

Abstract
Pro-inflammatory activation of vascular endothelium leading to increased surface expression of adhesion molecules and neutrophil (PMN) sequestration and subsequent activation is paramount in the development of acute lung (ALI) and organ injury in injured patients. We hypothesize that α-enolase, which accumulates in injured patients primes PMNs and causes pro-inflammatory activation of endothelial cells leading to PMN-mediated cytotoxicity.
Methods
Proteomic analyses of field plasma samples from injured vs. healthy patients was used for protein identification. Human pulmonary microvascular endothelial cells (HMVECs) were incubated with α-enolase or thrombin, and ICAM-1 surface expression was measured by flow cytometry. A two-event in vitro model of PMN cytotoxicity HMVECs activated with α-enolase, thrombin, or buffer was used as targets for lysophosphatidylcholine-primed or buffer-treated PMNs. The PMN priming activity of α-enolase was completed, and lysates from both PMNs and HMVECs were immunoblotted for protease activated receptor-1 (PAR-1) and PAR-2 and co-precipitation of α-enolase with PAR-2 and plasminogen/plasmin.
Results
α-enolase increased 10.8-fold in injured patients (p<0.05). Thrombin and α-enolase significantly increased ICAM-1 surface expression on HMVECs, which was inhibited by anti-proteases, induced PMN adherence, and served as the first event in the two-event model of PMN cytotoxicity. α-enolase co-precipitated with PAR-2 and plasminogen/plasmin on HMVECs and PMNs and induced PMN priming, which was inhibited by tranexamic acid, and enzymatic activity was not required. We conclude that α-enolase increases post-injury and may activate pulmonary endothelial cells and prime PMNs through plasmin activity and PAR-2 activation. Such pro-inflammatory endothelial activation may predispose to PMN-mediated organ injury.
Keywords: Organ injury, proteomics, red blood cells, transfusion, plasma, serine proteases
Introduction
Pro-inflammatory activation of vascular endothelium results in the release of chemokines and the increased surface expression of adhesion molecules including intercellular adhesion molecule-1 (ICAM-1) in the endothelial beds of end-organs, which may predispose individuals to PMN-mediated organ injury (1;2). Such endothelial pro-inflammatory activation results in PMN sequestration in the lung and in other organs, including the liver (1;2). Vascular chemokine release induces phenotypic alterations of neutrophils (PMNs) in the lung capillaries such that they become captured, via selectin-mediated interactions with the pulmonary endothelium, and then firmly adherent through binding of the PMN β2-integrin CD11b/CD18 and ICAM-1 (2). These sequestered, primed PMNs are hyper-reactive such that mediators that normally do not induce activation of the microbicidal arsenal cause activation of the respiratory burst and release of proteins from granules leading to PMN-mediated endothelial cell damage, capillary leak, and organ damage (3;4).
Although a life-saving intervention, the transfusion of stored red blood cells (RBCs) has been linked to increased morbidity and mortality in the critically ill (5;6). Injured patients, irrespective of the injury mechanism, are prone to ALI and multiple organ failure (MOF), which are thought to be the result of at least two clinical insults (5;6). In a prospective clinical study, controlled for the amount transfused, transfusion of stored RBCs, versus fresh, was an independent predictor for the development of ALI/MOF in injured patients (5;6). Although some of the pro-inflammatory mediators responsible for ALI/MOF have been identified, others have remained elusive and potential candidates include intracellular proteins that have protease activity similar to thrombin and accumulate during routine storage (7). Thrombin is a multifunctional serine protease involved in hemostasis, fibrinolysis, and pro-inflammatory activation of innate immunity through stimulation of proteinase-activated receptors (PARs), specifically PAR-1, PAR-3, and PAR-4, through cleavage of the N-terminal exodomain of the receptor, releasing the tethered ligand (8). Neutrophils (PMNs) and endothelial cells are reported to express PAR-2; however, thrombin does not activate the PAR-2 receptor, although other serine proteases, including plasmin, have this capacity (9;10).
α-enolase is an intracellular glycolytic lyase, phosphopyruvate hydratase, which converts 2-phospho-D-glycerate to phosphoenolypyruvate (11;12). It is also ubiquitously expressed on the surface of many cells including neutrophils, T cells, B cells, monocytes, and vascular endothelium and serves as a plasminogen receptor as well as being involved in the conversion of plasminogen to plasmin (11–13). α-enolase has been implicated in many pro-inflammatory states including malignancy, vasculitis, inflammatory bowel disease, and anti-centrosome/ANCA-related conditions (12). Furthermore, α-enolase-mediated increases in plasmin production on the surface of monocytes lead to migration and invasion of these cells into the inflamed lung (14). Because of its linkage to plasmin generation and its ability to activate intracellular signaling cascades, we hypothesize that α-enolase, which accumulates in injured patients, primes PMNs and causes pro-inflammatory activation of endothelial cells leading to PMN-mediated cytotoxicity.
