

Abstract
Organisms depend upon complex intercellular communication to initiate, maintain, or suppress immune responses during infection or disease. Communication occurs not only between different types of immune cells, but also between immune cells and nonimmune cells or pathogenic entities. It can occur directly at the cell–cell contact interface, or indirectly through secreted signals that bind cell surface molecules. Though secreted signals can be soluble, they can also be particulate in nature and direct communication at the cell–particle interface. Secreted extracellular vesicles are an example of native particulate communication, while viruses are examples of foreign particulates. Inspired by communication at natural immunological interfaces, biomimetic materials and designer molecules have been developed to mimic and direct the type of immune response. This review describes the ways in which native, biomimetic, and designer materials can mediate immune responses. Examples include extracellular vesicles, particles that mimic immune cells or pathogens, and hybrid designer molecules with multiple signaling functions, engineered to target and bind immune cell surface molecules. Interactions between these materials and immune cells are leading to increased understanding of natural immune communication and function, as well as development of immune therapeutics for the treatment of infection, cancer, and autoimmune disease.
I. INTRODUCTION
The immune system is capable of preventing or limiting infection by foreign pathogens or abnormal self cells. However, this ability is compromised in infection, cancer, and autoimmunity. Intervention can be successful in improving immune function, best illustrated by the success of vaccines for infectious diseases. Vaccination has eradicated smallpox and greatly reduced the incidence of polio, measles, rubella, tetanus, diphtheria, and Haemophilus influenzae type b.1 However, vaccines do not exist for other infectious diseases such as human immunodeficiency virus (HIV), which infects 2.6 million new people globally each year.2,3 Vaccine design for this and other diseases remains a challenge. The lack of knowledge of the type of immune effector response required for protection and the functional conditions for optimal activity of effector cells limits rational vaccine development.
Vaccine and immunotherapies are also being investigated for the treatment of cancer.4,5 Cancer is the second leading cause of death in the United States, afflicting more than 1.6 million new people each year.6 It has been established that cancer progresses by exploiting immune check point pathways to escape detection.7 Tumor antigens can be poorly immunogenic, which prevents immune cells from initiating a tumor specific response. In addition, the immunosuppressive tumor environment can also influence the type of immune response.
Though immune activation is the goal for protection against infection and cancer, uncontrolled immune activity also leads to disease. Autoimmune diseases, such as rheumatoid arthritis and type 1 diabetes, affect 23.5 million people in the United States.8 In this case, the immune system's tolerance to self is disrupted due to a combination of genetic and environmental factors.9 The underlying mechanisms that cause this are not completely understood. It is thus critical to identify therapeutic interventions that can help restore a tolerant state in the body.
In all these diseases, immune responses in the body are initiated between cells, cell and infectious agent, or cell and endogenous molecules. During infection, pathogens activate the innate components of the immune system that help in controlling the infection. When the infection is not contained, an adaptive immune response is initiated. During such immune responses, contact based interfaces are often formed between immune cells including antigen presenting cells (APCs), B cells, T cells, and phagocytes.10 This interface sustains contact dependent receptor engagement, polarization of molecules in partner cells, and confined release of secreted products to partner cells.11 Contact independent mechanisms also contribute to the stimulation or suppression of cells through secretion of signaling molecules by the source cells. This indicates that cell based interactions provide opportunities to direct immune responses.
Immunoengineering is an emerging field that seeks to understand and manipulate the immune system. Such fundamental knowledge of the immune system is necessary to create molecules, materials, and strategies that can alter immune responses in a disease setting. Native, biomimetic, and synthetic components can be engineered to achieve such responses through the formation of new immune–material interfaces. How these classes of materials can be used to tune immune responses is described in this review. Native materials, such as extracellular vesicles (EVs), are an emerging class of materials ripe for engineering. These particles contain immune-active molecules and also display functional surface molecules that can be used for targeting and interacting with cells.12 Biomimetic materials are inspired from nature to recapitulate a natural immune function. These particulates are capable of interacting with cells and eliciting specific cellular responses by manipulating parameters such as geometry, presentation of ligands, and incorporation of adjuvants. Though natural materials and biomimicry are valuable tools, the ability to create designer molecules with novel combinations of functions can access signaling and interactions that are not possible otherwise. These synthetic molecules may be more cost effective with advantages of precise control, enabling highly specific and tunable interactions with targets.
II. EXTRACELLULAR VESICLES
EVs are nano- or micronsized structures released by a wide variety of cells.13 Nanovesicles, called exosomes, are 30–100 nm and are produced by extracellular budding of the peripheral membrane of multivesicular bodies or endosomes. Microvesicles, called microparticles (MPs) or ectosomes, are 0.1–1 μm in size and are released from the plasma membrane of cells.14,15 EVs are released by most cells and their composition depends on the cell type and its physiological state.12,16,17 Their membranes are enriched in phosphatidylserine (PS) and contain cytoplasmic proteins, lipid-raft interacting proteins. EVs also encapsulate ribonucleic acid (RNA). The biogenesis and content of exosomes and microparticles are distinct, and have been well reviewed.17 Overall, EVs are important modes of communication between cells.12,17–22 Communication can take place through ligands present on the surface of EVs, transfer of EV contents to cells, or EV uptake through endocytic processes. By such modes of communication, EVs possess the ability to regulate immune responses. They are known to enhance or suppress immune responses and thus have therapeutic potential. For example, EVs from APCs carry major histocompatibility complex (MHC) class I, MHC class II, and costimulatory molecules that can activate CD4+ or CD8+ T cells.20,23,24 On the other hand, EVs from diseased cells, such as cancer cells, can transfer their content to APCs and stimulate or suppress the immune response.25,26 EVs are currently being explored for use as therapeutics, diagnostics, and vaccines for cancer and viral infection.
A. Exosomes
1. Stimulatory exosomes
Exosomes have therapeutic potential by promotion of immune responses, as seen in Fig. 1(a). Exosomes from bone marrow derived dendritic cells (BM-DCs) pulsed with tumor peptide were shown to eradicate tumors or delay tumor growth in mouse mastocytoma or mammary carcinoma models.27 This antitumor effect was attributed to the activation of CD8+ T cells. Tumor cell derived exosomes can also be used to transfer their antigen to dendritic cells (DC) to initiate an immune response, but exosomes derived from DCs pulsed with tumor peptide were much more effective in inducing a specific cytotoxic T cell (CTL) response than tumor cell exosomes.28 In mice deprived of DCs, stimulatory exosomes were unable to initiate CTL responses, highlighting the role of DCs in this immune response. The enhanced immunogenic capacity of DC-derived exosomes compared to tumor-derived exosomes was attributed to the presence of costimulatory molecules on DC-derived exosomes. Furthermore, exosomes from DCs containing both B and T cell epitopes were shown to be superior to those containing only T cell epitopes in protecting against tumor growth, by activating specific CTL responses.29,30 It was demonstrated that intravenously injected exosomes are taken up by DCs and marginal zone (MZ) B cells and transported into the T and B cell splenic zones. Activation and interaction of B cells with CD4+ T cells was shown to enhance antitumor responses. Similar to antitumor examples, exosomes from DCs that have been exposed to Toxoplasma gondii or Leishmania antigens lead to protection against the respective infections.31,32 Exosomes from DCs were also shown to promote strong immune responses by binding toll like receptor (TLR) ligands that activated DCs and increased the ability of these bystander DCs to activate natural killer (NK) cells.33
Fig. 1.

Stimulatory and suppressive effects of EVs: (a) exosomes and (b) microparticles, on the immune system. EVs can specifically interact with immune cells or can have an indirect effect through antigen presentation from dendritic cells.
Exosomes from red blood cells (RBCs) have also been shown to be proinflammatory in nature and drive T cell proliferation in an APC dependent manner.34 Exosomes from NK cells contain proteins that exert cytotoxic activity against tumor cell lines.35 Activated T cells release exosomes that result in the proliferation and cytokine production of T cells in the presence of IL-2.36
Exosomes, as discussed above, have been shown to exert immune responses through MHC-complexes. This suggests that it may be required to use autologous exosomes for therapeutic applications. However, there is some evidence where exosomes derived from human mesenchymal cells were tolerated in immune-competent mice.37 This aspect needs to be addressed to better direct therapies involving exosomes.
2. Suppressive exosomes
Exosomes can also suppress immune activation. Contrary to antitumorigenic effects of tumor derived exosomes (TEX), there is also evidence TEX can suppress antigen specific or antitumor responses. Transfer of plasma-derived exosomes from mice bearing ovalbumin (OVA) expressing tumors was able to suppress antigen specific responses in a delayed hypersensitivity mouse model.38 In addition, TEX contain Fas ligand (FasL), tumor necrosis factor related apoptosis inducing ligand (TRAIL) or programmed cell death ligand 1. Ligation of these ligands promotes CD8+ T cell apoptosis.39–42 TEX have the ability to interfere with T cell receptor (TCR) signaling by inhibition of CD3ζ chain expression in T cells.43,44 They also block the NKG2D dependent cytotoxic activity of NK cells.45,46
TEX can also promote regulatory T cells (Tregs) by enhancing their function or making them resistant to apoptosis. Tregs coincubated with TEX expressed higher levels of suppressive molecules such as CTLA-4, IL-10, and TGF-β1.47 They also block the differentiation of DCs and monocytes through a TGF-β1 dependent mechanism.48 TEX can modulate T cells in the tumor environment through adenosine production by delivery of surface presented CD73 to CD39+ cells.49 These examples highlight TEX as one mechanism by which tumors subvert immune response and also indicate their potential as biomarkers for tumor progression.
Exosomes released by various immune cell types also have a suppressive effect. Exosomes released by Tregs containing miRNA Let-7d resulted in suppression and prevention of systemic inflammation in a T cell deficient mouse model when exosome-mediated miRNA was transferred to Th1 cells.50 The mechanism employed for the transfer of miRNA between immune cells has not been elucidated yet. Similarly, exosomes released by T cells modulate endothelial cell responses such as vascular endothelial growth factor signaling, tube formation, and gene expression in a CD47 dependent manner.51 These vesicles can also modulate TCR signaling through CD47 expression and have implications in tumor angiogenesis. In addition, exosomes released from macrophages suppress endothelial cell migration by controlling integrin trafficking.52 This can help control inflammation by balancing leukocyte recruitment through modulation of integrin expression.
