

Abstract
Cyanobacteria are diverse photosynthetic microbes with the ability to convert CO2 into useful products. However, metabolic engineering of cyanobacteria remains challenging because of the limited resources for modifying the expression of endogenous and exogenous biochemical pathways. Fine-tuned control of protein production will be critical to optimize the biological conversion of CO2 into desirable molecules. Messenger RNA (mRNA) are labile intermediates that play critical roles in determining the translation rate and steady state protein concentrations in the cell. The majority of studies on mRNA turnover have focused on the model heterotrophic bacteria Escherichia coli and Bacillus subtilis. These studies have elucidated many RNA modifying and processing enzymes and have highlighted the differences between these Gram-negative and Gram-positive bacteria, respectively. In contrast, much less is known about mRNA turnover in cyanobacteria. We generated a compendium of the major ribonucleases (RNases) and provide an in-depth analysis of RNase III-like enzymes in commonly studied and diverse cyanobacteria. Furthermore, using targeted gene deletion, we genetically dissected the RNases in Synechococcus sp. PCC 7002, one of the fastest growing and industrially attractive cyanobacterial strains. We found that all three cyanobacterial homologs of RNase III and a member of the RNase II/R family are not essential under standard laboratory conditions, while homologs of RNase E/G, RNase J1/J2, PNPase, and a different member of the RNase II/R family appear to be essential for growth. This work will enhance our understanding of native control of gene expression and will facilitate the development of an RNA-based toolkit for metabolic engineering in cyanobacteria.
Keywords: mRNA, RNA, Ribonuclease, photosynthesis, cyanobacteria, synthetic biology, biofuels, comparative genomics
INTRODUCTION
The flow of information from DNA to mRNA to protein is highly regulated to generate the appropriate quantities of enzymes and structural proteins required for growth and metabolism. Transcriptional control of gene expression has been well studied in many organisms. However, there is little concordance between transcript abundance and protein abundance (Abreu et al. 2009). This disconnect can arise due to posttranscriptional regulation of mRNA and protein molecules (e.g. degradation or dilution). Among the many factors involved in posttranscriptional regulation in prokaryotes, the process of mRNA turnover is among the least understood. Much of our knowledge on mRNA turnover has been gained from studies of Escherichia coli and Bacillus subtilis, model Gram-negative and Gram-positive bacteria, respectively (Anderson and Dunman 2009). These studies have shed light on many of the ribonucleases and RNA modifying enzymes involved in RNA processing. However, less is known about mRNA turnover in non-model bacteria including cyanobacteria. These photosynthetic Gram-negative bacteria play a major role in the global carbon cycle and are becoming an attractive platform for renewable chemical production. A detailed understanding of mRNA turnover dynamics is crucial for implementing effective metabolic engineering strategies to turn cyanobacteria into green chemical factories. Further, a better understanding of mRNA turnover will facilitate studies of basic physiology including photosynthesis and carbon fixation. A better understanding of mRNA turnover begins by identifying the relevant enzymes involved.
Bacterial mRNA decay is facilitated by exoribonucleases, endoribonucleases, and RNA modifying enzymes (Fig. 1). In E. coli, mRNA degradation has been shown to proceed in the 5′-3′ direction, despite the absence of a specific 5′-3′ exoribonuclease in its genome. The directionality of degradation is initiated through a series of internal cleavages by RNase E, an endoribonuclease that targets single-stranded RNA (ssRNA) (Misra and Apirion 1979; Mackie 2013; Apirion 1973). For a subset of mRNA substrates, cleavage by RNase E is modulated by the presence of a 5′-triphosphate. Thus, 5′-monophosphate formation through removal of the 5′-pyrophosphate by RppH, a NUDIX hydrolase, enhances the subsequent rate of mRNA degradation for these substrates (Celesnik et al. 2007; Deana et al. 2008). However, rapid cleavage of substrates containing a 5′-triphosphate by RNase E suggests that direct entry and cleavage in a 5′-end-independent manner may be a major route for mRNA degradation in E. coli (Anupama et al. 2011; Kime et al. 2010). Similarly, removal of secondary structure by RNase III, an enzyme that cleaves a subset of structured RNA hairpins, can also facilitate mRNA processing by RNase E (Court et al. 2013). RNase E is a component of the degradosome, a multi-enzyme complex that is responsible for the majority of mRNA turnover in E. coli and other bacteria (Py et al. 1996; Carpousis et al. 1994; Miczak et al. 1996). The C-terminal scaffold domain of RNase E recruits PNPase, enolase, and an ATP-dependent RNA helicase, RhlB, to the degradosome (Vanzo et al. 1998). RNase G, an RNase E paralog that lacks the scaffold domain and is not recruited to the degradosome, appears to play a minor role in mRNA degradation compared to RNase E (Ow et al. 2003). Following primary cleavage by RNase E, three 3′-5′ exoribonucleases, RNase II, RNase R, and PNPase process the majority of mRNA in E. coli (Carpousis et al. 2009). Poly(A) polymerase can also increase the rate of mRNA decay by providing a template for PNPase (Mohanty et al. 2008). In contrast to E. coli, the majority of mRNAs in B. subtilis are processed by the 5′-3′ exoribonucleases, RNase J1 and RNase J2 (RNase J1/J2) (Condon 2010). The genome of B. subtilis does not encode an RNase E homolog, but instead contains the functionally analogous protein, RNase Y (Shahbabian et al 2009). RNase III, a type of endoribonuclease that cleaves dsRNA, is found in bacteria, plants, fungi, and animals. Members of the RNase III family have been divided into four classes based on their domain architecture (Olmedo and Guzmán 2008). Class 1 contains a single RNase III domain and a double-stranded RNA binding domain (dsRBD). Class 2 (e.g. Drosha) is composed of two RNase III domains, a dsRBD, and a poly-proline region. Class 3 (e.g. Dicer) contains two RNase III domains, a dsRBD, a PAZ domain, a helicase domain, and a DUF domain. Class 4 (Mini-III) lacks the dsRBD and is involved in 23S ribosomal RNA maturation in B. subtilis (Redko et al. 2008).
Fig. 1.
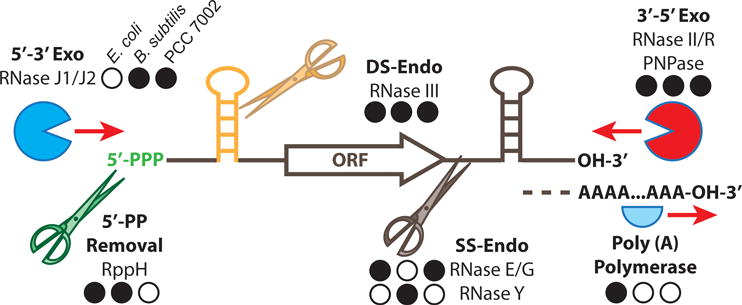
Schematic diagram of ribonucleases and RNA processing enyzmes in bacteria
Exoribonucleases process RNA to oligonucleotides from the 3′ and 5′ directions. Endoribonucleases process single stranded (SS) or double stranded (DS) RNA molecules, releasing additional targets for processing by exoribonucleases. RNA modifying enzymes can alter the rates of RNA processing by additional ribonucleases. Enzymes with homologs in E. coli, B. subtilis and Synechococcus sp. PCC 7002 (PCC 7002), respectively, are shown with a closed back circle. An open black circle indicates absence of a homolog in the corresponding strain.
Cyanobacteria are morphologically, ecologically, and genetically diverse photosynthetic microbes. Compared to E. coli and B. subtilis, relatively little is known about the components and mechanisms involved in mRNA turnover in cyanobacteria. Preliminary bioinformatic analysis revealed that the cyanobacterium Synechocystis sp. PCC 6803 (PCC 6803) contains homologs of E. coli and B. subtilis RNA degrading machinery including RNase E, RNase E/G, RNase J1/J2, and RNase II/R (Condon and Putzer 2002) (Fig. 1). In addition, more recent work identified the presence of the Class 4 RNase III, Mini-III, in some cyanobacterial genomes (Redko et al. 2008). It has been long thought that that cyanobacteria do not contain a multicomponent mRNA degradosome complex based on the low conservation between the cyanobacterial and E. coli RNase E scaffold domains and the inability to co-purify cyanobacterial degradosome components (Rott et al. 2003; Kaberdin et al. 1998). However, a conserved arginine rich peptide at the C-terminus of RNase E was shown to facilitate specific interactions with PNPase in two diverse cyanobacterial species, PCC 6803 and Anabaena PCC 7120 (PCC 7120) (Zhang 2014). Since cyanobacteria appear to have components similar to both E. coli and B. subtilis, and the homologs of the common RNases are often found as small gene families, the essential components of the cyanobacterial mRNA turnover machinery are not readily apparent. Therefore, in this study, we aimed to examine the RNases in commonly studied cyanobacteria and identify the major players in mRNA turnover in Synechococcus sp. PCC 7002 (PCC 7002).