Materials and Methods
All chemicals, unless otherwise specified, were obtained from Sigma-Aldrich (St. Louis, MO). All solutions and buffers were made from sterile water for human injection, United States Pharmacopeia (USP) purchased from Baxter Healthcare (Deerfield, NY), followed by sterile filtering with Nalgene MF75 series disposable sterilization filter units purchased from Fisher Scientific (Pittsburgh, PA). Human and yeast (enzymatically inactive) α-enolase were purchased from US Biological (Swampscott, MA) and Sigma-Aldrich. Antibodies to PAR-1 and PAR-2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Human pulmonary microvascular endothelial cells, media, and all related reagents were obtained from Lonza Corporation (San Diego, CA). A fluorescein isothiocyanate-linked antibody (clone YN1/1.7.4) to ICAM-1/CD54 was purchased from Beckman Coulter (Brea, CA). Tranexamic acid was purchased from the Pharmacy at University of Colorado Hospital, Aurora, CO.
Proteomics
Proteomics on the field blood (plasma) from 2 patients with blunt trauma who later developed ALI/MOF and the plasma from 4 healthy control donors were completed using 2-dimensional gel electrophoresis and mass spectroscopy with computer analyses of the resultant peptides specific for α-enolase (7).
ICAM-1 Surface Expression
Human pulmonary microvascular endothelial cells (HMVECs) activation was assessed as previously described (4). Briefly, HMVECs were grown to 90% confluence in 12-well plates. The cells were then incubated for 6 hours with α-enolase (0.1–50 μg/mL) ± 10% human plasma with 10 U/ml of heparin, thrombin (0.1–10 U/ml), or media control. The cells were then washed and incubated with a FITC-labeled monoclonal antibody to ICAM-1. ICAM-1 expression was measured by flow cytometry. Inhibition of thrombin-induced and α-enolase-induced ICAM-1 expression was determined via pre-incubation with leupeptin [1 mM] and benzenesulfonyl fluoride hydrochloride (AEBSF) [2.5 mM].
A Two-Event in vitro model of PMN-Mediated Cytotoxicity
A two-event in vitro model of PMN-mediated pulmonary endothelial injury was performed as previously described (4). HMVECs were incubated with α-enolase (50 μg/mL) or thrombin (5 U/ml) for 6 hours followed by the addition of PMNs. PMN adherence to the activated HMVECS was measured by myeloperoxidase content in the lysate (% MPO) (4). An in vitro two-event model of ALI was demonstrated by the addition of lysophosphatidylcholines (lyso-PCs) to activate the adherent PMNs and the number of viable HMVECs/mm2 was counted by microscopy as described (4).
PMN isolation and priming assays
PMNs were isolated from heparinized whole blood drawn after informed consent was obtained from healthy donors under a protocol approved by the Colorado Multiple Institutional Review Board at the University Of Colorado Denver School Of Medicine, as previously described (15). Briefly, isolated PMNs (3.75 × 106 cells) were incubated with buffer (Krebs-Ringers phosphate with 2% dextrose (KRPD) controls, thrombin (1–10 U/ml), α-enolase (1–50 μg/mL) ± 10% plasma and 10 U/ml of heparin, for 30 minutes at 37°C. PMNs were activated with 1 μM formyl-Met-Leu-Phe (fMLF), and the maximal rate of superoxide dismutase-inhibitable superoxide anion production was measured by the reduction of cytochrome c at 550 nm (15). Inhibition of PMN priming with α-enolase or thrombin was determined by the addition of 2.5 mM AEBSF, an antibody to plasminogen [1μg/ml], or 10–100 μg/ml of tranexamic acid to the reaction mixture in selected experiments.
PAR Immunoreactivity and co-precipitation with α-enolase
The proteins from HMVECs and PMNs were separated by SDS-PAGE and transferred to nitrocellulose. The membranes were then immunoblotted with antibodies to PAR-1 and PAR-2 as previously described (16).
Statistics
Statistics were calculated using the GB Stat 2.0. Repeat ANOVA was performed followed by Neuman-Keuls post-hoc analysis, which was dependent upon the equality of variance. Statistical significance was determined at the p<0.05 level (7).
