

Abstract
High-valent cobalt-oxo intermediates are proposed as reactive intermediates in a number of cobalt complex-mediated oxidation reactions. Herein we report the spectroscopic capture of low-spin (S = 1/2) Co(IV)-oxo species in the presence of redox-inactive metal ions, such as Sc3+, Ce3+, Y3+, and Zn2+ and investigation of their reactivity in C-H bond activation and sulfoxidation reactions. Theoretical calculations predict that the binding of Lewis-acidic metal ions to the cobalt-oxo core increases the electrophilicity of the oxygen atom, resulting in the redox tautomerism of a highly unstable [(TAML)CoIII-(O•)]2− species to a more stable [(TAML)CoIV-(O)(Mn+)] core. The present report supports the proposed role of the redox-inactive metal ions in facilitating formation of high-valent metal-oxo cores as a necessary step for oxygen evolution in chemistry and biology.
Keywords: Cobalt, Oxo ligand, Lewis acid stabilization, Redox tautomerization, Oxygenation
High-valent metal-oxo complexes have been implicated as active oxidants in oxygenation and water oxidation reactions by a number of biological and chemical catalysts.1 Although high-valent terminal metal-oxo complexes of groups 3-8 are ubiquitous, isolation of metal-oxo species to the right of group 8 (also known as the “oxo wall” for C4v symmetry) has only been achieved for heavy transition metals like iridium2 and platinum.3 For example, high-valent terminal metal-oxo complexes of the lighter analogues like cobalt, nickel, and copper still remain elusive, although they are often invoked as highly reactive transient intermediates in metal complex-catalyzed C-H bond activation and O-O bond formation reactions.1e,1g,4,5 Theory suggests that they should be powerful oxidants, perhaps even more reactive than the related iron-oxo species.1,6 The predicted high reactivity of the [M(O)]n+ (M = Co, Ni, and Cu) species is attributed to a relatively weak metal-oxo bond due to the population of the metal-oxo π* orbitals, such that their electronic structure is best described as intermediate between [M(n+1)+-O•] and [M(n+2)+=O].7 It is argued that the radical character resulting from significant spin density on the oxygen atom would make the [M(O)]n+ species particularly powerful hydrogen atom abstracting agents and therefore extremely difficult to trap.
Some of us recently reported the stabilization of an S = 3/2 cobalt(IV)-oxo species in the presence of a redox-inactive Lewis acidic metal ion (e.g., Sc3+ ion);8 however, others have claimed it to be a Co(III)-OH instead.9 In this study, we report the extension of the same methodology in stabilizing low-spin (S = 1/2) [CoIV-(O)(Mn+)](n+2)+ intermediates (Mn+ = Sc3+, Ce3+, Y3+, and Zn2+). Notably, a similar strategy was used recently to stabilize a [CuII-(NTs•)(Sc)]4+ core, with copper in an unusual formal oxidation state of +3.10 Thus, although the oxo-wall still holds (as terminal cobalt(IV)-oxo species by itself are still elusive), the present study firmly establishes the wider application of the strategy of utilizing redox-innocent Lewis-acids in trapping short-lived intermediates responsible for the oxo- and nitrene-transfer reactions mediated by mid-to-late transition metals (Scheme 1).
Scheme 1.

We have employed a tetraamido macrocyclic ligand (TAML), which has found application in the stabilization of a variety of high-valent iron(IV or V)-oxo and manganese(V)-oxo complexes.11 In contrast to the previously employed TMG3tren ligand,8 which enforced a trigonal bipyramidal geometry at the CoIV center stabilizing a S = 3/2 ground state, TAML can provide access to a square pyramidal cobalt(IV)-oxo center that would be a prerequisite for the targeted S = 1/2 state. Reactions of equimolar amounts of deprotonated TAML and cobalt(III) acetylacetonate in tetrahydrofuran afforded the previously reported purple [(TAML)CoIII]− anion,12 which is obtained as a lithium salt Li[(TAML)CoIII]•3(H2O) (1) containing three co-crystallized molecules of water (Supporting Information (SI), Tables S1-S2 and Figures S1-S2). Complex 1 is paramagnetic with S = 1 (μeff = 3.1 Bohr magnetons) ground state, as expected for a Co(III) ion in the observed square planar geometry. Cyclic voltammetry (CV) of 1 at 25 °C in CH3CN reveals a reversible oxidation wave centered at 1.00 V vs saturated calomel electrode (SI, Figure S3); coulometric measurements show that the oxidation corresponds to a 1e– process. The reversibility of this oxidation wave at room temperature suggests that a formal Co(IV) state is thermally and kinetically accessible.