RESULTS
Genomic analysis of major mRNA processing enzymes in model cyanobacteria
First, we sought to identify and catalog the major mRNA degrading enzymes in commonly studied cyanobacterial strains (Table 1). From primary sequence alignments and putative annotations, we were able to identify multiple homologs of E. coli RNase E/G, RNase III, RNase II/R, and PNPase as well as homologs of B. subtilis RNase J1/J2 (Mathy et al. 2007; Britton et al. 2007) and Mini-III (Redko et al. 2008). A single protein with homology to the N-terminal catalytic domains of RNase E and RNase G was identified in each of the model cyanobacteria strains. RNase G resembles the N-terminal catalytic region of RNase E and does not contain the C-terminal scaffolding domain. The C-terminal domain of the cyanobacterial homologs was variable in length and exhibited low sequence homology to the RNase E scaffold domain. Thus, we classified them as RNase E/G homologs. In each model species, we identified only a single protein with homology to RNaseJ1/J2. This contrasts the model case in which B. subtilis contains multiple RNase J1/J2 enzymes (Even et al. 2005). The primary sequences of RNase II and RNase R are highly similar and it remains to be determined whether the substrate specificity of these enzymes can be determined from sequence information alone. For this reason, we grouped the multiple cyanobacterial homologs of the RNase II/R family identified in the model organisms. The cyanobacterial species investigated contain proteins with homology to known 5′-3′ exoribonucleases, 3′-5′ exoribonucleases, dsRNA specific endoribonucleases, and ssRNA specific exoribonucleases, making it difficult to determine a priori which mechanisms of mRNA decay are dominant. Further, it is not known how these enzymes function together during mRNA degradation and which components are essential for the process. Therefore, we aimed to characterize each of the identified enzyme classes in PCC 7002, a fast-growing strain that is being used as a platform for synthetic biology and renewable chemical production (Begemann et al. 2013).
Table 1.
Ribonuclease homologs in commonly studied cyanobacteria
| Genome | Enzyme Class | ||||
|---|---|---|---|---|---|
| RNase E/G | RNase III | RNase J1/J2 | RNase II/R | PNPase | |
| Acaryochloris marina MBIC11017 | AM1_1705 |
AM1_1717 AM1_5197* |
AM1_3444 |
AM1_4216 AM1_E0095 AM1_D0003 AM1_E0127 AM1_B0417 AM1_4810 AM1_A0095 |
AM1_5920 AM1_4121 |
| Anabaena sp. PCC 7120 | alr4331 |
all4107 alr0280 alr1158* |
all3678 |
all4450 alr1240 |
all0136 all4396 |
| Arthrospira platensis NIES-39 | NIES39_J05380 |
NIES39_Q02990 NIES39_N00330 |
NIES39_L00100 |
NIES39_D02610 NIES39_E01890 |
NIES39_R00610 NIES39_L00330 NIES39_O05010 |
| Cyanobacterium sp. UCYN-A | UCYN_00380 |
UCYN_03680 UCYN_00750* |
UCYN_00320 |
UCYN_07880 UCYN_00190 UCYN_09960 |
UCYN_04860 UCYN_03170 |
| Cyanothece sp. BH68, ATCC 51142 | cce_0277 |
cce_3135 cce_1800* |
cce_0295 |
cce_5063 cce_5061 cce_0113 cce_3346 cce_3157 |
cce_1653 cce_0623 cce_3461 |
| Gloeobacter violaceus PCC 7421 | gvip208 |
gvip371 glr4180* |
glr1019 |
gll1616 gll1448 |
gvip129 glr4144 gll3149 |
| Microcystis aeruginosa NIES-843 | MAE_15200 |
MAE_31570 MAE_60800 MAE_28410* |
MAE_62670 |
MAE_23880 MAE_46880 |
MAE_53390 |
| Prochlorococcus marinus pastoris CCMP 1986 | PMM1501 |
PMM1603 PMM0949* |
PMM1652 |
PMM0868 PMM0847 |
PMM1191 |
| Synechococcus elongatus PCC 7942 | Synpcc7942_0878 |
Synpcc7942_1645 Synpcc7942_2120* |
Synpcc7942_1846 |
Synpcc7942_1911 Synpcc7942_1124 Synpcc7942_1546 |
Synpcc7942_2440 |
| Synechococcus sp. PCC 7002 | SYNPCC7002_A0788 |
SYNPCC7002_A0061 SYNPCC7002_A2542 SYNPCC7002_A0384* |
SYNPCC7002_A1273 |
SYNPCC7002_A1543 SYNPCC7002_A0574 |
SYNPCC7002_A1066 |
| Synechocystis sp. PCC 6803 | slr1129 |
slr1646 slr0346 slr0954* |
slr0551 |
sll1910 sll1290 |
slr1984 sll1043 |
| Thermosynechococcus elongatus BP-1 | tlr0597 | Tlr0428* | tlr1220 |
tlr1873 tlr0652 |
tll2193 |
| Trichodesmium erythraeum IMS101 | Tery_0483 |
Tery_2839 Tery_2144 Tery_1105 Tery_2312* |
Tery_0589 |
Tery_3730 Tery_1511 |
Tery_1541 Tery_2502 Tery_4663 |
Mini-III
Genetic analysis of ribonucleases in PCC 7002
In order to determine which of the identified RNase homologs are essential in PCC 7002, we attempted to replace each of the coding regions of the RNase E/G, RNase III, RNase J1/J2, RNase II/R, and PNPase homologs with an antibiotic resistant cassette via homologous recombination and targeted gene deletion strategies as described in “Materials and Methods”. Online Resources 1 and 2 describe all plasmids and oligonucleotides used in this study. Since many cyanobacteria including PCC 7002 (Murphy et al. 1990) contain multiple copies of the chromosome, deletion of essential genes often results in a heterozygous strain containing both mutant and WT alleles. However, it is possible that conditions permissible for full segregation of the mutant allele can be identified. In this study, we are only reporting whether we were able to fully segregate the mutant allele in standard laboratory conditions (Table 2). Full segregation indicates that the gene is not essential for survival, whereas the inability to segregate the mutant indicates that it is required for growth in the conditions tested. We were not able to isolate fully segregated mutants of RNase E/G (Δ0788), RNase J1/J2 (Δ1273), one RNase II/R homolog (Δ0574), or PNPase (Δ1066) (Online Resource 3), indicating that these genes are required under laboratory conditions. Due to the inherent instability of partially segregated mutants and difficulty in accurately assessing the physiology of these strains, we focused our efforts in characterizing the physiology of mutants that could be fully segregated (Table 2), RNase III and the second RNaseII/R homolog (A1543).
Table 2.
Summary of strains used in this study
| Strain | Resistance Marker | Enzyme Class | Segregated | Growth Defect | Reference |
|---|---|---|---|---|---|
| WT PCC7002 | – | n/a | n/a | n/a | PCC |
| Single Mutants | |||||
| Δ0788 | Km | RNase E/G | No | nd | This work |
| Δ0061 | Sp | RNase III | Yes | − | This work |
| Δ2542 | Km | RNase III | Yes | − | This work |
| Δ0384 | Gm | RNase III | Yes | − | This work |
| Δ1273 | Km | RNAse J1/J2 | No | nd | This work |
| Δ0574 | Sp | RNAse II/R | No | nd | This work |
| Δ1543 | Sp | RNAse II/R | Yes | + | This work |
| Δ1066 | Gm | PNPase | No | nd | This work |
| Double Mutants | |||||
| Δ0061/2542 | Sp/Km | RNase III | Yes | − | This work |
| Δ0061/0384 | Sp/Gm | RNase III | Yes | + | This work |
| Δ2542/0384 | Km/Gm | RNase III | Yes | − | This work |
| Triple Mutants | |||||
| Δ0061/2542/0384 | Sp/Km/Gm | RNase III | Yes | + | This work |
| Genetic Complementation | |||||
| Δ0061+/2542/0384 | Sp/Km/Gm/Amp | RNase III | Yes | − | This work |
| Δ1543+ | Sp/Gm | RNase II/R | Yes | − | This work |
n/a, not applicable
Growth defect compared to the WT observed (+) or not (−).