Results
Proteomics
Proteins identified in two patients’ plasma drawn in the field prior to the initiation of resuscitation post blunt-trauma were compared to the plasma of healthy controls (4) employing the identical anticoagulant, ethylenediaminetetraacetic acid (EDTA). The two patients were female and suffered blunt trauma with injury severity scores (ISS) of 35 and 43, base deficits of −10 and −6 mEq/L, and lysis at 30 minutes after achieving maximal amplitude (Ly30) of 0 on thromboelastography, respectively. Both patients developed multiple organ failure. α-enolase increased by 15.5±2.8-fold in the injured patients vs. healthy controls as well as a number of other proteins vs. healthy control plasmas (n=4; p<0.05) (Table 1). In addition, previously published data demonstrated that α-enolase increased in the supernatant of stored (day 42) vs. fresh (day 1) RBC units demonstrated an increase in α-enolase by 4.4-fold (n=5, p<0.005) as well as increasing to concentrations as high as 1.5 μg/ml in severely injured patients with ISS≥35 (7;17).
Table 1.
Selected plasma proteins that increase with injury (field plasma) based on proteomic analysis
| Protein | Fold-increase over normal plasma (p<0.05) |
|---|---|
| Von Willebrand factor | 19.9±4.6 |
| Heparin Cofactor 2 | 2.0±0.1 |
| Actin | 21.9±6.9 |
| α-enolase | 15.5±2.8 |
| Glyceraldehyde-3-phosphate dehydrogenase | 17.5±0.5 |
| Fetuin B | 9.5±0.5 |
| α-actinin-1 | 3.0±0.1 |
ICAM-1 Surface Expression
As compared to media-treated controls, thrombin increased ICAM-1 surface expression on HMVECs by 2.1±0.3 fold and 1.8±0.4 fold at concentrations of 5 U/ml and 10 U/ml, respectively (n=5, p<0.05) (Fig. 1). At 50 μg/ml α-enolase also increased ICAM-1 surface expression by 2.8±0.2-fold vs. media-treated controls (n=5 p<0.05) (Fig. 1). The positive control LPS [2μg/ml] induced a 2.5±0.3-fold increase in ICAM-1 surface expression (n=5 p<0.05) (Fig. 1). Both α-enolase- and thrombin-induced surface expression of ICAM-1 was inhibited by pretreatment with protease inhibitors: AEBSF and leupeptin (60±8% and 73±7% for AEBSF and 54±5% and 62±8% for leupeptin, respectively). Furthermore, if FBS was excluded from the HMVEC media, α-enolase did not elicit an increase in ICAM-1 surface expression vs. media-treated HMVECs: Media: 450±64 vs. α-enolase 462±81MFI (n=3). However, because these data rely on the recruitment of bovine plasminogen from FBS, analogous experiments were completed with the addition of 10% human plasma with 10 U/ml of heparin. In these experiments α-enolase at 1 μg/ml elicited a 1.78±0.2-fold increase in ICAM-1 vs. media-treated control HMVECs (p<0.05, n=3).
Figure 1. α-enolase increases the surface expression of ICAM-1 on HMVECs.

The mean fluorescent intensity (MFI) of intercellular adhesion molecule-1 (ICAM-1) surface expression is depicted as a function of treatment group. As compared to media-treated control HMVECs, the positive control endotoxin (LPS [2μg/ml]) caused significantly increased ICAM-1 surface expression on HMVECs on both panels. Panel A demonstrates the effect of α-enolase on ICAM-1 surface expression which was significantly increased at 50 μg/ml (*=p<0.05 vs. media-treated HMVECs, n=5). Panel B shows the effect of thrombin stimulation on the surface expression of ICAM-1 on HMVECs. In comparison to media-treated controls thrombin significantly increased ICAM-1 surface expression at 5–10 U/ml (*=p<0.05 vs. media treated HMVECs, n=5).
Two-event Model of PMN-mediated HMVEC cytotoxicity
Along with increased ICAM-1 expression, α-enolase [50 μg/ml], thrombin [5 U/ml], and the positive control LPS [2 μg/ml] induced significant PMN adhesion compared to media control: Media: 3.1±1.5%, α-enolase [50 μg/ml]: 14.4±4.7%*, thrombin 5 U/ml: 18.8±7.5%*, LPS [2 μg/ml]: 35.5±0.7* (n=5, *=p<0.05). When lysophosphatidylcholines (lyso-PCs), PMN priming agents shown to precipitate PMN cytotoxicity in vitro and in vivo, were added to HMVECs which were activated with α-enolase, thrombin or LPS and co-cultured with PMNs the number of viable HMVEC was significantly reduced (Fig. 2 last bar in each groups) (3;4). Importantly, the addition of PMNs or lyso-PCs alone did not affect the number of viable HMVECs in any treatment group (Fig. 2 or for HMVECs activated with LPS and treated with lyso-PCs data not shown) (3;4).
