

Summary
Despite being surrounded by diverse nutrients, mammalian cells preferentially metabolize glucose and free amino acids. Recently, Ras-induced macropinocytosis of extracellular proteins was shown to reduce a transformed cell’s dependence on extracellular glutamine. Here, we demonstrate that protein macropinocytosis can also serve as an essential amino acid source. Lysosomal degradation of extracellular proteins can sustain cell survival and induce activation of mTORC1, but fails to elicit significant cell accumulation. Unlike its growth-promoting activity under amino acid-replete conditions, we discovered that mTORC1 activation suppresses proliferation when cells rely on extracellular proteins as an amino acid source. Inhibiting mTORC1 results in increased catabolism of endocytosed proteins and enhances cell proliferation during nutrient-depleted conditions in vitro and within vascularly compromised tumors in vivo. Thus, by preventing nutritional consumption of extracellular proteins, mTORC1 couples growth to availability of free amino acids. These results may have important implications for the use of mTOR inhibitors as therapeutics.
Graphical Abtract
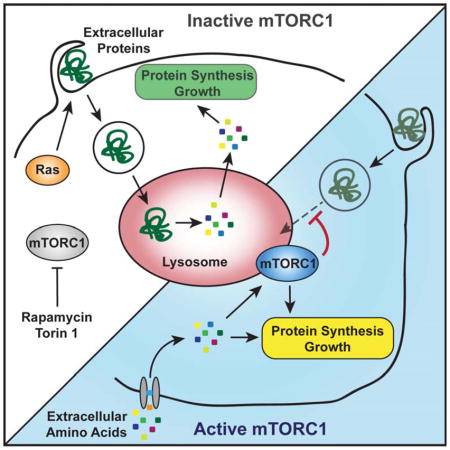
Introduction
To grow and divide, cells require a continuous supply of nutrients that support energy production and macromolecular synthesis (Cairns et al., 2011). Despite being surrounded by diverse bioenergetic substrates, mammalian cells preferentially metabolize low molecular weight nutrients such as glucose and amino acids. However, proteins are the most abundant organic constituents of body fluids, their combined amino acid content exceeding the amount of monomeric amino acids in human plasma by several orders of magnitude. Thus, extracellular proteins have the potential to function as important alternative nutrients, if accessible to cells.
The cells of metazoan organisms are instructed by external cues to engage in nutrient uptake. Growth factor signaling pathways not only stimulate cell cycle progression, but also promote nutrient uptake and initiate anabolic metabolism, thereby ensuring sufficient availability of building blocks for the synthesis of macromolecules to increase cellular mass (Thompson, 2011). Several growth factor-directed signaling pathways enhance cellular uptake of low molecular weight nutrients by increasing expression or surface presentation of metabolite transporters. For instance, both the PI3-kinase/Akt and Ras signaling pathways enhance glucose uptake (Manning and Cantley, 2007; Pylayeva-Gupta et al., 2011; Yun et al., 2009). Ras GTPases can also trigger internalization of extracellular macromolecules through macropinocytosis, an evolutionarily conserved, non-selective form of endocytosis (Bar-Sagi and Feramisco, 1986; Mercer and Helenius, 2009). Macropinocytosis allows unicellular amoeboid eukaryotes to live on extracellular macromolecules, but whether it functions in nutrient acquisition of metazoan cells is not well understood (Amyere et al., 2002). It was recently shown that by promoting macropinocytosis, oncogenic K-Ras signaling could reduce the dependence of proliferating cancer cells on exogenous glutamine supply (Commisso et al., 2013). This suggested that catabolism of extracellular proteins could provide anaplerotic substrates that allow mammalian cells to sustain mitochondrial bioenergetics and suppress apoptosis.
While growth factors regulate nutrient uptake, intracellular nutrient levels can inform the cellular signaling state. The kinase mammalian target of rapamycin complex 1 (mTORC1) is a central regulator that coordinates cellular nutrient levels with inputs from growth factor signaling to stimulate anabolic metabolism and growth (Ma and Blenis, 2009; Shimobayashi and Hall, 2014). mTORC1 activity strictly depends on sufficient levels of intracellular amino acids, which induce recruitment of mTORC1 to lysosomal membranes (Sancak et al., 2010). There, mTORC1 can be activated by further inputs from growth factor signaling. Activated mTORC1 phosphorylates multiple targets that concertedly enhance the generation of biomass. For instance, through phosphorylation of S6 kinase (S6K) and 4E binding protein (4E-BP), mTORC1 increases 5′ cap-dependent protein translation (Ma and Blenis, 2009). Conversely, through inhibition of Unc51-like kinase 1/2 (Ulk1/2), mTORC1 suppresses autophagy, thereby preventing degradation of cellular matter (He and Klionsky, 2009; Mizushima, 2010). By these means, mTORC1 promotes cell growth in response to an environment that provides favorable growth signals as well as ample nutrient supply.
Here, we show that mTORC1 suppresses the ability of mammalian cells to utilize extracellular proteins as a source of amino acids to support proliferation. Even in Ras mutant cells with constitutively activated macropinocytosis, mTORC1 impedes degradation of proteins internalized from the environment. Inhibition of mTORC1 elevates lysosomal catabolism of extracellular proteins and promotes proliferation of cells both during deprivation of amino acids in vitro and within poorly vascularized tumor regions in vivo. By preventing nutritional utilization of extracellular proteins, mTORC1 activity thus couples cell growth to the supply of free amino acids.
Results
Ras-Directed Macropinocytosis of Extracellular Proteins Can Sustain Cell Viability and Proliferation during Essential Amino Acid Starvation
We first asked whether the amino acids or hexose sugars contained in proteins could support cellular metabolism by determining whether physiological levels of extracellular proteins rescued viability and/or proliferation of cells cultured in the absence of glucose or different classes of amino acids. To investigate whether activation of K-Ras signaling altered a cell’s ability to metabolize extracellular proteins, we compared immortalized mouse embryonic fibroblasts (MEFs) harboring wild type K-Ras or a constitutively active K-RasG12D allele. Albumin is the major protein in plasma and interstitial fluids, its levels ranging from 2 – 5%. To mimic the protein-rich composition of extracellular fluids, culture medium was therefore supplemented with 10% dialyzed fetal bovine serum ± 3% albumin. When placed in glucose-free medium, both wild type and K-RasG12D MEFs ceased to proliferate, regardless of the presence of 3% albumin (Fig. 1A). When cells were subjected to non-essential amino acid (NEAA) starvation, albumin supplementation modestly improved viability. When cells were placed in medium lacking a single NEAA, glutamine, albumin supplementation allowed K-RasG12D MEFs to increase in cell numbers, albeit to a minor extent.