The phenotypes of partially segregated mutants were not determined (nd).
Diversity of RNase III homologs in cyanobacteria
The well-studied E. coli RNase III (Rnc) contains the RNase III signature motif (ERLEFLGDS) within the N-terminal RNase III catalytic domain and a C-terminal dsRBD. The E. coli genome encodes a single RNase III homolog that is not essential for growth under standard conditions; mutants are non-motile and have a temperature sensitivity at 45°C (Apirion and Watson 1978, 1975). In contrast, B. subtilis contains an RNase III that is essential for growth (Durand et al. 2012) and a short, RNase III-like homolog lacking the C-terminal dsRBD (YazC, Mini-III) (Redko et al. 2008). We were able to identify homologs of RNase III and Mini-III in the genomes of cyanobacteria representing all of the diverse morphological subgroups (Shih et al. 2013) (Online Resource 4). Typically, a single Mini-III homolog was accompanied by one or more RNase III homologs. However we identified several genomes that completely lacked either the full-length RNase III or the Mini-III, but not both (Online Resource 4). PCC 7002 contains two RNase III homologs (A0061 and A2542) and a single Mini-III homolog (A0384). The functions of these enzymes in PCC 7002 are not known. Multiple sequence alignments of RNase III and Mini-III sequences from E. coli, B. subtilis, PCC 7002, PCC 7120 and a single RNase III homolog lacking the dsRBD from Leptolyngbya sp. PCC 6406 (PCC 6406) were used to build a phylogenetic tree to compare the relationships between these proteins (Fig. 2a). The RNase III and Mini-III homologs could be resolved into distinct clades. Within the RNase III clade, A2542 from PCC 7002 branches more closely to Rnc than the other homolog, A0061. Alr0280 from PCC 7120 has been shown to be active in cleaving dsRNA substrates (Gao et al. 2013) and clustered with Rnc and A2542. A0061 clusters with Alr2542, which was shown not to have ribonuclease activity when incubated with the same substrate as Alr0280 (Gao et al. 2013). Interestingly, the RNase III-like protein from PCC 6406 (Lyn2207) that resembles a Mini-III since it lacks the dsRBD branched within the full-length RNase III clade. We therefore attempted to determine whether there were other features that distinguished the canonical RNase III domain from the Mini-III RNase III domain. Sequence alignments revealed differences between the RNase III signature motif. All of the proteins that branched more closely with Rnc, including the PCC6406 truncated protein exhibited an E. coli-like signature motif (ERLEFLGDA), whereas all of the Mini-III proteins contained a unique motif (AALAYLGDA). Based on a sequence conservation logo generated from multiple sequence alignments from 65 and 64 RNase III and Mini-III sequences, respectively, our results indicate that classification based on the signature motif might be more robust than the presence/absence of the dsRBD (Fig. 2a and Online Resource 4).
Fig. 2.
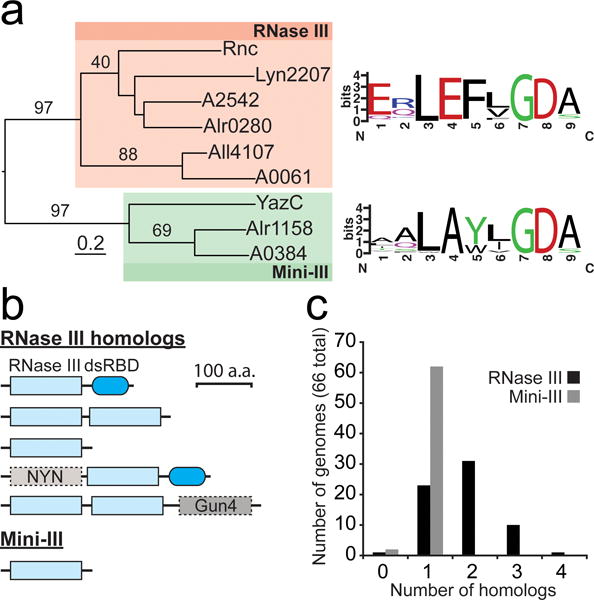
Diversity and distribution of RNase III homologs in cyanobacteria
(a) Phylogenic relationship between RNase III and Mini-III homologs in Anabaena sp. PCC 7120 (Alr0280, Alr4107, and Alr1158), Leptolyngbya sp. PCC 6406 (Lyn2207), PCC 7002 (A2542, A0061, and A0384), E. coli (Rnc), and B. subtilis (YazC). The RNase III homologs all contained the typical domain structure with a single, N-terminal RNase III domain and a C-terminal dsRBD, with the exception of Lyn2207, containing only a single RNase III domain. Conservation logos (weblogo.berkeley.edu) corresponding to the RNase III signature motif generated from multiple sequence alignments of RNase III and Mini-III homologs in 65 and 64 cyanobacterial genomes, respectively. (b) Domain architecture of RNase III and Mini-III homologs in cyanobacteria. (c) Number of RNase III and Mini-III homologs in 66 diverse cyanobacterial genomes. Additional details can be found in “Materials and Methods”. RNase III homologs in each analyzed genome are listed in Online Resource 4.
Analysis of 66 cyanobacterial genomes representing the five morphological subclades revealed RNase III homologs with diverse domain structures and unknown function (Fig. 2b and Online Resource 4). Most of the 66 genomes analyzed contained a single Mini-III homolog, whereas the number of RNase III homologs varied from zero to four per genome (Fig. 2c). The atypical domain structures (Fig. 2b) lacking the dsRBD, containing tandem RNase III domains, or with additional domain extensions were found in organisms containing at least three RNase III homologs (Online Resource 4).
Genetic deletion of RNase III and Mini-III homologs in PCC 7002
RNase III is essential in B. subtilis, but not in E. coli. Since the functions of RNase III and Mini-III homologs in PCC 7002 are not known, we aimed to determine whether they could be genetically deleted individually and in combination. Using targeted gene deletion, we were able to replace the open reading frame of each of the RNase III (A0061 and A2542) homologs and the Mini-III homolog (A0384) (Fig. 3). Insertion of the antibiotic resistances cassette into the locus of interest was checked with PCR using flanking primers (Fig. 3b, d, and f). Full segregation of the single mutants (Δ0061, Δ2542, and Δ0384), double mutants (Δ0061/2542, Δ0061/0384, and Δ2542/0384), and triple mutant (Δ0061/2542/0384) was confirmed by PCR using gene specific primers (Fig. 3g and Table 2). These results indicate that RNase III activity is dispensable in PCC 7002.
Fig. 3.