In agreement with the electrochemical data, 1 is readily oxidized in the presence of cerium ammonium nitrate (CAN) in acetone at 5 °C to form a metastable blue species (2-Ce) with a half-life (t1/2) of 20 min (SI, Figure S4, inset). The blue color is associated with a band at 600 nm (ε = 7200 M−1 cm−1) with a shoulder near 730 nm (SI, Figure S4). We tentatively assign these bands to be ligand to metal charge transfer (LMCT) in origin, which presumably arise from transitions from the amide nitrogens of TAML to the Co(IV) center in 2-Ce; as expected, these bands are significantly blue-shifted in the corresponding Co(III) complex 1 (λmax = 510 nm (ε = 4000 M−1 cm−1) and 650 (ε = 1200 M−1 cm−1). Interestingly, treatment of 1 with iodosylbenzene (PhIO) in the presence of Sc(CF3SO3)3 also generates a blue species (2-Sc) with absorption features indistinguishable from 2-Ce, but 2-Sc is slightly more stable with a t1/2 of 35 min (Figure 1). Similarly, treatment of 1 with PhIO in the presence of other redox-inactive metal ions, such as Y3+ and Zn2+, results in the generation of the characteristic absorption band at 600 nm corresponding to the formation of 2-Y and 2-Zn, respectively. It is notable that reactions of 1 with PhIO in the absence of redox-inactive metal ions or in the presence of weaker Lewis acidic metal ions,13 such as Mg(CF3SO3)2, Ca(CF3SO3)2, and Sr(CF3SO3)2, does not form the 2-M species, thereby establishing the importance of the Lewis-acidity of the redox-inactive metal ions in trapping the 2-M intermediates. Nevertheless, the absorption features of the 2-M (M = Sc3+, Ce3+, Y3+, and Zn2+) complexes are all identical irrespective of the nature of the redox-inactive metal ions.
Figure 1.

UV-vis spectra of 2-Sc (gray line) observed in the reaction of 1 (black line, 0.25 mM) and PhIO (3.0 equiv) in the presence of Sc(CF3SO3)3 (20 equiv) in acetone at 5 °C. Inset shows the experimental (gray line) and simulated (black line) X-band EPR spectrum of 2-Sc (0.5 mM) at 10 K. For simulation parameters see text.
The nature of these blue species can be established by a variety of spectroscopic techniques; characterizations were performed mainly on 2-Sc, as it was the most stable species among the trapped 2-M complexes. Thus, cold spray ionization time-of-flight mass spectrometry (CSI-TOF MS) of 2-Sc reveals two high-resolution ion peaks at a mass-to-charge (m/z) ratio of 785.1010 and 803.1042, whose mass and isotope distribution patterns correspond to respective formulations of [(TAML)Co(16O)(Sc)(CH3CN)2(CH3OH)2(CF3SO3)] (calculated m/z of 785.1057) and [(TAML)Co(16O)(Sc)(CH3CN)4(CF3SO3)] (calculated m/z = 803.1064) (SI, Figure S5). Upon introduction of 18O into 2-Sc using PhI18O, mass shifts from 785.1057 to 787.1015 and from 803.1042 to 805.1021 occur (SI, Figure S5 inset), thereby demonstrating that an oxygen atom from PhIO is incorporated into 2-Sc.