Figure 2. Thrombin and α-enolase serve as the first event in a two-event model of PMN-mediated HMVEC cytotoxicity.

The number of viable HMVECs/mm2 is shown as a function of treatment group. Media treated controls are the solid bar in the left group. Treatment alone with endotoxin (LPS [2μg/ml]), thrombin, α-enolase, lysophosphatidylcholines (LPCs, a PMN priming agent), or PMNs did not cause significant HMVEC toxicity. In addition HMVECs activated with LPS, thrombin, or α-enolase and co-cultured with PMNs did not demonstrate a significant decrease in viable HMVECs. However, HMVECs activated with LPS, thrombin, or α-enolase, co-cultured with PMNs and then incubated with LPCs demonstrated significant decrease in viable HMVECs (last bar on the right in each group) (*=p<0.05 vs. media-treated HMVECs, n=5).
PMN Priming with Thrombin and α-enolase
PMNs incubated with α-enolase [10–50 μg/ml] significantly primed the fMLF-activated respiratory burst when incubated with 10% plasma for 30 minutes at 37°C compared to PMNs incubated with 10% plasma alone (p<0.05, n=7) (Fig. 3, panel A). At lesser concentrations [1–5 μg/ml] α-enolase did not cause significant PMN priming nor did shorter incubation times of 5–10 min. Moreover, α-enolase priming of the PMN oxidase was abrogated when the plasma was omitted (buffer control 1.5±0.4 vs. α-enolase without plasma 1.5±0.6, n=4), by an antibody to plasminogen was included in the reaction mixture, or by pre-treatment with AEBSF [2.5 mM] by 64±7% (p<.05, n=5) without effecting the fMLF-activation of the respiratory burst. Pretreatment of PMNs with 10–100 μg/ml tranexamic acid also inhibited the α-enolase priming by 38±10–90±7%, respectively (p<0.05, n=7). Importantly, both enzymatically active and inactive α-enolase yielded identical priming activity (results not shown).
Figure 3. α-enolase and thrombin prime the PMN oxidase.

In both panels the maximal rate of superoxide anion production (O2− nmol/min) is depicted as a function of treatment group. Panel A demonstrates that α-enolase at concentration of 10–50 μg/ml caused significant priming of the formyl-Met-Leu-Phe (fMLF)-activated respiratory burst (*=p<0.05 vs. buffer-primed/fMLF-activated PMNs, n=6). Concentrations of α-enolase of 1 μg/ml did not cause PMN priming. Panel B shows that thrombin at concentrations of 1–10 U/ml significantly primed the fMLF-activated respiratory burst whereas lesser concentration of thrombin did not (*=p<0.05 vs. buffer primed/fMLF-activated PMNs, n=6).
PMNs incubated for 30 min at 37° C with thrombin at a concentration of 1–10 U/ml significantly primed the fMLF-activated respiratory burst: buffer-treated controls (p<0.05, n=7) (Fig. 2, panel B). Lower concentrations [0.1–0.5 U/ml] of thrombin and shorter incubation times did not induce PMN priming, and the exclusion of plasma did not affect the priming activity of thrombin (data not shown). Furthermore, pre-incubation of PMNs with AEBSF [2.5 mM] abrogated the thrombin-induced PMN priming by 100±8% (p<.05, n=5) without affecting fMLF-activation of the respiratory burst. Importantly, thrombin priming was identical whether 10% heparinized plasma [10 U/ml] was included or not (data not shown).
PAR Immunoreactivity in PMNs and HMVECs
Whole cell lysates immunoblotted for PAR-2 demonstrated that PMNs contained PAR-2 receptors (Fig. 4, panel A). Identical experiments were performed on HMVECs, which demonstrated the presence of immunoreactivity for both PAR-1 and PAR-2 in HMVEC lysates (Fig. 4, panel B), similar to previous results (18).
Figure 4. PMNs and HMVECs contain PAR-2 immunoreactivity.
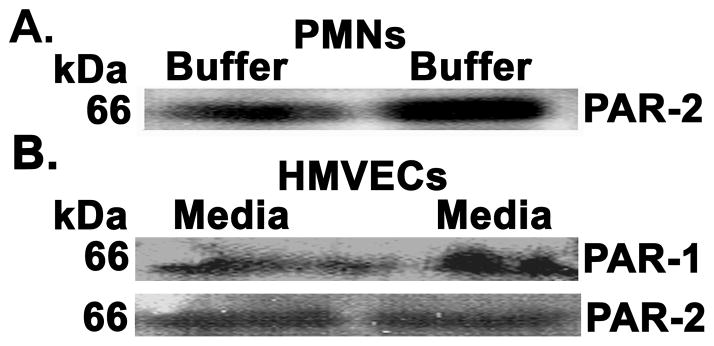
To determine if both PMNs and HMVECs contain the PAR-2 receptor isolated PMNs (panel A) and HMVECs (panel B) were lysed, the proteins separated and immunoblotted for the PAR-2 receptor in PMNs and both PAR-1 and PAR2 in HMVECs. PMNs demonstrate immunoreactivity for PAR-2 (Panel A) and HMVECs contain immunoreactivity for both PAR-1 and PAR-2 (panel B). These data represent identical results for two separate experiments.