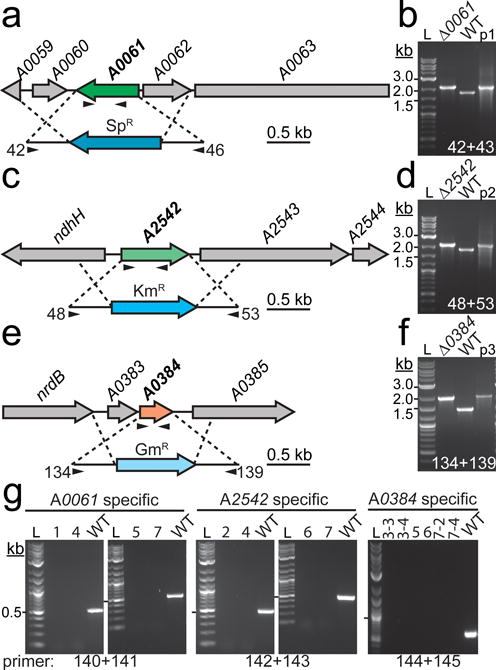
Genetic deletion and segregation analysis of RNase III homologs in PCC 7002
Targeted gene replacement via homologous recombination was used to replace the open reading frames of (a) A0061, (c) A2542, and (e) A0384 with three different antibiotic resistance cassettes. Insertion of the antibiotic resistance cassette into the correct genetic locus was confirmed by PCR using primers flanking the WT loci (b, d, and f). Arrows in (a), (c), and (d) below the gene and insertion cassette indicate approximate location of gene specific and flanking primers, respectively. The plasmid (p1, pJCC249; p2, pJCC250; p3, pJCC251) used to transform the mutant was used as a positive size control and WT genomic DNA was used as a negative size control. WT sized bands were not observed in the mutant strains, indicating that the regions of interest are homozygous for the mutation. (g) Gene specific primers were used to confirm segregation of the insert by PCR. In this case, WT genomic DNA served as the positive control. Lanes: 1, Δ0061; 2, Δ2542; 3, Δ0384; 4, Δ0061/2542; 5, Δ0061/0384; 6, Δ2542/0384; 7, Δ0061/2542/0384. The 0.5 kb size band is indicated with a hash mark next to the 2-log DNA ladder (L). Oligonucleotides used in cloning and segregation analysis are listed in Online Resource 2. Refer to “Materials and Methods” for additional details about strain construction and segregation analysis.
Physiology of RNase III deletion mutants in PCC 7002
To determine the effect of each RNase III and Mini-III deletion on the physiology of PCC 7002, we compared the growth of each strain on solid medium and in liquid culture (Fig. 4). Growth of the single mutants was not different than the WT on solid (Fig. 4a) or liquid medium (Fig. 4b). In contrast, the Δ0061/0384 (Fig. 4c) and Δ0061/2542/0384 (Fig. 4d) strains exhibited slightly reduced growth compared to the WT. In liquid medium (Fig. 4c and d), the reduced growth was more apparent during the early growth phase, whereas on solid medium (Fig. 4a) growth was not observed in the most dilute patches. We also compared growth of Δ0384 strains in an atmosphere containing ambient (~0.04 %) or enriched (0.4%) CO2 concentrations (Fig. 5). Growth of all strains was significantly improved in the enriched CO2 atmosphere. However, the colony size of the Δ0061/0384 and Δ0061/2542/0384 strains appeared smaller than the WT, indicating that the growth is still impacted under these conditions.
Fig. 4.
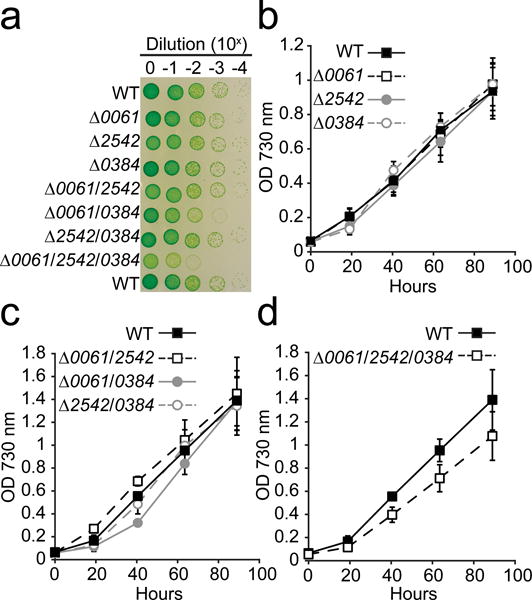
Growth of RNase III deletion mutants in liquid and on solid medium
(a) Cells suspended at an OD 730 nm = 0.05 (Tecan) were tenfold serially diluted four times in Media A+, spotted on solid medium, and grown for 65 h prior to being photographed. A representative of triplicate plates is shown. Liquid cultures of (b) single, (c) double, and (d) triple RNase III mutants were inoculated at an OD 730 nm = 0.05 (Tecan) and grown in 12 well culture plates on a shaking platform (150 rpm). Error bars represent s.d. of three separate cultures. All growth experiments were carried out in ambient CO2 at 38°C with a light intensity of 150 μmol photons m−2 s−1. Additional experimental details can be found in “Materials and Methods”.
Fig. 5.
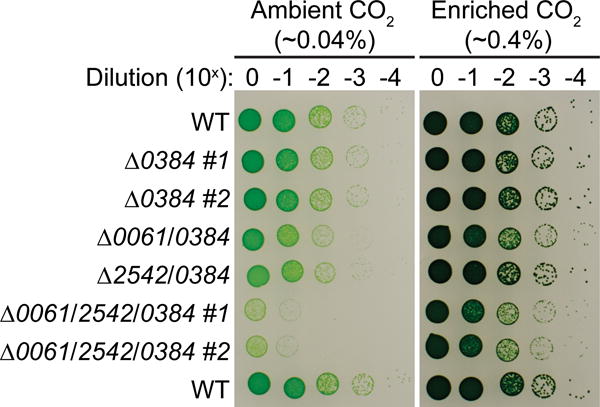
Growth of Δ0384 strains in varying CO2 concentrations
Strains harboring the Δ0384 mutation were spotted on to solid medium in triplicate following serial dilutions of cells suspended at an initial OD 730 nm = 0.05 (Tecan). For the single and triple mutants, two biological replicates were spotted on each plate (#1 and #2). Plates were grown in environmental chambers (38°C, 120 μmol photons m−2 s−1) containing different CO2 concentrations (ambient, ~0.04%; enriched, ~0.4%) for 86 h and then photographed.
Genetic complementation of the RNase III triple-mutant
Since the single mutants do not exhibit a growth phenotype, it is unlikely that the reduced fitness of the Δ0061/0384 and Δ0061/2542/0384 strains is due to polar effects resulting from the gene deletions. To confirm that the reduced growth is directly related to the absence of the RNase III genes, we performed genetic complementation of the most severe mutant lacking all RNase III homologs (Fig. 6). Analysis of double and triple RNase III mutants indicates that growth defects are only observed in strains lacking A0061 and A0384 (Fig. 4 and 5). Thus, we hypothesized that insertion of either of these genes could restore wild-type growth ability to the triple-mutant. The A0384 gene is in an operon, so we chose to insert the native A0061 gene into the genome of the RNase III triple mutant by targeting it to the glpK pseudogene, which has previously been used as a neutral insertion site (Begemann et al. 2014). Full segregation of the A0061 gene in the glpK locus was confirmed by PCR (Fig. 6a). We then compared the growth of the resulting strain, Δ0061+/2542/0384, with the WT and triple-mutant on solid (Fig. 6b) and in liquid (Fig. 6c) media. We calculated doubling times during exponential growth in liquid media for the WT (3.18 ± 0.21 h; n = 6), Δ0061/2542/0384 (3.95 ± 0.28 h; n = 6), and Δ0061+/2542/0384 (3.27 ± 0.11 h; n = 6) strains. The growth characteristics of the Δ0061+/2542/0384 strain were equivalent to the WT strain on plates and in liquid culture, supporting the hypothesis that the growth defect of the Δ0061/2542/0384 mutant strain is specifically due to the absence of RNase III activity.
Fig. 6.
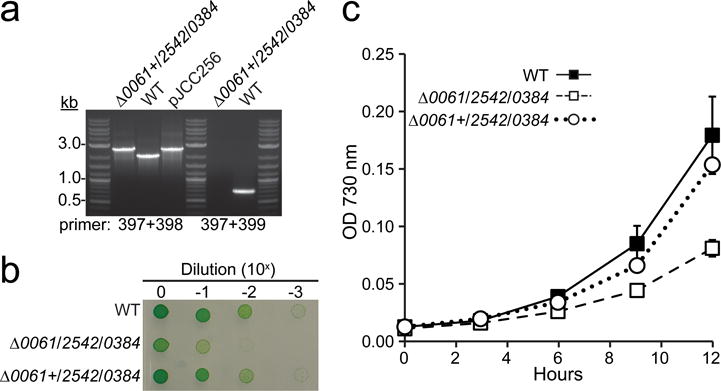
Genetic complementation of the RNase III triple mutant alleviates growth defect
The native A0061 gene was targeted to the glpK pseudogene as a neutral site in the Δ0061/2542/0384 genetic background to generate the Δ0061+/2542/0384 strain. (a) Full segregation of the A0061 gene in the glpK locus was confirmed by PCR using flanking (397+398) and gene specific primers (397+399). WT cells were used as the negative size control and the plasmid used in the transformation was used as the positive control (pJCC256). (b) Overnight cultures grown with bubbling in air (38°C, 200 μmol photons m−2 s−1) were suspended to an OD 730 nm = 0.5 (1 cm cuvette). Tenfold serial dilutions were plated in quadruplicate on media A+ (7.5 μl/spot) and grown for 48 h (120 μmol photons m−2 s−1) prior to being photographed. A representative image is shown. (c) For liquid growth, strains were diluted to an OD 730 nm = 0.01 (1 cm cuvette) and grown in test tubes bubbled for 12 h at a light intensity of 200 μmol photons m−2 s−1. Error bars represent s.d. for three biological replicates.