In contrast to 1, which is EPR silent, the X-band EPR spectrum of 2-Sc at 5.0 K shows a highly rhombic signal at gx = 2.57, gy = 2.16, and gz = 2.03 (Figure 1, inset). Additionally, 59Co hyperfine pattern is observed at gy according to an Ay component of the hyperfine coupling tensor of 52 × 10–4 cm–1. The observed large g-anisotropy (Δg = gz-gx) of 0.54 is consistent with a cobalt-centered radical (S = 1/2) resulting from a low-spin Co(IV) configuration14 in the ground state of 2-Sc, further corroborated by X-ray absorption and DFT studies (see below). Spin quantification of the EPR signal can account for 70% of the total cobalt spins present in solution, thereby showing that 2-Sc represents the major product of the reaction of 1 with PhIO in the presence of Sc3+ ion. EPR spectra of other 2-M complexes were also measured and found to be identical to 2-Sc.15
All our attempts to collect resonance Raman spectra proved unsuccessful owing to the photodecomposition of 2-Sc under laser irradiation. Hence, structural evidence for the nature of 2-Sc was derived from X-ray absorption spectroscopic (XAS) studies at the Co K edge (Figure 2; SI, Figures S6-S7). A frozen acetone solution of 2-Sc exhibits a K edge at 7721.5 eV, an energy value 0.9 eV higher than that associated with its cobalt(III) precursor 1, thereby supporting metal-centered oxidation to the Co(IV) state. The principal feature of the inner-sphere scattering peaks in the extended X-ray absorption fine structure (EXAFS) spectra of 2-Sc at R’ ~1.40 Å can be best fitted (SI, Table S3, fit 11) by considering two shells; 0.7 O/N scatterer at 1.67 Å (assigned to the CoIV-O unit) and a further shell of 4.3 O/N scatterers at 1.86 Å (corresponding to the N-donors of TAML). Fitting was also performed by considering only one shell of 5 O/N scatterers at 1.86 Å (SI, Table S3, fit 3); the quality of the fit was significantly improved by incorporating an additional O/N shell (SI, Table S3, fit 4). While the outer shell features can be satisfactorily accounted for by several single scattering paths involving 9C at 2.80 Å, 1Sc at 3.38 Å, 7C at 3.10 Å, and 4O at 4.17Å, the fit could be satisfactorily improved by introducing multi-scattering paths involving elements of the supporting TAML ligand. Based on the 0.9 eV blue shift of the K-edge of 2-Sc relative to 1 and the presence of oxygen and scandium scatterers at 1.67 Å and 3.38 Å, 2-Sc is proposed to have one Sc3+ ion directly bound to the S = 1/2 [(TAML)CoIV-(O)]2− core. Notably, the determined Co-O distance of 1.67 Å for 2-Sc in EXAFS is significantly shorter than the Co-O distances of 1.79 Å and 1.85 Å determined for [(α2-P2W17O61CoIV)2O]14– (by X-ray diffraction)16 and [(TMG3tren)CoIV-(O)(Sc)(CF3SO3)3)]2+ (by EXAFS)8 complexes, respectively, where the formal oxidation state of the cobalt centers is also +4. This may reflect a higher single bond character of the Co-O bonds in [(α2-P2W17O61Co)2O]14– and [(TMG3tren)CoIV-(O)(Sc(CF3SO3)3)]2+ than in 2-Sc.
Figure 2.

X-ray absorption spectroscopy (XAS) at the cobalt K edge. Left, XANES spectra of 1 (black line) and 2-Sc (red line); top right, EXAFS data of 2-Sc on a wave vector scale as back-Fourier-transformation (black solid line) and simulation (dotted blue line); bottom right, Fourier-transformed EXAFS spectrum of 2-Sc (black solid line) and simulation (blue dotted line). For details see Supporting Information