Co-Precipitation of α-enolase and plasminogen/plasmin with the PAR-2 Receptor
HMVECs were incubated with α-enolase [50 μg/ml] for 5–30 min, the cells were lysed, and PAR-2 was immunoprecipitated as previously described (16). The immunoprecipitate was probed for α-enolase which co-precipitated with PAR-2 first beginning at 5 min with maximal at 15–30 min (Fig. 5, panel A). The PAR-2 immunoprecipitate was then probed for β-arrestin-1 which maximally co-precipitated with PAR-2 at 5 min (Fig. 5, panel B). Lastly, because α-enolase does not contain a protease domain and is a known cell surface receptor for plasminogen these PAR-2 immunoprecipitates were probed with an antibody that recognizes plasmin (13;19). Plasmin co-precipitated with α-enolase and PAR-2 in HMVECs beginning at 5 min and was still present at 15 min.
Figure 5. α-enolase causes occupancy, activation, and co-precipitation of the PAR-2 receptor on HMVECs with β-arrestin-1 and plasmin.

HMVECs were incubated with media or α-enolase [50 μg/ml] for 5–30 min, the cells were lysed, PAR-2 was immunoprecipitated, and the immunoprecipitate was blotted for α-enolase, β-arrestin-1 or plasmin. For each panel a bar graph is included which illustrates the mean±SEM of the densitometry of the bands of interest. Panel A demonstrates that α-enolase co-precipitated with the PAR-2 receptor beginning first at 5 min and continuing through 30 min. Panel B demonstrates the treatment of HMVEC with α-enolase induced co-precipitation of β-arrestin-1 beginning at 1 min and persisting through 30 min. Panel C depicts α-enolase elicited co-precipitation of plasmin with the PAR-2 receptor beginning at 5 min and continuing through 15 min. This figure represents identical results from two separate experiments using cells from different donors.
Discussion
The role of proteins, which accumulate during blood storage and circulate following traumatic injury, in the development of ALI or organ failure remains undefined. The presented data demonstrate that both α-enolase and thrombin cause PMN priming, pro-inflammatory activation of pulmonary vascular endothelium, increased ICAM-1 surface expression leading to PMN adherence, and PMN-mediated cytotoxicity of endothelial cells indicating a possible role for these proteins in the development of ALI/organ injury. In addition, these cellular effects could be inhibited by protease antagonists and priming of PMNs and α-enolase induced pro-inflammatory activation of HMVECs occurred via activation of PAR-2 through plasmin for which α-enolase is a known cell surface receptor (13). α-enolase activated PAR-2 as demonstrated by co-precipitation with β-arrestin-1. The possible sources of plasminogen/plasmin in these experiments were fetal bovine serum [10%]FINAL in the media for HMVECs and plasma [10%]FINAL for the observed PMN priming. Importantly, both pro-inflammatory HMVEC activation and PMN priming were not present when plasma or FBS were deleted and priming occurred whether or not the α-enolase was active. In addition, the concentration of α-enolase employed is these experiments may seem relatively high; however, recent data demonstrated that α-enolase was present at concentrations of 1.5 μg/ml in the field plasma of the upper quartile of injured patients with higher ISS, and identical concentrations of α-enolase with 10% human plasma cause pro-inflammatory activation of HMVECs (17). Furthermore, recombinant proteins may not be as active as native proteins as demonstrated for CD40 ligand (20).
α-enolase does not contain protease activity, is a cellular receptor, and has an important role in plasmin activation and resistance to degradation (11;13). Furthermore, it is involved in monocyte-mediated lung injury through increased surface expression of α-enolase on the surface of monocytes leading to increased plasmin activity, monocyte transmigration into the acutely inflamed lung (14). Bacterial and fungal pathogens employ α-enolase to recruit plasminogen, which becomes activated upon the cell surface allowing for cellular invasion and infection; thus, α-enolase is a virulence factor in this context (21–24). Furthermore, the α-enolase:plasminogen interaction may be inhibited by monoclonal antibodies specific for α-enolase, as well as aminocaproic acid or traneexamic acid, the latter two inhibit by binding to the surface lysine groups or Kringle domains (14;21–25). Plasminogen becomes activated to plasmin on cellular surfaces due to recruitment of cellular proteases including the matrix metalloproteases, and plasmin has the capacity to cleave PAR receptors including PAR-2 (26).