Genetic deletion of A1543, an RNase II/R homolog, alters growth of PCC 7002
A blast search using E. coli RNase II and RNase R as the query resulted in the identification of two RNase II/R homologs (A0574 and A1543) in PCC 7002 and multiple homologs in other cyanobacterial strains (Table 1). While most genomes contained two homologs, some genomes contained more than three RNase II/R homologs. For instance, the genome of Acaryochloris marina MBIC 11017 contained seven sequences homologous to RNase II/R (Table 1). Moreover, several of these proteins exhibited unique domain architectures such as the presence of an C-terminal dsRBD in place of the typical S1 domain. Like PCC 7002, PCC 6803 contains two RNase II/R homologs (Sll1290 and Sll1910). Recently, the function of Sll1290 was reported to exhibit RNase II, and not RNase R, activity (Matos et al. 2012). In addition, Sll1290 was shown to be essential in PCC6803 (Rott et al. 2003). A phylogenetic analysis comparing the RNase II and RNase R sequences from E. coli to the homologs in PCC 7002 and PCC 6803 indicates that A0574 is more closely related to Sll1290 (Fig. 7a). We also found that A0574 is likely essential for growth in PCC 7002 since we were not able to obtain a fully segregated mutant (Online Resource 3). A1543 branched more closely to Sll1910 (Fig. 7a), a protein previously reported to confer sensitivity to the carbonic anhydrase inhibitor, acetazolamide (Beuf et al. 1995; Bedu et al. 1990). In contrast to A0574, we were able to replace the A1543 gene with a SpR cassette (Fig. 7b). Full segregation of the resulting strain was confirmed by PCR using gene specific and flanking primers (Fig. 7c). The Δ1543 strain exhibited reduced growth when cultured in liquid medium in 12 well plates (Online Resource 5a and 5b) and appeared slightly chlorotic. Comparison of absorption spectra indicates that the altered color of the Δ1543 strain is due to reduced phycocyanin content compared to the WT (Online Resource 5c). Since increasing the CO2 concentration improved the growth of the RNase III strains, we tested whether the addition of 10 mM HCO3− to the liquid medium would improve the growth of the Δ1543 strain (Online Resource 5b). Although the addition of HCO3− resulted in increased clumping in the cultures, we did see an improvement in growth of the Δ1543 strain. Genetic complementation of the Δ1543 strain was accomplished by introducing the native gene into the glpK neutral site. The initial construct for complementation contained the native gene and ~500 bp of upstream sequence. However, this construct failed to rescued the growth defect of the Δ1543 strain (not shown). Thus, we generated an additional construct that included ~700 bp upstream and ~80 bp downstream intergenic regions thought to contain the native promoter and terminator sequences. Segregation of the native A1543 with additional upstream and downstream regions in the glpK locus was confirmed by PCR (Fig. 7d). Growth of the WT, Δ1543, and Δ1543+ strains were compared in liquid (Fig. 7e) and on solid (Fig. 7f) media. Full restoration of growth was observed in both growth regimes, supporting the hypothesis that the A1543 gene confers fitness to PCC 7002. However, the molecular mechanisms leading to the growth defect remain to be identified.
Fig. 7.
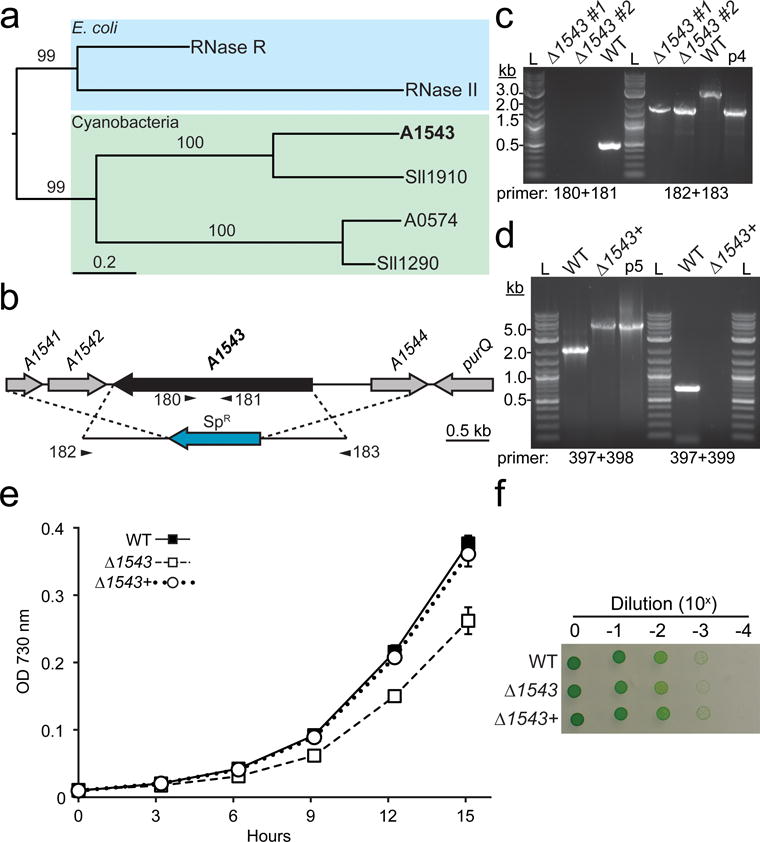
Generation and physiological characterization of a Δ1543 mutant in PCC 7002
(a) Phylogenetic relationship between RNase II/R homologs in E. coli (RNase R and RNase II), PCC 7002 (A1543 and A0574), and Synechocystis sp. PCC6803 (Sll1910 and Sll1290). Scale bar indicates the number of substitutions and values above branches indicate support from 100 bootstrap replicates. (b) The open reading from of A1543 was replaced with a SpR cassette via targeted homologous recombination. Gene specific and flanking primers used in segregation analysis are shown below the gene and the integration cassette, respectively. (c) Segregation of the mutation was confirmed using gene specific and flanking primers. Two independent Δ1543 clones were analyzed (#1, and #2). WT genomic DNA was used as a control. The plasmid used to generate the Δ1543 mutant (p4, pJCC254) was used as a positive size control. L, 2-log ladder. (d) To genetically complement the Δ1543 mutant strain, the native A1543 gene, including the ~700 bp upstream and ~80 bp downstream intergenic regions, was targeted to the glpK neutral site in the Δ1543 background. Segregation of the glpK locus in the resulting strain (Δ1543+) was confirmed using flanking and gene specific primers. Growth of the WT, Δ1543, and Δ1543+ strains in liquid bubbling culture (200 μmol photons m−2 s−1) (e) and solid medium (120 μmol photons m−2 s−1) in ambient CO2 (f). Error bars represent s.d. for three biological replicates. Growth on solid medium was performed in quadruplicate; a representative image is shown.
DISCUSSION
Compared to E. coli and B. subtilis, very little is known about the components and mechanisms of the mRNA decay pathway in cyanobacteria. Genomic analysis shows that cyanobacteria share certain components of the mRNA degradation machinery with E. coli, and other components with B. subtilis. Moreover, a high level of ribonuclease diversity exists among the many cyanobacteria for which genomic information is available. However, the cellular functions and requirement of many of these ribonucleases have not yet been determined and it is clear from this study that predictions based purely on comparisons with E. coli and B. subtilis are not sufficient. This study provides a foundation to studying the function of ribonucleases and RNA modifying enzymes in the cyanobacterium PCC 7002, a model platform for the photosynthetic production of renewable chemicals.