In our effort to understand the role of redox-inactive metal ions in the stabilization of the S = 1/2 Co(IV)-oxo core, density functional theory (DFT) calculations were performed on 2-Sc as well as on the putative [(TAML)CoIV(O)]2− (2) species in the absence of Sc3+ ion (SI, Tables S4-S5 and Figures S8-S9). Notably, the calculated bond lengths of Co-O @ 1.700 Å, Co-N @ 1.855 Å, and Co-Sc @ 3.683 Å for 2-Sc in the doublet ground state are in good agreement with the EXAFS derived distances of 1.67, 1.86, and 3.38 Å, respectively, which validates our theoretical methods. Notably, a six coordinate scandium geometry is assumed in the calculation (with three bound triflates and two hydroxides; SI, Figure S8) based on the crystal structure of [(TMC)FeIV-(O)(Sc)(CF3SO3)4(OH)].17 Furthermore, although a doublet ground state is predicted for both 2 and 2-Sc, analysis of the Mulliken spin densities reveals significant differences in their electronic structures (SI, Table S5). While the electronic structure of 2 can be best represented as [(TAML)CoIII-(O•)]2− with 70% of the unpaired-electron spin being localized on the oxygen and only 24% on Co, 2-Sc possesses a [(TAML)CoIV-(O)(Sc)]+ ground state with the unpaired spin being predominantly (78%) localized on the cobalt center. Thus, the binding of Sc3+ ion to the [(TAML)CoIII-(O•)]2− core initiates a redox tautomerization event, whereby one-electron is transferred from the Co(III) center to the “oxo” ligand thereby stabilizing a [(TAML)CoIV-(O)(Sc)]+ core in 2-Sc. Such a Lewis-acid induced valence tautomerism is not unprecedented in the literature; for example, Zn2+ is recently shown to bind to a MnV(O) corrolazine complex to induce interconversion to a valence tautomer characterized as a MnIV(O) corrolazine π-cation-radical.18
The reactivity of 2-Sc was then investigated in hydrogen atom (H-atom) abstraction and oxygen atom transfer (OAT) reactions. In the H-atom abstraction reaction, addition of xanthene to the solution of 2-Sc at 5 °C regenerated 1 with an isosbestic point at 530 nm (Figure 3a; SI, Figure S10). The first-order rate constant, determined by pseudo-first-order fitting of the kinetic data for the decay of 2-Sc, increases linearly with increasing xanthene concentration, thereby giving a second-order rate constant of 2.3 × 10−1 M−1 s−1 at 5 °C (SI, Figure S11). A kinetic isotope effect (KIE) value of 13 was determined when xanthene-d2 was used in the reaction (SI, Figure S11). Such a large KIE value suggests that the H-atom abstraction by 2-Sc occurs via tunnelling, which is typical for metal-oxo species-mediated H-atom abstraction reactions.1 The second-order rate constants were also determined for the oxidation of other substrates by 2-Sc, and a linear correlation between the reaction rates and the C-H bond dissociation energies (BDE)19 of the substrates was obtained, leading us to propose that the H-atom abstraction from the C-H bonds of substrates by 2-Sc is the rate-determining step (r.d.s.) in the C-H bond activation reaction (Figure 3b; SI, Table S6 and Figure S12). Product analysis of the reaction mixture revealed the formation of xanthone as a major product (30(5)% yield based on the amount of 2-Sc used) (Figure 3a; SI, Table S7). Also, the source of oxygen in the xanthone product turned out to be the cobalt-oxo species by carrying out an 18O-labeling experiment using 18O-labeled 2-Sc (SI, Figure S13).
Figure 3.
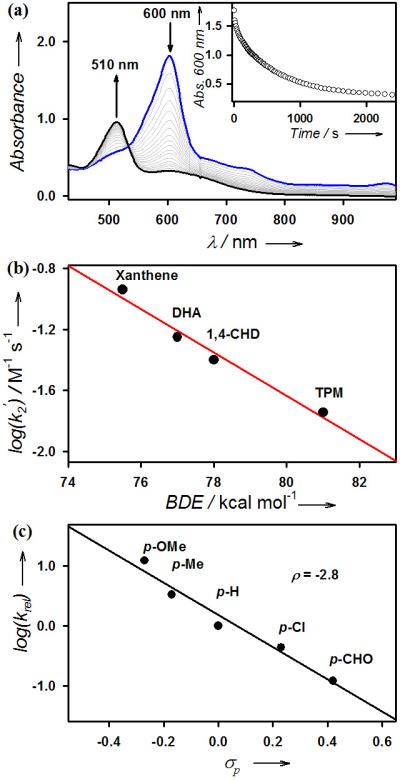
(a) UV-vis spectral changes observed in the reaction of 2-Sc (0.25 mM) and xanthene (20 equiv) in acetone at 5 °C. Inset shows the time course monitored at 600 nm. (b) Plot of log k2’ against the C-H BDE of substrates for the reactions of 2-Sc. Second-order rate constants, k2, were determined and then adjusted for reaction stoichiometry to yield k2′ based on the number of equivalent target C-H bonds of substrates (see data in SI, Table S6 and Figure S12). (c) Plots of log (krel) against Hammett σp constants for para-substituted thioanisoles in acetone at 5 °C. (see data in SI, Table S8 and Figure S14).
