

Abstract
Background
The airway epithelial cell plays a central role in coordinating the pulmonary response to injury and inflammation. Here, transforming growth factor-β (TGFβ) activates gene expression programs to induce stem cell-like properties, inhibit expression of differentiated epithelial adhesion proteins and express mesenchymal contractile proteins. This process is known as epithelial mesenchymal transition (EMT); although much is known about the role of EMT in cellular metastasis in an oncogene-transformed cell, less is known about Type II EMT, that occurring in normal epithelial cells. In this study, we applied next generation sequencing (RNA-Seq) in primary human airway epithelial cells to understand the gene program controlling Type II EMT and how cytokine-induced inflammation modifies it.
Results
Generalized linear modeling was performed on a two-factor RNA-Seq experiment of 6 treatments of telomerase immortalized human small airway epithelial cells (3 replicates). Using a stringent cut-off, we identified 3,478 differentially expressed genes (DEGs) in response to EMT. Unbiased transcription factor enrichment analysis identified three clusters of EMT regulators, one including SMADs/TP63 and another NF-κB/RelA. Surprisingly, we also observed 527 of the EMT DEGs were also regulated by the TNF-NF-κB/RelA pathway. This Type II EMT program was compared to Type III EMT in TGFβ stimulated A549 alveolar lung cancer cells, revealing significant functional differences. Moreover, we observe that Type II EMT modifies the outcome of the TNF program, reducing IFN signaling and enhancing integrin signaling. We confirmed experimentally that TGFβ-induced the NF-κB/RelA pathway by observing a 2-fold change in NF-κB/RelA nuclear translocation. A small molecule IKK inhibitor blocked TGFβ-induced core transcription factor (SNAIL1, ZEB1 and Twist1) and mesenchymal gene (FN1 and VIM) expression.
Conclusions
These data indicate that NF-κB/RelA controls a SMAD-independent gene network whose regulation is required for initiation of Type II EMT. Type II EMT dramatically affects the induction and kinetics of TNF-dependent gene networks.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1707-x) contains supplementary material, which is available to authorized users.
Keywords: Epithelial mesenchymal transition, Transforming growth factor β, Nuclear factor κB, RNA-Seq, Generalized linear modeling, Transcription factor enrichment
Background
The airway mucosa consists of highly polarized, differentiated epithelial cell types, whose primary role is to restrict fluid loss and limit inhaled particulate access to the internal milieu [1]. Exposure to aero-allergens, respiratory viruses or reactive oxygen stress, induces anchorage-dependent cell death (anoikis). During the process of anoikis, epithelial sloughing and disruption of the basement membrane releases sequestered extracellular matrix-associated epithelial growth factors, transforming growth factor (TGF), epidermal growth factor (EGF) and fibroblast growth factor (FGF). These epithelial growth factors activate gene expression programs in resident stem cell population (basal cells) to initiate epithelial repair and regeneration.
TGFβ-stimulation of normal epithelial cells activates a de-differentiation program to promote cellular regeneration and extracellular matrix formation [1–3], referred to as Type II epithelial mesenchymal transition (EMT; [4]). Type II EMT induces loss of apical-basal polarity through cytoskeletal reorganization and dissolution of tight junctions. Concomitantly the cells express mesenchymal smooth muscle cell actin and intermediate filament vimentin (VIM) enhancing motility, and secrete collagen (Col1A), fibronectin (FN1), and matrix metalloproteinases (MMPs) promoting extracellular matrix deposition. Collectively this response promotes airway remodeling with expansion of the smooth muscle cell layer, repair of the epithelial surface and fibrosis [4, 5]. In this way, the airway epithelium plays a primary role in the resolution of injury or, if unresolved, the pathogenesis of chronic airway disease [6].
EMT is a dynamic and reversible epigenetic reprogramming event that plays a role in embryogenesis, organ homeostasis in response to acute injury, or cancer progression/metastasis, referred to as Types I, II, or III EMT, respectively [4]. Using Type III EMT as a model of cancer progression and metastases, the molecular signaling of how TGFβ1 initiates EMT has been studied extensively. Ligand induced activation of the transmembrane serine/threonine kinase TGFβ receptor type II (TGFβRII) recruits and phosphorylates TGFβRI to signal through Smad-dependent, “canonical” and Smad-independent “noncanonical” pathways [7]. In the canonical pathway, phosphorylated Smad2/3 binds to Smad4 and the complex then translocates to the nucleus. The noncanonical signaling pathways involve downstream PI3K/Akt, Ras small GTPases, Wnt/β-catenin, ERK, p38, and JNK. Collectively, the canonical and noncanonical pathways converge on a core set of transcription factors that function to initiate and maintain EMT: SNAIL (SNAI) 1/2, zinc finger E-box binding (ZEB) 1/2, Twist 1/2, Goosecoid and others [8].
The core EMT transcription factors mediate a coordinated series of gene activation/repression and chromatin reprogramming events to shift cells to the mesenchymal phenotype. The activated Smad 2/3/4 trimer binds to Smad-binding elements in the regulatory regions of SNAIL, JunB and c-Jun, activating their expression by recruiting coactivators including the cAMP-response element binding protein (CBP)/p300 histone acetyltransferases [9]. SNAIL, in turn, represses ECad and zona occludin-1 genes by recruiting the polycomb complex, producing silencing histone modifications [10–12]. Smad signaling also increases expression of ZEB1/2, resulting in MMP and collagen 1A expression [8]. ZEB interacts with lysine-specific demethylase (LSD1), a protein involved in histone demethylation and chromatin reprogramming in EMT [13, 14]. Together these proteins coordinate both the repression of epithelial related genes and activation of mesenchymal genes.
Because of the temporal interplay of diverse signaling programs required to initiate and maintain EMT reprogramming, the EMT is highly modified by the state of cellular transformation and concomitant activation of extracellular signaling pathways. Oncogenic mutations in K-ras, activation of Wnt signaling, ROS stress and activation of insulin-like growth factor pathways that cross-talk with the TGFβ pathway modify the expression of the EMT program [15]. As a result, the EMT program can be modulated by extracellular matrix interactions [16], and, of interest here, pro-inflammatory monocyte derived cytokines. TNF is a prototypical monokine [16, 17], whose actions trigger activation of p38 MAPK and JNK, essential components of the noncanonical TGFβ signaling pathways [18, 19], and induce EMT in K-ras transformed epithelial cells through the actions of NF-κB on the Twist EMT core transcription factor [16, 20]. However, the role of NF-κB signaling in the EMT of normal epithelial cells is not known.
In this study we sought to examine the gene program of Type II EMT and to identify how this process was modulated by interaction with the innate signaling pathway. A well-established model of TGFβ-induced EMT was applied to primary immortalized human small airway epithelial cells (hSAECs) to identify the gene expression networks responsible [5], and understand how activation of the innate response was modulated by EMT. Surprisingly, we observed that TGFβ produced a gene expression program that was significantly enriched in NF-κB-dependent genes identified by comparison to TNF dependent genes and to RelA enriched target genes in public ChIP-Seq data. Moreover, Type II EMT produces profound rewiring of the TNF gene program, skewing the pathway towards expression of integrin signaling to maintain the EMT state. We demonstrate that inhibiting NF-κB/RelA via gene silencing or by inhibition of the IKK regulatory kinase blocked TGFβ-induced EMT. These data indicate that NF-κB/RelA gene expression program is a major regulator of TGFβ-induced Type II EMT.
Methods
hSAEC culture and EMT transformation
An immortalized human small airway epithelial cell (hSAEC) line was established by infecting primary hSAECs with human telomerase (hTERT) and cyclin dependent kinase (CDK)-4 retrovirus constructs [21]. The immortalized hSAECs were grown in SAGM small airway epithelial cell growth medium (Lonza, Walkersville, MD) in a humidified atmosphere of 5 % CO2. For induction of EMT, hSAECs were TGFβ stimulated for 15 days (10 ng/ml, PeproTech, Rocky Hill, NJ). The small molecule inhibitor of IKK, BMS345541 was purchased from Sigma Aldrich and used at 10 μM [22].
Fluorescence microscopy
hSAECs were incubated in the absence or presence of TGFβ (10 ng/mL) for 15 days, re-plated on glass cover slips pretreated with rat tail collagen (Roche Applied Sciences) and fixed with 4 % paraformaldehyde in PBS. Afterwards, the fixed cells were stained with Alexa Fluor® 568 phalloidin for cytoplasmic distribution of F-actin (shown in red color) and also counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) for nuclear staining (shown in blue color). The cells were visualized with a Nikon fluorescence confocal microscope at a magnification of 63× [5, 23].
Quantitative real time PCR (Q-RT-PCR)
Reverse transcription was performed on 1 μg of total RNA with random primers, utilizing SuperScript III first-strand synthesis system for RT-PCR (Life technologies, Invitrogen) under conditions recommended by the manufacturer. Equivalent amounts of RNA were assayed by quanitative (Q) Q-PCR. The PCR reaction consisted of SYBR Green® PCR Master Mix (Bio-Rad), template cDNA and assay primers (Table 1) in a total reaction volume of 20 μl. Thermal cycling (50 °C, 2 min; 95 °C, 10 min; and 40 cycles at 95 °C, 15 S; 60 °C, 1 min) was performed using a MyiQ Single Color Real-Time PCR Detection System (Bio-Rad). Threshold cycle numbers (Ct) were defined as fluorescence values, generated by SYBR green binding to double stranded PCR products, exceeding baseline. Relative transcript levels were quantified as a comparison of measured Ct values for each reaction [24], normalized using cyclophilin as an internal control.
Table 1.
PCR Primers for genes in Q-RT-PCR analysis
| Sequence (5’- 3’) | ||
|---|---|---|
| Primer Set | Forward | Reverse |
| hCol1A | CCAGAAGAACTGGTACATCAGCA | CGCCATACTCGAACTGGAATC |
| hFN1 | CGGTGGCTGTCAGTCAAAG | AAACCTCGGCTTCCTCCATAA |
| hIL-6 | CTGGATTCAATGAGGAGACTTGC | TCAAATCTGTTCTGGAGGTACTCTAGG |
| hSNAIL1 | GCGCTCTTTCCTCGTCAGG | GGGCTGCTGGAAGGTAAACTCT |
| hTwist1 | TCTCGGTCTGGAGGATGGA | CAATGACATCTAGGTCTCCG |
| hVIM | GCTCAATGTTAAGATGGCCCTT | TGGAAGAGGCAGAGAAATCCTG |
| hZEB1 | GATGATGAATGCGAGTCAGATGC | ACAGCAGTGTCTTGTTGTTGT |
| hPPIA | CCCACCGTGTTCTTCGACATT | GGACCCGTATGCTTTAGGATGA |
| mIL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| mSNAIL1 | CACACGCTGCCTTGTGTCT | GGTCAGCAAAAGCACGGTT |
| mZEB1 | ACCGCCGTCATTTATCCTGAG | CATCTGGTGTTCCGTTTTCATCA |
| mPPIA | GAGCTGTTTGCAGACAAAGTTC | CCCTGGCACATGAATCCTGG |
RNA extraction and qualification
Total cellular RNA was extracted using either RNAqueous™ phenol-free total RNA isolation kits (Life Technologies, CA) or Quick-RNA MiniPrep kits (ZYMO Research) according to the manufacture’s recommendations. RNA was quantified spectrophotometrically using a NanoDrop ND-1000 (NanoDrop Technologies, DE). Quality of the purified RNA was assessed by visualization of 18S and 28S RNA bands using an Agilent BioAnalyzer 2100 (Agilent Technologies, CA). The resulting electropherograms were used in the calculation of the 28S/18S ratio and the RNA Integrity Number.