Exosomes can play a role in promoting tolerance. Combination treatment of immature DC-derived exosomes and rapamycin before and after transplant prolonged the survival of cardiac allografts.53 Placental exosomes display FasL and inhibit T cell signaling to reduce immune rejection of the fetus.54 Similar tolerance was observed to exosomes from human mesenchymal stem cells (MSC) delivered to immune-competent mice.37 This suggests that, based on the exosome source and requirement of the application, therapies can be identified for suppression or inducing tolerance.
B. Microparticles
1. Stimulatory microparticles
Though MPs are often compared to exosomes, the difference in their composition has a distinct effect on immune function. Under certain conditions, MPs can be immune-stimulatory. Neutrophils release MPs depending on the condition of stimuli, which dictates their composition and the outcome with target cells.55 MPs were found to be upregulated in inflamed conditions and further activate endothelial cells through a stress induced signaling pathway, as shown in Fig. 1(b).56 In inflammatory conditions such as atherosclerotic lesions, the interaction of leukocytes and platelets is critical. MPs from stimulated neutrophils caused activation of resting platelets through engagement of macrophage-1 (Mac-1) receptor on MPs and sustained thrombus formation.57 MPs from mycobacteria-infected macrophages in two different studies were found to be proinflammatory.58,59 They induced cytokine release and cell migration,59 and presented antigen to naïve T cells.58 Similarly, endothelial, macrophage, T cell, and DC-derived MPs have all been implicated in promoting inflammation depending upon the activation state of the source cell.60–62 MPs play a critical role in promoting inflammation, unlike exosomes that have the ability to initiate specific effector functions.
2. Suppressive microparticles
Like exosomes, MPs can also be suppressive in nature. Neutrophil MPs have PS on their surface, which, upon engagement, inhibits the transcriptional activity of NFκB and modulates activity of macrophages and DCs.63 Release of TGF-β1 by macrophages on exposure to neutrophil MPs further enhances immune-modulatory effects. Similar PS interactions of neutrophil MPs with IL-2/IL-12 treated neutrophils skewed the cytokine profile toward anti-inflammatory by inhibiting production of IFN-γ and TNF-α.64 MPs are also released by platelets when stored for transfusion purposes and have been shown to downregulate and modify immune cells like macrophages and DCs.65
MPs from apoptotic cells also have an influence on immune cells. Apoptosis induced platelet MPs have been shown to promote M2 differentiation of macrophages,66 while MPs from T cells undergoing apoptosis cause apoptosis in macrophages upon uptake.67 Moreover, chemoattractants released by apoptotic MPs mediate the migration of macrophages toward them.68
Intercellular communication through EVs has advantages of specific function based on the cell source and context, as summarized in Fig. 1. EVs also possess good stability due to their lipid composition and have the advantage of preventing complement activation.69 However, there are some practical challenges for use of EVs in therapeutic applications. These include isolation of sterile vesicles and scale up of EV production in sufficiently large quantities.
III. BIOMIMETIC MATERIALS
Biomimetic strategies represent a way to recreate interactions of cells with other cells, pathogens, or molecules with the ability to tune the degree of complexity. This involves understanding key features of cellular communication such as antigenic components, cell entry mechanisms, surface ligands, and tolerization. This section highlights biomimetic strategies based on these features. Some designs seek to recapitulate the structure and chemistry of cell–cell, cell–particle, or cell–molecule interactions, with the hypothesis that function will follow structure. Other approaches are inspired by the natural function but design to achieve function directly, not necessarily with the same structure used in the native system.
A. Cell mimics
Cell mimics are created by using cellular membranes to cloak nanoparticles (NPs) or preparing cell ghosts through a combination of hypotonic treatment and extrusion processes.70,71 Extraction of cellular membranes has been incorporated in various applications. Cellular membranes contain diverse antigenic profiles and specific surface molecules that enable targeting. These membranes can be utilized to alter immune responses by their incorporation on NPs. Cancer cell membrane coated NPs retain their membrane bound tumor antigens, as depicted in Fig. 2(a).70 These particles, in combination with an adjuvant, were shown to induce anticancer immune responses such as stimulation of CTLs. Moreover, these coated particles were shown to use the homotypic binding mechanism, by which adhesive domains on tumor cells form multicellular aggregates, and target cancer cells in vitro.
Fig. 2.

Examples of cell mimic strategies. (a) Cancer cell membrane coated nanoparticles prepared through hypotonic and disruption technique. These coated particles retained the source cell's associated antigens and found applications in delivering tumor-associated antigens to APCs or homotypically targeting cancer cells. Reprinted with permission from Fang et al., Nano Lett. 14, 2181 (2014). Copyright 2014, American Chemical Society. (b) Nanoghosts prepared from MSCs through hypotonic treatment and homogenization. They exhibit MSC surface molecules, targeting abilities and are capable of inducing cancer cell death when loaded with a drug. Reprinted with permission from Toledano Furman et al. Nano Lett. 13, 3248 (2013). Copyright 2013, American Chemical Society. (c) Un-natural killer cells were prepared by functionalizing leukocytes with liposomes coated with E-selectin and TRAIL. They induced apoptosis of circulating cancer cells by activating the death receptor, TRAIL. Reprinted with permission from Mitchell et al., Proc. Natl. Acad. Sci. 111, 930 (2014). Copyright 2014, National Academy of Sciences.
Similarly, MSC membranes can also be used for cancer targeting. MSCs are known for their hypoimmunogenicity and ability to target multiple types of cancer through surface interactions.71 Nanoghosts derived from MSC membranes, as seen in Fig. 2(b), retained the surface molecules of source MSCs. These nanoghosts did not exhibit any immunogenicity in vivo. When loaded with a drug, nanoghosts were shown to inhibit tumor growth of human prostate cancer.
Membrane isolation and cloaking was further extended to RBCs. In autoimmune diseases such as type II, type III, and type IV immune hypersensitivity, autoantibodies against RBC membrane components are produced in the body.72 Nanoparticles that present natural RBC membrane and its associated surface antigens that are involved in antibody-mediated RBC clearance were prepared. RBC cloaked nanoparticles were shown to intercept the autoreactive antibodies of type II immune hypersensitivity reaction. These particles acted as “antibody decoys” that bind to anti-RBC antibodies, protecting circulating RBCs. In another application, nanosponges made of RBC membrane coated nanoparticles provided a platform to absorb pore-forming toxins that are common in bacterial infections and lyse host RBCs.73
In contrast, “un-natural killer cell” mimics were made by functionalizing leukocytes with liposomes presenting E-selectin (ES) and TRAIL, as shown in Fig. 2(c).74 ES and TRAIL are often used to target cancerous cells, and NK cells that take part in immunosurveillance present TRAIL on their surfaces. ES and TRAIL coated liposomes, which mimic NK cells bound leukocytes via selectins on the cell surface under shear flow conditions. These liposomes induced apoptosis of circulating cancer cells in mice. The high compressive force experienced by the cancer cell upon collision with the liposome functionalized leukocytes induced flattening and binding of ligands on the cell surfaces.
Similarly, membrane receptors were immobilized on an artificial, cell-like protocell to prevent infection. Protocells bearing the entry receptor of henipavirus in lipid bilayers were supported on silica particles.75 The protocells specifically and renewably inactivated henipavirus envelope glycoprotein pseudovirus particles. A protein, Ephrin-B2, a protein on the surface of the particle allowed fusion of the viral envelope with the protocell. The protocells were hypothesized to disarm the virus by deactivating a protein that causes the virus to enter the cell. Such biomimetic combination approaches, containing both antigenic information and natural surface properties of cells, could lead to new therapeutics for disease intervention.
In another application, biomimetic leukopolymersomes were assembled from block copolymers and functionalized with sialyl Lewis X (selectin) and an antibody against intercellular adhesion molecule 1 (ICAM-1, integrin).76 These polymersomes bound exclusively to inflamed HUVECs. By optimizing the ratios of selectin and integrin receptor mimics on the surface of the polymersomes, selective binding to inflamed sites and mimicry of leukocyte adhesion was achieved. These can be used to selectively target sites of inflammation and has applications in imaging and drug delivery.
Like synthetic polymers, proteins can also be engineered to possess properties that enable their applications in immunotherapies. Platelets have been implicated in inflammatory processes such as arterial thrombosis that leads to myocardial infarction.77 They can be a useful target at the site of inflammation and when combined with antithrombotic can help reduce the incidence of arterial thrombosis. Multifunctional protein nanomicelles were fabricated from elastin like polypeptides with two components, a single-chain antibody that targets the ligand-induced binding site of activated glycoprotein IIb/IIIa receptors on platelets and the active domain of thrombomodulin.78 These micelles bound activated platelets and inhibited thrombus formation in an in vivo model.
Cell mimetic nanoparticles produced through membrane coating recreate the cellular interface to enable complex cellular processes initiated at the membrane. These particles have the physical properties of the underlying synthetic nanomaterial and are able to avoid opsonization, delay uptake by mononuclear phagocyte system, have long circulation times, bind to target cells, and deliver a therapeutic load.71,72,79 In addition to the abovementioned applications, this technique is also being extended to develop vaccines for bacterial infections.80 The prospects of this membrane coating application are exciting, but the scaling of this technique and immunogenicity needs to be addressed. Synthetic versions can address the issue of scalability, but in vivo testing of these still needs to be pursued.
B. Pathogen mimicking
Pathogens such as bacteria and viruses evade and manipulate the immune system to induce favorable interactions with target cells. Particulate forms of antigen and codelivery of danger signals and antigen have been identified as critical parameters for pathogen mimics for vaccine development. Nanoparticles composed of multilamellar lipid vesicles with antigen entrapped and expressed on the surface were shown to elicit a strong humoral response.81 This strong response was attributed to the formation of germinal B cell centers near the NP depot that facilitated B cell responses. In addition, activation of follicular T cells supported the induction of strong humoral response.
In another study, polyelectrolyte multilayer capsules composed of dextran sulfate and poly-l-arginine with surface-bound CpG adjuvant were engineered to mimic pathogens.82 These CpG coated and OVA encapsulated capsules induced antibody, Th1 and CTL responses. In a similar approach, malaria antigenic triepitope peptide incorporated on a polypeptide layer-by-layer microparticle was shown to induce neutralizing antibodies and malaria-specific T cell responses in vivo.83 These examples suggest understanding the synergistic effect of various pathogen structures and composition can enhance the effectiveness of vaccines. Lipid enveloped PLGA micro- and nanoparticles modified with adjuvants monophosphoryl lipid A and α-galactosylceramide in the layers and OVA antigen on the surface were created to mimic pathogens.84 These particles were shown to elicit strong immune responses such as antibody titers and CD8+ T cell stimulation at doses lower than conventional doses of alum or protein-adjuvant solutions.