2-Sc also showed a high reactivity in OAT reaction with thioanisole at 5 °C, thereby producing thioanisole oxide quantitatively and regenerating 1 with an isosbestic point at 530 nm (SI, Table S7 and Figure S14). The source of oxygen in the sulfoxide product was found to be 2-Sc on the basis of the result of 18O-labeling experiment (SI, Figure S15), consistent with the cobalt-oxo assignment of 2-Sc. Moreover, as observed in the oxidation of sulfides by metal-oxo species,20 the electrophilic character of the Co-O-Sc3+ core in 2-Sc was established by a large negative ρ value of –2.8 in the oxidation of para-substituted thioanisoles (Figure 3c; SI, Table S8). Finally, the effect of redox-inactive metal ions on the reactivities of 2-M was investigated in the oxidation of thioanisole. As we have shown in the case of nonheme iron(IV)-oxo and manganese(IV)-oxo complexes,21 the OAT reactivity of 2-M becomes greater with the increasing Lewis acidity13 of the redox-inactive metal ions in 2-M (SI, Figure S16).
In summary, we report the trapping of reactive low-spin S = 1/2 [(TAML)CoIV-(O)(Mn+)](n-2)+ (Mn+ = Sc3+, Ce3+, Y3+, and Zn2+) complexes in the reaction of [(TAML)CoIII]− with CAN/H2O or with PhIO/Mn+ mixtures. In the absence of Mn+ ions, no high-valent cobalt-oxo cores were accessible. The binding of Lewis-acidic metal ions presumably increases the electrophilicity of the oxygen atom of the cobalt-oxo core, resulting in the redox tautomerism of a highly unstable [(TAML)CoIII-(O•)]2− species to a more stable [(TAML)CoIV-(O)(Mn+)](n-2)+ core. Our particular interest in low-spin cobalt(IV)-oxo species pertains to their proven potential as active oxidants in C-H bond activation and water oxidation reactions.4 Very recently, a heterogeneous cobalt-oxide catalyst for water oxidation has been proposed to contain alkaline metals (albeit not detected by X-ray absorption spectroscopy)4f as part of Co3O4 cubane moieties.4c,e The alkaline metal ion in this species could facilitate access to cobalt species with high oxidation states, similar to the Lewis-acidic metal ions described in this manuscript. Similarly, the potential role of Ca2+ in biological oxygen evolution by facilitating high oxidation states at manganese has been suggested by structural and electrochemical comparison between a [Mn3CaO4]6+ cubane and a related Mn4O4 cubane.22 The present work supports the above hypotheses, and therefore, provides an opportunity to elucidate elementary steps in cobalt complex-mediated alkane and water oxidation reactions, using the high purity of the Lewis-acid bound low-spin cobalt(IV)-oxo species, with its various prominent spectroscopic signatures, as a stoichiometric reagent.
Supplementary Material
Acknowledgment
The authors gratefully acknowledge research support of this work by the NRF of Korea through CRI (2-2012-1794-001-1 to W.N.), GRL (2013-4213-001-1 to W.N.) and NFSC (grants 21173211 and 21233008 to Y.W.). K.R. acknowledges financial support of this work from the Cluster of Excellence “Unifying Concepts in Catalysis” (EXC 314/1), Berlin and Cost Action CM1305. XAS data were obtained on NSLS beamline X3B (Brookhaven National Laboratory), with support from NIH Grant P30-EB-009998 and the U.S. Department of Energy. We thank Dr. Erik R. Farquhar for help with XAS data collection and analysis.