Library construction and sequencing
Poly-A+ RNA was selected from total RNA (1 μg) using oligo dT-attached magnetic beads. Bound RNA was fragmented by incubation at 94 °C for eight (8) minutes in 19.5 μl of fragmentation buffer (Illumina). First and second strand synthesis, adapter ligation and amplification of the library were performed using the Illumina TruSeq RNA Sample Preparation kit under conditions prescribed by the manufacturer (Illumina). Samples were tracked using “index tags” incorporated into the adapters. Library quality was evaluated using an Agilent DNA-1000 chip on an Agilent 2100 Bioanalyzer. Quantification of library DNA templates was performed using qPCR and a known-size reference standard. Cluster formation of the library DNA templates was performed using the TruSeq PE Cluster Kit v3 (Illumina) and the Illumina cBot workstation using conditions recommended by the manufacturer. Template input was adjusted to obtain a cluster density of 700–900 K/mm2. Paired end 50 base sequencing by synthesis was performed using TruSeq SBS kit v3 (Illumina) on an Illumina HiSeq 1000 using protocols defined by the manufacturer.
GLM modeling and pathway analysis
As shown in Table 2, the experimental design has six sample groups and requires a two factor analysis for time of TNF stimulation (“Time”) and presence of TGFβ stimulation (“Transformed”). For this purpose we employed the Generalized Linear Modeling (GLM) capabilities in the R Bioconductor package edgeR [25] to perform the data modeling phase of the analysis within our data analysis pipeline shown in Fig. 2b. EdgeR was developed to analyze differential count data arising from designed experiments such as RNA-Seq with single and multiple experimental factors and small numbers of replicates (here n = 3). It includes important statistical methods such as scaled normalization using trimmed means, the negative binomial distribution to describe read count variability, estimates of gene specific dispersion parameters by conditional maximum likelihood using Empirical Bayes methods, exact tests for differential gene expression analysis (DEGA) of one factor experimental designs and GLM for DEGA on multiple factor designs.
Table 2.
Abbreviations and descriptions of the sample groups for differential gene expression (DGE) analysis
| Code | Description |
|---|---|
| CE | Control (C) untransformed Epithelial cells (E) |
| CM | Control (C) TGFβ transformed Mesenchymal cells (M) |
| T1E | TNFα treated (T) E cells 1 h post treatment |
| T1M | TNFα treated (T) M cells 1 h post treatment |
| T12E | TNFα treated (T) E cells 12 h post treatment |
| T12M | TNFα treated (T) M cells 12 h post treatment |
Fig. 2.
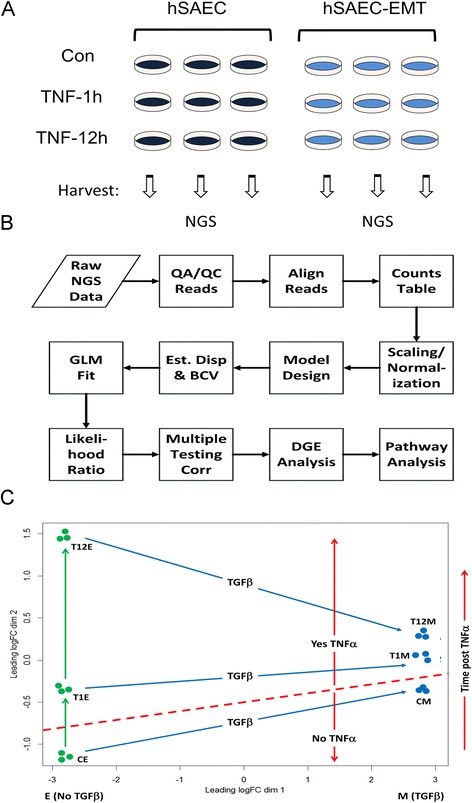
Experimental perturbation and RNA-Seq analysis. a Experimental design. Triplicate biological replicates were subjected to no treatment (hSAEC) or TGFβ treatment (hSAEC-EMT). Cells were stimulated with TNFα for 0, 1 or 12 h prior to RNA extraction. b Flow diagram for RNA-Seq GLM data analysis. Abbreviations: NGS, next generation sequencing; QA/QC, quality assurance/quality control; GLM, generalized linear model; DGE, differential gene expression. c Multidimensional unsupervised clustering analysis of RNA-Seq data sets. Shown is the similarity of each replicate based on TGF treatment or time post TNF and their clustering by group. Abbreviations: CE, control (unstimulated) epithelial cells; CM, control mesenchymal cells
The raw NGS analysis passed eleven separate quality analyses by FastQC [26]. Reads were aligned using TopHat2 [27], a fast splice junction mapper that aligns RNA-Seq reads using the ultra-high-throughput short read aligner Bowtie2 [28], using the Burrows-Wheeler index method. The alignment across all 18 samples produced a mean Overall Read Alignment Rate of 96.4 ± 0.4 % and a mean Concordant Pair Alignment Rate of 88.0 ± 0.2 %, confirming that the read alignment was excellent and that data was of high quality. (The results of QA/QC of RNA Seq are provided at Additional file 1).
As shown in Fig. 2b, edgeR used the resulting RNA-Seq read count data to build the counts table in edgeR to begin DGEA. Our experimental design has six groups of cell types (Table 2) each comprised of three samples, thereby giving a total of 18 sample columns with 23,710 rows of read counts, each row representing a unique gene in the count table.
Data filtering by removing unresponsive (i.e., uninformative) genes, was performed to improve the power of the study by reducing effects of multiple testing correction. We use this step to eliminate seemingly unresponsive genes (rows) from the count table using our rule that to be considered responsive, at least one of the six groups has to have a significant number of counts in at least two of its three samples. This filtering step culled out 7,346 rows resulting in a final read count table having 16,364 genes with a mean library size of 4.54E^07 across the 18 samples. This reduced read count table resulted in a mean scaling factor of 1.002 (range: 0.7985 – 1.0990).
Before creating a model design, we identified the model factors (Transformed. Time) and levels to edgeR as [N.0 h, N.1 h, N.12 h, Y.0 h, Y.1 h, Y.12 h] where N = no TGFβ transformation, Y = transformation with TGFβ and hr = time post TNFα treatment in hours. From these factors and levels we can identify the important contrasts that we would like to explore in our DEGA as shown in Table 3. We then designed the statistical model that was most appropriate for guiding the analysis in edgeR. This approach allowed us to optimize the information content while minimizing type I and type II errors, so we can then select those genes most suitable for pathways analysis.
Table 3.
Compared sample group pairs (contrasts) in the DGE analysis
| Contrasts |
| CM–CE |
| T1E–CE |
| T12E–CE |
| T12E–T1E |
| T1M–T1E |
| T12M–T12E |
| T1M–CM |
| T12M–CM |
| T12M–T1M |
The edgeR GLMs are non-linear models requiring iterative fitting and dispersion estimation to allow it to account for variations in gene abundance between RNA samples. EdgeR uses the Cox-Reid profile-adjusted likelihood method in estimating dispersions and the read count data is modeled using the negative binomial distribution because it accounts for overdispersion, which the Poisson model does not. This is important in RNA-Seq experiments because the observed variance (i.e., dispersion) in the read count data is often greater than that predicted from statistical theory. The tagwise dispersion was estimated in a gene-wise fashion in three steps using empirical Bayes shrinkage via a weighted likelihood method. Our estimated common dispersion was 0.01548079 which suggests that our sequencing quality is very good.
We next performed the GLM fit using the statistical design then performed the GLM Likelihood Ratio Test (LRT) for each of the contrasts in Table 3. Using each of the LRT outputs, we then performed Bonferroni FWER [29] and Benjamini and Hochberg FDR [30] multiple testing corrections on the DGE for each contrast using the edgeR topTags method. For further analysis such as pathway analysis, gene selection stringency is a key issue so we used a 2× absolute fold change cutoff before p-value filtering. This was a very strong response field and was amenable to a very high level of stringency so we focused on the Bonferonni corrected results with a FWER p-value cutoff of 0.00001 to insure that genes chosen for further analysis were reliably responsive to our experimental conditions and to keep the number of genes in the downstream analytical pool reasonable. Differentially expressed genes are shown in Additional files 2, 3, 4, 5, 6, 7, 8, and 9.
Venn diagrams were created to examine the intersections and relative complements of groups of contrasts (treatment pairs compared) that were deemed biologically interesting using locally developed tools. Subsets of genes identified as interesting were explored using QIAGEN’s Ingenuity® Pathways Analysis suite (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) for pathways, networks, and functional analyses.
Transcription factor enrichment analysis
ChIP-X data were downloaded from the website http://amp.pharm.mssm.edu/lib/chea.jsp in April 2014. The ChIP-X data consist of 148 transcription factors (TFs) and their respective target genes (TGs) derived from ChIP-chip, ChIP-Seq, ChIP-PET or DamID experiments in 237 publications [31]. Since TFs in different cell lines and treatments may result in different TF binding data, we separated experiments on the same TF, resulting in 345 combinations of TF and experiments performed on human, mouse and rat cells. Among the 345 TF binding experiments, 148 were performed on human. Here we refer to the ChIP-X data as the ChEA ChIP-X data set.
We also downloaded the uniform histone and TF binding peaks from the Encyclopedia of DNA Elements (ENCODE) Consortium website (http://ftp.ebi.ac.uk/pub/databases/ensembl/encode/integration_data_jan2011/byDataType/signal/jan2011/bigwig/) to serve as a second data set. As described above, we separated binding peaks of the same TF from different experiments, which results in 1,169 distinct ChIP-Seq experiments. The target genes corresponding to the TF binding peaks were identified according to PAVIS criteria by using our own scripts [32]. Here the coordinates of the peaks were mapped based on the hg19 refFlat.txt file downloaded from UCSC on Feb. 2012.
Differentially expressed genes (DEGs) between epithelial cells and mesenchymal cells were identified with FPKM fold change ratio > = 2 or < = − 2 and q-value < =0.01 from the RNA-Seq data. 3,487 differentially expressed genes were identified among the 23,284 genome-wide genes considered. Among the DEGs, 2010 genes are downregulated and 1477 are upregulated.
To determine if the transcriptional program regulated by a specific transcription factor from the above 148 transcription factors was activated in the EMT process or not, the hypergeometric probability distribution was used as the background distribution. The enrichment for each transcription factor regulated target gene (TG) was calculated by p-value = 1-hygecdf (k-1, N, K, n) using the Matlab build-in function hygecdf. The number of overlapping genes between the DEGs and the target genes for the specific transcription factor is k; N is the number of genome-wide genes: 23,284; K, the number of the TGs of the TF in the ChIP-data; n, the number of DEGs. For multiple testing corrections, Bonferroni FWER was then performed by multiplying the p-value with the number of TF binding experiments, i.e., 345 TF and experimental combinations used in the analysis based on the ChEA ChIP-X data and 1,169 that based on ENCODE ChIP-Seq data as the corrected p-value. Enrichment fold ratio was calculated by k/n/(K/N). If the corrected p-value by Bonferroni correction was smaller than 0.01 and fold ratio is greater than 1.5, the corresponding transcription factor was considered significantly enriched. For the TF enrichment analysis on ENCODE ChIP-Seq data, a Benjamini-Hochberg correction was also performed to estimate the false discovery rate (FDR) and q-value.
Hierarchical clustering analysis
TF and target genes were subjected to hierarchical clustering analysis using the Euclidean distance measure and the Ward. D2 linkage function in Spotfire (TIBCO). Variance, Skewness, and Kurtosis of dendrogram results were compared to 1000 random permutations of the data.
Comparison of type II and type III EMT programs
We compared DEGs in Type II EMT with a publicly available dataset from a model of Type III EMT (GSE177708, NCBI Gene Expression Omnibus). Affymetrix microarrays were used to measure gene expression in human adenocarcinoma cell line A549 after TGFβ induction of EMT. Genes in the two datasets were matched by Entrez ID, and only genes that were common to both datasets, and detected to be expressed in the microarray, were kept for further analysis.
Results
Induction of the EMT program
We have previously validated a model of Type II EMT using a continuously replicating line of human small airway epithelial cells (hSAECs) immortalized with human telomerase (hTERT) and CDK4 expression [21]. These cells show a stable epithelial morphology and differentiated cytokeratin isoforms after over 100 population doublings, express the stem cell marker p63 and high levels of p16INK4a, and have an intact p53 checkpoint pathway [21]. In the absence of TGFβ stimulation, hSAECs assumed a normal cuboidal morphology with perinuclear cytoplasmic distribution of F-actin, detected by fluorescence microscopy using confocal microscopy after staining with Alexa568-conjugated phalloidin (Fig. 1a). In response to chronic TGFβ stimulation, the cells acquired an elongated shape with markedly induced F-actin staining. This morphological change of enhanced front-rear polarity and actin rearrangement into cytosolic stress fibers are characteristic morphological changes of EMT [5, 33].