An alternative to synthetic approaches is the type III bacterial protein secretion system (T3SS). T3SS delivers bacterial virulence effector proteins into the cytosol of host cells, which stimulates antigen-specific T cells. However, these systems need live bacteria. Inspired by the T3SS mechanism, bacterial minicells were engineered to deliver proteins through encoded Salmonella typhimurium T3SS.85 Bacterial minicells are capable of protein synthesis and metabolic activities, but are incapable of undergoing cell division. In addition to having an immune adjuvant capability, these engineered nanoparticles were shown to deliver an antigen to murine lymphoma RMA cell line in vitro and stimulate production of antigen-specific CD8+ response in vivo. This platform provides an effective strategy to prime immune cells ex vivo for immunotherapy.
Synthetic particles have been successfully used to mimic pathogens for vaccination. This has led to the identification of materials for encapsulation of antigen and tuning immune responses through choice of danger and adjuvant signal that achieve immunization at lower doses than soluble forms of antigen. However, critical features of such immune responses like type of antibodies, degree of affinity maturation of antibodies and optimal T cell response needs to be addressed to achieve effective vaccination.86,87 For these purposes, it might be beneficial to engineer systems like Salmonella T3SS system that retain their natural ability of delivery antigens for processing.
C. Artificial antigen presentation
Artificial antigen presenting cells (aAPCs) can be used for both adoptive and active immunotherapy. These aAPCs should be able to create a physical interface with T cells to present antigen, costimulatory factors, and stimulate cytokine release.88 aAPCs can activate and expand T cells ex vivo, which are then adoptively transferred into a patient.89,90 They can also be injected in vivo to achieve T cell activation. RAFTsomes are liposomes derived from enriched MHC class II lipid rafts of membranes from DCs that were stimulated with antigens.91 They elicit CD4+ T cell priming, as shown in Fig. 3(a), antibody production, and antigen specific responses in vivo. This shows that membrane microdomains enriched in MHC-peptide complexes alone, without costimulatory factors, can achieve an immune response.
Fig. 3.

Artificial antigen presenting cells. (a) Raftosomes contain peptide-MHC II enriched lipid rafts obtained from DCs that were stimulated with antigens. They were capable of stimulating T cells and priming immune responses (Ref. 91). (b) Nano-aAPCs were synthesized by coupling soluble MHC-Ig dimer (signal 1) and B7.1-Ig (signal 2) to the surface of paramagnetic iron oxide, dextran-coated particle. Reprinted with permission from Perica et al., Nanomed. Nanotechnol., Biol. Med. 10, 119 (2014). Copyright 2014, Elsevier. (c) Bull's eye janus particles prepared by microcontact printing. Three-dimensional confocal fluorescence images depict the native and reverse bull's eye particles that display patterns of anti-CD3 and fibronectin. Reprinted with permission from Chen et al., ACS Appl. Mater. Interfaces 6, 18435 (2014). Copyright 2014, American Chemical Society.
Polystyrene bead aAPCs have been functionalized with MHC class I tetramers and costimulatory molecules.92 When coupled with melanoma associated self-antigen and injected intravenously and subcutaneously, they elicited specific CTL responses and delayed tumor progression in a melanoma model of naïve and antigen-primed mice. However, biosafety and organ toxicity is a concern with these systems.
Nanoscale iron–dextran and quantum dot particles coupled with MHC-Ig dimers and anti-CD28, as depicted in Fig. 3(b), were effective in T cell stimulation.93 These nanoparticles induced antigen specific T cell responses in vitro and antitumor activity in vivo. This was attributed to the use of MHC-Ig dimers with a flexible hinge region and the ability of MHC-dimers to enhance TCR and MHC interactions. Unlike micro-aAPCs, these nano-aAPCs were shown to localize in the lymph nodes and distribute away from the injection site. In an extension of this study, these iron–dextran paramagnetic nano-aAPC were shown to facilitate magnetic-field induced T cell activation.94 The magnetic field caused aggregation of these paramagnetic aAPC, which was associated with increased TCR clusters and T cell activation in vitro. This technique was suitable to activate naïve T cells, antigen-specific T cells and such activated T cells were shown to inhibit tumor growth in a melanoma adoptive immunotherapy in vivo model. These studies are the first demonstration of nanoscale aAPC particles with applications in immunotherapy through in vivo administration of nano-aAPC or adoptive immunotherapy through magnetic-field induced nano-aAPC based antigen specific T cell expansion.
Janus particles have also been used as aAPCs to activate T cells. Micron-sized silica particles with a “bull's eye” pattern, shown in Fig. 3(c), that resembles the immunological synapse were fabricated.95 Anti-CD3 was enriched in the central domain and surrounded by fibronectin molecules for adhesion to mimic the native pattern. A reversed pattern with fibronectin as a central domain surrounded by anti-CD3 was also fabricated. The reverse bull's eye pattern enhanced polyclonal T cell activation over the native bull's eye, which performed slightly better than particles coated with fibronectin. This enhanced activation was attributed to a combination of spatial organization and increased surface coverage by anti-CD3.
While Janus particles with the bull's eye pattern captured the importance of ligand spatial organization to enhance interactions between aAPCs and T cells, geometry of aAPCs has also been identified as a crucial parameter for such interactions. Ellipsoidal aAPCs displaying MHC-Ig dimers and anti-CD28 were shown to activate CD8+ T cells in vitro better than spherical aAPCs.96 This improved activation was demonstrated to be dependent on the shape of aAPCs and not differences in the density of the ligands on the surface. These ellipsoidal aAPCs were effective in increasing the survival of mice compared to spherical aAPCs in a subcutaneous melanoma tumor model.
The previous aAPC systems primarily incorporate antigen recognition and costimulation but not cytokine release. PLGA micro-aAPCs with peptide-MHC, costimulatory ligands and encapsulated IL-2 were fabricated to replicate all the key features of APCs.97 CD4+ and CD8+ T cells were stimulated to a greater extent when cocultured with these aAPCs than aAPCs that lack encapsulated IL-2 but were supplemented with the cytokine exogenously. Paracrine delivery of IL-2 upon aAPC contact with T-cell resulted in increased accumulation of synaptic IL-2 and increased proliferation of CD8+ T cells in vitro than exogenous administration of IL-2. Furthermore, these T cell responses were found to be dependent on the sustained release of the cytokine and proximity between aAPCs and T cells.
aAPCs can also be used to activate T cells with chimeric antigen receptors (CARs). Adoptive transfer of T cells with CARs that recognize specific tumor antigens has been shown to be effective in tumor immunotherapy.98 However, this method requires aAPCs to be tuned to the specific antigen recognized by CARs. A universal aAPC was developed with K562 lymphoblast cells expressing a ligand directed toward a conserved extracellular domain on CARs.99 This contributed toward activation of CAR modified T cells, independent of their antigen specificity, but preserved their antigen specificity. These strategies and design parameters provide insight for the development of targeted therapies that circumvent the need for natural APCs to initiate immune responses.
D. Specific ligand immobilization
APCs present antigen to T cells within an immunological synapse that forms between the two cells. Adhesion molecules on these cells aid in the formation of this synapse.100 Synthetic peptides LABL and cIBR have been developed to bind receptors ICAM-1 and lymphocyte function associated antigen (LFA-1) on DCs and T cells, respectively.101 Functionalization of LABL peptides on NP inhibited binding of T cells to DCs through blockade of ICAM-1, as depicted in Fig. 4(a). Similarly, cIBR-NP pretreated T cells interacted with DCs to a lesser extent than untreated T cells. These peptide-functionalized nanoparticles can be used to interfere with the maturation of DCs or T cell expansion.
Fig. 4.

Specific ligand immobilization. (a) cIBR conjugated NP bind LFA-1 or LABL conjugated NP bind ICAM-1 on DCs and block T cell association and activation. Reprinted with permission from Chittasupho et al., ACS Nano 5, 1693 (2011). Copyright 2011, American Chemical Society. (b) Anti-CD3 coated nanoparticles take advantage of receptor clustering upon T cell activation with antigen and induce strong signaling upon binding the cluster (Ref. 102).
T cells expand when activated with antigen and costimulatory signals. Activation of T cells is mediated through the TCR-CD3 complex. On naïve T cells, TCRs are present in nanoclusters on the cell surface that oligomerize into microclusters upon activation.102 Anti-CD3 functionalized quantum dots exploited this difference in TCR clustering to selectively activate antigen-experienced T cells as shown in Fig. 4(b). The sensitivity of TCR clusters in antigen-experienced cells is greater than naïve T cells, which initiates this response. These nanoparticles enhanced antigen-specific T cell responses in vivo and exhibited increased recall response upon challenge. This strategy could also be extended to other clustered receptors such as CD20 on B cells.
CD200R is expressed on immune cells and is an inhibitory immune receptor. It associates with its ligand CD200, a known immune-modulatory protein.103 CD200 functionalized polystyrene microbeads bound CD200R on macrophages and reduced activation and inflammatory cytokine release. These particles also reduced inflammation in vivo. This strategy could be applied to other types of biomaterials to reduce the foreign material immune response.
E. Peripheral tolerance induction
Apoptotic cells and their debris are removed from the body by immune cells without activation of the immune system.104 When these antigens are processed by APCs in the absence of an inflammatory signal, tolerance is induced. This processing is a key mechanism through which peripheral tolerance is maintained. Apoptotic protein/peptide antigens covalently coupled to splenocytes (Ag-SPs), pictured in Fig. 5(a), were shown to induce antigen-specific T-cell tolerance.105 Upon intravenous administration, Ag-SPs initiated IL-10 production by MZ macrophages. IL-10 production was found to regulate the expression of PD-L1 on MZ macrophages, which was critical for tolerance induction. Treg cell induction was also confirmed. In a recent phase I clinical trial for multiple sclerosis, myelin peptide antigens were coupled to autologous peripheral blood mononuclear cells.106 A reduction in myelin-specific autoreactive T cell response was observed.
Fig. 5.

Peripheral tolerance induction. (a) Autologous splenocytes treated with ethylene carbodiimide in the presence of exogenous antigen results in apoptotic antigen bearing debris. This apoptotic debris induces T cell tolerance (Ref. 106). (b) Antigen can be fused to a single chain variable fragment (scFv) specific to proteins present on RBCs. scFv bound RBCs are cleared subsequently upon aging and tolerizes the immune system to the antigen on the RBC debris (Ref. 107). (c) Polymer microparticles conjugated to antigen induce T cell tolerance. Macrophages that take up these particles through the scavenger receptor MARCO play a key role in this response (Ref. 108).