Figure 1. Physiological Levels of Extracellular Proteins Provide Nutritional Benefits for Wild Type and K-Ras Mutant Cells.

(A) Cell numbers of wild type and heterozygous K-RasG12D MEFs at day 3 of culture in medium ± 3% albumin lacking different nutrients as indicated [glucose, non-essential amino acids (NEAA), glutamine, essential amino acids (EAA), leucine]. The dashed line indicates starting cell numbers. # below detection limit. * p < 0.05, ** p < 0.01, *** p < 0.001. (B) Growth curve of wild type and K-RasG12D MEFs in leucine-free medium ± 3% albumin. (C) Growth curve of wild type MEFs expressing myristoylated Akt1 (myr- Akt1) in leucine-free medium ± 3% albumin.
Data are represented as mean ± STDEV (n = 3). See also Fig. S1.
In contrast to the above results, addition of 3% albumin caused a striking rescue of cell survival in medium lacking all essential amino acids (EAAs) (Fig. 1A, S1A). When starved for a single EAA, leucine, which is the most abundant amino acid in both albumin and the mammalian proteome, viability of wild type MEFs was completely restored by albumin supplementation (Fig. 1A). K-RasG12D MEFs were able to sustain proliferation in leucine-free medium containing 3% albumin, with cell numbers increasing over several consecutive days (Fig. 1B). Because growing cells must acquire leucine from exogenous sources to support protein synthesis, these results suggest that K-RasG12D MEFs could derive leucine from extracellular proteins. However, we noted that albumin supplementation of amino acid-deficient medium could sustain only limited cell proliferation as compared to amino acid-replete medium (Fig. 1A).
Besides Ras, PI3-kinase/Akt signaling is a second key pathway that instructs cells to engage in nutrient uptake (Manning and Cantley, 2007). However, expressing myristoylated Akt 1 or PTEN shRNA did not improve the response of cells to albumin supplementation of leucine-free medium (Fig. 1C, S1B). In contrast, inducing the expression of constitutively active K-RasG12V or H-RasG12V supported proliferation under these conditions (Fig. S1C, D), consistent with what we observed in MEFs harboring an endogenous K-RasG12D allele. The capacity of constitutively activated Ras signaling to sustain moderate levels of proliferation in medium that is protein-rich but amino acid-deficient is consistent with the shared ability of Ras GTPases to enhance macropinocytosis.
One mechanism by which oncogenic Ras signaling has been proposed to sustain cell viability during starvation is increased autophagy to recover nutrients from catabolism of intracellular macromolecules (Pylayeva-Gupta et al., 2011). To examine the role of autophagy in cell growth that utilizes extracellular proteins as a nutrient source, we determined requirements for the autophagy initiator kinases Ulk1/2. Wild type and Ulk1/2 double knockout (DKO) MEFs expressing K-Ras G12V or H-RasG12V could sustain proliferation in leucine-free medium when 3% albumin was added as an exogenous amino acid source (Fig. S1E, F). Loss of Ulk1/2 did not impair extracellular protein-dependent cell growth. Instead, Ulk1/2 DKO MEFs proliferated at significantly higher rates than wild type controls. Thus, not only are the autophagy initiator kinases Ulk1/2 dispensable for the utilization of extracellular proteins as nutrients, but loss of Ulk1/2-dependent autophagy actually improves the ability of Ras mutant cells to proliferate under these nutritional conditions.
We next asked whether albumin supported cell proliferation in medium containing reduced amounts of all EAAs. When EAAs were supplied at 5% of the levels present in complete medium, wild type and K-RasG12D MEFs ceased to proliferate and lost viability over time (Fig. 2A). Addition of 3% albumin improved survival of wild type MEFs, but did not support their proliferation. In contrast, albumin supplementation caused a significant increase in proliferation of K-RasG12D MEFs. As the proteins provided in 10% dialyzed serum did not suffice to promote cell proliferation during EAA starvation, we determined the concentration of albumin required for proliferation under these conditions. Wild type MEFs cultured in low-EAA medium displayed improved viability in response to increasing albumin levels (1 – 3%) (Fig. S2A). K-RasG12D MEFs could sustain proliferation when at least 1% albumin was added, and their proliferation rate increased progressively as the albumin concentration was raised to 3% (Fig. 2B). Thus, albumin must be supplied at physiological levels to support cell survival and growth in EAAdeficient medium. This may explain, why the protein-poor media formulations commonly used in cell culture do not suffice for MEFs to proliferate during EAA starvation.
Figure 2. Macropinocytosis and Lysosomal Degradation of Extracellular Proteins Supports Growth of Ras Mutant Cells during EAA Starvation.

(A) Growth curve of wild type and K-RasG12D MEFs in amino acid-deficient medium containing EAAs at 5% of the levels in complete medium ± 3% albumin. (B) Growth curve of K-RasG12D MEFs in amino aciddeficient medium containing 5% EAAs supplemented with indicated albumin concentrations. (C) Uptake and intracellular degradation of albumin in K-RasG12D MEFs, assessed by fluorescently labeled BSA and DQ-BSA. Protease inhibitors: 2 μM pepstatin A, 2 μM E-64, 10 μM leupeptin. Scale bars = 10 μm. (D) Growth curve of K-RasG12D MEFs in leucine-free medium ± 3% albumin and protease inhibitors as in (C). (E) Cell numbers of K-RasG12D MEFs at day 3 of culture in leucine-free medium ± 3% albumin and 25 μM EIPA. The dashed line indicates starting cell numbers. *** p < 0.001.
Data are represented as mean ± STDEV (n = 3). See also Fig. S2.