Fig. 1.
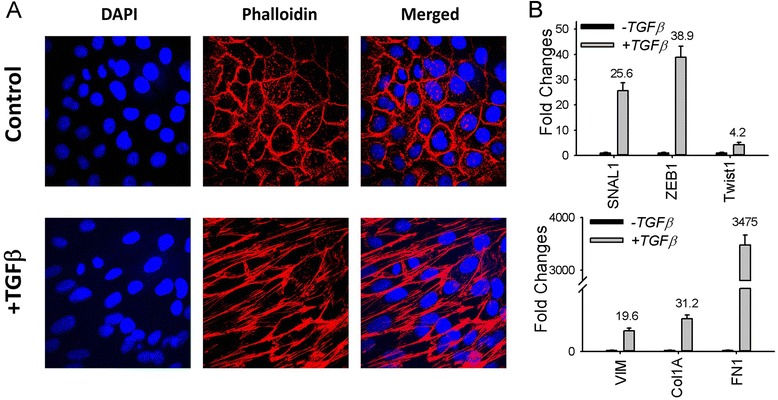
Induction of Type II EMT. a Primary human small airway epithelial cells (hSAECs) were incubated in the absence or presence of TGFβ (10 ng/mL) for 15 days. Cells were fixed, stained with Alexa568-conjugated phalloidin (for distribution of F-actin, shown in red color) and DAPI (a nuclear DNA stain, shown in blue color), and examined by confocal microscopy. b Total RNA was extracted, purified, and reverse-transcribed. The expression of core transcription factors (SNAIL1, Twist1, and ZEB1, upper panel) and mesenchymal markers (Vimentin (VIM), collagen 1A (Col1A), and fibronectin (FN1), lower panel) were examined by Q-RT-PCR. Shown is fold-change mRNA abundance normalized to cyclophilin. Data are from three independent experiments
To demonstrate the induction of the Type II EMT gene program, the steady state expression of mesenchymal genes and EMT-associated transcription factors were assessed by Q-RT-PCR. TGFβ-treatment induced the expression of FN1, Col1A and VIM by 3,475-, 31- and 20-fold, respectively (Fig. 1b). Similarly, the EMT core transcription factors SNAIL1, ZEB1 and Twist1 were induced by 26-, 39- and 4-fold, respectively (Fig. 1b). Together these data suggest that TGFβ induces morphological and gene signatures of stable Type II EMT in hSAECs.
Analysis of the EMT gene program
To understand the core EMT program and how TNF signaling modulates its expression, hSAECs were subjected to TGFβ induced reprogramming, and stimulated in the absence or presence of TNF for 1 or 12 h. Each experimental condition was reproduced in triplicate, representing 18 samples subjected to RNA-Seq analysis (Fig. 2a). The schema used in RNA sequence analysis is described in Fig. 2b.
Multidimensional scaling (MDS) was used as an unsupervised approach to visualize the level of similarity of the 18 RNA samples (Fig. 2c). MDS is a method similar to principal component analysis that enables high level representation of RNA Seq data based on similarities in expression of 500 genes with the largest fold change difference in the data set. In this analysis, distances were calculated as leading log-fold-changes between each sample pair. From Fig. 2c it is readily apparent that MDS differentiates the RNA seq expression patterns of the mesenchymal state from that of the epithelial cells in the first dimension and that of TNFα in the second dimension. Although TNF induces very strong effect on the epithelial cells in the second dimension, the relative response of the mesenchymal cells is highly compressed in the second dimension. Finally, the differences in fold change between replicates in the same group are very small relative to the inter-group responses. These analyses indicated that we had a robust and reproducible dataset and that chronic TGFβ treatment partially appeared to mimic acute stimulation of epithelial cells with TNF.
Generalized linear modeling was used to identify genes that were differentially expressed in the data set; contrasts were used to compare the differences between treatment groups. The results of generalized linear modeling (GLM) are provided in Additional files 2–10. We first focused on the comparison of the mesenchymal vs epithelial states. This comparison identified 3,487 differentially expressed genes (DEGs) that met a fold change cut-off of >2 with a highly significant, Bonferroni corrected pvalue (p < 0.00001). This group of DEGs was subjected to Ingenuity Pathway Analysis (IPA), where canonical pathways were ranked by the enrichment of genes in each process. Here we noted the rank ordered appearance of “cell death and survival”, “organismal injury and abnormalities”, and “cellular development” were influenced by the TGFβ-induced EMT (c.f. Fig. 3a, 3b).
Fig. 3.
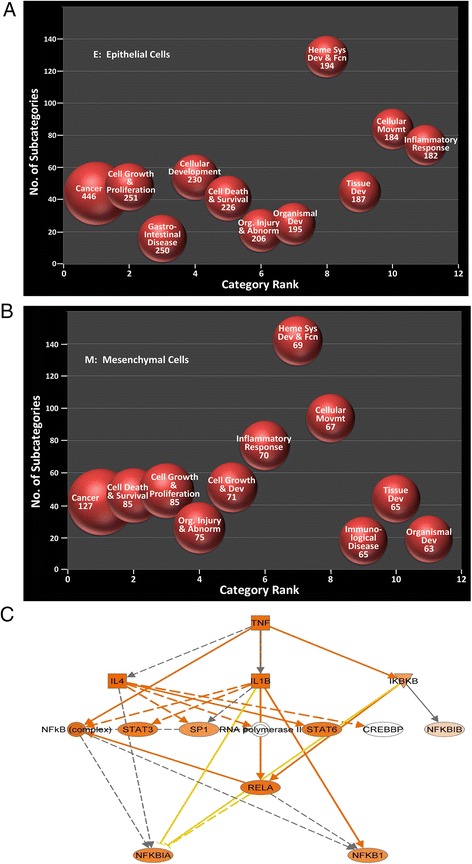
Functional analysis of differentially expressed genes in EMT. a Ingenuity pathway analysis (IPA) is shown for genes expressed in control hSAECs. X axis, category rank; Y axis, number of subcategories in each pathway. b IPA for genes in hSAEC-EMT. Note the cell death and survival and organismal injury and abnormality become the 2nd and 4th ranked pathway. c Enriched TNF signaling pathway identified by IPA in DEG data set. Intensity of red hue is proportional to upregulation by EMT
Analysis of canonical pathways further showed that NF-κB/RelA-signaling pathway was significantly enriched in a network with SP1 and STATs (Fig. 3c), factors known to modulate RelA-dependent signaling [34, 35].
Transcription factor enrichment of the EMT
An unbiased transcription factor (TF) enrichment analysis was performed on the DEGs. In this analysis, 345 distinct TF experiments of ChIP-chip, ChIP-Seq, ChIP-PET or DamID were tested for TF enrichment using hypergeometric probability distribution [31]. Here, we only consider the enriched TFs from 148 DNA binding experiments performed on human cells. Of these, 47 distinct TF experiment datasets were significantly enriched for the 3,487 DEGs in EMT. The enriched TFs are shown in Table 4, ranked by enrichment fold ratio and associated with the number of upregulated genes. Many of these TFs are linked to EMT in the literature. Of these, SMADs were among the top highly enriched TFs, consistent with their known role in mediating canonical TGFβ signaling [16]. Also included in the top list was tripartite motif containing 28 (TRIM28), an epigenetic modulator regulating histone H3 modifications on E-cadherin and N-cadherin promoters [36]. SOX2 promotes tumor metastasis by stimulating EMT via regulation of the WNT/β-catenin signal network [37]. ESR1 and ESR2, reported to suppress EMT [38, 39], were also significantly enriched. The CLOCK TF binds the E-box of PER2 and regulates this and other circadian rhythm genes, which are reported to be involved in EMT [40].
Table 4.
Significantly enriched human transcription factors for upregulated genes in the EMT process. The enriched transcription factors (TFs) were inferred by comparing ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF with the upregulated DEGs in the EMT process. The enriched TFs were sorted according to the enriched fold ratios
| Human TFs | PMID | # total TGs | # DETGs | Fold ratio | Corrected p-value | Cell lines |
|---|---|---|---|---|---|---|
| TRIM28 | 21343339 | 85 | 21 | 3.9 | 1.8E-05 | HEK293 |
| ESR1 | 21235772 | 213 | 36 | 2.7 | 4.8E-04 | MCF-7 |
| CLOCK | 20551151 | 398 | 58 | 2.3 | 2.3E-05 | 293 T |
| ESR2 | 21235772 | 392 | 57 | 2.3 | 9.1E-07 | MCF-7 |
| SMAD3 | 21741376 | 1074 | 144 | 2.1 | 1.4E-06 | HESC |
| SOX2 | 20726797 | 2060 | 275 | 2.1 | <3.45E-14 | SW620 |
| RELA | 24523406 | 1147 | 152 | 2.1 | <3.45E-14 | A549 |
| BACH1 | 22875853 | 1183 | 150 | 2 | <3.45E-14 | HELA-AND-SCP4 |
| GATA1 | 19941826 | 700 | 84 | 1.9 | 3.8E-14 | K562 |
| TP53 | 18474530 | 734 | 88 | 1.9 | 4.4E-06 | U2OS |
| GATA2 | 21666600 | 634 | 75 | 1.9 | 2.1E-06 | HMVEC |
| PAX3-FKHR | 20663909 | 975 | 115 | 1.9 | 4.8E-05 | RHABDOMYOSARCOMA |
| SMAD4 | 21741376 | 2032 | 237 | 1.8 | 2.4E-08 | HESC |
| DROSHA | 22980978 | 441 | 51 | 1.8 | <3.45E-14 | HELA |
| ZNF217 | 24962896 | 1400 | 160 | 1.8 | 8.7E-03 | MCF7 |
| NFE2L2 | 22581777 | 508 | 58 | 1.8 | 4.7E-11 | LYMPHOBLASTOID |
| EOMES | 21245162 | 861 | 98 | 1.8 | 3.7E-03 | HESC |
| SMAD2 | 18955504 | 1643 | 184 | 1.8 | 4.1E-06 | HaCaT |
| SMAD3 | 18955504 | 1643 | 184 | 1.8 | 3.8E-12 | HaCaT |
| NR3C1 | 21868756 | 1023 | 111 | 1.7 | 3.8E-12 | MCF10A |
| DNAJC2 | 21179169 | 803 | 87 | 1.7 | 6.1E-06 | NT2 |
| EGR1 | 20690147 | 5216 | 549 | 1.7 | 2.4E-04 | ERYTHROLEUKEMIA |
| SOX2 | 21211035 | 2830 | 288 | 1.6 | <3.45E-14 | LN229_GBM |
| GATA2 | 19941826 | 1727 | 175 | 1.6 | <3.45E-14 | K562 |
| MYC | 19915707 | 1793 | 177 | 1.6 | 8.5E-08 | AK7 |
| AR | 19668381 | 2958 | 291 | 1.6 | 5.0E-07 | PC3 |
| ATF3 | 23680149 | 1995 | 193 | 1.5 | 8.8E-13 | GBM1-GSC |
| SMAD4 | 21799915 | 2302 | 220 | 1.5 | 4.3E-07 | A2780 |
| GATA1 | 21571218 | 2388 | 228 | 1.5 | 7.2E-08 | MEGAKARYOCYTES |
PMID pubmed article number; # total TGs number of ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF; #DETGs number of differentially expressed genes
The tumor suppressor TP53 is known to regulate EMT [41, 42]. We observed that p53-related TP63, IRF1, TFAP2C, CTNNB1, AHR1, ELF5, FOXM1 and FOXO3 were also exclusively enriched TFs for downregulated DEGs (Table 5). Among these, TP63 attenuates EMT in prostate cells [43] and alternatively spliced isoform ΔNp63α inhibits EMT in human bladder cancer cells [44]. IRF1 is a tumor suppressor modulated by miR-23, promoting TGF-beta-induced EMT in lung cancer [45]. TFAP2C governs the luminal epithelial phenotype in mammary development and carcinogenesis [46]. AHR1 was reported to inhibit TGFβ-induced EMT [47]; ELF5 inhibits Type I EMT in mammary gland development and Type III EMT in breast cancer by transcriptionally repressing SNAIL2 [48]. Constitutive activation of CTNNB1 stabilized mesenchymal phenotypes of epithelial cells [49]. Of relevance to the hypothesis developed in this work, we noted that NF-κB/RelA was among the top significantly enriched TFs, with ChIP-Seq binding sites for 152 of the 1477 upregulated genes in the EMT data set, indicating that NF-κB/RelA dependent genes may play an important role in Type II EMT.