A modular biomolecular approach was developed to induce immunological tolerance by exploiting apoptotic cell carriers. A targeted antigen that binds to the protein glycophorin-A on the surface of mouse erythrocytes was engineered.107 This antigen induced anergy when processed by APCs following erythrocyte-apoptosis, as shown in Fig. 5(b). In a mouse model of autoimmune diabetes, deletion of autoreactive T cells was achieved when β-cell antigen was bound to erythrocytes through a designed antibody construct. This technique could also be applied to other antigens for tolerization.
Particle based tolerogenic approaches are also being pursued. Encephalitogenic antigen peptide covalently coupled to polystyrene or biodegradable PLGA microparticles, as depicted in Fig. 5(c), were shown to induce T cell tolerance in a mouse model of experimental autoimmune encephalomyelitis.108 MZ macrophages internalized these particles through scavenger receptor, macrophage receptor with collagenous structure (MARCO), and played a key role in regulating the response. This tolerance induction depended both on T cell anergy and activity of Tregs. In a related approach, nanoparticles were coated with different peptide-MHC complexes to blunt the polyspecific autoimmune responses in a type 1 diabetes model.109 Nanoparticles coated with disease-relevant peptide-MHC complexes were also shown to expand cognate “autoregulatory” T cells. These antigen experienced CD8+ autoreactive T cells were found to suppress the activation and recruitment of other noncognate specificities. This technique can be extended to other diseases by incorporating relevant antigen.
Extending the particle strategy, synthetic nanoparticles encapsulating protein or peptide antigens and tolerogenic immunomodulator rapamycin were shown to induce tolerance toward the antigen.110 These nanoparticles were effective in models such as allergic hypersensitivity disorder, multiple sclerosis, and hemophilia A. Rapamycin has been established to induce tolerance even in the presence of inflammation. Similarly, these nanoparticles retained their tolerogenic potential even when coadministered in the presence of antigen and TLR agonists. This technique can address the issue of anaphylaxis, where antigen administration during inflammation often leads to disease exacerbation.
A biodegradable PLGA nanoparticle platform was developed by an emulsion process using poly(ethylene-co-maleic acid) as a surfactant.111 These particles were investigated for their ability to couple to antigen, and safety and protection in a mouse multiple sclerosis model. The incorporation of the surfactant into the PLGA particle synthesis was shown to mitigate the disease, relapses, and minimize epitope spreading during intravenous administration. These particles had superior performance than commercially available nanoparticles, but the mechanism has not yet been established. These antigen-coupled particles were demonstrated to be safe and not induce an anaphylactic reaction. This biodegradable nanoparticle platform can be extended to other diseases by changing the antigenic epitopes.
Biomimetic materials take advantage of the physicochemical properties of nano- and micromaterials. The nanomaterials, owing to their size, can enable targeting of specific cell types such as those in the lymph nodes.112 These materials can also be tuned to mimic cells or pathogens while retaining a key advantage of synthetic materials, mass production. They represent an exciting area where engineering principles can be applied to biologically inspired design to achieve immunologically active materials.
IV. DESIGNER MOLECULES
In contrast to biomimetic particles presenting native membrane or ligands, designer immunomodulatory molecules have features not seen in natural immune molecules. They have the potential to induce immune function that is not possible with a single natural molecule or entity, through creative design involving novel signal combinations and alternative spatial or temporal presentation. These structures have precise molecular control and can be used to study immune function or develop new immune therapeutics or vaccines. Antibody–drug conjugates are not covered here as often their use is not for immunomodulation, but rather for targeting or pharmacokinetic benefits.113 Designer antibodies, toll-like receptor agonists, and carbohydrate molecules, as well as approaches to design molecules present on living cells, are described.
A. Antibodylike molecules
Antibodies are widely engineered molecules but much of their modification has been to achieve new or higher affinity binding to targets. This section describes antibodylike molecules that have been designed to achieve better immune function. Synthetic antibody mimics (SyAMs) are medium sized (7 kDa) molecules that have both the targeting and effector function of antibodies.114 Like antibodies, they are capable of forming a three-part complex with a target cell (prostate cancer in this work) and an immune effector cell, as shown in Fig. 6. Unlike antibodies, SyAMs have bivalent Fc receptor (FcR) binding as well as bivalent target binding. Modeling of the three-part complex supported experimental results that bivalent FcR binding significantly enhanced phagocytosis of target cells more than bivalent target cell binding. Importantly, bivalent FcR binding did not result in nonspecific activation of effector cells in the absence of target cells. Since SyAMs are ∼1/20th the size of antibodies and are synthesized chemically, they have delivery, stability, and immunogenicity advantages over traditional antibodies.
Fig. 6.
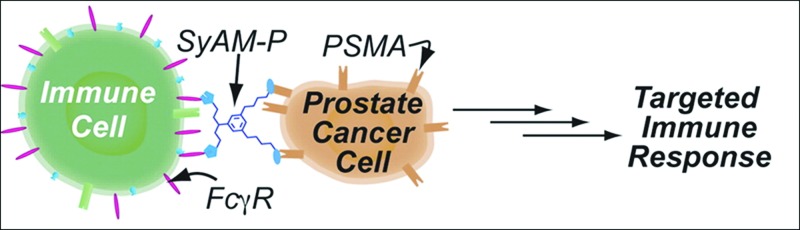
Synthetic antibody mimic. SyAM displays a pair of PSMA targeting motifs to bind prostate cancer cells and a pair of FcγRI targeting motifs to bind immune cells. This simultaneous binding elicits selective phagocytosis of cancer cells. Reprinted with permission McEnaney et al., J. Am. Chem. Soc. ▪, ▪ (2014). Copyright 2014, American Chemical Society.
While SyAMs operate as independent immune effector molecules, antibody-recruiting molecules (ARMs) are synthetic small molecules that bind antigens and enhance host antibody binding to cells bearing those antigens.115 ARMs bind target cells on one end and display a recruiting antigen on the other end that binds known native or supplemented antibodies. They can engage both complement dependent cytotoxicity and antibody dependent cellular cytotoxicity to enhance the native immune response for applications in cancer and bacterial and viral infections, including HIV (Ref. 116) and prostate cancer117 most recently. Additionally, they can be used as tools to study human antibodies including variation across people, subtypes, and antigens, as well as to identify antigens with the greatest therapeutic potential.117,118 The history and varied use of ARMs has been recently reviewed.115 The breadth of targeting capabilities and the ability to utilize endogenous antibodies makes this small molecule platform amenable to scale up and translation.
Unlike SyAMs and ARMs that replicate a part of or recruit antibodies for effector function, antibody functionalized polyisocyanides are a class of polymers being explored for antibody display to exert immune function. Polymerization of isocyanopeptides leads to a helical backbone stabilized through hydrogen bonding between side chains.119 This helical β-sheet like structure forms a rodlike polymer. This polymer exhibited semistiffness and well-defined stereoregularity based on its persistence length. These filamentous polymers were functionalized with anti-CD3 antibodies and activated T cells through CD3 ligation seven fold more efficiently than PLGA spherical counterparts displaying the same number of antibodies and spacing.120 This enhanced interaction was attributed to the structural architecture of the polymer that enabled superior receptor–ligand interactions. In an extension of this study, both anti-CD3 and anti-CD28 were functionalized on the polymer.121 This combination of ligands activated effector CD8+ and memory CD4+ T cells. The CD8+ T cells exhibited cytotoxic activities like production of granzyme B and degranulation marker CD107a. This highlights a unique molecular design factor, semiflexibility, which enables excellent mimicking of DCs. Further, these polymers have potential in applications requiring antigen specific immune responses.
B. Toll-like receptor ligands
TLRs are a family of receptors that recognize different chemical species and play an important role in activation of innate immune responses. In native pathogens, multiple TLR agonists are displayed with particular spatial and temporal patterns. Molecular strategies are being developed to present synthetic TLR agonists under controlled spatial and temporal conditions. To spatially control presentation to two TLRs, TLR 2 (lipoteichoic acid) and TLR 9 (CpG DNA) agonists were conjugated via an α, ω-heterotelechelic polyethylene glycol linker.122 In in vitro stimulation assays with APCs, the connected agonists induced greater immune stimulation of both polarizing cytokines and T cell adhesion proteins, compared to either agonist alone, mixtures of unconjugated agonists, or lipopolysaccharide. Temporal control was achieved by photocaging. Soluble TLR7 and TLR8 agonists Imiquimod and Resiquimod were synthesized in photocaged forms.123 The caged agonists were inactive until application of UV light, which induced their activation of APCs, as shown in Fig. 7(a). Combinations of these approaches and inclusion of other TLR agonists will enable more complex stimulation patterns for basic study and mimicry of natural pathogens for vaccination.
Fig. 7.

Toll-like receptor agonists. (a) Deprotection of the photocaged agonist in the presence of light causes TLR7/8 activation and subsequent signaling. Reprinted with permission from Ryu et al., J. Am. Chem. Soc. 136, 10823 (2014). Copyright 2014, American Chemical Society. (b) Synergistic combination of agonists on the surface of a tumor cell, LTA for TLR2/6 and CpG-oligonucleotides for TLR9, causes enhanced stimulation in immune cells. Reproduced with permission from Tom et al., Chem. Commun. 49, 9618 (2013). Copyright 2013, the Royal Society of Chemistry.
TLR3 recognizes double stranded viral RNA, and its synthetic analog polyinosinic–polycytidylic acid (poly I:C). Poly I:C is synthesized enzymatically and is not well defined, containing a range of molecular weights and unpaired strands. A method was developed to design and synthesize double stranded synthetic oligoribonucleotides (dsORNs) of defined length using solid phase synthesis.124 The ORNs consist of an alignment region and either a poly-I or poly-C tail. The alignment regions of two ORNs were designed to be complementary and hybridize, leaving the tails exposed on either end. Self-assembly of the ORNs through hybridization and formation of double I:C helices in the tails resulted in long double stranded structures. The lengths of the alignment and tail regions were found to be critical to self-assembly of stable structures and subsequent TLR3 activation. The optimized dsORNs activated TLR3 in vitro and stimulated immune responses in mice and nonhuman primates in vivo. Future work will investigate the use of dsORNs as adjuvants in vaccine formulations.