Pro-inflammatory activation of pulmonary vascular endothelium by thrombin and its effects on PMN priming and margination have already been reported (27). The presented data has extended this work to demonstrate that thrombin-activated HMVECs may serve as the first event in an in vitro model of PMN-mediated cytotoxicity of endothelial cells. This modeling is dependent upon activation of the PMN oxidase, the second event, in PMNs that are adherent to activated endothelium, the first event as previously published (4).
Post-injury ALI, as well as MOF, is a pro-inflammatory condition that is the result of at least two, if not multiple, clinical events (1;28). The first event (injury) leads to activation of the pulmonary endothelium and PMN priming and PMN pulmonary sequestration (1;3;28). The second event may be due to infection or other pro-inflammatory mediators that serve to activate the sequestered PMNs leading to endothelial injury and the development of ALI or organ injury (1;3;28). Blood transfusion and more specifically the storage age RBC units are independent predictors of ALI and MOF development (6;29). Moreover, despite the decreasing incidence of post-injury MOF, which correlates with decreased RBC transfusions, a significant number of injured patients still succumb to post-injury ALI/ARDS in which agents which activate microvascular endothelium may play a role (29;30). Much of the data regarding the pro-inflammatory potential of RBC units has focused on lipid mediators, and with the ability to rapidly characterize the proteome of injured patients and the components used for resuscitation novel protein mediators are being recognized.
Proteomic analysis of plasma from patients who have suffered blunt trauma and subsequently developed ALI/MOF, has identified a number potential pro-inflammatory mediators responsible for lung injury, specifically α-enolase and thrombin as well as concomitant decreases in anti-proteases (31;32). Recently, identical proteomic analyses of the plasma from rats that underwent hemorrhagic shock demonstrated that α-enolase significantly increased; moreover, these identical proteins are also found in stored, but not fresh, PRBCs, which may be used to resuscitate these injured patients (7;31). The function of α-enolase in conjunction with plasminogen in PMN-mediated lung injury and organ dysfunction is largely unexplored; while thrombin has been implicated in the activation of endothelial cells resulting in decreased barrier function and fibrin deposition, a hall mark of ALI/ARDS (33–35).
The presented data demonstrate a pro-inflammatory role of α-enolase in a complex with plasminogen in solution leading to plasmin activation and to pro-inflammatory activation of pulmonary endothelial cells resulting in PMN adherence as the first event in a two-event model of PMN cytotoxicity. This step is independent of a decrease in endothelial cell barrier function. Pre-incubation of HMVECs with protease inhibitors or tranexamic acid inhibited of thrombin- and α-enolase:plasmin-induced pro-inflammatory activation of HMVECs most likely on different PARs: PAR-2 for α-enolase:plasmin and PAR-1,-3, or -4 for thrombin (9).
In summary, many mediators have been implicated in the development of ALI/MOF post-injury; however, the role of circulating proteins in the development of ALI or MOF has not been fully elucidated. α-enolase, which accumulates in the plasma of injured patients and in the supernatant of stored RBCs, binds plasminogen, activates plasmin, and elicits pro-inflammatory endothelial activation. Such endothelial activation may predispose patients to PMN-mediated ALI and end organ damage. Thus, α-enolase represents a masquerading mediator: a protein with a well-described intracellular function which demonstrates extracellular pro-inflammatory activity.
Acknowledgments
These studies were funded by Bonfils Blood Center and grant #P50 GM049222 from NIGMS, NIH.
Footnotes
There are no relevant conflicts of interest.