Table 5.
Significantly enriched human TFs for the downregulated genes in the EMT process. The enriched transcription factors (TFs) were inferred by comparing ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF with the downregulated DEGs in the EMT process. The enriched TFs were sorted according to the enriched fold ratios
| Human TFs | PMID | # total TGs | # DETGs | Fold ratio | Corrected p-value | Cell lines |
|---|---|---|---|---|---|---|
| TP63 | 17297297 | 34 | 14 | 3.9 | 1.1E-04 | HaCaT |
| TRIM28 | 21343339 | 85 | 22 | 3 | 7.4E-04 | HEK293 |
| ESR2 | 21235772 | 392 | 98 | 2.9 | <3.45E-14 | MCF-7 |
| CTNNB1 | 24651522 | 138 | 30 | 2.5 | 6.5E-04 | LGR5+ INTESTINAL STEM CELL |
| SOX2 | 20726797 | 2060 | 433 | 2.4 | <<3.45E-141.0E-16 | SW620 |
| IRF1 | 21803131 | 326 | 67 | 2.4 | 5.9E-09 | MONOCYTES |
| TFAP2C | 20629094 | 1125 | 229 | 2.4 | <3.45E-14 < 3.45E-14 < 3.45E-14 | MCF7 |
| ESR1 | 21235772 | 213 | 43 | 2.3 | 4.1E-05 | MCF-7 |
| ZNF217 | 24962896 | 1400 | 271 | 2.2 | <3.45E-14 | MCF7 |
| FOXM1 | 23109430 | 253 | 47 | 2.2 | 1.4E-04 | U2OS |
| NR1H3 | 23393188 | 574 | 104 | 2.1 | 8.6E-11 | ATHEROSCLEROTIC-FOAM |
| CDX2 | 20551321 | 383 | 69 | 2.1 | 1.2E-06 | CACO-2 |
| BACH1 | 22875853 | 1183 | 211 | 2.1 | <3.45E-14 | HELA-AND-SCP4 |
| AHR | 22903824 | 621 | 109 | 2 | 1.9E-10 | MCF7 |
| FOXO3 | 23340844 | 650 | 114 | 2 | 5.9E-11 | DLD1 |
| AR | 21909140 | 258 | 45 | 2 | 1.5E-03 | LNCAP PROSTATE CANCER CELL LINES |
| TP53 | 16413492 | 276 | 48 | 2 | 7.8E-04 | HCT116 |
| SMAD2 | 18955504 | 1643 | 285 | 2 | <3.45E-14 | HaCaT |
| SMAD3 | 18955504 | 1643 | 285 | 2 | <3.45E-14 | HaCaT |
| TP53 | 18474530 | 734 | 122 | 1.9 | 3.9E-10 | U2OS |
| RELA | 24523406 | 1147 | 187 | 1.9 | <3.45E-14 | A549 |
| TP63 | 23658742 | 3355 | 541 | 1.9 | <3.45E-14 | EP156T |
| ARNT | 22903824 | 912 | 147 | 1.9 | 2.2E-11 | MCF7 |
| SMAD4 | 19686287 | 348 | 56 | 1.9 | 1.4E-03 | HaCaT |
| TCF4 | 18268006 | 392 | 63 | 1.9 | 3.9E-04 | LS174T |
| CLOCK | 20551151 | 398 | 62 | 1.8 | 1.3E-03 | 293 T |
| PAX3-FKHR | 20663909 | 975 | 144 | 1.7 | 3.1E-08 | RHABDOMYOSARCOMA |
| TP63 | 22573176 | 3867 | 570 | 1.7 | <3.45E-14 | HFKS |
| GATA1 | 19941826 | 700 | 103 | 1.7 | 1.9E-05 | K562 |
| SMAD3 | 21741376 | 1074 | 158 | 1.7 | 4.8E-09 | HESC |
| CTNNB1 | 20460455 | 900 | 130 | 1.7 | 1.1E-06 | HCT116 |
| FOXA2 | 19822575 | 2791 | 402 | 1.7 | <3.45E-14 | HepG2 |
| SMAD4 | 21741376 | 2032 | 290 | 1.7 | <3.45E-14 | HESC |
| ELF5 | 23300383 | 920 | 130 | 1.6 | 4.2E-06 | T47D |
| GATA2 | 19941826 | 1727 | 241 | 1.6 | 5.2E-12 | K562 |
| TRIM28 | 17542650 | 680 | 94 | 1.6 | 1.2E-03 | NTERA2 |
| GATA2 | 21666600 | 634 | 87 | 1.6 | 3.6E-03 | HMVEC |
| AR | 19668381 | 2958 | 398 | 1.6 | <3.45E-14 | PC3 |
| HNF4A | 19761587 | 816 | 108 | 1.5 | 1.7E-03 | HUMAN INTESTINAL CELL LINE CACO-2 |
| AR | 22383394 | 1690 | 221 | 1.5 | 4.3E-08 | PROSTATE_CANCER |
PMID pubmed article number; # total TGs number of ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF; #DETGs number of differentially expressed genes
The depleted TFs based on the ChEA ChIP-X data set include ELF1, GABP, KDM6A, EST1, ETS1 (Tables 6, 7), suggesting that these TFs may be involved in the reversal of EMT, termed MET. GABP has a predicted binding site on miR200c, an important effector of EMT in immortalized mammary epithelial cells, MCF12A [41]. KDM6A was reported to regulate the expression of TFs critical for stem cell differentiation. EMT is a process of dedifferentiation and gain of stemness, which may explain the depletion of this TF in Type II EMT. EST1 and ETS1 have not yet been reported to be involved in MET. Although our TF enrichment results may be affected by limited experiments in the public databases of human epithelial cells, this analysis has generated valuable hypotheses on the TFs involved in EMT, most of which are supported by the literature.
Table 8.
Gene list in each of the six clusters obtained from hierarchical clustering analysis of 249 genes upon stimulation by TNFα in epithelial and mesenchymal state. Each cluster corresponds to that in Fig. 6b
| Cluster A | Cluster B | Cluster C | Cluster D | Cluster E | Cluster F | |
|---|---|---|---|---|---|---|
| 1 | RNF19B | DUSP6 | SNCAIP | PIM3 | PLAUR | SLC2A6 |
| 2 | IRF1 | EGR1 | TNFAIP2 | CLDN1 | MAFF | SYNPO |
| 3 | GBP1 | DUSP5 | CSF1 | SERPINB1 | NKX3-1 | TLR2 |
| 4 | TNFSF15 | SLITRK6 | DRAM1 | STARD5 | IL6 | COL12A1 |
| 5 | BTG2 | MAT2A | IKBKE | KRT6B | IL8 | FEZ1 |
| 6 | SPRR2D | NAB2 | SLC39A8 | GBP2 | PTX3 | PID1 |
| 7 | HCAR3 | CYP1A1 | DHRS3 | TNF | MGLL | |
| 8 | HCAR2 | FAM110C | SPRR1A | CXCL3 | SERPINE2 | |
| 9 | IL1RN | KIAA1551 | ITGB8 | CXCL2 | LBH | |
| 10 | TNFAIP8 | FBXW7 | ANKRD33B | NFKBIZ | TNC | |
| 11 | FOSB | SGK223 | PI3 | CSF2 | MMP9 | |
| 12 | NR4A1 | EMP1 | CDC42EP4 | IL1A | IL7R | |
| 13 | NCOA7 | SPRY2 | LYPD3 | IL1B | LAMB3 | |
| 14 | SERPINB2 | TIPARP | LPAR6 | NFKBIA | PIK3IP1 | |
| 15 | ARL5B | SRF | PRSS8 | NFKBIE | NAMPT | |
| 16 | ITPKC | TGIF1 | DDX58 | NUAK2 | ITGA5 | |
| 17 | GATA6 | FOSL1 | C1orf226 | LIF | DNER | |
| 18 | SDC4 | GADD45A | ISG15 | TNFAIP3 | GPR176 | |
| 19 | RAP2B | CXCL16 | TNFAIP6 | TGM2 | ||
| 20 | OVOL1 | SLPI | JPH2 | |||
| 21 | PIM1 | OAS1 | COL27A1 | |||
| 22 | HBEGF | TGM1 | STC2 | |||
| 23 | EREG | MUC1 | FSTL3 | |||
| 24 | TRIB1 | PSTPIP2 | SOX4 | |||
| 25 | EGR3 | SCNN1G | DUSP1 | |||
| 26 | PHLDA2 | SRD5A1 | ST3GAL1 | |||
| 27 | MAP3K14 | S100A8 | FADS3 | |||
| 28 | LRIG1 | MMP28 | BHLHE40 | |||
| 29 | KRT15 | PRODH | RHOB | |||
| 30 | HAS3 | IFIH1 | SPRY4 | |||
| 31 | IFFO2 | ASS1 | AKAP12 | |||
| 32 | AKR1B10 | GCLC | ANGPTL4 | |||
| 33 | CKB | LRRC8D | DLC1 | |||
| 34 | CALML3 | OAS3 | THBS1 | |||
| 35 | SLC2A1 | UBE2L6 | SEMA7A | |||
| 36 | AQP3 | IFI6 | NNMT | |||
| 37 | IL6R | TAP1 | ICAM1 | |||
| 38 | SESN3 | PSMB10 | XBP1 | |||
| 39 | ZNF462 | HLA-F | CXCR7 | |||
| 40 | DST | CDKN1C | MSC | |||
| 41 | HR | ALDH2 | CXCL1 | |||
| 42 | C1orf116 | SQRDL | CSF3 | |||
| 43 | WEE1 | STAP2 | ZC3H12A | |||
| 44 | GJB2 | PRKAG2 | INHBA | |||
| 45 | TSC22D1 | SGPP2 | SERPINA1 | |||
| 46 | CYP1B1 | C1R | EDN1 | |||
| 47 | BCL10 | TRANK1 | G0S2 | |||
| 48 | ALDH1A3 | APOL6 | LOC284454 | |||
| 49 | PHLDA1 | PARP14 | BMP2 | |||
| 50 | ERRFI1 | CXCL10 | DKK3 | |||
| 51 | TCF4 | BIRC3 | DKK1 | |||
| 52 | STK40 | TYMP | SMAD7 | |||
| 53 | AP5B1 | CD74 | PTPRE | |||
| 54 | FAM214A | SERPINA3 | F2R | |||
| 55 | OAS2 | TNFRSF12A | ||||
| 56 | WFDC2 | TNFRSF10D | ||||
| 57 | SFTPD | ZBED2 | ||||
| 58 | RHCG | NEDD9 | ||||
| 59 | MX1 | EMP3 | ||||
| 60 | PSMB9 | GFPT2 | ||||
| 61 | BBOX1 | MRAS | ||||
| 62 | IFI27 | AFAP1L1 | ||||
| 63 | SLC6A9 | IL1R1 | ||||
| 64 | IFIT3 | TLR4 | ||||
| 65 | MLPH | |||||
| 66 | CCBE1 | |||||
| 67 | IGFL1 | |||||
| 68 | HRH1 | |||||
| 69 | AOX1 | |||||
| 70 | MARCKSL1 | |||||
| 71 | CHST2 | |||||
| 72 | SERPINE1 | |||||
| 73 | KCNJ12 | |||||
| 74 | MCAM | |||||
| 75 | PRSS23 |
Table 6.