The TLRs that recognize nucleic acids (TLR3, 7, 8, 9) can also play key roles in autoimmune disease through recognition of self DNA or RNA. However, direct inhibition of these TLRs can compromise the ability to fight viral and bacterial infections. Synthetic nucleic-acid scavenging polymers (NASPs) were screened and several candidates identified that can inhibit TLR activation by binding a variety of nucleic acid types and inhibiting nucleic acid interaction with TLRs.125 Generation 3-polyamidoamine dendrimer on a 1,4-diaminobutane core (PAMAM-G3) was found to be the most promising because, unlike some others studied, it did not inhibit nucleic-acid independent T cell activation or TLR recognition of encapsulated nucleic acids such as those in viruses.126 NASPs have the potential to serve as therapeutics for nucleic acid related autoinflammation with less effect on protective immune function during infection.
Taking cues from molecular strategies, cell surfaces have also been modified to direct immune stimulation through TLRs. Lewis lung carcinoma cell surface proteins were chemically modified with a polymeric linker to incorporate lipoteichoic acid (TLR-2/6 agonist) and CpG-oligonucleotides (TLR 9 agonist) as depicted in Fig. 7(b).127 This synergistic combination of agonists induced enhanced immune stimulation in a murine macrophage reporter cell line and BM-DCs in vitro. This could enable the design of effective vaccines by combining multiple danger signals and antigens.
C. Carbohydrate ligands
Carbohydrates are on the surface of all cells, self, and foreign, and their recognition and impact on subsequent immune responses is critical to understand. Dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) is an example of an immunologically important carbohydrate. It is a lectin on DCs that has activity in antigen presentation, T cell activation, and also can be exploited by HIV and other pathogens to aid infection. In order to better understand the ligands and function of DC-SIGN, a synthetic noncarbohydrate glycomimetic from a shikimic acid scaffold was designed.128 The glycomimetic not only binds DC-SIGN but it also acts as a functional agonist, undergoing internalization and signaling. Only synthetic antagonists have been made prior. Molecules like this, and those designed along similar principles, will not only be valuable probes in the understanding of the complicated roles of lectins in immunity, but also in the design of potentially therapeutic agonists and antagonists.
Ebola virus utilizes DC-SIGN to induce uptake in DCs. To inhibit this, a nanoparticle was designed to competitively bind DC-SIGN.129 To achieve a size and carbohydrate density similar to viruses, a combination synthetic-recombinant approach was used. Mannose glycodendrons were made synthetically and attached via click chemistry to recombinant Qβ viruslike particles bearing non-natural alkyne-containing amino acids on their surface. The resulting nanoparticles bear up to 1620 glycans each, the highest density reported with quasisymmetry. The constructs were able to bind DC-SIGN and block a model Ebola infection in vitro. Though future in vivo work will determine if infection can be prevented in animals, this work highlights immunological applications for controlled multivalent carbohydrate structures. Recent reports have also described fullerenes and ferritin cages as nanoparticles on which carbohydrate moieties can be immobilized.130,131
CD22 is an immune-inhibitory lectin that colocalizes with the B cell receptor (BCR). It has been difficult to decouple BCR endocytosis versus signaling. To address this, a set of synthetic multivalent, defined BCR antigen polymers have been created.132 One contains only BCR stimulatory antigen (dinitrophenol, DNP) and the other contains both DNP and an inhibitory sialylated CD22 ligand. These polymer antigens revealed that both types of ligands induced BCR internalization but do so at different rates. The combination polymer with inhibitory ligands is internalized faster and depletes the cell surface of BCRs more quickly, while the stimulatory-only ligands increase BCR surface duration to allow for more stimulation. Future work with these antigens and newly designed ones will not only contribute basic understanding to BCR endocytosis and signaling but also inform antigen design for both vaccination and autoimmunity.
D. Cell surface engineering
Molecular design is also being applied to the cell surface for new immune-therapies. Metabolic oligosaccharide engineering was applied to the Helicobacter pylori surface.133 Cells metabolically incorporate unnatural sugars containing biorthogonal azides into cellular glycans. Phosphine probes conjugated to immune stimulants, such as DNP, were targeted to these azide-functionalized glycans through Staudinger ligation as seen in Fig. 8(a). This technique was shown to elicit cytotoxicity via effector cells. It might be useful to selectively label pylori ex vivo to direct antibodies to its surface prior to in vivo injection for vaccination or to enhance the response against an active infection. It has been established that anti-DNP antibodies exist naturally in humans and this strategy can have potential for pylori killing.134
Fig. 8.

Cell surface engineering. (a) The glycans of H. pylori are selectively labeled with azide-containing sugar. The azide undergoes Staudinger ligation with an immune stimulant. This surface modification enables the immune system to trigger a response resulting in H. pylori death. Reprinted with permission from Kaewsapsak et al., ChemBioChem 14, 721 (2013). Copyright 2013, Wiley. (b) Mucin domain of CXC3CL1 was combined with a glycosylphosphatidylinositol anchor and integrated into the cell membranes of Chinese hamster ovary and endothelial cells. The CXCL10 chemokine head enabled the recruitment of leukocytes to the tumor microenvironment. Reprinted from Ref. 135: Muenchmeier et al., PLoS One 8, e72749 (2013).
In another study, a novel CXCL10-mucin-glycosylphosphatidylinositol (GPI) fusion protein was designed to enhance the recruitment of NK cells, whose infiltration is poor at tumor sites. The fusion protein was demonstrated to integrate into the cell membranes of Chinese hamster ovary or endothelial cells as seen in Fig. 8(b). This fusion protein was shown to take advantage of the GPI anchor and integrate into any cell membrane. Fusion protein incorporated endothelial cells were shown to recruit NK cells under physiologic flow in vitro. This fusion protein was also found to recruit NK cells to a subcutaneous tumor when injected at the tumor site in vivo.135
Glycocalyx engineering is another technique to mediate cell responses. Modification of cancer cells, allogeneic hematopoietic stem cells and xenogeneic porcine cells with synthetic sialylated glycopolymers protected them from NK cell cytotoxicity.136 The engagement of the receptor Siglec-7 on NK cells by these glycopolymers induced an inhibitory signal and decreased NK cell activity. It suggests hypersialylation to be a potential mechanism by which tumor cells confer resistance. These polymers can increase understanding of Siglec-7 based mechanisms in cancer biology and lead to new therapeutics targeting this receptor.
A one step modification of cell surfaces using sortase A has been recently employed.137 Sortase A was used to conjugate LPTEG-tagged probes to glycines present naturally on the cell surface. This technique was shown to effectively redirect immune responses based on the domains sortase-tagged on the surface of cells. Sortase-tagged conjugation of single domain antibodies on activated T cells redirected cytotoxicity toward cells expressing the antigen. Similarly, parasites sortagged with antibodies were shown to target cells that bear the relevant antigen.
Most of the cellular engineering approaches require extensive ex vivo processing, which limits the applicability of these techniques. Retention of molecules introduced on the surface is critical and the normal degradation or internalization of cell membrane components will affect the density of engineered molecules in the membrane.138 In addition, these modifications should also be compatible with in vivo factors such as protein adsorption that can affect the efficacy of cell-engineered molecules. These challenges limit the translation of these approaches into clinic but they can still be valuable for basic understanding.
The designer molecules described can be precisely tailored for specificity and in turn control the type of immune response. However, issues such as immunogenicity to these molecules can arise.139 These molecules can also result in adverse effects like autoimmunity or anaphylaxis.140 In addition, directing specific responses toward cells through these molecules can also result in systemic inflammatory reactions like tumor lysis syndrome or cytokine storm. Though these molecules hold promise for the future, thorough in vivo testing needs to be carried out.
V. CONCLUSION
Although immune-engineering is a relatively new field, a wide variety of materials and molecules have been produced to interact with the immune system in various ways. Extracellular vesicles, biomimetic materials, and designer molecules all have different building blocks, physicochemical properties, and immunological function. What they have in common is the ability to generate an immunological molecular interface that induces either activation or suppression of immune cell functions. The number, type, and arrangement of signals present in the interface all influence the outcome. Biomimetic materials generally present combinations of these properties as they are observed in natural immune interfaces between cells, cells and particles, or cells and molecules. Designer molecules present these properties in ways that may not exist in native immune systems. As products of cells, these properties cannot be controlled in EVs.
Artificial vesicles are being explored to exercise control over exosomelike functionalities by manipulating the type of signals required. Liposomes derived from lipids of intestine-derived exosome like nanoparticles were shown to induce similar reduction in natural killer T cell (NKT) cytokine production as the native structures.141 The native structures were shown to induce NKT cell anergy through interaction between prostaglandin E2 on the native structures and its associated receptor on NKT. Incorporating such lipids on liposomes can help retain native functionality but allow control over physicochemical properties. In another synthetic EV approach, liposomes were prepared by coupling MHC-cytomegalovirus (CMV) peptide complex and costimulatory Fab fragments and encapsulating iron oxide NP. Like exosomes, these liposomes were shown to activate antigen-specific CD8+ cells through APCs when incubated with peripheral blood mononuclear cells from CMV-positive individuals in vitro.142 Physical methods like serial extrusion can also be used to prepare exosome-like nanovesicles. These nanovesicles were shown to retain the counter-receptor LFA-1 and thus mimic the targeting ability of the source macrophage cell. The targeting ability enabled delivering a therapeutic load to activated endothelial cells in vitro and when injected intravenously in vivo in a subcutaneous tumor model.143
Though these strategies represent ways in which exosomelike mimetics can be engineered retaining concerned surface molecules, it is also crucial to impart good functionality. Exosomes contain various candidate membrane proteins that can be incorporated into mimetics for enhancing immune functionality.144 Tetraspanins like CD9, CD63, CD81, and CD82 are enriched on exosomes and play a role in binding to target cells. Similarly, other proteins like CD55 and CD59 are found in exosomes from diverse cell types and protect exosomes from complement-mediated lysis. Incorporation of tetraspanins in addition to antigenic ligands may enhance binding of exosome-mimetics like liposomes to target cells. Inclusion of CD55 and CD59 on liposomes may hamper their lysis and improve circulation.
As potential immune therapeutics, each class of materials described in this review has advantages and disadvantages. EVs have the benefit of complexity and multifunctionality. They contain proteins and RNA of different types, both on the membrane and inside. These molecules are displayed or contained in a manner that preserves their activity, but there is less control over EV properties, contents, and function. The cell type and state or environment of the cell can be selected to modulate EVs. Because cells produce EVs, no synthesis processes are required. However, separation, purification, and sterilization of EVs are significant challenges, as well as scale-up of production.