Footnotes
Supporting information for this article, including Experimental section, Tables S1 - S8 and Figures S1 - S17, is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.a Nam W. Acc. Chem. Res. 2007;40:465. doi: 10.1021/ar700027f. and references therein. [DOI] [PubMed] [Google Scholar]; b Abu-Omar MM, Loaiza A, Hontzeas N. Chem. Rev. 2005;105:2227. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]; c Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr. Acc. Chem. Res. 2007;40:484. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gunay A, Theopold KH. Chem. Rev. 2010;110:1060. doi: 10.1021/cr900269x. [DOI] [PubMed] [Google Scholar]; e Hohenberger J, Ray K, Meyer K. Nat. Commun. 2012;3:720. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]; f Borovik AS. Chem. Soc. Rev. 2011;40:1870. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ray K, Heims F, Pfaff FF. Eur. J. Inorg. Chem. 2013;22-23:3784. [Google Scholar]; h de Visser SP, Rohde J-U, Lee Y-M, Cho J, Nam W. Coord. Chem. Rev. 2013;257:381. [Google Scholar]; i Hammarström I, Hammes-Schiffer S. Acc. Chem. Res. 2009;42:1859. doi: 10.1021/ar900267k. [DOI] [PubMed] [Google Scholar]; j Rivalta I, Brudvig GW, Batista VS. Curr. Opin. Chem. Biol. 2012;45:767. doi: 10.1016/j.cbpa.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hay-Motherwell RS, Wilkinson G, Hussain-Bates B, Hursthouse MB. Polyhedron. 1993;12:2009. [Google Scholar]
- 3.Poverenov E, Efremenko I, Frenkel AI, Ben-David Y, Shimon LJW, Leitus G, Konstantinovski L, Martin JML, Milstein D. Nature. 2008;455:1093. [Google Scholar]
- 4.a Chang CJ, Loh Z-H, Shi C, Anson FC, Nocera DG. J. Am. Chem. Soc. 2004;126:10013. doi: 10.1021/ja049115j. [DOI] [PubMed] [Google Scholar]; b Kanan MW, Nocera DG. Science. 2008;321:1072. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]; c Nam W, Kim I, Kim Y, Kim C. Chem. Commun. 2001:1262. [Google Scholar]; d Fukuzumi S, Mandal S, Mase K, Ohkubo K, Park H, Benet-Buchholz J, Nam W, Llobet A. J. Am. Chem. Soc. 2012;134:9906. doi: 10.1021/ja303674n. [DOI] [PubMed] [Google Scholar]; e Symes MD, Surendranath Y, Lutterman DA, Nocera DG. J. Am. Chem. Soc. 2011;133:5174. doi: 10.1021/ja110908v. [DOI] [PubMed] [Google Scholar]; f Kanan MW, Yano J, Surendranath Y, Dincă M, Yachandra VK, Nocera DG. J. Am. Chem. Soc. 2010;132:13692. doi: 10.1021/ja1023767. [DOI] [PubMed] [Google Scholar]; g Zhang M, de Respinis M, Frei H. Nat. Chem. 2014;6:362. doi: 10.1038/nchem.1874. [DOI] [PubMed] [Google Scholar]; h Nguyen AI, Hadt RG, Solomon EI, Tilley TD. Chem. Sci. Advance article. 2014 doi: 10.1039/c4sc00108g. DOI: 10.1039/C4SC00108G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Pfaff FF, Heims F, Kundu S, Mebs S, Ray K. Chem. Commun. 2012:3730. doi: 10.1039/c2cc30716b. [DOI] [PubMed] [Google Scholar]; b Nagataki T, Tachi Y, Itoh S. Chem. Commun. 2006:4016. doi: 10.1039/b608311k. [DOI] [PubMed] [Google Scholar]; c Yao S, Driess M. Acc. Chem. Res. 2012;45:276. doi: 10.1021/ar200156r. [DOI] [PubMed] [Google Scholar]
- 6.a Schröder D, Holthausen MC, Schwarz H. J. Phys. Chem. B. 2004;108:14407. [Google Scholar]; b Gherman BF, Tolman WB, Cramer CJ. J. Comput. Chem. 2006;27:1950. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]; c Pierpont AW, Cundari TR. Inorg. Chem. 2010;49:2038. doi: 10.1021/ic901250z. [DOI] [PubMed] [Google Scholar]
- 7.Limberg C. Angew. Chem. Int. Ed. 2009;48:2270. doi: 10.1002/anie.200805977. [DOI] [PubMed] [Google Scholar]
- 8.Pfaff FF, Kundu S, Risch M, Pandian S, Heims F, Pryjomska-Ray I, Haack P, Metzinger R, Bill E, Dau H, Comba P, Ray K. Angew. Chem. Int. Ed. 2011;50:1711. doi: 10.1002/anie.201005869. [DOI] [PubMed] [Google Scholar]