Significantly depleted human TFs for the upregulated genes in the EMT process
| Human TFs | PMID | # total TGs | # DETGs | Fold ratio | Corrected p-value | Cell lines |
|---|---|---|---|---|---|---|
| ELF1 | 17652178 | 99 | 0 | Inf | <3.45E-14 | JURKAT |
| GABP | 17652178 | 655 | 9 | 4.6 | 2.2E-08 | JURKAT |
| EST1 | 17652178 | 636 | 9 | 4.5 | 6.4E-08 | JURKAT |
| ETS1 | 20019798 | 1446 | 28 | 3.3 | 2.8E-14 | JURKAT |
| KDM6A | 18722178 | 410 | 9 | 2.9 | 7.4E-03 | U937_AND_SAOS2 |
| FOXP3 | 21729870 | 1300 | 43 | 1.9 | 6.1E-05 | TREG |
| VDR | 23849224 | 2029 | 69 | 1.9 | 9.1E-08 | CD4+ |
| GABP | 19822575 | 2430 | 92 | 1.7 | 7.2E-07 | HepG2 |
| HOXC9 | 25013753 | 1858 | 72 | 1.6 | 1.7E-04 | NEUROBLASTOMA BE2-C |
The depleted transcription factors (TFs) were inferred by comparing ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF with the upregulated DEGs in the EMT process. Depleted TFs were sorted according to the depleted fold ratios
PMID pubmed article number; # total TGs number of ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF; #DETGs number of differentially expressed genes
Table 7.
Significantly depleted human TFs for the downregulated genes in the EMT process. The depleted transcription factors (TFs) were inferred by comparing ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF with the downregulated genes in the EMT process. The depleted TFs were sorted according to the depleted fold ratios
| Human TFs | PMID | # total TGs | # DETGs | Fold ratio | corrected P-value | Cell Lines |
|---|---|---|---|---|---|---|
| GABP | 17652178 | 655 | 16 | 3.5 | 2.8E-09 | JURKAT |
| KDM6A | 18722178 | 410 | 14 | 2.5 | 2.2E-03 | U937_AND_SAOS2 |
| ETS1 | 20019798 | 1446 | 59 | 2.1 | 3.3E-10 | JURKAT |
| EST1 | 17652178 | 636 | 28 | 2 | 3.3E-03 | JURKAT |
| GABP | 19822575 | 2430 | 132 | 1.6 | 4.4E-08 | HepG2 |
PMID pubmed article number; # total TGs number of ChIP-Seq or ChIP-chip identified target genes (TGs) of each TF; #DETGs number of differentially expressed genes
Further comparison of the TF enrichment analysis indicated that the 30 enriched TFs for upregulated DEGs significantly overlapped with the 40 TFs associated with downregulated DEGs (p-value = 4.3e-13), with 18 TFs in common (Tables 4 and 5). A similar finding was observed with depleted TFs in the EMT DEGs (Tables 6 and 7), suggesting that many transcription factors are bimodal, i.e., they both inhibit or activate the target genes depending on the chromatin environment.
Topographical map of EMT regulated transcription factors and target genes
We next sought to map the relationships between TFs and target genes controlling EMT. In this analysis, hierarchical clustering identified three significant TF clusters and two DEG clusters (Fig. 4). The gene clusters correspond to genes that were either up- or downregulated by EMT. Conversely, we noted that each TF is associated with both up- and down-regulated target genes, indicating bimodal behavior consistent with the above TF enrichment analysis. The TF cluster A contains TP63 and Smad2/3. Both Smad 2 and 3 regulate relatively fewer DEGs, some of which are coregulated by EGR and SOX2. TF cluster B contains RelA, BTB and CNC homology 1 (BACH1), globin transcription factor (GATA1/2), and MYC proteins; TF cluster B regulates a distinct group of DEGs from those regulated by cluster A, but also a bimodal fashion. Cluster C is the largest group of TFs containing estrogen receptor (ESR1/2), forkhead box 03 (FOXO3), TRIM28 and others, and is associated with the fewest up- or downregulated DEGs.
Fig. 4.
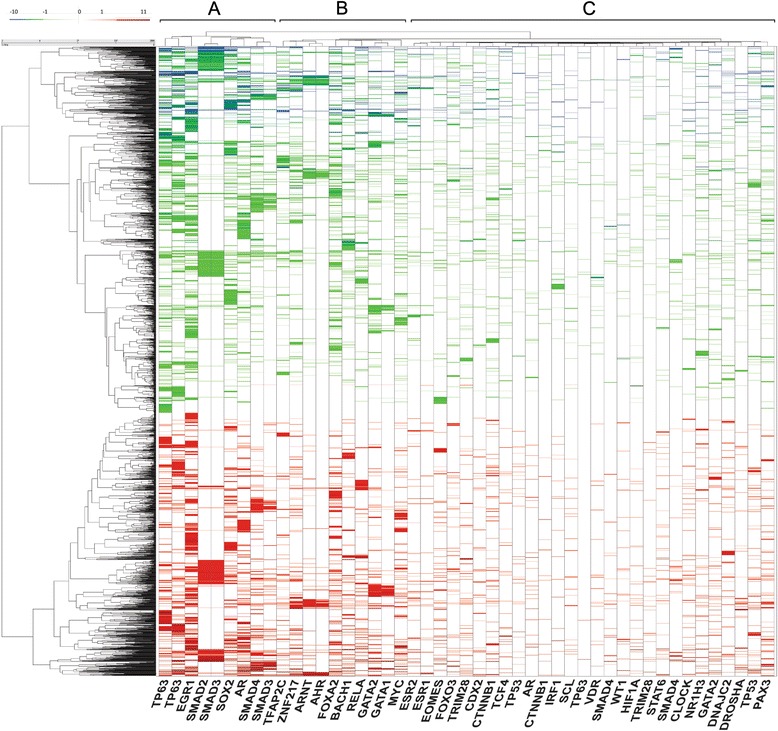
Transcription factor-target gene topological analysis. Hierarchical clustering analysis was conducted on 109 transcription factors (gene names of the enriched transcription factors from 47 experiments are shown in columns) and DEGs associated with EMT using the Euclidean distance measure and the Ward. D2 linkage function. Variance, Skewness, and Kurtosis dendrogram results were compared to 1000 random permutations of the data. Three significant clusters, labeled A, B and C are identified at top. The three clusters were highly significant (p < 0.001)
Type II EMT program controls biological functions distinct from than that of Type III EMT
Because Type II EMT occurs in primary epithelial cells without background oncogenic K-ras mutations characteristic of epithelial tumors [50], we hypothesized that Type II EMT induces distinct gene expression profiles. Differences in gene regulation between Type II EMT and Type III EMT may help identify pathways that are specifically involved in fibrosis (Type II EMT) vs invasion and metastases (Type III EMT). We therefore compared the TGFβ-induced gene expression patterns in primary hSAEC with that of an established human epithelial alveolar carcinoma cell, A549. A549 cells are K-Ras activated, keratin positive epithelial cells with features of the lower airways [51, 52]. For this purpose, we compared our Type II model dataset with a publicly available Type III model dataset (GSE177708, NCBI Gene Expression Omnibus). Affymetrix microarrays were used to measure gene expression in Human adenocarcinoma cell line A549 after TGFβ induction of EMT. Genes in the two datasets were matched by Entrez ID, and only genes that were common to both sets, and detected to be expressed in the microarray, were kept for analysis.
We identified 137 genes upregulated in Type II- but not in Type III-EMT, and 124 genes upregulated in Type III- but not in Type II EMT. Fig. 5 (top) illustrates the top 10 canonical pathways for genes uniquely induced in Type II EMT. In this plot, both the p value and ratio of enrichment are presented for each pathway. Here, lipid metabolism and thrombin signaling pathways were the most statistically significant. By contrast, Fig. 5 (bottom) displays the top 10 canonical pathways for genes uniquely induced in Type III EMT. Plasma membrane estrogen receptor signaling, ErbB receptor signaling and glycipan pathways were the most highly significant, consistent with the effect of Type III EMT on cellular motility, invasion and metastasis. Together these data suggest that the induction of Type II EMT produces a cellular biology state distinct from that seen in Type III EMT.
Fig. 5.
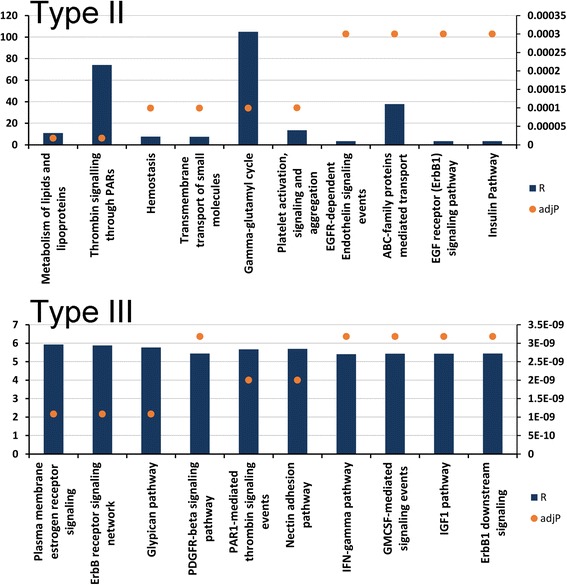
Unique pathways for Type II and Type III EMT. Shown are unique pathways associated with TGFβ-induced Type II EMT (top) and Type III EMT (bottom). Primary y-axis is the Ratio of Enrichment (R) while secondary y-axis is adjusted P-value as calculated by the hypergeometric test. The test is applied to evaluate the significance of enrichment (R), given as p-value (adjP). The value R is calculated by R = k/ke, where k = number of genes in our set present in a given pathway, and ke = expected value of k based on the reference set present in the same pathway
Modulation of the TNF response by Type II EMT
NF-κB is an inducible transcription factor that is regulated by a rapid release from cytoplasmic IκBα stores, followed by a slower release from 105 kDa NFκB2 complexes referred to as the canonical and noncanonical pathways respectively [53, 54], producing sequential waves of NF-κB dependent gene expression [55, 56]. Our mechanistic studies have found that the noncanonical pathway is coupled to the canonical pathway activation at a fixed time interval. This coupling is due to a feed-forward pathway mediated by inducible expression of the rate-limiting signaling adapter, TRAF1, that triggers activation of the noncanonical pathway [57]. Recently we observed that that the EMT state produces profound changes in canonical-noncanonical pathway coupling through transcriptional reprogramming of TRAF1 and NFκB2 promoters [5, 58]. As a result, the “coupling constant” e.g., the lag time between activation of the canonical and the noncanonical NF-κB program, was markedly reduced by EMT.
We therefore hypothesized that TGFβ-induced EMT produced profound differences in the response to activating the TNF signaling pathway. We first compared the TGFβ-regulated gene network with that induced by TNF stimulation. Of the 3,487 DEGs detected in EMT, 547 were also regulated by TNF, indicating that the two signaling pathways are highly overlapping (Fig. 6a). The expression patterns of the TNF-dependent EMT regulators were examined. To reduce noise, the data set was filtered to only the more robustly expressed 249 genes and subjected to hierarchical clustering, resulting in 6 major clusters (A-F, Fig. 6b). Cluster A represented early genes activated in hSAECs and silenced in EMT; Cluster B represented genes downregulated by TNF in hSAECs and not expressed in EMT; Cluster C represented late genes activated in hSAECs and whose expression was potentiated in EMT; Cluster D represented late genes activated in hSAECs and silenced in EMT; Cluster E represented early genes in hSAECs and potentiated by EMT; finally Cluster F represented genes not activated by TNF in hSAECs, but strongly upregulated by TNF in EMT in a late expression pattern. TF enrichment analysis identified 13 shared transcription factors controlling these 6 clusters including androgen receptor (AR), GATA1/2, MYC, NANOG, NR0B1, NR1H3, RelA, SMAD2/3, STAT3, TP-53 and −63 (not shown). These are members from all three of the major TF clusters associated with the EMT program (Fig. 4).
Fig. 6.