In E. coli, RNase E is an essential ssRNA endonuclease that initiates much of the RNA degradation in E. coli and is part of the mRNA degradosome. The E. coli RNase E contains an N-terminal catalytic domain and a C-terminal scaffolding domain that functions in recruiting degradosome components. The RNase E scaffolding domain confers enhanced cellular fitness (Leroy et al. 2002), but is not essential for viability in E. coli (Kido et al. 1996; Vanzo et al. 1998). A0788 and other cyanobacterial homologs typically have a C-terminal extension of the catalytic domain that does not resemble RNase E, suggesting that cyanobacteria do not contain a degradosome, though recent evidence suggests that the cyanobacterial RNase E interacts with PNPase (Zhang et al. 2014). We were unable to generate a fully segregated mutant of the PCC 7002 RNase E homolog, A0788, which suggests that this protein is essential in cyanobacteria (Online Resource 4); whether a degradosome is essential for function in cyanobacteria remains to be determined. B. subtilis does not contain an RNase E homolog, but instead contains the endoribonuclease RNase Y (Shahbabian et al. 2009). Though a majority of cyanobacterial genomes do not contain an RNase Y homolog, a BLAST search revealed the presence of RNase Y homologs in a single strain. The genome Tolypothrix bouteillei VB521301 encodes a single RNase E/G homolog and two proteins that share ~45% identity with RNase Y.
Based on previous work showing that RNase III is dispensable in E. coli, but required in B. subtilis, it could not be predicted whether RNase III activity is essential in cyanobacteria. In addition, most cyanobacterial species contain multiple homologs of RNase III, including Mini-III, a truncated version lacking the dsRBD (Fig. 2). The PCC 7002 genome encodes two RNase III homologs and a single Mini-III homolog. We were able to delete each of these genes in all single, double and triple combinations, indicating that none of these are essential for growth in laboratory conditions (Fig. 3). Reduced growth of strains containing both Δ0061 and Δ0384 mutations could be due to redundant functions of the encoded proteins (Fig. 4 and 5). An RNase III homolog in the heterocyst producing strain PCC 7120 that is closely related to A2542 in PCC 7002 was implicated in non-coding RNA mediated gene regulation (Gao et al. 2013). Non-coding RNAs have been found to regulate many processes associated with photosynthesis in cyanobacteria (Georg et al. 2014; Sakurai et al. 2012; Eisenhut et al. 2012; Duhring et al. 2006). For example, Sakurai et al. (2012) found that a cis-encoded antisense RNA can stabilize the psbA2 mRNA, encoding the photosystem II D1 protein, by inhibiting processing by RNase E. Therefore, we were surprised that the Δ2542 strain did not exhibit a growth phenotype, even when in combination with Δ0061, which encodes the other full length RNase III homolog (Fig. 4 and 5). However, we did observe a slight increase in photosystem II oxygen evolution activity in the Δ0061/2542 strain, but only at light intensities over 50× higher (8250 vs 150 μmol photons m2 s−1) than typical growth conditions (Online Resource 6).
Multiple RNase II/R homologs could be identified in the genomes of diverse cyanobacteria (Table 1). However, little is known about the functions of these enzymes in cyanobacteria. PCC 7002 contains two proteins, A1543 and A0574, with homology to RNase II/R. We were able to fully segregate a Δ1543 mutant, but the Δ0574 strain could not be fully segregated. The ortholog of A0574, Sll1290 in PCC6803 (67% identity) was recently shown to have RNase II activity (Matos et al. 2012); thus, it is possible that A0574 also shares this activity. A mutant of the A1543 ortholog of in PCC6803, A1910, was isolated in a screen for resistance to the carbonic anhydrase inhibitor acetazolamide and renamed Zam (Beuf et al. 1995; Bedu et al. 1990). However, the mechanism for the observed resistance to acetazolamide was not elucidated. There has been substantial work on a yeast homolog of A1543, Ssd1, a protein that is involved in translational repression through binding and sequestering of mRNAs into subcellular processing bodies (Li et al. 2013), but it remains to be determined whether a similar function exists in cyanobacteria.
CONCLUSIONS
Ribonucleases play critical roles in posttranscriptional regulation of gene expression. However, our knowledge of mRNA turnover in photosynthetic microbes is limited. We find that genetic comparisons between E. coli and B. subtilis, models for mRNA turnover, is not sufficient to understand RNA metabolism in cyanobacteria. Using genomic analysis and genetic tools, we have defined the essential RNases in PCC 7002 and have laid the foundation for future investigations to elucidate the functional roles of these critical enzymes.
MATERIALS AND METHODS
Bioinformatics analysis
Cyanobacterial ribonuclease were identified based on similarity to E. coli and B. subtilis sequences using BLASTP with an E-value threshold cutoff of 10−5 on the Department of Energy Joint Genome Institute Integrated Microbial Genomes (JGI-IMG) database. For RNAse III, multiple sequence alignments were performed using MUSCLE on the EMBL-EBI web platform (McWilliam et al. 2013) with default parameters and ClustalW output format. The RNAse III phylogenic tree was constructed with the maximum likelihood method and the default substitution method on Phylogeny.fr (Dereeper et al. 2008) using the “A la Carte” mode with the built-in curation “remove gap positions” setting. Branch support was performed using 100 bootstrap replicates and the tree was rooted to the midpoint. For the RNase R analysis, the “A la Carte” setting was used to generate multiple sequence alignments MUSCLE (v3.7), curation with Gblocks, and the phylogenic reconstruction with the maximum likelihood analysis and 100 bootstrap replicates. RNase III signature motifs were aligned with MUSCLE (v3.8) and the resulting alignments were used to generate the sequence conservation logos using Weblogo (v2.8.2) (Crooks et al. 2004). RNase III domain organization was analyzed using the sequence search function and the default E-value cutoff equal to 1.0 on the Pfam database (Punta et al. 2012). The cyanobacterial genomes used for analysis of the RNase III domain organization, signature motif, and frequency are listed in Online Resource 4.
Growth and physiological measurements of strains
Strains were maintained on A+ media (Stevens 1973) solidified with 1% (w/v) Bacto-Agar (Fisher). For selection and routine maintenance of mutants, solid media was supplemented with antibiotics (Kan, 100 μg/ml; Spec, 100 μg/ml; Gent, 30 μg/ml; Amp, 2 μg/ml). All growth and physiological assays were performed in media without the addition of antibiotics. For precultures used in solid and liquid growth experiments, glass culture tubes (2 × 15 cm) containing 20 ml media were inoculated with fresh (< 1 week old) plate grown cells and bubbled with air or supplemented with 0.4% CO2 at a light intensity of 150 μmol photons m−2 s−1 in a temperature controlled chamber at 38°C. After approximately 3 days of growth, precultures were diluted to an optical density (OD) at 750 nm = 0.05 and transferred to 12 well culture plates (3 ml/well) for liquid growth assays or tenfold serially diluted in A+ media and spotted onto solid media (7.5 μl/spot). OD measurements were performed on a Tecan M2000 plate reader using Costar 96 well transparent plates and a volume of 200 μl. A light intensity of 150 μmol photons m−2 s−1 was used in all experiments unless indicated otherwise. Growth measurements for Figures 6 and 7 were performed in glass culture tubes bubbled with air at a light intensity of 200 μmol photons m−2 s−1. For these experiments, optical densities were measured on a Genesys 20 spectrophotometer (Thermo Scientific) in a 1 cm cuvette cells and were initially diluted to an OD 730 = 0.01. Oxygen evolution was measured on a Clark-type electrode at 30°C at various light intensities achieved using neutral density filters.