To characterize how cells derive leucine from extracellular proteins, cellular albumin uptake was investigated using constitutively fluorescent albumin and intracellular albumin proteolysis using a self-quenched albumin that emits fluorescence upon degradation (DQ-BSA) (Reis et al., 1998). After 1.5 h label uptake, K-RasG12D MEFs displayed multiple intracellular structures marked by both albumin and DQBSA (Fig. 2C). Adding lysosomal protease inhibitors did not perturb albumin internalization, but caused a strong decrease in DQ-BSA fluorescence. We next asked whether lysosomal albumin degradation was required to support growth during leucine starvation. Indeed, lysosomal protease inhibitors or the lysosomal acidification inhibitor chloroquine suppressed proliferation of K-RasG12D MEFs in leucine-free medium containing 3% albumin (Fig. 2D, S2B). Ras GTPases can promote cellular uptake of macromolecules through macropinocytosis (Bar-Sagi and Feramisco, 1986). Consistently, K-RasG12D MEFs displayed higher macropinocytic activity than wild type controls (Fig. S2C). To investigate whether this endocytic pathway contributed to albumin-dependent growth, cells were treated with the Na+/H+ exchange inhibitor EIPA, which blocks macropinocytosis but not other endocytic pathways (West et al., 1989). Cell proliferation in leucine-free medium + 3% albumin was strongly decreased by EIPA treatment, but restored when free leucine was added to culture medium (Fig. 2E). Together, these data show that macropinocytic uptake and lysosomal degradation of extracellular proteins can provide nutritional benefits during EAA starvation.
Catabolism of Extracellular Proteins Induces Lysosomal Recruitment and Activation of mTORC1
Cells can sense EAAs through the mTORC1 pathway, which integrates amino acid levels with inputs from growth factor signaling to promote cell growth (Ma and Blenis, 2009; Shimobayashi and Hall, 2014). When cells are placed in EAA-free medium, the ability of mTORC1 to phosphorylate downstream targets is repressed. To test if catabolism of extracellularly provided proteins could restore mTORC1 activity in the absence of EAAs, mTORC1 was inactivated by subjecting MEFs to 1 h EAA starvation. Fresh medium containing EAAs or different concentrations of albumin was then added and mTORC1 re-activation assessed by Western blotting against phosphorylated S6K1. Supplementing EAA-free medium with 1 – 3% albumin caused a concentration-dependent increase in S6K1 phosphorylation in both wild type and K-RasG12D MEFs, which was completely suppressed by the mTOR inhibitor torin 1 (Fig. S3A). Thus, physiological levels of albumin can activate the mTORC1 pathway. Although wild type and K-RasG12D MEFs exhibited comparable mTORC1 activation in response to EAAs, K-RasG12D MEFs displayed higher mTORC1 activity upon addition of 3% albumin (Fig. 3A). Similarly, H-RasG12V expression increased mTORC1 activity in response to albumin (Fig. S3B). Conversely, elevating PI3-Kinase/Akt signaling through shRNA-mediated depletion of PTEN increased mTORC1 activation by EAAs but not albumin (Fig. S3C). These data suggest that Ras signaling enhances mTORC1 activation in response to albumin stimulation through the amino acid-sensing branch rather than the growth factor-regulated branch of the pathway.
Figure 3. Lysosomal Degradation of Internalized Proteins Activates the mTORC1 Pathway.
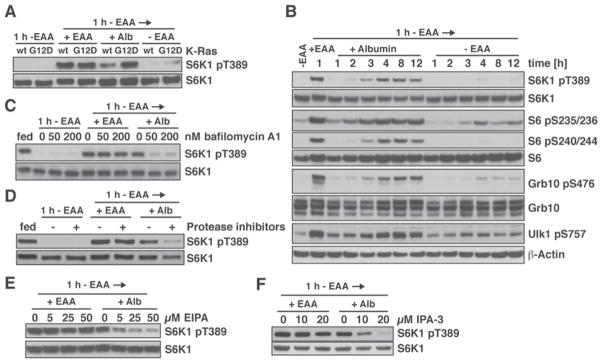
(A) Comparison of mTORC1 activation in wild type and K-RasG12D MEFs, analyzed by Western Blotting (WB). MEFs were starved of EAAs for 1 h, then placed in medium containing EAAs or 3% albumin, or in fresh EAA-free medium for 4 h. (B) Time course of mTORC1 activation in K-RasG12D MEFs by stimulation with 3% albumin after 1 h EAA starvation, analyzed by WB. (C) – (F) Effects of inhibiting lysosomal function or macropinocytosis on albumin-dependent mTORC1 activation, analyzed by WB. K-RasG12D MEFs were starved of EAAs for 1 h, then placed in medium containing EAAs or 3% albumin, or in fresh EAA-free medium for 3 h. (C) bafilomycin A1, (D) lysosomal protease inhibitors (10 μM pepstatin A, 20 μM E-64), (E) EIPA (Na+/H+ exchange inhibitor) or (F) IPA-3 (PAK1 inhibitor) were added at the onset of starvation. See also Fig. S3.
Extracellularly provided EAAs are rapidly taken up by cells through transmembrane transporters and activate mTORC1 within minutes (Nicklin et al., 2009). To examine the kinetics of mTORC1 pathway activation by extracellular proteins, mTORC1 re-activation in response to albumin stimulation was followed over time. Re-addition of EAAs caused rapid phosphorylation of mTORC1 targets such as S6K1, Grb10 and Ulk1 as well as the S6K target ribosomal protein S6 (Fig. 3B, S3D) (Kang et al., 2013; Nicklin et al., 2009). In contrast, in response to albumin, mTORC1-dependent phosphorylation of these proteins resumed only after 2 h and reached maximal levels after 4 h. Thus, extracellular proteins activate the mTORC1 pathway more slowly than EAAs, suggesting that distinct cellular processes are involved in mTORC1 activation by these nutrients.