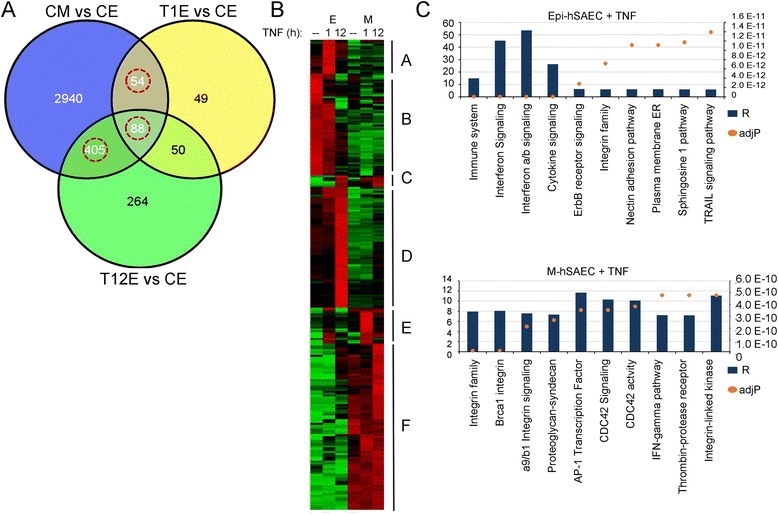
TGF treatment induces a core of TNF regulated genes. a Venn diagram of the overlap of TGF regulated genes (CMvsCE) with those induced by TNF at 1 h (T1EvsCE) and 12 h (T12EvsCE) in hSAECs. The overlapping genes between TGF state (CMvsCE) and both or either of TNF 1 h or 12 h were considered for further analysis. This gene set amounts to 547 genes. b. Heat map of TNF regulated EMT genes. a clustered image heatmap of a normalized matrix was created that correlates gene expression pattern of 249 genes to the time course in HSAECs upon stimulation by TNFα and TGFβ. For each gene, mean and standard deviation were calculated from their expression values across the time course and were normalized to unstimulated HSAECs. Z-score transformation was applied for each gene [5]. Hierarchical clustering was performed using an average-linkage clustering algorithm across the time points in the absence or presence of stimulants. The cluster tree of genes is represented on the y-axis, and time-points and stimulants are shown on the x-axis. Each block of red or green represents a high positive or negative correlation between the gene expression and the stimulant under a specific time point. The list of genes in each cluster are shown in Table 8. (C) Top 10 pathways uniquely associated with TNF-induced genes in epithelial cells (top) and mesenchymal cells (bottom). Primary y-axis is the Ratio of Enrichment (R); secondary axis is adjusted P value (described in Fig. 5)
The hierarchical clustering analysis suggested that the TNF pathway elicits distinct biological functions in epithelial cells versus that after Type II EMT. We therefore examined the function of genes activated by TNF in hSAECs but not activated after EMT, and those activated after EMT but not in hSAECs. From this analysis, interferon signaling and immune system activation were prominent in hSAECs (Fig. 6c, top). By contrast, integrin signaling was upregulated by TNF in the EMT background (Fig. 6c, bottom). Integrin αV signaling is particularly important because other studies have demonstrated that integrin signaling drives the maintenance of the EMT state [59], providing an additional cross-talk mechanism for how cytokine signaling reinforces EMT.
Analysis of TNF-dependent genes common to both epithelial and mesenchymal states, revealed that NFKBIE/IκBα is found in Cluster E. NFKBIE/IκBα is a prototypical early-response gene that we showed earlier was potentiated by EMT. In contrast, TNFAIP1/Naf1, a prototypic late gene under noncanonical NF-κB pathway control is found in Cluster F consistent with our earlier observations [5, 55–57]. Together we conclude that TGFβ-induced EMT produces complex modifications of the TNF response program through both potentiation and inhibition of distinct gene subnetworks.
RelA is essential for TGFβ-induced Type II EMT
Together, the findings that TGFβ induces a gene expression profile similar to that of TNF (Fig. 2), a significant fraction of TGFβ-regulated genes are NF-κB target genes (Table 4), and that TGFβ induces a core of TNF-induced NF-κB-regulated genes (Fig. 6a) led us to investigate the relationship of NF-κB activation with EMT.
Earlier studies have shown that TGFβ is coupled to NF-κB activation in carcinoma cells by oncogenic transformation induced expression of the TGFβ-associated kinase, TAK1 [60]. Whether TGFβ is coupled to NF-κB activation in primary cells without TAK1 amplification is not known. We therefore measured the relative abundance of nuclear RelA in response to TGFβ-stimulation. We observed a 2.5-fold induction of nuclear RelA 1 h after TGFβ stimulation (Fig. 7a, top panel). We noted that this induction was weaker than the 5-fold induction induced by the prototypical TNFα ligand; together there was no additive effect of the two ligands (Fig. 7a, lower panel).
Fig. 7.
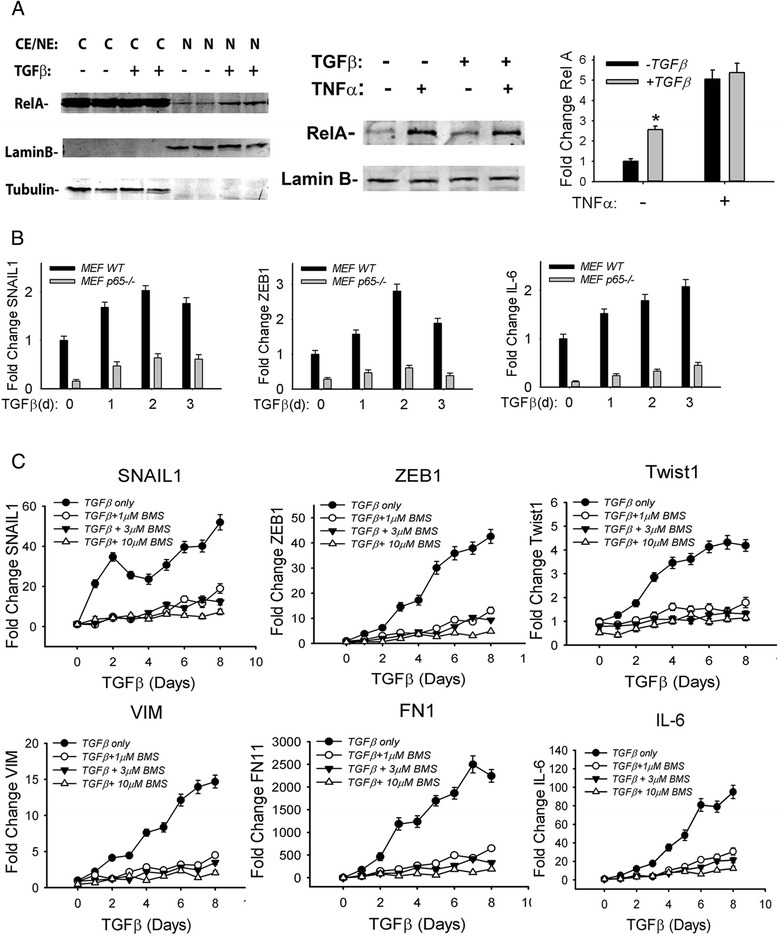
Requirement of NFκB signaling for TGFβ induced Type II EMT. a Cytoplasmic and nuclear extracts from hSAECs with or without EMT were isolated. 100 μg of cytoplasmic or nuclear extracts were processed for Western blot using anti-RelA Ab (upper panel). Lamin B and tubulin were detected as loading control Abs respectively. Low left panel: epithelial cells or mesenchymal cells were stimulated with 25 μg/ml TNF for 1 h and processed for RelA Western blot. Lower right panel: Quantification of nuclear RelA from Odyssey infrared imager. * P < 0.05 compared with control epithelial cells. b Wild-type (WT) or RelA−/− mouse embryonic fibroblasts (MEF) were incubated with TGFβ for 0, 1, 2, 3 days respectively. Total RNA was extracted and expression of EMT core transcription factors (SNAIL1 and ZEB1) and NF-κB-dependent genes (IL-6) were measured by Q-RT-PCR. The data shown is from n = 3 independent experiments. c Epithelial cells were stimulated with TGFβ for various times in the absence or presence of a small molecule IκB kinase inhibitor (BMS-345541, 1, 3, and 10 μM respectively). The expression of SNAIL1, Twist1, VIM, ZEB1, FN1 and IL-6 were measured by Q-RT-PCR. The data shown are from n = 3 experiments
Two strategies were used to determine the functional requirement of RelA in TGFβ-induced EMT. First, the induction of EMT was examined in RelA−/− mouse embryonic fibroblasts (MEFs). Here, the expression of TGFβ-induced SNAIL1, IL-6 and ZEB1 were completely blocked in RelA−/− MEFs compared to RelA WT MEFs (Fig. 7b). Second, we measured the effect of IKK inhibition on a time course experiment of TGFβ stimulation in epithelial cells. For this purpose, we used a highly selective, allosteric inhibitor of IKK (BMS-345541); this compound does not affect the JNK, MAPK or Jak-STAT pathways [22]. We observed that TGFβ stimulation produced a complex pattern of SNAIL1 expression, with a rapid peak of 30-fold induction after 2 days, followed by a decline, with a second peak of 40-fold induction after 5 days of treatment (Fig. 7c). By contrast, in epithelial cells treated with BMS-345541 at concentrations of 1, 3 and 10 μM respectively, SNAIL1 mRNA expression was significantly reduced at all time points measured. The patterns of ZEB1 and Twist1 showed a monotonic increase in expression peaking ~6 days after treatment at 40-fold and 3-fold inductions relative to untreated hSAECs, respectively. Similar to the effect on SNAIL1, BMS-345541 significantly reduced ZEB1 expression and completely blocked Twist1 expression (Fig. 7c). Similar patterns of inducible expression and BMS inhibition were observed for the mesenchymal cytoskeletal genes, VIM, FN1 and IL-6 (Fig. 7c). Together these data indicate that IKK-NF-κB/RelA activation is a necessary component of the TGFβ-induced EMT program.
Discussion
Type II EMT is a dynamic process that mediates airway response to injury, stimulating basal epithelial cells to promote re-epithelialization and extracellular matrix remodeling. EMT transcriptional reprogramming is a coordinated intracellular response triggered by epithelial growth factors (TGF, EGF) signaling via tyrosine kinase-coupled receptor growth factors to converge on a core set of transcription factors including SNAIL1, ZEB1, Twist1, and others. EMT is further modulated by intercellular signaling cross-talk through innate inflammatory cytokines, extracellular matrix and ROS stress. In this study, we employed a well-established model of TGFβ-induced Type II EMT in primary telomerase-immortalized hSAECs to understand the gene expression programs responsible for EMT in normal cells and how they are modified by the innate response. Surprisingly we observe that TGFβ influences a 3,487 member gene network, 547 of which are also controlled by TNF. By comparison to genome-wide ChIP-Seq data sets, we observe that this core pathway is enriched in genes under NF-κB/RelA control. We infer distinct biological functions compared to Type III EMT, demonstrate re-routing of the TNF pathway and experimentally demonstrate the functional role of NF-κB activation in Type II EMT. Finally, we note that the response of the TNF network is markedly affected by the EMT state, with distinct arms of the response being silenced, while others are enhanced. These studies are the first to our knowledge that demonstrate the unique TGFβ signatures in inducing Type II EMT and the requirement of NF-κB in this process.
TGFβ-induced Type II EMT
TGFβ activates cells by TGFβ receptor type II (TGFβRII), a transmembrane serine- threonine kinase that recruits and phosphorylates TGFβRI on the cell membrane [7]. Activated TGFβRI triggers two inter-related pathways, known as the canonical (SMAD-dependent) and noncanonical (ERK/MAPK-dependent) pathways, that converge on a core set of transcription factors responsible for coordinated suppression of epithelial genes and induction of mesenchymal genes. In the canonical pathway, the activated TGFβRI/II complex phosphorylates cytoplasmic Smads 2/3 that bind to Smad4. This trimeric Smad complex binds to regulatory sequences in the E cadherin promoter, leading its repression and subsequent loss of adherens junctions [12]. In our study, we have found that SNAIL1 expression is the most rapidly induced member of the core EMT regulators (Figs. 1b and 7c). Based on its rapid appearance, we suggest that SNAIL1 expression is one of the early triggers of Type II EMT. The absence of SNAIL1 in the public ChIP-Seq datasets prevented its enrichment and target gene analysis.