References
- 1.Partrick DA, Moore FA, Moore EE, Barnett CC, Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz. 1996 May;4(2):194–210. [PubMed] [Google Scholar]
- 2.Smith CW. 3. Adhesion molecules and receptors. J Allergy Clin Immunol. 2008 Feb;121(2 Suppl):S375–S379. doi: 10.1016/j.jaci.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 3.Kelher MR, Masuno T, Moore EE, Damle S, Meng X, Song Y, Liang X, Niedzinski J, Geier SS, Khan SY, Gamboni-Robertson F, Silliman CC. Plasma from stored packed red blood cells and MHC class I antibodies causes acute lung injury in a 2-event in vivo rat model. Blood. 2009 Feb 26;113(9):2079–87. doi: 10.1182/blood-2008-09-177857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyman TH, Bjornsen AJ, Elzi DJ, Smith CW, England KM, Kelher M, Silliman CC. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol. 2002 Dec;283(6):C1592–C1603. doi: 10.1152/ajpcell.00540.2001. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg JA, McGwin G, Jr, Vandromme MJ, Marques MB, Melton SM, Reiff DA, Kerby JD, Rue LW., III Duration of red cell storage influences mortality after trauma. J Trauma. 2010 Dec;69(6):1427–31. doi: 10.1097/TA.0b013e3181fa0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zallen G, Offner PJ, Moore EE, Blackwell J, Ciesla DJ, Gabriel J, Denny C, Silliman CC. Age of transfused blood is an independent risk factor for postinjury multiple organ failure. Am J Surg. 1999 Dec;178(6):570–2. doi: 10.1016/s0002-9610(99)00239-1. [DOI] [PubMed] [Google Scholar]
- 7.Dzieciatkowska M, Silliman CC, Moore EE, Kelher MR, Banerjee A, Land KJ, Ellison M, West FB, Ambruso DR, Hansen KC. Proteomic analysis of the supernatant of red blood cell units: the effects of storage and leucoreduction. Vox Sang. 2013 Oct;105(3):210–8. doi: 10.1111/vox.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005 Feb;26(1):1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- 9.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000 Sep 14;407(6801):258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 10.Molino M, Woolkalis MJ, Reavey-Cantwell J, Pratico D, Andrade-Gordon P, Barnathan ES, Brass LF. Endothelial cell thrombin receptors and PAR-2. Two protease-activated receptors located in a single cellular environment. J Biol Chem. 1997 Apr 25;272(17):11133–41. doi: 10.1074/jbc.272.17.11133. [DOI] [PubMed] [Google Scholar]
- 11.Diaz-Ramos A, Roig-Borrellas A, Garcia-Melero A, Lopez-Alemany R. alpha-Enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol. 2012;2012:156795. doi: 10.1155/2012/156795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pancholi V. Multifunctional alpha-enolase: its role in diseases. Cell Mol Life Sci. 2001 Jun;58(7):902–20. doi: 10.1007/PL00000910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miles LA, Ellis V. Alpha-enolase comes muscling in on plasminogen activation. Thromb Haemost. 2003 Oct;90(4):564–6. [PubMed] [Google Scholar]
- 14.Wygrecka M, Marsh LM, Morty RE, Henneke I, Guenther A, Lohmeyer J, Markart P, Preissner KT. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood. 2009 May 28;113(22):5588–98. doi: 10.1182/blood-2008-08-170837. [DOI] [PubMed] [Google Scholar]
- 15.Silliman CC, Clay KL, Thurman GW, Johnson CA, Ambruso DR. Partial characterization of lipids that develop during the routine storage of blood and prime the neutrophil NADPH oxidase. J Lab Clin Med. 1994 Nov;124(5):684–94. [PMC free article] [PubMed] [Google Scholar]
- 16.McLaughlin NJ, Banerjee A, Kelher MR, Gamboni-Robertson F, Hamiel C, Sheppard FR, Moore EE, Silliman CC. Platelet-activating factor-induced clathrin-mediated endocytosis requires beta-arrestin-1 recruitment and activation of the p38 MAPK signalosome at the plasma membrane for actin bundle formation. J Immunol. 2006 Jun 1;176(11):7039–50. doi: 10.4049/jimmunol.176.11.7039. [DOI] [PubMed] [Google Scholar]
- 17.Gonzales E, Moore EE, Moore HB, Chapman MP, Silliman CC, Banerjee A. Alpha-enolase, a glycolotic enzyme elevated in hemorrhagic shock, potentiates fibrinolysis, and is reversible by tranexamic acid. Shock. 2014;41(supplement 2):48–49. [Google Scholar]
- 18.Hirano K, Kanaide H. Role of protease-activated receptors in the vascular system. J Atheroscler Thromb. 2003;10(4):211–25. doi: 10.5551/jat.10.211. [DOI] [PubMed] [Google Scholar]