Biomimetic materials are diverse and have a variety of benefits and challenges. These materials provide the opportunity to mimic natural compositions of cells through cloaking, or chemically decorate the surface with one or more ligands, in an isotropic or anisotropic way through the employment of patterning techniques. In addition, the spatial organization of these ligands can be altered over time as in a cell. The example of a protocell designed to inactivate henipavirus achieves this by taking advantage of the protocell's membrane fluidity for dynamic ligand presentation.75 As seen in the section on peripheral tolerance mechanisms, these materials can also be tuned to replicate natural mechanisms through biomolecular approaches. Though these materials can be tuned for varied applications, it still remains a challenge to understand how they can achieve a specific response such as controlled immune-suppression in autoimmune diseases, effective antitumor response through cancer vaccination by prevention of Treg induction in tumor microenvironment, or eliciting sustained levels of high-affinity antibodies for vaccination.
Designer molecules are synthetic. This gives them advantages of being well-defined chemically, which can lead to better reproducibility and regulatory potential. It also allows for fine control of the architecture of molecules to tune immune function. However, the possible designs are, in theory, infinite so high-throughput synthesis and screening methods may be needed to efficiently produce new functional molecules. For those that do have therapeutic potential, more synthesis steps may be required, but purification and sterilization may be easier than cell derived products.
As with any material to be introduced in the body, immunogenicity is always a concern. For immune-stimulatory materials such as vaccines, which are administered one or few times, there is a risk of overstimulation or cytokine storm.145 For immune suppressive materials that may be used to treat autoimmune or other chronic inflammatory states, repeated administration could lead to antibodies against the material. The risk of either of these outcomes could depend significantly on the immune and disease status of the individual and may vary widely. Additionally, there could be unintended consequences of immune signaling or targeting by these materials. In the body, many immune molecules have multiple functions and balance is maintained via feedback and control mechanisms. Even natural materials, like EVs, are likely to have functions that are not yet known. Cancer immune therapy trials have revealed that not all patients respond in the same manner.146 While some have durable disease remission, others have severe immune-related side effects. Careful assessment of immune tuning materials must be performed in suitable in vivo models to identify risk.
In the case of vaccination, dose sparing of adjuvants can help reduce side effects. Lipid coated PLGA MP or NP represents a promising strategy to address this. These particles can mimic pathogens effectively by encapsulating hydrophilic antigen in the core and entrapping hydrophobic antigen or adjuvants on the lipid envelope.84 Such incorporation facilitated a 12-fold increase in antibody titers compared to lipid-enveloped particles. Contrary to this, immunotherapy involving treatment with immune-cell activating adjuvant without administration of antigen is being explored for cancer. Pluronic-stabilized poly(propylene sulfide) and pyridyl sulfide NPs (30 nm) were prepared by encapsulating paclitaxel (PXL, TLR 4 agonist) or conjugating CpG (TLR 9 agonist) to the surface.112 These NPs when delivered to the tumor draining lymph node (TDLN) were able to induce DC maturation and achieve antitumor responses by inducing antigen-specific CD8+ T cells in the tumor. CpG-NPs were found to bias a Th1 immune response whereas PXL-NP reduced Treg numbers in the TDLN. Similarly, the polymer composition associated with the antigen altered the type of T cell response initiated by the lymph node APCs. OVA encapsulated PLGA NP but not N-trimethyl chitosan-tri-polyphosphate were found to induce FoxP3 in activated T cell via expression of retinaldehyde dehydrogenase enzymes in cervical lymph node dendritic cells in vitro.144 Furthermore, immune-modifying particles derived from carboxylated biodegradable poly(lactic-co-glycolic) were shown to reduce inflammatory monocyte mediated pathology in various disease models including experimental autoimmune encephalomyelitis when injected intravenously in vivo.147 These highly negatively charged particles were taken up by inflammatory monocytes via the scavenger receptor MARCO. Subsequently, these monocytes were sequestered in the spleen through apoptosis and thus alleviating inflammation. The relationship between the particle uptake and apoptosis induction is not yet known. These studies are exciting as they showcase the ability of the immune system to reshape the environment without the need for multiple signals. This also highlights the need to understand how material properties can be tuned to achieve immune responses without the need for antigen.
As the field of immune-engineering grows, so will opportunities to manipulate immune interfaces with natural or synthetic materials. Advances in understanding of immune cell function and signaling in health and disease will create new sources of bioinspiration and new ideas for therapeutic interventions. These will be coupled with improving chemical synthesis and molecular control, nano- and microparticle fabrication, and practical scale up and production.
ACKNOWLEDGMENTS
The authors acknowledge funding from the National Institutes for Health (AI101047) and Georgia Tech Emory Center for Regenerative Engineering & Medicine.
References
- 1. Plotkin S. A., Nat. Med. 11, S5 (2005). 10.1038/nm1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rappuoli R. and Aderem A., Nature 473, 463 (2011). 10.1038/nature10124 [DOI] [PubMed] [Google Scholar]
- 3. Germain R. N., Immunity 33, 441 (2010). 10.1016/j.immuni.2010.09.014 [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg S. A., Yang J. C., and Restifo N. P., Nat. Med. 10, 909 (2004). 10.1038/nm1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vonderheide R. H. and Nathanson K. L., Nat. Med. 19, 1098 (2013). 10.1038/nm.3317 [DOI] [PubMed] [Google Scholar]
- 6.American Cancer Society, Cancer Facts & Figures 2013 (American Cancer Society, Atlanta 2013).
- 7. Malmberg K.-J., Cancer Immunol. Immunother. 53, 879 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Autoimmune Diseases Coordinating Committee, NIH Publication 05-5140, The Autoimmune Diseases Coordinating Committee, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, 2005. Progress in Autoimmune Disease Research, Report to Congress.
- 9. Smilek D. E., Ehlers M. R., and Nepom G. T., Dis. Models Mech. 7, 503 (2014). 10.1242/dmm.015099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fooksman D. R. et al. , Annu. Rev. Immunol. 28, 79 (2010). 10.1146/annurev-immunol-030409-101308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Angus K. L. and Griffiths G. M., Curr. Opin. Cell Biol. 25, 85 (2013). 10.1016/j.ceb.2012.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcus M. E. and Leonard J. N., Pharmaceuticals 6, 659 (2013). 10.3390/ph6050659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vlassov A. V., Magdaleno S., Setterquist R., and Conrad R., Biochim. Biophys. Acta, Gen. Subj. 1820, 940 (2012). 10.1016/j.bbagen.2012.03.017 [DOI] [PubMed] [Google Scholar]
- 14. Gasser O. and Schifferli J. A., Blood 104, 2543 (2004). 10.1182/blood-2004-01-0361 [DOI] [PubMed] [Google Scholar]
- 15. Cocucci E., Racchetti G., and Meldolesi J., Trends Cell Biol. 19, 43 (2009). 10.1016/j.tcb.2008.11.003 [DOI] [PubMed] [Google Scholar]
- 16. György B., Hung M. E., Breakefield X. O., and Leonard J. N., Annu. Rev. Pharmacol. Toxicol. 55, 439 (2014). 10.1146/annurev-pharmtox-010814-124630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ling Z. L., Combes V., Grau G. E., and King N. J. C., Front. Immunol. 2, 67 (2011). 10.3389/fimmu.2011.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parolini I. et al. , J. Biol. Chem. 284, 34211 (2009). 10.1074/jbc.M109.041152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montecalvo A. et al. , Blood 119, 756 (2012). 10.1182/blood-2011-02-338004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Segura E., Guérin C., Hogg N., Amigorena S., and Théry C., J. Immunol. 179, 1489 (2007). 10.4049/jimmunol.179.3.1489 [DOI] [PubMed] [Google Scholar]
- 21. Calzolari A. et al. , J. Cell Sci. 119, 4486 (2006). 10.1242/jcs.03228 [DOI] [PubMed] [Google Scholar]
- 22. Robbins P. D. and Morelli A. E., Nat. Rev. Immunol. 14, 195 (2014). 10.1038/nri3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raposo G., Nijman H. W., Stoorvogel W., Liejendekker R., Harding C. V., Melief C. J., and Geuze H. J., J. Exp. Med. 183, 1161 (1996). 10.1084/jem.183.3.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kolowos W., Gaipl U. S., Sheriff A., Voll R. E., Heyder P., Kern P., Kalden J. R., and Herrmann M., Scand. J. Immunol. 61, 226 (2005). 10.1111/j.1365-3083.2005.01551.x [DOI] [PubMed] [Google Scholar]
- 25. Escudier B. et al. , J. Transl. Med. 3, 10 (2005). 10.1186/1479-5876-3-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whiteside T. L., Biochem. Soc. Trans. 41, 245 (2013). 10.1042/BST20120265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zitvogel L., Regnault A., Lozier A., Wolfers J., Flament C., Tenza D., Ricciardi-Castagnoli P., Raposo G., and Amigorena S., Nat. Med. 4, 594 (1998). 10.1038/nm0598-594 [DOI] [PubMed] [Google Scholar]