Other work has shown that the activated Smad 2/3/4 trimer also binds to Smad-binding elements in the regulatory regions of JunB and c-Jun genes, inducing their interaction with other coactivators including the EP300 coactivator [9]. This result indicates that the activities of the EMT core TFs are modulated by the EMT state, producing a bi-modal behavior, both activating and inhibiting gene expression. To this end, we note that over half of the TFs identified by enrichment analysis associated with upregulated DEGs in EMT are also associated with the downregulated genes in EMT. We note that these TFs include Smad2/3, RelA, SOX2 and others. Of relevance here, our earlier inference of RelA modulator-target gene triplets showed 6 patterns of modulator interaction with RelA involved in activation and inhibition of specific target genes within biologically relevant pathways [35]. In extension of these studies, we predicted that FN1 and FOXP1 function as modulators of RelA action in the setting of EMT [61]. These latter predictions suggest that the target genes and behavior of RelA are influenced by the EMT. It will be of interest to develop systems-wide maps of the effects of TGFβ on expression of modulators for the NFκB/RelA and other EMT-regulated TFs.
Activated TGFβRI complex triggers a Smad-independent (“noncanonical”) pathway through the PI3K/Akt, Ras small GTPases, Wnt/β-catenin, ERK, p38, and JNK kinases [7]. Although the actions of Smads are dominant on EMT, signaling through the noncanonical pathway is required for the full expression of EMT. Recent findings in non-small cell lung cancer cell lines indicate that phospho-Erk1/2 mediates a decrease in ECad and an increase in FN1 expression [62]. Also, inhibition of JNK, p38 and Akt activities without affecting Smad phosphorylation blocks TGFβ1 induced α-SMA, SNAIL1 and col1A in primary alveolar epithelial cells [63]. Together, these data indicate that Smad signaling is necessary but not sufficient for EMT. In the TF-TG topology map of EMT (Fig. 4), we note that the core TFs are clustered into three major clusters. We suggest that the TGs associated with Cluster A, containing Smad 2/3/4, TP63 and SOX2, are genes downstream of the canonical TGFβ signaling pathway.
Transcription factors controlling Type II EMT
We computed the over-representation of 148 TF targets from publically available ChIP-chip, ChIP-Seq, ChIP-PET or DamID data [38] using a hypergeometric probability distribution to tentatively identify regulators of the DEGs in EMT. 18 of the 30 enriched transcription factors in the EMT upregulated genes overlapped with the 40 transcription factors regulating the EMT downregulated genes (p-value = 4.3e-13), indicating that these 18 had bimodal activities in a target gene dependent manner. Many of have been associated with EMT. In addition to Smad and TP63 proteins discussed above, NF-E2-related factor-2 (NFEL2l2/Nrf2) is negatively regulated by E-cadherin expression, whose downregulation may promote Nrf2 nuclear translocation, resulting in the enhanced resistance of transformed cells to ROS stress [64]. The transcriptional repressor BACH1 has been shown to be upregulated in metastable Type III EMT [65]. We note that RelA and BACH1, GATA1, GATA2 and TFAP2C are enriched in the upregulated genes, whereas only TFAP2C is enriched in the downregulated gene set. This observation suggests to us that the activation mode of RelA is modulated by association of distinct coactivators, discussed below. Further dissection of this transcription network will be of interest. For example, the implication of the histone acetyltransferase, CLOCK, in the EMT program and depletion of KDM6A have not been reported to our knowledge and merits further investigation.
TNF and TGFβ cross-talk
TGFβ-induced EMT is modulated by a variety of signals including cytokine stimulation, morphogen signaling, and ECM interactions. Of relevance here, studies of transformed alveolar basal epithelial cells show that TGFβ-induced EMT is accelerated by the presence of members of the proinflammatory TNF/ IL-1 superfamily of cytokines [16, 17]. TNF/ IL-1 signal through highly conserved death domain containing receptors activating downstream Ras GTPase, p38 MAPK and JNK pathways, shared components of the noncanonical TGFβ signaling pathway [7, 58]. By contrast to this modulatory role our observations in normal epithelial cells indicate that the TNF signature is a major component of TGFβ-induced EMT (Fig. 5a). Pathway analysis of the DEGs in EMT indicates enrichment of TNFR signaling pathways (Fig. 3d) and we observe statistically significant enrichment of RelA target genes mapping to ChIP-Seq datasets (Table 4).
Requirement of NF-κB in Type II EMT
NF-κB is a master regulator of airway inflammation and cell fate determination. Our previous genome-wide analysis using whole genome-wide RNA-Seq and ChIP-Seq approaches have shown that this transcription factor regulates a ~4,000 member gene network mediating anti-apoptosis, inflammation, and adaptive immunity [34, 66]. In studies of cancer-associated Type III EMT, NF-κB is required for IGF-induced EMT by directly inducing SNAIL1 [67]. NF-κB has also been shown to upregulate ZEB1/2 and Twist1 [20], explaining, in part, how the IL-1/TNF superfamily of cytokines mediates Type III EMT. Our studies indicate that SNAIL1 is the earliest upregulated core transcription factor in Type II EMT and whose significant induction precedes that of ZEB1 and Twist1, leading us to suggest that SNAIL1 is an initial trigger of EMT.
Our study here shows that chronic TGFβ stimulation induces nuclear translocation of NF-κB/RelA. Other studies have shown that TGFβ is a potent activator of NF-κB/RelA in Ras-transformed cell lines. However, the relevance of these findings to normal epithelial cells is confounded by the observations that K-ras is a potent activator of NF-κB [68], and that oncogenic transformation induces expression of the TGFβ-associated kinase, TAK1, coupling NF-κB to TGFβRI [60]. Our western blot quantification indicates that TGFβ treatment in primary epithelial cells weakly induces NF-κB/RelA translocation at levels significantly less than the prototypical TNF monokine (Fig. 7a).
NF-κB/RelA translocation is controlled by two distinct pathways controlling sequential waves of NF-κB dependent gene expression [55, 56]. The canonical pathway liberates cytoplasmic NF-κB/RelA from IκBα complexes in response to IKKα∙IKKβ [54], whereas the noncanonical cross-talk pathway liberates cytoplasmic NF-κB/RelA from NFκB2 complexes in response to IKKα∙NIK. Our experiments using various doses of the allosteric IKK inhibitor indicate that expression of the core EMT genes is significantly inhibited at very low concentrations of BMS-345541. At this 1 μM concentration, BMS is relatively selective for IKK, over that of other related MAPKs [22]. This behavior, together with our observations that RelA knockout cells are also defective in TGFβ-induced SNAI1 and ZEB expression, support our conclusion that the IKK-NFκB pathway is required for type II EMT. More work will be required to identify whether the NF-κB pathway activated by TGFβ is via the canonical, noncanonical or a combination of the two pathways.
EMT induced alterations in the TNF signaling program
Our earlier genome-wide microarray studies discovered that tonic cellular stimulation by TNF produces sequential waves of NF-κB-dependent gene expression, with each wave controlling distinct biological functions [58, 59]. The most rapidly inducible, “early” genes were a group encoding cytokines, important in paracrine signaling, whereas late genes were those involved in antigen processing and cell surface presentation of MHCI complexes [56]. Mechanistically, the early genes are under canonical pathway control, and the late genes by noncanonical pathways [55, 56]. Recently we observed that that the EMT state produces both profound accentuation of early gene expression and changes in canonical-noncanonical pathway coupling resulting in shortening the coupling interval between them [5, 58]. Although the mechanisms are still under investigation, part of this effect was through transcriptional reprogramming of NFκB-dependent promoters, including that of TRAF1 and NFκB2 [5, 58].
In this study we observe that the EMT state markedly changes the TNF program in unexpected ways. Although a cluster of early and late genes (Clusters E, C, respectively Fig. 6b) are potentiated by EMT, our data suggest that this is not a uniform behavior. Interestingly, large subgroups of early and late genes (Clusters A, D, Fig. 6b) are largely silenced in the EMT state. Conversely, a large cluster of genes are activated in a late gene expression pattern in mesenchymal cells that is not activated in normal epithelial cells (Cluster F, Fig. 6b). Pathway analysis shows that the TNF program in normal hSAECs is coupled to interferon signaling and innate immunity. By contrast, the TNF program is re-routed by Type II EMT to elicit the integrin signaling program, an essential pathway controlling maintenance of EMT [59].
How might the TNF signaling pathway be affected so dramatically by EMT? One explanation for this complex behavior is through contextual effects of NF-κB associated modulators. In a recent study, we conducted a probabilistic inference of modulator activity on RelA activity using experimentally determined protein-protein interactions integrated with ChIP-Seq and a compendium of microarray data. The action identified 8349 modulator-RelA-target gene triplets, whose target genes were regulated in six distinct expression modes [35]. Using this approach, a preliminary inference of EMT-regulated NF-κB/RelA modulators identified FOXP1, a tumor suppressor gene involved in cardiac development; FN1 and CCDC80, interacting gene involved in extracellular matrix formation; ACTN1, a gene important in formation of actin polymers; and DOCK10, a Rho GTPase associated with cytoskeletal motility, as key modulators of the NF-κB pathway in EMT [61]. These functions may suggest how the EMT state may modulate the activity of the ubiquitous NF-κB/RelA in a cell-state dependent manner. More investigation will be required to determine the effect of EMT on the expression of RelA modulators. Moreover, these data suggest that the biological consequences of activating the TNF program are distinct in the normal vs EMT state.
Differences in Type II vs III EMT gene expression programs
TGFβ-induced Type II EMT leads to disruption of mucosal barrier function by inducing the loss of apical polarity, reduced epithelial cadherin and disruption of epithelial adherens junctions [6], express α-SMA stress fibers and intermediate filament VIM, to produce ECM through secretion of Col1A and FN1, and to increase expression of MMPs to promote airway remodeling. This cellular biology is a distinct biological phenomenon from that of Type III induced in a primary transformed cell background, associated with acquisition of motility and resistance to chemotherapeutics.
Conclusions
Analysis of the gene program triggered by TGFβ has led to a new understanding of the Type II EMT process. First, we have identified a core set of TNF-inducible genes enriched in experimentally determined NF-κB/RelA binding sites. Second, we demonstrate that activation of NF-κB/RelA is required for initiation of TGFβ-induced core EMT TFs and mesenchymal genes. Third, we provide evidence that the gene program induced by EMT in normal epithelial cells confers distinct biological properties than that induced in mesenchymal cells. Finally, we demonstrate that the evolution of the TNF pathway is distinct in the EMT state, altering innate immunity and reinforcing EMT via integrin αV signaling. These data have implications for the effect of EMT on airway mucosal response in chronic respiratory disease.
Availability of supporting data
The RNA-Seq sequence data have been deposited in the NCBI GEO (Gene Expression Omnibus) database under the accession number GSE61220.
Acknowledgements
Research support was provided by funds from the Sealy Center for Molecular Medicine, UL1TR000071 UTMB CTSA (ARB), NIEHS P30 ES006676 (ARB), Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (CPRIT Grant No. RP140113, PI - Rathindra Bose, co-PI – B. Montgomery Pettitt, to XL, AK and ARB) and NSF/DMS Collaborative Grant No. 1361411 (to ARB, M. Kimmel and H. Levine).
Additional files
QA/QC of RNA Seq.
Result of GLM for comparison of DEGs in E vs M state (no TGFβ treatment vs plus TGFβ).
Result of GLM for comparison of DEGs in E state: 1 h TNF vs Control.
Result of GLM for comparison of DEGs in E state 12 h TNF vs Control.
Result of GLM for comparison of DEGs in E state 12 h TNF vs 1 h TNF.
Result of GLM for comparison of DEGs after 1 h TNF E state vs 1 h TNF M state.
Result of GLM for comparison of DEGs in 12 h TNF E state vs 12 h TNF M state.
Result of GLM for comparison of DEGs in M state: 1 h TNF vs Control.
Result of GLM for comparison of DEGs in M state 12 h TNF vs Control.
Result of GLM for comparison of DEGs in M state 12 h TNF vs 1 h TNF.