We next asked whether macropinocytosis and lysosomal proteolysis were required for mTORC1 activation by extracellular proteins. The lysosomal acidification inhibitors bafilomycin A1 and chloroquine did not perturb mTORC1 activation by EAAs (Fig. 3C, S3E). In contrast, even low concentrations of either inhibitor strongly suppressed albumin-dependent mTORC1 activation. Similarly, protease inhibitors blocked mTORC1 activation in response to stimulation with albumin (Fig. 3D). To establish the relevance of macropinocytosis, the effects of pharmacological inhibitors of macropinocytosis including the Na+/H+ exchange inhibitor EIPA, the Pak1 inhibitor IPA-3 and the actin inhibitors jasplakinolide and cytochalasin D (Mercer and Helenius, 2009) were investigated. All these inhibitors strongly reduced mTORC1 activation by addition of albumin but not by re-addition of EAAs (Fig. 3E, F, S3F, G).
Amino acids signal to mTORC1 by inducing its translocation to lysosomal membranes, where it can be activated by further inputs from growth factor signaling (Sancak et al., 2010). To determine whether extracellular proteins similarly regulated mTORC1 by inducing its recruitment to lysosomes, K-RasG12D MEFs were subjected to 1 h EAA starvation, re-fed with EAAs or 3% albumin, and the subcellular localization of mTOR kinase was monitored by immunofluorescence. mTOR was distributed throughout the cytoplasm in EAA-starved cells. Upon albumin addition, mTOR localized to punctate structures that were marked by the lysosomal membrane protein LAMP2, similar to the pattern observed upon re-addition of free EAAs (Fig. 4A). Inhibiting lysosomal proteolysis with bafilomycin A1 completely blocked movement of mTOR to lysosomal membranes in response to albumin but not to EAAs (Fig. 4B).
Figure 4. Lysosomal Degradation of Internalized Proteins Induces Lysosomal Recruitment of mTOR.

(A) Lysosomal recruitment of mTOR by extracellular proteins or EAAs, analyzed by immunofluorescence against mTOR and the lysosomal marker LAMP2. K-RasG12D MEFs were starved of EAAs for 1 h, then placed in medium containing EAAs or 3% albumin, or in fresh EAA-free medium for 2 h. (B) Consequences of inhibiting lysosomal proteolysis on lysosomal recruitment of mTOR by extracellular proteins in K-RasG12D MEFs treated and analyzed as in (A). 200 nM bafilomycin A1 was added at the onset of EAA starvation. (C) Consequences of RagA/B knockdown on lysosomal recruitment of mTOR in K-RasG12D MEFs treated and analyzed as in (A). Scale bars = 10 μm. See also Fig. S4.
EAAs induce movement of mTORC1 to lysosomal membranes through a mechanism that requires the lysosome-associated Rag GTPases (Kim et al., 2008; Sancak et al., 2008). In contrast, glutamine can activate mTORC1 in a Rag-independent mechanism (Jewell et al., 2015). To investigate whether mTORC1 activation by lysosomal proteolysis of albumin depended on Rag GTPases, the consequences of shRNA-mediated knockdown of RagA and RagB was determined. Depletion of RagA/B resulted in diffuse localization of mTOR throughout the cytosol, regardless whether medium contained albumin or EAAs (Fig. 4C). Consistently, neither albumin nor EAAs could induce mTORC1-dependent phosphorylation of S6K1 in RagA/B knockdown cells (Fig. S4). Thus, both extracellular proteins and EAAs activate mTORC1 through a Rag-dependent mechanism.
mTORC1 Suppresses Cell Growth that Relies on Extracellular Proteins as Nutrients
To examine the signaling pathways downstream of Ras that contribute to a cell’s ability to utilize extracellular proteins as an amino acid source, we determined the effects of inhibitors against MEK1/2, PI3-kinase, tyrosine kinases and mTOR on proliferation of K-RasG12D MEFs in leucine-free medium + 3% albumin. To our surprise, mTOR inhibition did not retard cell proliferation in leucine-free medium supplemented with albumin. Rather, each of the mTOR inhibitors, including mTOR/PI3-kinase dual inhibitors, enhanced proliferation under these conditions (Fig. 5A left panel, Fig. S5A), although they effectively blocked mTOR signaling activity (Fig. 5B). In contrast, inhibition of MAP kinase, PI3-kinase or tyrosine kinase signaling modestly decreased cell proliferation in leucine-free medium (Fig. 5A left panel). The effects of these kinase inhibitors were different in leucine-containing medium, where mTOR inhibition was particularly effective at suppressing cell proliferation (Fig. 5A right panel). We next examined concentration-dependent effects of mTOR inhibitors on cell growth. Raising concentrations of torin 1 from 50 to 300 nM progressively improved cell proliferation in leucine-free medium + 3% albumin, suggesting that proliferation is inversely correlated with mTOR signaling activity when cells use albumin as a source of leucine (Fig. 5C). In contrast, cell proliferation was significantly higher when free leucine was provided extracellularly, but decreased with increasing doses of torin 1 (Fig. S5B). Similarly, torin 1 treatment induced several carcinoma cell lines harboring activating Ras mutations to proliferate in leucine-free medium + 3% albumin, while it strongly decreased their proliferation in leucinecontaining medium (Fig. S5C). As albumin provides a mixture of all proteinogenic amino acids, we also examined whether it could support survival / growth of cells in medium lacking other single EAAs (isoleucine, lysine, or arginine). Indeed, physiological levels of albumin rescued cell viability, and inhibition of mTOR signaling by torin 1 induced cells to robustly proliferate in medium lacking these EAAS (Fig. S5D).
Figure 5. mTORC1 Signaling Is a Negative Regulator of Extracellular Protein-Dependent Growth.

(A) Cell numbers of K-RasG12D MEFs at day 3 of culture in leucine-containing or leucine-free medium + 3% albumin and following inhibitors: MEK1/2 (1 μM PD0325901, 50 μM PD98059), PI3-kinase (25 μM LY294002, 2 μM wortmannin), tyrosine kinases (50 μM genistein), mTOR (50 nM rapamycin, 250 nM torin 1), mTOR/PI3-kinase (0.5 μM BEZ235, 0.5 μM GDC0980). Dashed lines indicate starting cell numbers. (B) mTOR, PI3-kinase and MAP kinase pathway activity in K-RasG12D MEFs cultured for 1 day in leucine-free medium + 3% albumin, analyzed by WB. Inhibitors were as in (A) (C) Growth curve of K-RasG12D MEFs in leucine-free medium ± 3% albumin and indicated concentrations of torin 1. (D) Growth curve of K-RasG12D MEFs expressing shRNA against Raptor, Rictor or control in leucine-free medium ± 3% albumin. (E) Bright field images of K-RasG12D MEFs expressing shRNA against Raptor, Rictor or control at day 4 of culture in leucine-free medium ± 3% albumin. Scale bars = 50 μm.
