/nmeth.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 25.Shteynberg D, Deutsch EW, Lam H, Eng JK, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics. 2011;10:M111 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shteynberg D, Mendoza L, Sun Z, Moritz RL, Deutsch EW. PTMProphet: statistical analysis of post-translational modification localization for shotgun proteomics datasets. 2014 in preparation. [Google Scholar]
- 27.Pedrioli PG, Raught B, Zhang XD, Rogers R, et al. Automated identification of SUMOylation sites using mass spectrometry and SUMmOn pattern recognition software. Nat Methods. 2006;3:533–539. doi: 10.1038/nmeth891. [DOI] [PubMed] [Google Scholar]
- 28.Zieske LR. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J Exp Bot. 2006;57:1501–1508. doi: 10.1093/jxb/erj168. [DOI] [PubMed] [Google Scholar]
- 29.Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 30.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.Li XJ, Zhang H, Ranish JA, Aebersold R. Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal Chem. 2003;75:6648–6657. doi: 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- 32.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 33.Marzolf B, Deutsch EW, Moss P, Campbell D, et al. SBEAMS-Microarray: database software supporting genomic expression analyses for systems biology. BMC Bioinformatics. 2006;7:286. doi: 10.1186/1471-2105-7-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trudgian DC, Mirzaei H. Cloud CPFP: a shotgun proteomics data analysis pipeline using cloud and high performance computing. J Proteome Res. 2012;11:6282–6290. doi: 10.1021/pr300694b. [DOI] [PubMed] [Google Scholar]
- 35.Rauch A, Bellew M, Eng J, Fitzgibbon M, et al. Computational Proteomics Analysis System (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J Proteome Res. 2006;5:112–121. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- 36.Shifman MA, Li Y, Colangelo CM, Stone KL, et al. YPED: a web-accessible database system for protein expression analysis. J Proteome Res. 2007;6:4019–4024. doi: 10.1021/pr070325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9:429–434. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farrah T, Deutsch EW, Hoopmann MR, Hallows JL, et al. The state of the human proteome in 2012 as viewed through PeptideAtlas. J Proteome Res. 2013;12:162–171. doi: 10.1021/pr301012j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griss J, Foster JM, Hermjakob H, Vizcaino JA. PRIDE Cluster: building a consensus of proteomics data. Nat Methods. 2013;10:95–96. doi: 10.1038/nmeth.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 41.Vizcaino JA, Deutsch EW, Wang R, Csordas A, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolstencroft K, Haines R, Fellows D, Williams A, et al. The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud. Nucleic Acids Res. 2013;41:W557–561. doi: 10.1093/nar/gkt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones AR, Eisenacher M, Mayer G, Kohlbacher O, et al. The mzIdentML data standard for mass spectrometry-based proteomics results. Mol Cell Proteomics. 2012;11:M111 014381. doi: 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walzer M, Qi D, Mayer G, Uszkoreit J, et al. The mzQuantML Data Standard for Mass Spectrometry-based Quantitative Studies in Proteomics. Mol Cell Proteomics. 2013;12:2332–2340. doi: 10.1074/mcp.O113.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griss J, Jones AR, Sachsenberg T, Walzer M, et al. The mzTab Data Exchange Format: communicating MS-based proteomics and metabolomics experimental results to a wider audience. Mol Cell Proteomics. 2014 doi: 10.1074/mcp.O113.036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slagel J, Mendoza L, Shteynberg D, Deutsch EW, Moritz RL. Processing shotgun proteomics data on the Amazon Cloud with the Trans-Proteomic Pipeline. Mol Cell Proteomics. 2014 doi: 10.1074/mcp.O114.043380. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Apweiler R, Bairoch A, Wu CH, Barker WC, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashburner M, Ball CA, Blake JA, Botstein D, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellis MJ, Gillette M, Carr SA, Paulovich AG, et al. Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer discovery. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang B, Wang J, Wang X, Zhu J, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knudsen GM, Chalkley RJ. The effect of using an inappropriate protein database for proteomic data analysis. PloS one. 2011;6:e20873. doi: 10.1371/journal.pone.0020873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reiter L, Claassen M, Schrimpf SP, Jovanovic M, et al. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2405–2417. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farrah T, Deutsch EW, Omenn GS, Campbell DS, et al. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10:M110 006353. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gillet LC, Navarro P, Tate S, Rost H, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111 016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]