- 9.Lacy DC, Park YJ, Ziller JW, Yano J, Borovik AS. J. Am. Chem. Soc. 2012;134:17526. doi: 10.1021/ja304525n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kundu S, Miceli E, Farquhar E, Pfaff FF, Kuhlmann U, Hildebrandt P, Braun B, Greco C, Ray K. J. Am. Chem. Soc. 2012;134:14710. doi: 10.1021/ja306674h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Tiago de Oliveira F, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Jr., Bominaar EL, Münck E, Collins TJ. Science. 2007;315:835. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]; b Chanda A, Shan X, Chakrabarti M, Ellis WC, Popescu DL, Tiago de Oliveira F, Wang D, Que L, Jr., Collins TJ, Münck E, Bominaar EL. Inorg. Chem. 2008;47:3669. doi: 10.1021/ic7022902. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Collins TJ, Gordon-Wylie SW. J. Am. Chem. Soc. 1989;111:4511. [Google Scholar]
- 12.Ghosh A, Ramidi P, Pulla S, Sullivan SZ, Collom SL, Gartia Y, Munshi P, Biris AS, Noll BC, Berry BC. Catal. Lett. 2010;137:1. [Google Scholar]
- 13.a Fukuzumi S, Ohkubo K. Chem.–Eur. J. 2000;6:4532. doi: 10.1002/1521-3765(20001215)6:24<4532::aid-chem4532>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; b Fukuzumi S, Ohkubo K. J. Am. Chem. Soc. 2002;124:10270. doi: 10.1021/ja026613o. [DOI] [PubMed] [Google Scholar]
- 14.McAlpin JG, Surendranath Y, Dincă M, Stich TA, Stoian SA, Casey WH, Nocera DG, Britt RD. J. Am. Chem. Soc. 2010;132:6882. doi: 10.1021/ja1013344. [DOI] [PubMed] [Google Scholar]
- 15.The EPR spectrum (Figure 1 inset) of 2-Sc also reveals a minor sharp signal (<5% yield) at g = 2.00, which corresponds to the product formed as a result of the thermal decay of 2-Sc (SI, Figure S17).
- 16.B.-Damatoy D, Shimon LJW, Weiner L, Schreiber RE, Jiménez-Lozano P, Poblet JM, de Graaf C, Neumann R. Inorg. Chem. 2014;53:1779. doi: 10.1021/ic402962c. [DOI] [PubMed] [Google Scholar]
- 17.Fukuzumi S, Morimoto Y, Kotani H, Naumov P, Lee Y-M, Nam W. Nat. Chem. 2010;2:756. doi: 10.1038/nchem.731. [DOI] [PubMed] [Google Scholar]
- 18.Leeladee P, Baglia RA, Prokop KA, Latifi R, de Visser SP, Goldberg DP. J. Am. Chem. Soc. 2012;134:10397. doi: 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]
- 19.Luo Y-R. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- 20.a Taki M, Itoh S, Fukuzumi S. J. Am. Chem. Soc. 2002;124:998. doi: 10.1021/ja016023a. [DOI] [PubMed] [Google Scholar]; b Arias J, Newlands CR, Abu-Omar MM. Inorg. Chem. 2001;40:2185. doi: 10.1021/ic000917z. [DOI] [PubMed] [Google Scholar]; c McPherson LD, Drees M, Khan SI, Strassner T, Abu-Omar MM. Inorg. Chem. 2004;43:4036. doi: 10.1021/ic0498945. [DOI] [PubMed] [Google Scholar]; d Kumar A, Goldberg I, Botoshansky M, Buchman Y, Gross Z. J. Am. Chem. Soc. 2010;132:15233. doi: 10.1021/ja1050296. [DOI] [PubMed] [Google Scholar]
- 21.a Chen J, Lee Y-M, Davis KM, Wu X, Seo MS, Cho K-B, Yoon H, Park YJ, Fukuzumi S, Pushkar YN, Nam W. J. Am. Chem. Soc. 2013;135:6388. doi: 10.1021/ja312113p. [DOI] [PubMed] [Google Scholar]; b Park J, Morimoto Y, Lee Y-M, Nam W, Fukuzumi S. Am. Chem. Soc. 2011;133:5236. doi: 10.1021/ja200901n. [DOI] [PubMed] [Google Scholar]
- 22.a Kanady JS, Tsui EY, Day MW, Agapie T. Science. 2011;333:733. doi: 10.1126/science.1206036. [DOI] [PubMed] [Google Scholar]; b Cox N, Rapatskiy L, Su J-H, Pantazis DA, Sugiura M, Kulik L, Dorlet P, Rutherford AW, Neese F, Boussac A, Lubitz W, Messinger J. J. Am. Chem. Soc. 2011;133:3635. doi: 10.1021/ja110145v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.