Data are represented as mean ± STDEV (n = 3). See also Fig. S5.
mTOR kinase is present in two distinct complexes: mTORC1, which regulates growth in response to nutrients and growth factor signaling, and mTORC2, which is a component of the PI3-kinase signaling pathway (Shimobayashi and Hall, 2014). To dissect their role in regulating albumin-dependent growth, the essential mTORC1 component, Raptor, or the essential mTORC2 component, Rictor, was depleted in K-RasG12D MEFs through shRNA-mediated knockdown (Fig. S5E). Rictor knockdown did not enhance cell proliferation during leucine deprivation, regardless of the presence of 3% albumin (Fig. 5D, E). In contrast, Raptor knockdown caused a dramatic increase in proliferation of K-RasG12D MEFs in leucine-free medium supplemented with albumin, with cells undergoing almost one population doubling per day. Thus, mTORC1 is a negative regulator of cell growth that relies on extracellular proteins as an amino acid source.
mTORC1 Inhibition Enhances Lysosomal Degradation of Internalized Albumin
We reasoned that the recovery of leucine from extracellular proteins constituted the rate-limiting step for cell growth in leucine-free medium. This suggested that mTORC1 suppresses extracellular protein-dependent growth by restricting a cell’s access to the amino acid content of extracellular proteins, conceivably by blocking either their endocytosis or lysosomal degradation. To distinguish these possibilities, we first tested whether mTORC1 inhibition increased internalization of extracellular macromolecules. However, neither torin 1 nor Raptor knockdown elevated cellular uptake of fluorescently labeled dextran or albumin, indicating that mTORC1 signaling does not inhibit bulk internalization of extracellular macromolecules (Fig. S6A–C).
We next examined whether mTORC1 inhibition affected lysosomal degradation of internalized proteins. K-RasG12D MEFs were placed in medium containing DQ-BSA and, to mark lysosomes, lysotracker ± torin 1, and the fluorescent signal generated by lysosomal proteolysis of DQ-BSA was followed over time. mTOR inhibition caused a dramatic increase in the fluorescence dequenching of DQ-BSA, indicative of its enhanced degradation (Fig. 6A, B). Similarly, inhibiting mTORC1 by Raptor knockdown increased cellular DQ-BSA fluorescence (Fig. S6D). Blocking lysosomal proteolysis with chloroquine or lysosomal protease inhibitors abrogated DQ-BSA degradation in torin 1-treated cells (Fig. S6E). Thus, mTORC1 negatively regulates lysosomal degradation of proteins that were taken up from the environment.
Figure 6. mTORC1 Suppresses Lysosomal Degradation of Internalized Proteins.

(A) Time-course of lysosomal DQ-BSA degradation in K-RasG12D MEFs in the presence or absence of 250 nM torin 1. (B) Quantification of DQ-BSA fluorescence of cells shown in (A). (C) Lysosomal degradation of DQ-BSA in wild type and K-RasG12D MEFs after 6 h DQ-BSA uptake in the presence or absence of 250 nM torin 1. (D) Quantification of DQ-BSA fluorescence of cells shown in (C).
Data are represented as mean ± STDEV (n ≥ 5 fields of view with > 20 cells each). * p < 0.05, *** p < 0.001. Scale bars = 20 μm. See also Fig. S6.
To further investigate how Ras and mTORC1 signaling regulated lysosomal proteolysis of internalized proteins, we compared the effects of mTORC1 inhibition and K-Ras activation. Inhibiting mTORC1 with torin 1 in wild type MEFs caused a significant increase in DQ-BSA fluorescence (Fig. 6C, D). Similarly, MEFs harboring the constitutively active K-RasG12D mutation displayed higher DQ-BSA degradation than wild type MEFs. Combining torin 1 treatment with constitutive K-Ras activation had synergistic effects and increased DQ-BSA proteolysis almost four times more than either manipulation alone. Thus, K-Ras enhances a cell’s ability to take up and degrade extracellular proteins, whereas mTORC1 specifically restricts their lysosomal degradation.
One mechanism by which mTORC1 signaling might impede degradation of internalized proteins is to suppress expression of genes involved in endocytosis or lysosomal biogenesis (Shimobayashi and Hall, 2014). To address whether mTORC1 inhibition enhanced DQ-BSA degradation through secondary effects of transcriptional changes, we determined the time frame during which mTORC 1 inhibitors exerted their effect. Pre-treating K-RasG12D MEFs with torin 1 for 6 h or 16 h did not increase the rate of DQ-BSA degradation (Fig. S6F). Moreover, when transcription was blocked by actinomycin D or triptolide, the torin 1-induced increase in DQ-BSA fluorescence was unaffected (Fig. S6G). Thus, lysosomal proteolysis of internalized proteins is an immediate cellular response to mTORC1 inhibition. Autophagic engulfment and degradation of intracellular constituents is suppressed by mTORC1 under nutrient-replete conditions by inhibition of the autophagy initiator kinases Ulk1/2 (He and Klionsky, 2009; Mizushima, 2010). As endocytosis and autophagy are both vesicular trafficking pathways that deliver macromolecules to the lysosome, we examined the consequences of Ulk1/2 deletion on DQ-BSA proteolysis. Ulk1/2 wild type and DKO MEFs expressing K-RasG12V degraded DQ-BSA at similar rates and responded to torin 1 treatment with comparable increase in DQ-BSA proteolysis (Fig. S6H). Moreover, torin 1 enhanced proliferation of Ulk1/2 DKO MEFs in leucine-free medium containing 3% albumin (Fig. S6I). Thus, mTORC1 suppresses lysosomal proteolysis of extracellular proteins by a mechanism that is distinct from its regulation of autophagy.