Footnotes
Bing Tian, Xueling Li, Mridul Kalita, and Steven G. Widen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BT- developed cell system, conducted biological assays, wrote manuscript. JY- conducted biological assays, wrote manuscript. ARB- experimental design, wrote manuscript. SGW-NGS sequencing, analysis, wrote manuscript. BAL- modeling of NGS data, bioinformatics, wrote manuscript. TGW- NGS sequencing, analysis, wrote manuscript. MK, AK, XL, SB, BD- bioinformatics, wrote manuscript. All authors read and approved the final manuscript.
Authors’ information
Bing Tian, Xueling Li, Mridul Kalita, Steven G. Widen Joint first authors
Thomas G. Wood, Bruce A. Luxon, Allan R. Brasier Joint senior authors
Contributor Information
Bing Tian, Email: bitian@utmb.edu.
Xueling Li, Email: xuli@utmb.edu.
Mridul Kalita, Email: mrkalita@utmb.edu.
Steven G. Widen, Email: sgwiden@utmb.edu
Jun Yang, Email: junyang@utmb.edu.
Suresh K. Bhavnani, Email: subhavna@utmb.edu
Bryant Dang, Email: brdang@utmb.edu.
Andrzej Kudlicki, Email: askudlic@utmb.edu.
Mala Sinha, Email: masinha@utmb.edu.
Fanping Kong, Email: fakong@utmb.edu.
Thomas G. Wood, Email: tgwood@utmb.edu
Bruce A. Luxon, Email: baluxon@utmb.edu
Allan R. Brasier, Email: arbrasie@utmb.edu
References
- 1.Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8(4):432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 2.Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J. 2004;23(2):327–333. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 3.Hippensteil S, Opitz B, Schmeck B, Suttorp N. Lung epithelium as a sentinel and effector system in pneumonia - molecular mechanisms of pathogen recognition and signal transduction. Respir Res. 2002;7(1):97. doi: 10.1186/1465-9921-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalita M, Tian B, Gao B, Choudhary S, Wood TG, Carmical JR, Boldogh I, Mitra S, Minna JD, Brasier AR: Systems approaches to modeling chronic mucosal inflammation. BioMed Research International 2013, in press (Application of Systems Biology and Bioinformatics Methods in Biochemistry and Biomedicine) [DOI] [PMC free article] [PubMed]
- 6.Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18(5):684–692. doi: 10.1038/nm.2737. [DOI] [PubMed] [Google Scholar]
- 7.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 8.Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25(1):76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 9.Boxall C, Holgate ST, Davies DE. The contribution of transforming growth factor-beta and epidermal growth factor signalling to airway remodelling in chronic asthma. Eur Respir J: Off J Euro Soc Clinic Res Physiol. 2006;27(1):208–229. doi: 10.1183/09031936.06.00130004. [DOI] [PubMed] [Google Scholar]
- 10.Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Munoz I, Di Croce L, Helin K, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008;28(15):4772–4781. doi: 10.1128/MCB.00323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2(2):84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 12.Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11(8):943–950. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18(8):867–874. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446(7138):882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 15.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camara J, Jarai G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair. 2010;3(1):2. doi: 10.1186/1755-1536-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/S0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/S0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 20.Li C-W, Xia W, Huo L, Lim S-O, Wu Y, Hsu JL, Chao C-H, Yamaguchi H, Yang N-K, Ding Q, et al. Epithelial–mesenchymal transition induced by TNF-α requires NF-κB–mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72(5):1290–1300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64(24):9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 22.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, et al. BMS-345541 Is a Highly Selective Inhibitor of IκB Kinase That Binds at an Allosteric Site of the Enzyme and Blocks NF-κB-dependent Transcription in Mice. J Biol Chem. 2003;278(3):1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 23.Tian B, Zhao Y, Kalita M, Edeh CB, Paessler S, Casola A, Teng MN, Garofalo RP, Brasier AR. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J Virol. 2013;87(12):7075–7092. doi: 10.1128/JVI.03399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.FastQC: A quality control tool for high throughput sequence data. [http://www.bioinformatics.babraham.ac.uk/projects/fastqc/]
- 27.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. [DOI] [PMC free article] [PubMed]
- 28.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. J R Stat Soc Ser B Methodol. 1995;57(1):298–300.
- 30.Dunn OJ. Multiple comparisons among means. J Am Stat Assoc. 1961;56(293):52–64.
- 31.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26(19):2438–44. [DOI] [PMC free article] [PubMed]
- 32.Huang W, Loganantharaj R, Schroeder B, Fargo D, Li L. PAVIS: a tool for peak annotation and visualization. Bioinformatics. 2013;29(23):3097–9. [DOI] [PMC free article] [PubMed]
- 33.Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci. 2012;125(Pt 19):4417–22. [DOI] [PubMed]
- 34.Yang J, Mitra A, Dojer N, Fu S, Rowicka M, Brasier AR: A probabilistic approach to learn chromatin architecture and accurate inference of the NF-kappaB/RelA regulatory network using ChIP-Seq. Nucleic acids research 2013;41(15):7240-59. [DOI] [PMC free article] [PubMed]
- 35.Li X, Zhao Y, Tian B, Jamaluddin M, Mitra A, Yang J, et al. Modulation of gene expression regulated by the transcription factor NF-kappaB/RelA. J Biol Chem. 2014;289(17):11927–44. [DOI] [PMC free article] [PubMed]
- 36.Chen L, Munoz-Antonia T, Cress WD. Trim28 contributes to EMT via regulation of E-cadherin and N-cadherin in lung cancer cell lines. PLoS One. 2014;9(7), e101040. [DOI] [PMC free article] [PubMed]
- 37.Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T, et al. SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/beta-catenin signal network. Cancer Lett. 2013;336(2):379–89. [DOI] [PubMed]
- 38.Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol. 2007;9(4):470–8. [DOI] [PMC free article] [PubMed]
- 39.Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, et al. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17(4):319–32. [DOI] [PMC free article] [PubMed]
- 40.Hwang-Verslues WW, Chang PH, Jeng YM, Kuo WH, Chiang PH, Chang YC, et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci U S A. 2013;110(30):12331–6. [DOI] [PMC free article] [PubMed]
- 41.Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–23. [DOI] [PMC free article] [PubMed]
- 42.Wang Z, Jiang Y, Guan D, Li J, Yin H, Pan Y, et al. Critical roles of p53 in epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma cells. PLoS One. 2013;8(9), e72846. [DOI] [PMC free article] [PubMed]
- 43.Olsen JR, Oyan AM, Rostad K, Hellem MR, Liu J, Li L, et al. p63 attenuates epithelial to mesenchymal potential in an experimental prostate cell model. PLoS One. 2013;8(5), e62547. [DOI] [PMC free article] [PubMed]
- 44.Tran MN, Choi W, Wszolek MF, Navai N, Lee IL, Nitti G, et al. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem. 2013;288(5):3275–88. [DOI] [PMC free article] [PubMed]
- 45.Liu X, Ru J, Zhang J, Zhu LH, Liu M, Li X, et al. miR-23a targets interferon regulatory factor 1 and modulates cellular proliferation and paclitaxel-induced apoptosis in gastric adenocarcinoma cells. PLoS One. 2013;8(6), e64707. [DOI] [PMC free article] [PubMed] [Retracted]
- 46.Cyr AR, Kulak MV, Park JM, Bogachek MV, Spanheimer PM, Woodfield GW, White-Baer LS, O’Malley YQ, Sugg SL, Olivier AK et al.: TFAP2C governs the luminal epithelial phenotype in mammary development and carcinogenesis. Oncogene 2014;34(4):436-44. [DOI] [PMC free article] [PubMed]
- 47.Rico-Leo EM, Alvarez-Barrientos A, Fernandez-Salguero PM. Dioxin receptor expression inhibits basal and transforming growth factor beta-induced epithelial-to-mesenchymal transition. J Biol Chem. 2013;288(11):7841–56. [DOI] [PMC free article] [PubMed]
- 48.Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukacisin M, Romano RA, et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat Cell Biol. 2012;14(11):1212–22. [DOI] [PMC free article] [PubMed]
- 49.Stewart CA, Wang Y, Bonilla-Claudio M, Martin JF, Gonzalez G, Taketo MM, et al. CTNNB1 in mesenchyme regulates epithelial cell differentiation during Mullerian duct and postnatal uterine development. Mol Endocrinol. 2013;27(9):1442–54. [DOI] [PMC free article] [PubMed]
- 50.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–6. [DOI] [PubMed]
- 51.Foster KA, Oster CG, Mayer MM, Avery ML, Audus KL. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp Cell Res. 1998;243(2):359–66. [DOI] [PubMed]
- 52.Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer. 1976;17(1):62–70. [DOI] [PubMed]
- 53.Brasier AR: The NF- k B Signaling Network: Insights from systems approaches. In: Cellular Signaling And Innate Immune Responses To RNA Virus Infections. Edited by Brasier AR, Lemon SM, Garcia-Sastre A: American Society for Microbiology Press, Herndon, VA. 2008: 119–135.
- 54.Brasier AR. The NF-kappaB regulatory network. Cardiovasc Toxicol. 2006;6(2):111–30. [DOI] [PubMed]
- 55.Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, et al. RelA Ser276 phosphorylation is required for activation of a subset of NF-{kappa}B-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol. 2008;28(11):3623–38. [DOI] [PMC free article] [PubMed]
- 56.Tian B, Nowak DE, Brasier AR. A TNF-induced gene expression program under oscillatory NF-kappaB control. BMC Genomics. 2005;6:137–54. [DOI] [PMC free article] [PubMed]
- 57.Choudhary S, Kalita M, Fang L, Patel K, Tian B, Zhao Y, et al. Inducible TNF receptor associated factor 1 expression couples the canonical to the Non-canonical NF-κB pathway in TNF stimulation. J Biol Chem. 2013;288(20):14612-23. [DOI] [PMC free article] [PubMed]
- 58.Ijaz T, Pazdrak K, Kalita M, Konig R, Choudhary S, Tian B, et al. Systems biology approaches to understanding epithelial mesenchymal transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ J. 2014;7(1):13. [DOI] [PMC free article] [PubMed]
- 59.Mamuya FA, Duncan MK. aV integrins and TGF-beta-induced EMT: a circle of regulation. J Cell Mol Med. 2012;16(3):445–55. [DOI] [PMC free article] [PubMed]
- 60.Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, et al. TGF-[beta] and NF-[kappa]B signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013;32(12):1549–59. [DOI] [PMC free article] [PubMed]
- 61.Li X, Zhu M, Brasier AR, Kudlicki AS. Inferring genome-wide functional modulatory network: a case study on NF-kappaB/RelA transcription factor. J Comp Bio: J Comp Mol Cell Bio. 2015;22(4):300–12. [DOI] [PMC free article] [PubMed]
- 62.Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74(1):309–19. [DOI] [PMC free article] [PubMed]
- 63.Xi Y, Tan K, Brumwell AN, Chen SC, Kim YH, Kim TJ, et al. Inhibition of epithelial-to-mesenchymal transition and pulmonary fibrosis by methacycline. Am J Respir Cell Mol Biol. 2014;50(1):51–60. [DOI] [PMC free article] [PubMed]
- 64.Kim WD, Kim YW, Cho IJ, Lee CH, Kim SG. E-cadherin inhibits nuclear accumulation of Nrf2: implications for chemoresistance of cancer cells. J Cell Sci. 2012;125(Pt 5):1284–95. [DOI] [PubMed]
- 65.Thomson S, Petti F, Sujka-Kwok I, Mercado P, Bean J, Monaghan M, et al. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis. 2011;28(2):137–55. [DOI] [PMC free article] [PubMed]
- 66.Tian B, Brasier AR. Identification of an NF-kB dependent gene network. Recent Prog Horm Res. 2003;58:95–130. [DOI] [PubMed]
- 67.Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27(8):3165–75. [DOI] [PMC free article] [PubMed]
- 68.Bassères DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-κB subunit p65/RelA for K-Ras–induced lung tumorigenesis. Cancer Res. 2010;70(9):3537–46. [DOI] [PMC free article] [PubMed]