- 28. Hao S., Bai O., Yuan J., Qureshi M., and Xiang J., Cell Mol. Immunol. 3, 205 (2006). [PubMed] [Google Scholar]
- 29. Näslund T. I., Gehrmann U., Qazi K. R., Karlsson M. C. I., and Gabrielsson S., J. Immunol. 190, 2712 (2013). 10.4049/jimmunol.1203082 [DOI] [PubMed] [Google Scholar]
- 30. Näslund T. I., Gehrmann U., and Gabrielsson S., Oncoimmunology 2, e24533 (2013). 10.4161/onci.24533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beauvillain C., Ruiz S., Guiton R., Bout D., and Dimier-Poisson I., Microbes Infect. 9, 1614 (2007). 10.1016/j.micinf.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 32. Schnitzer J. K., Berzel S., Fajardo-Moser M., Remer K. A., and Moll H., Vaccine 28, 5785 (2010). 10.1016/j.vaccine.2010.06.077 [DOI] [PubMed] [Google Scholar]
- 33. Sobo-Vujanovic A., Munich S., and Vujanovic N. L., Cell. Immunol. 289, 119 (2014). 10.1016/j.cellimm.2014.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Danesh A., Inglis H. C., Jackman R. P., Wu S., Deng X., Muench M. O., Heitman J. W., and Norris P. J., Blood 123, 687 (2014). 10.1182/blood-2013-10-530469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lugini L. et al. , J. Immunol. 189, 2833 (2012). 10.4049/jimmunol.1101988 [DOI] [PubMed] [Google Scholar]
- 36. Wahlgren J., Karlson T. D. L., Glader P., Telemo E., and Valadi H., PLoS One 7, e49723 (2012). 10.1371/journal.pone.0049723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arslan F. et al. , Stem Cell Res. 10, 301 (2013). 10.1016/j.scr.2013.01.002 [DOI] [PubMed] [Google Scholar]
- 38. Yang C., Ruffner M. A., Kim S., and Robbins P. D., Eur. J. Immunol. 42, 1778 (2012). 10.1002/eji.201141978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J. W., Wieckowski E., Taylor D. D., Reichert T. E., Watkins S., and Whiteside T. L., Clin. Cancer Res. 11, 1010 (2005). [PubMed] [Google Scholar]
- 40. Hoffmann T. K., Dworacki G., Tsukihiro T., Meidenbauer N., Gooding W., Johnson J. T., and Whiteside T. L., Clin. Cancer Res. 8, 2553 (2002). [PubMed] [Google Scholar]
- 41. Andreola G. et al. , J. Exp. Med. 195, 1303 (2002). 10.1084/jem.20011624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huber V. et al. , Gastroenterology 128, 1796 (2005). 10.1053/j.gastro.2005.03.045 [DOI] [PubMed] [Google Scholar]
- 43. Taylor D. D., Gerçel-Taylor Ç., Lyons K. S., Stanson J., and Whiteside T. L., Clin. Cancer Res. 9, 5113 (2003). [PubMed] [Google Scholar]
- 44. Taylor D. D. and Gercel-Taylor C., Br. J. Cancer 92, 305 (2005). 10.1038/sj.bjc.6602316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reiners K. S. et al. , Mol. Ther. 21, 895 (2013). 10.1038/mt.2013.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clayton A., Mitchell J. P., Linnane S., Mason M. D., and Tabi Z., J. Immunol. 180, 7249 (2008). 10.4049/jimmunol.180.11.7249 [DOI] [PubMed] [Google Scholar]
- 47. Szajnik M., Czystowska M., Szczepanski M. J., Mandapathil M., and Whiteside T. L., PLoS One 5, e11469 (2010). 10.1371/journal.pone.0011469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu S. et al. , J. Immunol. 178, 6867 (2007). 10.4049/jimmunol.178.11.6867 [DOI] [PubMed] [Google Scholar]
- 49. Clayton A., Al-Taei S., Webber J., Mason M. D., and Tabi Z., J. Immunol. 187, 676 (2011). 10.4049/jimmunol.1003884 [DOI] [PubMed] [Google Scholar]
- 50. Okoye I. S., Coomes S. M., Pelly V. S., Czieso S., Papayannopoulos V., Tolmachova T., Seabra M. C., and Wilson M. S., Immunity 41, 89 (2014). 10.1016/j.immuni.2014.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaur S., Singh S. P., Elkahloun A. G., Wu W., Abu-Asab M. S., and Roberts D. D., Matrix Biol. 37, 49 (2014). 10.1016/j.matbio.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee H. D., Kim Y. H., and Kim D., Eur. J. Immunol. 44, 1156 (2014). 10.1002/eji.201343660 [DOI] [PubMed] [Google Scholar]
- 53. Li X. et al. , PLoS One 7, e44045 (2012). 10.1371/journal.pone.0044045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taylor D. D., Akyol S., and Gercel-Taylor C., J. Immunol. 176, 1534 (2006). 10.4049/jimmunol.176.3.1534 [DOI] [PubMed] [Google Scholar]
- 55. Dalli J., Montero-Melendez T., Norling L. V., Yin X., Hinds C., Haskard D., Mayr M., and Perretti M., Mol. Cell. Proteomics 12, 2205 (2013). 10.1074/mcp.M113.028589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mesri M. and Altieri D. C., J. Biol. Chem. 274, 23111 (1999). 10.1074/jbc.274.33.23111 [DOI] [PubMed] [Google Scholar]
- 57. Pluskota E., Woody N. M., Szpak D., Ballantyne C. M., Soloviev D. A., Simon D. I., and Plow E. F., Blood 112, 2327 (2008). 10.1182/blood-2007-12-127183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramachandra L. et al. , Infect. Immun. 78, 5116 (2010). 10.1128/IAI.01089-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walters S. B., Kieckbusch J., Nagalingam G., Swain A., Latham S. L., Grau G. E. R., Britton W. J., Combes V., and Saunders B. M., J. Immunol. 190, 669 (2013). 10.4049/jimmunol.1201856 [DOI] [PubMed] [Google Scholar]
- 60. Arteaga R. B. et al. , Am. J. Cardiol. 98, 70 (2006). 10.1016/j.amjcard.2006.01.054 [DOI] [PubMed] [Google Scholar]
- 61. Scanu A., Molnarfi N., Brandt K. J., Gruaz L., Dayer J.-M., and Burger D., J. Leukocyte Biol. 83, 921 (2008). 10.1189/jlb.0807551 [DOI] [PubMed] [Google Scholar]
- 62. Obregon C., Rothen-Rutishauser B., Gitahi S. K., Gehr P., and Nicod L. P., Am. J. Pathol. 169, 2127 (2006). 10.2353/ajpath.2006.060453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Eken C., Sadallah S., Martin P. J., Treves S., and Schifferli J. A., Immunobiology 218, 382 (2013). 10.1016/j.imbio.2012.05.021 [DOI] [PubMed] [Google Scholar]
- 64. Pliyev B. K., Kalintseva M. V., Abdulaeva S. V., Yarygin K. N., and Savchenko V. G., Cytokine 65, 126 (2014). 10.1016/j.cyto.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 65. Sadallah S., Eken C., Martin P. J., and Schifferli J. A., J. Immunol. 186, 6543 (2011). 10.4049/jimmunol.1002788 [DOI] [PubMed] [Google Scholar]
- 66. Vasina E. M., Cauwenberghs S., Feijge M. A. H., Heemskerk J. W. M., Weber C., and Koenen R. R., Cell Death Dis. 2, e210 (2011). 10.1038/cddis.2011.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Distler J. H. W., Huber L. C., Hueber A. J., Reich C. F. III, Gay S., Distler O., and Pisetsky D. S., Apoptosis 10, 731 (2005). 10.1007/s10495-005-2941-5 [DOI] [PubMed] [Google Scholar]
- 68. Tsai W.-H., Shih C.-H., Feng S.-Y., Li I.-T., Chang S.-C., Lin Y.-C., and Hsu H.-C., Cell. Physiol. Biochem. 33, 594 (2014). 10.1159/000358637 [DOI] [PubMed] [Google Scholar]
- 69. Clayton A., Harris C. L., Court J., Mason M. D., and Morgan B. P., Eur. J. Immunol. 33, 522 (2003). 10.1002/immu.200310028 [DOI] [PubMed] [Google Scholar]
- 70. Fang R. H., Hu C.-M. J., Luk B. T., Gao W., Copp J. A., Tai Y., O'Connor D. E., and Zhang L., Nano Lett. 14, 2181 (2014). 10.1021/nl500618u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Toledano Furman N. E., Lupu-Haber Y., Bronshtein T., Kaneti L., Letko N., Weinstein E., Baruch L., and Machluf M., Nano Lett. 13, 3248 (2013). 10.1021/nl401376w [DOI] [PubMed] [Google Scholar]
- 72. Copp J. A., Fang R. H., Luk B. T., Hu C.-M. J., Gao W., Zhang K., and Zhang L., Proc. Natl. Acad. Sci. 111, 13481 (2014). 10.1073/pnas.1412420111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hu C.-M. J., Fang R. H., Copp J., Luk B. T., and Zhang L., Nat. Nanotechnol. 8, 336 (2013). 10.1038/nnano.2013.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mitchell M. J., Wayne E., Rana K., Schaffer C. B., and King M. R., Proc. Natl. Acad. Sci. 111, 930 (2014). 10.1073/pnas.1316312111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Porotto M., Yi F., Moscona A., and LaVan D. A., PLoS One 6, e16874 (2011). 10.1371/journal.pone.0016874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Robbins G. P., Saunders R. L., Haun J. B., Rawson J., Therien M. J., and Hammer D. A., Langmuir 26, 14089 (2010). 10.1021/la1017032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Plow E. F. and Kelly P., in Antithrombotic Drug Therapy in Cardiovascular Disease ( Springer, New York, 2010), pp. 3–17. [Google Scholar]
- 78. Kim W., Haller C., Dai E., Wang X., Hagemeyer C. E., Liu D. R., Peter K., and Chaikof E. L., Angew. Chem. 127, 1481 (2014). 10.1002/ange.201408529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hu C.-M. J., Zhang L., Aryal S., Cheung C., Fang R. H., and Zhang L., Proc. Natl. Acad. Sci. 108, 10980 (2011). 10.1073/pnas.1106634108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gao W., Fang R., Thamphiwatana S., Luk B. T., Li J., Angsantikul P., Zhang Q., Hu C.-M. J., and Zhang L., Nano Lett. 15, 1403 (2015). 10.1021/nl504798g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Moon J. J., Suh H., Li A. V., Ockenhouse C. F., Yadava A., and Irvine D. J., Proc. Natl. Acad. Sci. 109, 1080 (2012). 10.1073/pnas.1112648109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. De Geest B. G., Willart M. A., Lambrecht B. N., Pollard C., Vervaet C., Remon J. P., Grooten J., and De Koker S., Angew. Chem. 124, 3928 (2012). 10.1002/ange.201200048 [DOI] [PubMed] [Google Scholar]
- 83. Powell T. J. et al. , Vaccine 31, 1898 (2013). 10.1016/j.vaccine.2013.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bershteyn A., Hanson M. C., Crespo M. P., Moon J. J., Li A. V., Suh H., and Irvine D. J., J. Controlled Release 157, 354 (2012). 10.1016/j.jconrel.2011.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Carleton H. A., Lara-Tejero M., Liu X., and Galán J. E., Nat. Commun. 4, 1590 (2013). 10.1038/ncomms2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Appay V., Douek D. C., and Price D. A., Nat. Med. 14, 623 (2008). 10.1038/nm.f.1774 [DOI] [PubMed] [Google Scholar]