mTORC1 Signaling Can Have Opposite Effects on Cell Proliferation depending on a Cell’s Source of Amino Acids
While it has been shown in many different systems that mTORC1 promotes growth when amino acids are abundant extracellularly, the above data indicated that mTORC1 could suppress cell growth that relies on the catabolism of extracellular proteins. This led us to investigate the impact of mTORC1 inhibition on growth of K-RasG12D MEFs in medium containing decreasing amounts of EAAs, but supplemented with 3% albumin as an alternative EAA source. Torin 1 treatment or Raptor knockdown strongly decreased cell proliferation when EAAs were abundant extracellularly (Fig. 7A, B). However, whereas the proliferation of control cells quickly dropped with decreasing EAA levels, cells treated with torin 1 or expressing Raptor shRNA displayed only slightly reduced proliferation over a 10-fold range of EAA concentrations. At low EAA concentrations, torin 1 treatment or Raptor knockdown significantly improved cell proliferation. These results suggest that mTORC1 signaling stimulates proliferation of K-RasG12D MEFs in amino acid-rich medium, but restricts it under conditions when extracellular proteins become a required source of essential amino acids.
Figure 7. mTORC1 Signaling Has Opposing Effects on Cell Proliferation in Nutrient-Rich and Nutrient-Depleted Conditions.

(A), (B) Cell numbers of K-RasG12D MEFs (A) ±250 nM torin 1, (B) expressing Raptor or control shRNA, at day 3 of culture in medium containing 3% albumin and indicated amounts of EAAs. (C) Proliferation of pancreatic tumor cells in control and rapamycin-treated KPC mice, analyzed by immunohistochemistry against Ki-67. Scale bars = 400 μm; scale bars in blow-ups = 50 μm. (D) Quantification of Ki-67-positive tumor cells in outer and inner tumor regions as shown in (C). (E) Volume increase of pancreatic tumors in control and rapamycin-treated KPC mice, quantified by 3d high-resolution ultrasound. (F) Growth curve of Raptor KO MEFs in leucine-containing or free medium + 3% albumin. (G) Cell numbers of wild type MEFs expressing Raptor or control shRNA at day 3 of culture in medium containing 3% albumin and indicated amounts of EAAs.
Data in (A), (B), (F), (G) are represented as mean ± STDEV (n = 3). Dashed lines indicate starting cell number. Data in (D), (E) are represented as mean ± SEM (n = 5). * p < 0.05, ** p < 0.01, *** p < 0.001. See also Fig. S7.
Rapamycin Enhances the Growth of K-Ras-Induced Pancreatic Tumors In Vivo
To ask whether mTORC1 inhibition could promote proliferation of K-Ras mutant cells in vivo, we examined the effects of mTORC1 inhibition on the development of pancreatic ductal adenocarcinoma (PDA), using a genetically engineered mouse model that expresses endogenous alleles of mutant K-Ras and p53 specifically in the pancreas (KPC) (Hingorani et al., 2005). In humans, K-Ras is mutated in the majority of pancreatic cancers; consistently, tumor cells of KPC mice were recently shown to internalize high molecular weight dextran through macropinocytosis, indicating that they have the potential to access extracellular proteins (Commisso et al., 2013). Moreover, the tumor microenvironment of pancreatic cancer is poorly vascularized and highly nutrient-depleted (Kamphorst et al., 2015), which raises the possibility that extracellular proteins could function as an important alternative nutrient source in this context.
KPC mice with established tumors of comparable size were treated over the course of 8 days with rapamycin, and cell proliferation was examined by Ki-67 staining of tumor tissue. Because mTORC1 inhibition enhanced cell growth in cultured cells specifically during amino acid starvation, we examined the effects of rapamycin on proliferation of tumor cells in interior, hypovascularized tumor regions that were negative for the endothelial marker CD31. Indeed, rapamycin treatment caused a striking increase in the number of Ki-67-positive cells in those tumor regions despite the absence of the mTORC1 downstream target, phosphorylated S6 (Fig. 7C, D, S7A). In contrast, rapamycin decreased the fraction of Ki-67-positive cells in outer, vascularized tumor regions with a concomitant decrease in phospho-S6. These data indicate that pancreatic cancer cells respond to rapamycin in vivo depending on the tumor microenvironment they reside in – while rapamycin decreases cell proliferation in outer, vascularized regions, it enhances proliferation in interior, hypovascularized regions. These findings support the idea that mTORC1 can function as a suppressor of cell growth during nutrient starvation. Strikingly, rapamycin treatment significantly accelerated tumor growth in KPC mice (Fig. 7E).
Genetic Ablation of mTORC1 Signaling Can Induce Extracellular Protein-Dependent Growth Independently of Ras Transformation
Finally, to determine if the role of mTORC1 in suppressing utilization of extracellular proteins as an amino acid source to support cell growth was restricted to Ras-transformed cells, we investigated the consequences of mTORC1 inhibition in cells harboring wild type Ras alleles. Wild type MEFs expressing Raptor shRNA or treated with mTOR inhibitors could robustly proliferate in leucine-free medium supplemented with 3% albumin, (S7B–D). To more stringently block mTOR signaling, Raptor or Rictor were genetically ablated from MEFs harboring conditional alleles (Fig. S7E) (Cybulski et al., 2012). While Raptor knockout cells displayed strongly decreased cell proliferation in nutrient-replete medium as compared to wild type controls, they could sustain proliferation in leucine-free medium + 3% albumin (Fig. 7F). In contrast, deletion of Rictor only modestly decreased cell proliferation in leucine-containing medium and did not result in growth of leucine-deprived cells in albumin-supplemented medium (Fig. S7F). The proliferation of wild type MEFs expressing control or Raptor shRNA was also examined in medium containing decreasing amounts of EAAs as well as 3% albumin as an alternative EAA source. Raptor knockdown impaired cell proliferation under EAA-replete conditions (Fig. 7G). However, the difference in cell proliferation between control and Raptor knockdown cells diminished when EAA levels were reduced, and at low EAA levels, Raptor knockdown enhanced proliferation.