- 19.Kang HJ, Jung SK, Kim SJ, Chung SJ. Structure of human alpha-enolase (hENO1), a multifunctional glycolytic enzyme. Acta Crystallogr D Biol Crystallogr. 2008 Jun;64(Pt 6):651–7. doi: 10.1107/S0907444908008561. [DOI] [PubMed] [Google Scholar]
- 20.Khan SY, Kelher MR, Heal JM, Blumberg N, Boshkov LK, Phipps R, Gettings KF, McLaughlin NJ, Silliman CC. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood. 2006 Oct 1;108(7):2455–62. doi: 10.1182/blood-2006-04-017251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergmann S, Rohde M, Chhatwal GS, Hammerschmidt S. Characterization of plasmin(ogen) binding to Streptococcus pneumoniae. Indian J Med Res. 2004 May;119( Suppl):29–32. [PubMed] [Google Scholar]
- 22.Chumchua V, Pornputtapong N, Thammarongtham C, Meksuriyen D. Homology modeling of Mycoplasma pneumoniae enolase and its molecular interaction with human plasminogen. Bioinformation. 2008;3(1):18–23. doi: 10.6026/97320630003018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Jacobs-Lorena M. Surface-expressed enolases of Plasmodium and other pathogens. Mem Inst Oswaldo Cruz. 2011 Aug;106(Suppl 1):85–90. doi: 10.1590/s0074-02762011000900011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pancholi V, Fontan P, Jin H. Plasminogen-mediated group A streptococcal adherence to and pericellular invasion of human pharyngeal cells. Microb Pathog. 2003 Dec;35(6):293–303. doi: 10.1016/j.micpath.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Kariko K, Malkowicz S, Li W, Kuo A, Barnathan E. Invasive neoplastic uroepithelial cells express high-levels of urokinase receptor and plasminogen receptor, alpha-enolase. Int J Oncol. 1993 Dec;3(6):1089–95. doi: 10.3892/ijo.3.6.1089. [DOI] [PubMed] [Google Scholar]
- 26.Domotor E, Bartha K, Machovich R, Adam-Vizi V. Protease-activated receptor-2 (PAR-2) in brain microvascular endothelium and its regulation by plasmin and elastase. J Neurochem. 2002 Mar;80(5):746–54. doi: 10.1046/j.0022-3042.2002.00759.x. [DOI] [PubMed] [Google Scholar]
- 27.Vercellotti GM, Wickham NW, Gustafson KS, Yin HQ, Hebert M, Jacob HS. Thrombin-treated endothelium primes neutrophil functions: inhibition by platelet-activating factor receptor antagonists. J Leukoc Biol. 1989 Jun;45(6):483–90. doi: 10.1002/jlb.45.6.483. [DOI] [PubMed] [Google Scholar]
- 28.Salzer WL, McCall CE. Primed stimulation of isolated perfused rabbit lung by endotoxin and platelet activating factor induces enhanced production of thromboxane and lung injury. J Clin Invest. 1990 Apr;85(4):1135–43. doi: 10.1172/JCI114545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sauaia A, Moore FA, Moore EE, Haenel JB, Read RA, Lezotte DC. Early predictors of postinjury multiple organ failure. Arch Surg. 1994 Jan;129(1):39–45. doi: 10.1001/archsurg.1994.01420250051006. [DOI] [PubMed] [Google Scholar]
- 30.Ciesla DJ, Moore EE, Johnson JL, Burch JM, Cothren CC, Sauaia A. A 12-year prospective study of postinjury multiple organ failure: has anything changed? Arch Surg. 2005 May;140(5):432–8. doi: 10.1001/archsurg.140.5.432. [DOI] [PubMed] [Google Scholar]
- 31.D’Alessandro A, Dzieciatkowska M, Peltz ED, Moore EE, Jordan JR, Silliman CC, Banerjee A, Hansen KC. Dynamic Changes in Rat Mesenteric Lymph Proteins Following Trauma Using Label-Free Mass Spectrometry. Shock. 2014 Sep 19; doi: 10.1097/SHK.0000000000000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dzieciatkowska M, D’Alessandro A, Moore EE, Wohlauer M, Banerjee A, Silliman CC, Hansen KC. Lymph is not a plasma ultrafiltrate: a proteomic analysis of injured patients. Shock. 2014 Dec;42(6):485–98. doi: 10.1097/SHK.0000000000000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belvitch P, Adyshev D, Elangovan VR, Brown ME, Naureckas C, Rizzo AN, Siegler JH, Garcia JG, Dudek SM. Proline-rich region of non-muscle myosin light chain kinase modulates kinase activity and endothelial cytoskeletal dynamics. Microvasc Res. 2014 Jul 27;95C:94–102. doi: 10.1016/j.mvr.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzales JN, Kim KM, Zemskova MA, Rafikov R, Heeke B, Varn MN, Black S, Kennedy TP, Verin AD, Zemskov EA. Low anticoagulant heparin blocks thrombin-induced endothelial permeability in a PAR-dependent manner. Vascul Pharmacol. 2014 Aug;62(2):63–71. doi: 10.1016/j.vph.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS. Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia--a review. Crit Care Med. 2006 Mar;34(3):871–7. [PubMed] [Google Scholar]