- 87. Mora-Solano C. and Collier J. H., J. Mater. Chem. B 2, 2409 (2014). 10.1039/C3TB21549K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sunshine J. C. and Green J. J., Nanomedicine 8, 1173 (2013). 10.2217/nnm.13.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Butler M. O. et al. , Int. Immunol. 22, dxq440 (2010). 10.1093/intimm/dxq440 [DOI] [Google Scholar]
- 90. Steenblock E. R., Wrzesinski S. H., Flavell R. A., and Fahmy T. M., 9, 451 (2009). [DOI] [PubMed] [Google Scholar]
- 91. Ding Q., Chen J., Wei X., Sun W., Mai J., Yang Y., and Xu Y., Pharm. Res. 30, 60 (2013). 10.1007/s11095-012-0849-7 [DOI] [PubMed] [Google Scholar]
- 92. Shen C., Cheng K., Miao S., Wang W., He Y., Meng F., and Zhang J., Immunol. Lett. 150, 1 (2013). 10.1016/j.imlet.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 93. Perica K. et al. , Nanomed. Nanotechnol., Biol. Med. 10, 119 (2014). 10.1016/j.nano.2013.06.015 [DOI] [Google Scholar]
- 94. Perica K., Tu A., Richter A., Bieler J. G., Edidin M., and Schneck J. P., ACS Nano 8, 2252 (2014). 10.1021/nn405520d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chen B., Jia Y., Gao Y., Sanchez L., Anthony S. M., and Yu Y., ACS Appl. Mater. Interfaces 6, 18435 (2014). 10.1021/am505510m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sunshine J. C., Perica K., Schneck J. P., and Green J. J., Biomaterials 35, 269 (2014). 10.1016/j.biomaterials.2013.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Steenblock E. R., Fadel T., Labowsky M., Pober J. S., and Fahmy T. M., J. Biol. Chem. 286, 34883 (2011). 10.1074/jbc.M111.276329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Park J. R. et al. , Mol. Ther. 15, 825 (2007). 10.1038/sj.mt.6300104 [DOI] [PubMed] [Google Scholar]
- 99. Rushworth D., Jena B., Olivares S., Maiti S., Briggs N., Somanchi S., Dai J., Lee D., and Cooper L. J. N., J. Immunother. 37, 204 (2014). 10.1097/CJI.0000000000000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Van Seventer G. A., Shimizu Y., Horgan K. J., and Shaw S., J. Immunol. 144, 4579 (1990). [PubMed] [Google Scholar]
- 101. Chittasupho C., Shannon L., Siahaan T. J., Vines C. M., and Berkland C., ACS Nano 5, 1693 (2011). 10.1021/nn102159g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lo Y.-C., Edidin M. A., and Powell J. D., J. Immunol. 191, 5107 (2013). 10.4049/jimmunol.1301433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kim Y. K., Que R., Wang S.-W., and Liu W. F., Adv. Healthcare Mater. 3, 989 (2014). 10.1002/adhm.201300532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gray M., Miles K., Salter D., Gray D., and Savill J., Proc. Natl. Acad. Sci. 104, 14080 (2007). 10.1073/pnas.0700326104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Getts D. R. et al. , J. Immunol. 187, 2405 (2011). 10.4049/jimmunol.1004175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lutterotti A. et al. , Sci. Transl. Med. 5, 188ra75 (2013). 10.1126/scitranslmed.3006168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kontos S., Kourtis I. C., Dane K. Y., and Hubbell J. A., Proc. Natl. Acad. Sci. 110, E60 (2013). 10.1073/pnas.1216353110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Getts D. R. et al. , Nat. Biotechnol. 30, 1217 (2012). 10.1038/nbt.2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tsai S. et al. , Immunity 32, 568 (2010). 10.1016/j.immuni.2010.03.015 [DOI] [PubMed] [Google Scholar]
- 110. Maldonado R. A. et al. , Proc. Natl. Acad. Sci. 112, E156 (2015). 10.1073/pnas.1408686111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hunter Z., McCarthy D. P., Yap W. T., Harp C. T., Getts D. R., Shea L. D., and Miller S. D., ACS Nano 8, 2148 (2014). 10.1021/nn405033r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Thomas S. N., Vokali E., Lund A. W., Hubbell J. A., and Swartz M. A., Biomaterials 35, 814 (2014). 10.1016/j.biomaterials.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 113. Alley S. C., Okeley N. M., and Senter P. D., Curr. Opin. Chem. Biol. 14, 529 (2010). 10.1016/j.cbpa.2010.06.170 [DOI] [PubMed] [Google Scholar]
- 114. McEnaney P. J., Fitzgerald K. J., Zhang A. X., E. F. Douglass, Jr. , Shan W., Balog A., Kolesnikova M. D., and Spiegel D. A., J. Am. Chem. Soc. 7, 1139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. McEnaney P. J., Parker C. G., Zhang A. X., and Spiegel D. A., ACS Chem. Biol. 7, 1139 (2012). 10.1021/cb300119g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Parker C. G. et al. , Chem. Sci. 5, 2311 (2014). 10.1039/c4sc00484a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sheridan R. T. C., Hudon J., Hank J. A., Sondel P. M., and Kiessling L. L., ChemBioChem 15, 1393 (2014). 10.1002/cbic.201402019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jakobsche C. E., Parker C. G., Tao R. N., Kolesnikova M. D., E. F. Douglass, Jr. , and Spiegel D. A., ACS Chem. Biol. 8, 2404 (2013). 10.1021/cb4004942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cornelissen J. J. L. M., Donners J. J. J. M., de Gelder R., Graswinckel W. S., Metselaar G. A., Rowan A. E., Sommerdijk N. A. J. M., and Nolte R. J. M., Science 293, 676 (2001). 10.1126/science.1062224 [DOI] [PubMed] [Google Scholar]
- 120. Mandal S. et al. , Chem. Sci. 4, 4168 (2013). 10.1039/c3sc51399h [DOI] [Google Scholar]
- 121. Mandal S., Hammink R., Tel J., Eksteen-Akeroyd Z. H., Rowan A. E., Blank K., and Figdor C. G., ACS Chem. Biol. 10, 485 (2014). 10.1021/cb500455g [DOI] [PubMed] [Google Scholar]
- 122. Mancini R. J., Tom J. K., and Esser-Kahn A. P., Angew. Chem. 126, 193 (2014). 10.1002/ange.201306551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ryu K. A., Stutts L., Tom J. K., Mancini R. J., and Esser-Kahn A. P., J. Am. Chem. Soc. 136, 10823 (2014). 10.1021/ja412314j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lan T. et al. , Org. Biomol. Chem. 11, 1049 (2013). 10.1039/c2ob26946e [DOI] [PubMed] [Google Scholar]
- 125. Lee J., Sohn J. W., Zhang Y., Leong K. W., Pisetsky D., and Sullenger B. A., Proc. Natl. Acad. Sci. 108, 14055 (2011). 10.1073/pnas.1105777108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Holl E. K., Shumansky K. L., Pitoc G., Ramsburg E., and Sullenger B. A., PLoS One 8, e69413 (2013). 10.1371/journal.pone.0069413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tom J. K., Mancini R. J., and Esser-Kahn A. P., Chem. Commun. 49, 9618 (2013). 10.1039/c3cc45468a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Prost L. R., Grim J. C., Tonelli M., and Kiessling L. L., ACS Chem. Biol. 7, 1603 (2012). 10.1021/cb300260p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ribeiro-Viana R., Sánchez-Navarro M., Luczkowiak J., Koeppe J. R., Delgado R., Rojo J., and Davis B. G., Nat. Commun. 3, 1303 (2012). 10.1038/ncomms2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kang Y. J., Yang H. J., Jeon S., Kang Y., Do Y., Hong S. Y., and Kang S., Macromol. Biosci. 14, 619 (2014). 10.1002/mabi.201300528 [DOI] [PubMed] [Google Scholar]
- 131. Luczkowiak J., Muñoz A., Sánchez-Navarro M., Ribeiro-Viana R., Ginieis A., Illescas B. M., Martín N., Delgado R., and Rojo J., Biomacromolecules 14, 431 (2013). 10.1021/bm3016658 [DOI] [PubMed] [Google Scholar]
- 132. Courtney A. H., Bennett N. R., Zwick D. B., Hudon J., and Kiessling L. L., ACS Chem. Biol. 9, 202 (2013). 10.1021/cb400532y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kaewsapsak P., Esonu O., and Dube D. H., ChemBioChem 14, 721 (2013). 10.1002/cbic.201300006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tra V. N. and Dube D. H., Chem. Commun. 50, 4659 (2014). 10.1039/c4cc00660g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Muenchmeier N. et al. , PLoS One 8, e72749 (2013). 10.1371/journal.pone.0072749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hudak J. E., Canham S. M., and Bertozzi C. R., Nat. Chem. Biol. 10, 69 (2014). 10.1038/nchembio.1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Swee L. K., Lourido S., Bell G. W., Ingram J. R., and Ploegh H. L., ACS Chem. Biol. 10, 460 (2014). 10.1021/cb500462t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cone R. E., Marchalonis J. J., and Rolley R. T., J. Exp. Med. 134, 1373 (1971). 10.1084/jem.134.6.1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sathish J. G. et al. , Nat. Rev. Drug Discovery 12, 306 (2013). 10.1038/nrd3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Geering B. and Fussenegger M., Trends Biotechnol. 2, 65 (2014). [DOI] [PubMed] [Google Scholar]
- 141. Deng Z.-B. et al. , J. Immunol. 190, 3579 (2013). 10.4049/jimmunol.1203170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. De La Peña H., Madrigal J. A., Rusakiewicz S., Bencsik M., Cave G. W. V., Selman A., Rees R. C., Travers P. J., and Dodi I. A., J. Immunol. Methods 344, 121 (2009). 10.1016/j.jim.2009.03.011 [DOI] [PubMed] [Google Scholar]
- 143. Jang S. C. et al. , ACS Nano 7, 7698 (2013). 10.1021/nn402232g [DOI] [PubMed] [Google Scholar]
- 144. Kooijmans S. A. A., Vader P., van Dommelen S. M., van Solinge W. W., and Schiffelers R. M., Int. J. Nanomed. 2012, 1525 (2012). 10.2147/ijn.s29661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ly L. V., Sluijter M., Versluis M., Luyten G. P. M., van der Burg S. H., Melief C. J. M., Jager M. J., and van Hall T., Cancer Res. 70, 8339 (2010). 10.1158/0008-5472.CAN-10-2288 [DOI] [PubMed] [Google Scholar]
- 146. Lesterhuis W. J., Haanen J. B. A. G., and Punt C. J. A., Nat. Rev. Drug Discovery 10, 591 (2011). 10.1038/nrd3500 [DOI] [PubMed] [Google Scholar]
- 147. Getts D. R. et al. , Sci. Transl. Med. 6, 219ra7 (2014). 10.1126/scitranslmed.3007563 [DOI] [PMC free article] [PubMed] [Google Scholar]