Discussion
mTORC1 Suppresses the Utilization of Extracellular Proteins as Nutrients
The above results demonstrate that in mammalian cells mTORC1 signaling suppresses lysosomal catabolism of proteins that were taken up from the environment. As a corollary, mTORC1 inhibition enhances cell proliferation that relies on extracellular proteins as nutrients, for instance in cultured cells deprived of EAAs or pancreatic cancer cells residing in poorly vascularized tumor regions. It is well known that the mTORC1 pathway is a potent stimulator of cell growth under nutrient-rich conditions, in part through enhancing translation (Ma and Blenis, 2009; Shimobayashi and Hall, 2014). However, the ability of mTORC1 to promote net protein synthesis strictly requires an exogenous source of amino acids. The present work indicates that by restricting amino acid recovery from extracellular proteins, mTORC1 couples cell growth to extracellular availability of free amino acids. This suggests that mTORC1 inhibition can promote growth under conditions when protein biosynthesis is limited by the acquisition of amino acids rather than the efficiency of translation. Whether mTORC1 stimulates or suppresses cell growth may therefore depend on a cell’s amino acid source.
Previous work showed that inhibition of mTORC1 could support cell survival in the absence of a source of extracellular EAAs. When cells are deprived of leucine in the absence of extracellular proteins, the ensuing inactivation of mTORC1 leads to de-repression of the autophagy initiation kinases Ulk1/2, which trigger the formation of autophagosomes to engulf intracellular constituents for subsequent delivery to the lysosome (He and Klionsky, 2009; Mizushima, 2010). Through this mechanism, autophagy supports cell survival during leucine deprivation. However, catabolism of intracellular proteins cannot lead to net acquisition of leucine (or other EAAs) required for cell growth and proliferation. Rather, autophagic degradation of intracellular proteins recovers sufficient EAAs for cells to engage in adaptive protein synthesis to sustain cell survival during limited periods of nutrient deprivation. The work presented here demonstrates that mammalian cells can utilize extracellular proteins as a source of EAAs that allows sustained cell viability. If cells catabolize sufficient amounts of extracellular proteins, as a result of activating mutations in Ras and/or suppression of mTORC1, they can even support net protein synthesis to increase in biomass and proliferate. The data presented here demonstrate that Ulk1/2 are dispensable for extracellular protein-dependent growth of Ras mutant cells. In fact, genetic deletion of Ulk1/2 enhances cell proliferation upon mTORC1 inhibition. This suggests that the degradation of intracellular constituents through autophagy as a result of mTORC1 inhibition can limit the rate at which amino acid-deprived cells can grow by taking up and degrading extracellular proteins.
Amino acids regulate mTORC1 activity by inducing its recruitment to lysosomal membranes (Sancak et al., 2010). The present work demonstrates that lysosomal degradation of endocytosed proteins regulates the mTORC1 pathway at the same step as amino acids that were imported from the environment in their monomeric form: both nutrients induce Rag-dependent recruitment of mTORC1 to lysosomal membranes, which permits its subsequent activation by growth factor signaling. Similarly, lysosomal catabolism of intracellular proteins that were delivered through autophagy leads to recruitment of mTORC1 to this organelle (Yu et al., 2010). Together, these data suggest that the regulation of mTORC1 activity at the lysosome constitutes a sensitive mechanism that allows cells to monitor amino acids recovered from proteins that were delivered through endocytosis or autophagy. Conversely, being activated at lysosomal membranes, mTORC1 may be well positioned to function as a regulator of cargo delivery to the lysosome by these membrane trafficking pathways. Notably, proteins that regulate endosomal trafficking have been recently emerging as a novel class of mTORC1 substrates (Hsu et al., 2011; Kim et al., 2015; Yu et al., 2011).
Implications for the Use of mTOR Inhibitors as Therapeutics
The present results may also shed light on the puzzling lack of efficacy of mTOR inhibitors as cancer therapeutics. There has been much effort over the last several years to target the mTORC1 pathway in cancer treatment. These studies have been motivated by the observation that human tumors often display elevated mTORC1 activity, commonly because of mutations in its upstream activator, the PI3- kinase pathway (Manning and Cantley, 2007). However, recent clinical trials showed only limited efficacy of rapamycin analogs (rapalogs) in a variety of solid tumors. The present work reveals an intrinsic weakness of mTOR inhibitors in cancer treatment. By enhancing catabolism of extracellularly derived proteins, mTOR inhibitors increase the use of extracellular proteins as alternative nutrients to support survival and sustain growth. This may be particularly important in nutrient-depleted tumor microenvironments or during metastasis, when tumor cells must adapt to novel metabolic niches. These findings predict that the growth promoting effects of mTOR inhibitors correlate with the capacity of cancer cells to take up sufficient extracellular proteins through endocytic pathways such as Ras-directed macropinocytosis. Consistently, treating KPC mice with rapamycin increases proliferation of pancreatic cancer cells that reside in poorly vascularized tumor regions and accelerates net tumor growth. This may explain the failure of mTOR inhibitors in the treatment of a variety of tumor types with activating mutations in Ras signaling, such as pancreatic cancer, in which K-Ras is the major driver oncogene, (Javle et al., 2010) or neurofibromatosis, which is caused by mutation in the Ras suppressor NF1.
One current explanation for the limited success of rapalogs is that they alleviate feedback repression of PI3-kinase/Akt signaling. This led to the development of active site mTOR inhibitors, which by targeting mTOR kinase inhibit mTORC1 and the Akt activator mTORC2, as well as dual mTOR/PI3- kinase inhibitors. The efficacy of these inhibitors in cancer therapy is currently being investigated, but the above data show that in cultured cells, the different classes of mTOR inhibitors share the caveat of promoting nutritional utilization of extracellular proteins. However, our work suggests that an alternative approach of combining mTOR inhibitors with inhibitors that block uptake or lysosomal degradation of extracellular proteins could achieve greater efficacy. When evaluating the role of mTORC1 signaling in tumorigenesis, it may also be worth considering insights from cancer genome sequencing efforts. Relatively few cancers were found to display mutations in mTOR kinase or direct pathway regulators that cause constitutive pathway activation. In contrast, mutations in the upstream PI3- kinase/Akt pathway are prevalent in human cancers (Manning and Cantley, 2007). This conceivably allows cancer cells to increase mTORC1 activity under nutrient-rich conditions, while not abrogating its regulation by amino acids, thus allowing tumor cells to adapt to nutrient deprivation.