Construction of mutants and segregation analysis
Mutants were generated by targeted gene deletion using homologous recombination as described (Clerico et al. 2007). Briefly, ~500–1500 bp regions upstream and downstream of the gene of interest and an appropriate antibiotic resistance cassette were amplified by PCR using primers that contained 5′ extensions with ~15–20 bp homology with the adjacent fragment. These fragments were assembled using the Gibson Assembly method (Gibson et al. 2009) into a circular plasmid using a pUC19 backbone that was generated by digestion with XbaI. For transformations, 1 ml of three-day old liquid preculture was incubated for ~12 hr with 1 μg plasmid in an environmental chamber with standard light condition and then plated on solid medium containing the appropriate antibiotic. After ~4–5 days, colonies were isolated and patched onto solid medium containing antibiotic. To facilitate segregation of the inserted sequence and removal of the WT sequence, cells were passaged several times on media containing antibiotic. Segregation was analyzed by PCR with GoTaq Green Master Mix (Promega) using primers flanking the insert and with gene specific primers. Full segregation was only concluded if the WT size band was absent from the flanking and gene specific PCR reactions. A list of plasmids used in this study can be found in Online Resource 1. All oligonucleotides used in cloning and segregation are listed in Online Resource 2.
Supplementary Material
Online Resource 1 Table of plasmids used in this study.
Online Resource 2 Oligonucleotides used in this study for generation of plasmids and for segregation analysis.
Online Resource 3 Segregation analysis of ribonucleases in PCC 7002.
Targeted gene deletion via homologous recombination was used to replace the entire ORF of A0788, A1273, A0574, and A1066 with an antibiotic resistance cassette (a, c, e, and g). Insertion of the cassette into each of these genes was confirmed by PCR using primer pairs flanking the insert (Left gel panel in b, d, f, and h). WT genomic DNA was used as the negative control and the plasmid (pJCC#) used to generate the strain was used as a positive size control. PCR using primers that amplify the WT gene (Right gel panel in b, d, f, and h) indicates that these strains are heterozygous at the locus of interest. Arrows and numbers below antibiotic resistance cassette and gene in a, c, e, and g indicate approximate regions of flanking and gene specific primers respectively. For the gene specific reaction shown in g and h, a flanking primer and gene specific primer combination was used to amplify a 1 kb band that should not be present in a fully segregated mutant. Plasmids used as the positive control are found in Online Resource 1. All primers used in segregation analysis are listed in Online Resource 2.
Online Resource 4 Analysis of RNase III-like sequences in diverse cyanobacterial genomes.
Online Resource 5 Growth and physiology of Δ1543 strains.
Growth of WT and Δ1543 strains in liquid medium (12-well tissue culture plates) with shaking (150 rpm) in the absence (a) and presence (b) of 10 mM NaHCO3−. Error bars are s.d. of three cultures. Cells were grown in standard conditions as described in “Materials and Methods”. (c) Absorption spectra of WT, Δ1543, and genetically complemented Δ1543+ strain.
Online Resource 6 Light-dependent oxygen evolution activity of RNase III mutants.
Oxygen evolution was measured as a function of light intensity on a Clark-type electrode in cell solutions suspended at a chlorophyll concentration of 5 μg/ml in A+ media containing 10 mM NaHCO3− as described in “Materials and Methods”.
Acknowledgments
We thank Dr. Bryant for providing the pSRA81 vector. We thank Dr. Himadri Pakrasi for providing access to the oxygen electrode used in physiological measurements. We thank Kymberleigh Romano, Alex Linz, and Alex Sanchez for their help with preliminary experiments. Funding for this work was provided by the Department of Energy (DE-SC0010329). GCG is the recipient of an NIH Biotechnology Training Fellowship (NIGMS – 5 T32 GM08349).
JCC and BFP conceived and designed the study. JCC and GCG performed experiments. JCC, GCG, and BFP analyzed and interpreted the data. JCC and BFP wrote the manuscript.
References
- Abreu RD, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Molecular Biosystems. 2009;5(12):1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, Dunman PM. Messenger RNA Turnover Processes in Escherichia coli, Bacillus subtilis, and Emerging Studies in Staphylococcus aureus. Int J Microbiol. 2009;2009:525491. doi: 10.1155/2009/525491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anupama K, Leela JK, Gowrishankar J. Two pathways for RNase E action in Escherichia coli in vivo and bypass of its essentiality in mutants defective for Rho-dependent transcription termination. Mol Microbiol. 2011;82(6):1330–1348. doi: 10.1111/j.1365-2958.2011.07895.x. [DOI] [PubMed] [Google Scholar]
- Apirion D. Degradation of RNA in Escherichia coli. A hypothesis. Mol Gen Genet. 1973;122(4):313–322. doi: 10.1007/BF00269431. [DOI] [PubMed] [Google Scholar]
- Apirion D, Watson N. Mapping and characterization of a mutation in Escherichia coli that reduces the level of ribonuclease III specific for double-stranded ribonucleic acid. J Bacteriol. 1975;124(1):317–324. doi: 10.1128/jb.124.1.317-324.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apirion D, Watson N. Ribonuclease III is involved in motility of Escherichia coli. J Bacteriol. 1978;133(3):1543–1545. doi: 10.1128/jb.133.3.1543-1545.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedu S, Peltier G, Sarrey F, Joset F. Properties of a Mutant from Synechocystis PCC6803 Resistant to Acetazolamide, an Inhibitor of Carbonic Anhydrase. Plant Physiol. 1990;93(4):1312–1315. doi: 10.1104/pp.93.4.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann MB, Zess EK, Walters EM, Schmitt EF, Markley AL, Pfleger BF. An organic acid based counter selection system for cyanobacteria. PLoS One. 2013;8(10):e76594. doi: 10.1371/journal.pone.0076594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuf L, Bedu S, Cami B, Joset F. A protein is involved in accessibility of the inhibitor acetazolamide to the carbonic anhydrase(s) in the cyanobacterium Synechocystis PCC 6803. Plant Mol Biol. 1995;27(4):779–788. doi: 10.1007/BF00020230. [DOI] [PubMed] [Google Scholar]
- Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, Mathy N, Tobin C, Daou R, Szyk J, Condon C. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol. 2007;63(1):127–138. doi: 10.1111/j.1365-2958.2006.05499.x. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, Luisi BF, McDowall KJ. Endonucleolytic Initiation of mRNA Decay in Escherichia coli. Molecular Biology of RNA Processing and Decay in Prokaryotes. 2009;85:91–135. doi: 10.1016/S0079-6603(08)00803-9. [DOI] [PubMed] [Google Scholar]
- Carpousis AJ, Vanhouwe G, Ehretsmann C, Krisch HM. Copurification of Escherichia coli RNAase E and PNPase: Evidence for a specific association between two enzymes important in RNA Processing and Degradation. Cell. 1994;76(5):889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- Celesnik H, Deana A, Belasco JG. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol Cell. 2007;27(1):79–90. doi: 10.1016/j.molcel.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerico EM, Ditty JL, Golden SS. Specialized techniques for site-directed mutagenesis in cyanobacteria. Methods Mol Biol. 2007;362:155–171. doi: 10.1007/978-1-59745-257-1_11. [DOI] [PubMed] [Google Scholar]
- Condon C. What is the role of RNase J in mRNA turnover? RNA Biol. 2010;7(3):316–321. doi: 10.4161/rna.7.3.11913. [DOI] [PubMed] [Google Scholar]
- Condon C, Putzer H. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res. 2002;30(24):5339–5346. doi: 10.1093/nar/gkf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court DL, Gan J, Liang YH, Shaw GX, Tropea JE, Costantino N, Waugh DS, Ji X. RNase III: Genetics and function; structure and mechanism. Annu Rev Genet. 2013;47:405–431. doi: 10.1146/annurev-genet-110711-155618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008;451(7176):355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–469. doi: 10.1093/nar/gkn180. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhring U, Axmann IM, Hess WR, Wilde A. An internal antisense RNA regulates expression of the photosynthesis gene isiA. Proc Natl Acad Sci U S A. 2006;103(18):7054–7058. doi: 10.1073/pnas.0600927103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Gilet L, Condon C. The essential function of B. subtilis RNase III is to silence foreign toxin genes. PLoS Genet. 2012;8(12):e1003181. doi: 10.1371/journal.pgen.1003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhut M, Georg J, Klahn S, Sakurai I, Mustila H, Zhang P, Hess WR, Aro EM. The antisense RNA As1_flv4 in the Cyanobacterium Synechocystis sp. PCC 6803 prevents premature expression of the flv4-2 operon upon shift in inorganic carbon supply. J Biol Chem. 2012;287(40):33153–33162. doi: 10.1074/jbc.M112.391755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even S, Pellegrini O, Zig L, Labas V, Vinh J, Brechemmier-Baey D, Putzer H. Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 2005;33(7):2141–2152. doi: 10.1093/nar/gki505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Gong Y, Xu X. RNase III-dependent down-regulation of ftsH by an artificial internal sense RNA in Anabaena sp. PCC 7120. FEMS Microbiol Lett. 2013;344(2):130–137. doi: 10.1111/1574-6968.12165. [DOI] [PubMed] [Google Scholar]
- Georg J, Dienst D, Schürgers N, Wallner T, Kopp D, Stazic D, Kuchmina E, Klähn S, Lokstein H, Hess WR, Wilde A. The Small Regulatory RNA SyR1/PsrR1 Controls Photosynthetic Functions in Cyanobacteria. Plant Cell. 2014;26(9):3661–3679. doi: 10.1105/tpc.114.129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Kaberdin VR, Miczak A, Jakobsen JS, Lin-Chao S, McDowall KJ, von Gabain A. The endoribonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc Natl Acad Sci U S A. 1998;95(20):11637–11642. doi: 10.1073/pnas.95.20.11637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido M, Yamanaka K, Mitani T, Niki H, Ogura T, Hiraga S. RNase E polypeptides lacking a carboxyl-terminal half suppress a mukB mutation in Escherichia coli. J Bacteriol. 1996;178(13):3917–3925. doi: 10.1128/jb.178.13.3917-3925.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kime L, Jourdan SS, Stead JA, Hidalgo-Sastre A, McDowall KJ. Rapid cleavage of RNA by RNase E in the absence of 5′ monophosphate stimulation. Mol Microbiol. 2010;76(3):590–604. doi: 10.1111/j.1365-2958.2009.06935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy A, Vanzo NF, Sousa S, Dreyfus M, Carpousis AJ. Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA. Mol Microbiol. 2002;45(5):1231–1243. doi: 10.1046/j.1365-2958.2002.03104.x. [DOI] [PubMed] [Google Scholar]
- Li L, Miles S, Melville Z, Prasad A, Bradley G, Breeden LL. Key events during the transition from rapid growth to quiescence in budding yeast require posttranscriptional regulators. Mol Biol Cell. 2013;24(23):3697–3709. doi: 10.1091/mbc.E13-05-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie GA. RNase E: at the interface of bacterial RNA processing and decay. Nat Rev Microbiol. 2013;11(1):45–57. doi: 10.1038/nrmicro2930. [DOI] [PubMed] [Google Scholar]
- Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129(4):681–692. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- Matos RG, Fialho AM, Giloh M, Schuster G, Arraiano CM. The rnb gene of Synechocystis PCC6803 encodes a RNA hydrolase displaying RNase II and not RNase R enzymatic properties. PLoS One. 2012;7(3):e32690. doi: 10.1371/journal.pone.0032690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600. doi: 10.1093/nar/gkt376. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miczak A, Kaberdin VR, Wei CL, Lin-Chao S. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci U S A. 1996;93(9):3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra TK, Apirion D. RNase E, an RNA processing enzyme from Escherichia coli. J Biol Chem. 1979;254(21):11154–11159. [PubMed] [Google Scholar]
- Mohanty BK, Giladi H, Maples VF, Kushner SR. Analysis of RNA Decay, Processing, and Polyadenylation in Escherichia coli and Other Prokaryotes. RNA Turnover in Bacteria, Archaea and Organelles. 2008;447:3–29. doi: 10.1016/S0076-6879(08)02201-5. [DOI] [PubMed] [Google Scholar]
- Murphy RC, Gasparich GE, Bryant DA, Porter RD. Nucleotide sequence and further characterization of the Synechococcus sp. strain PCC 7002 recA gene: complementation of a cyanobacterial recA mutation by the Escherichia coli recA gene. J Bacteriol. 1990;172(2):967–976. doi: 10.1128/jb.172.2.967-976.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow MC, Perwez T, Kushner SR. RNase G of Escherichia coli exhibits only limited functional overlap with its essential homologue, RNase E. Molecular Microbiology. 2003;49(3):607–622. doi: 10.1046/j.1365-2958.2003.03587.x. [DOI] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py B, Higgins CF, Krisch HM, Carpousis AJ. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996;381(6578):169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- Redko Y, Bechhofer DH, Condon C. Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol. 2008;68(5):1096–1106. doi: 10.1111/j.1365-2958.2008.06207.x. [DOI] [PubMed] [Google Scholar]
- Rott R, Zipor G, Portnoy V, Liveanu V, Schuster G. RNA polyadenylation and degradation in cyanobacteria are similar to the chloroplast but different from Escherichia coli. J Biol Chem. 2003;278(18):15771–15777. doi: 10.1074/jbc.M211571200. [DOI] [PubMed] [Google Scholar]
- Sakurai I, Stazic D, Eisenhut M, Vuorio E, Steglich C, Hess WR, Aro EM. Positive Regulation of psbA Gene Expression by cis-Encoded Antisense RNAs in Synechocystis sp. PCC 6803. Plant Physiol. 2012;160(2):1000–1010. doi: 10.1104/pp.112.202127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009;28(22):3523–3533. doi: 10.1038/emboj.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PM, Wu D, Latifi A, Axen SD, Fewer DP, Talla E, Calteau A, Cai F, Tandeau de Marsac N, Rippka R, Herdman M, Sivonen K, Coursin T, Laurent T, Goodwin L, Nolan M, Davenport KW, Han CS, Rubin EM, Eisen JA, Woyke T, Gugger M, Kerfeld CA. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc Natl Acad Sci U S A. 2013;110(3):1053–1058. doi: 10.1073/pnas.1217107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SEJ, Patterson COP, Meyers J. The production of hydrogen peroxide by blue-green algae: A survey. J Phycol. 1973;9:427–430. [Google Scholar]
- Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 1998;12(17):2770–2781. doi: 10.1101/gad.12.17.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Deng XM, Li FP, Wang L, Huang QY, Zhang CC, Chen WL. RNase E forms a complex with polynucleotide phosphorylase in cyanobacteria via a cyanobacterial-specific nonapeptide in the noncatalytic region. RNA. 2014;20(4):568–579. doi: 10.1261/rna.043513.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Resource 1 Table of plasmids used in this study.
Online Resource 2 Oligonucleotides used in this study for generation of plasmids and for segregation analysis.
Online Resource 3 Segregation analysis of ribonucleases in PCC 7002.
Targeted gene deletion via homologous recombination was used to replace the entire ORF of A0788, A1273, A0574, and A1066 with an antibiotic resistance cassette (a, c, e, and g). Insertion of the cassette into each of these genes was confirmed by PCR using primer pairs flanking the insert (Left gel panel in b, d, f, and h). WT genomic DNA was used as the negative control and the plasmid (pJCC#) used to generate the strain was used as a positive size control. PCR using primers that amplify the WT gene (Right gel panel in b, d, f, and h) indicates that these strains are heterozygous at the locus of interest. Arrows and numbers below antibiotic resistance cassette and gene in a, c, e, and g indicate approximate regions of flanking and gene specific primers respectively. For the gene specific reaction shown in g and h, a flanking primer and gene specific primer combination was used to amplify a 1 kb band that should not be present in a fully segregated mutant. Plasmids used as the positive control are found in Online Resource 1. All primers used in segregation analysis are listed in Online Resource 2.
Online Resource 4 Analysis of RNase III-like sequences in diverse cyanobacterial genomes.
Online Resource 5 Growth and physiology of Δ1543 strains.
Growth of WT and Δ1543 strains in liquid medium (12-well tissue culture plates) with shaking (150 rpm) in the absence (a) and presence (b) of 10 mM NaHCO3−. Error bars are s.d. of three cultures. Cells were grown in standard conditions as described in “Materials and Methods”. (c) Absorption spectra of WT, Δ1543, and genetically complemented Δ1543+ strain.
Online Resource 6 Light-dependent oxygen evolution activity of RNase III mutants.
Oxygen evolution was measured as a function of light intensity on a Clark-type electrode in cell solutions suspended at a chlorophyll concentration of 5 μg/ml in A+ media containing 10 mM NaHCO3− as described in “Materials and Methods”.