Macropinocytosis of Extracellular Macromolecules as a Strategy of Nutrient Acquisition in Eukaryotic Cells
The present work demonstrates that mammalian cells can tap extracellular proteins as a source of EAAs to sustain survival and proliferation. These data support and extend recent work showing that Ras-directed protein macropinocytosis could alleviate the dependence of transformed cells on extracellular glutamine supply (Commisso et al., 2013). Ras-induced macropinocytosis of extracellular proteins has been previously recognized to sustain growth of unicellular amoeboid eukaryotes such as Dictyostelium discoideum (Amyere et al., 2002). Taken together, these findings indicate that macropinocytosis constitutes an evolutionarily ancient strategy of nutrient acquisition in eukaryotic cells. Interestingly, macropinocytosis in metazoan cells is under control of growth factor signaling, and macropinocytosis induction is an immediate response of various mammalian cell types to serum stimulation (Amyere et al., 2002; Mercer and Helenius, 2009). This suggests that growth factors can instruct cells to take up not only low molecular weight nutrients such as glucose and amino acids, but also extracellular macromolecules. However, concerted inputs from growth factor signaling and amino acids result in mTORC1 activation, which limits catabolism of proteins internalized from the environment. Therefore, mTORC1 initiates anabolic cellular metabolism that utilizes free amino acids, while preventing degradation of extracellularly derived proteins. This mechanism conceivably prevents futile turnover of the proteins contained in body fluids, as long as monomeric amino acids are available.
Experimental Procedures
For detailed protocols, see Extended Experimental Procedures.
Cell Culture and Nutrient Starvation Experiments
Cell culture experiments were performed at 37 °C and 5% CO2 in DMEM/F12 with 10% dialyzed FBS (molecular weight cut-off 10,000; Gemini Biosystems), 100 U/mL penicillin, 100 μg/mL streptomycin, 5 mM glucose and 2 mM glutamine.
For proliferation assays, MEFs were plated in complete medium, 5 h later briefly rinsed and then cultured in starvation medium as indicated. For EAA titration experiments, EAAs were supplemented at indicated percentages of 1x MEM amino acid solution (M5550, Sigma). Cancer cell lines were cultured in complete medium for 1 day prior to starvation. Numbers of adherent cells, which are > 95% viable as assessed by Trypan Blue exclusion (data not shown), were determined using a Multisizer 4 Coulter Counter (Beckman).
For mTOR activation and localization experiments, cells were rinsed and then incubated in EAA starvation medium (DMEM/F12 lacking all amino acids except glutamine) for 1 h, and subsequently placed in medium supplemented with EAAs (1x MEM amino acid solution), albumin (3%, if not stated otherwise) or fresh EAA-free starvation medium for indicated periods of time.
Mouse Strains and Rapamycin Treatment
The KPC mouse model (LSL-K-RasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre) has been described previously (Hingorani et al., 2005). KPC mice develop advanced and metastatic PDA with 100% penetrance, recapitulating the histopathological and clinical features of human PDA. Mice were housed at a 12 h light / 12 h dark cycle. All procedures were conducted in accordance with the Institutional Animal Care and Use Committee at CSHL.
KPC mice with established tumors of comparable size (7 – 9 mm in diameter, n = 5) as determined by ultrasound (Vevo software, Visualsonics) were treated daily for 8 days by oral gavage with either 5 mg/kg Rapamune (Pfizer) or saline (vehicle control). The last Rapamune administration was given 4 h prior to the endpoint. Tumor volumes were measured from 3d ultrasound images taken on day 4 and 7 of treatment.
Statistical Analysis
P-values were calculated using a two-tailed unpaired t-test for proliferation and fluorescence microscopy experiments of cultured cells and using the Mann-Whitney nonparametric t-test for analysis of murine tumors.
Supplementary Material
Acknowledgments
We gratefully acknowledge Michael Hall and Scott Lowe for providing reagents. We thank Dea Filippini and the Molecular Cytology Core of MSKCC for technical assistance. We are grateful to members of the Thompson laboratory for critical discussions and comments on the manuscript. WP is the recipient of an EMBO Long-Term Fellowship and the Genentech Foundation Hope Funds for Cancer Research Fellowship. This work was performed with assistance from the MSKCC Molecular Cytology Core and CSHL Laboratory Animal Shared Resources, which are supported by Cancer Center Support Grants P30 CA008748 and 5P30CA045508. This work was supported by grants from NCI (P01 CA104838 for CBT), NIH (5P30CA45508-26, 5P50CA101955-07, 1U10CA180944-01, 5U01CA168409-3 and 1R01CA190092-01 for DAT) and DOD (W81XWH-13-PRCRP-IA for YP and DAT). DAT is an investigator of the Lustgarten Foundation for Pancreatic Cancer Research, is supported by the Cold Spring Harbor Laboratory Association and the David Rubinstein Center for Pancreatic Cancer Research at MSKCC. CBT is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. CBT also serves on the board of directors of Merck.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amyere M, Mettlen M, Van Der Smissen P, Platek A, Payrastre B, Veithen A, Courtoy PJ. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. International journal of medical microbiology : IJMM. 2002;291:487–494. doi: 10.1078/1438-4221-00157. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science (New York, NY) 1986;233:1061–1068. doi: 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski N, Zinzalla V, Hall MN. Inducible raptor and rictor knockout mouse embryonic fibroblasts. Methods in molecular biology (Clifton, NJ) 2012;821:267–278. doi: 10.1007/978-1-61779-430-8_16. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science (New York, NY) 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA, Abbruzzese JL. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC cancer. 2010;10:368. doi: 10.1186/1471-2407-10-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science (New York, NY) 2015;347:194–198. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer research. 2015;75:544–553. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science (New York, NY) 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nature cell biology. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Molecular cell. 2015;57:207–218. doi: 10.1016/j.molcel.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature reviews Molecular cell biology. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Virus entry by macropinocytosis. Nature cell biology. 2009;11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Current opinion in cell biology. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nature reviews Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis RC, Sorgine MH, Coelho-Sampaio T. A novel methodology for the investigation of intracellular proteolytic processing in intact cells. European journal of cell biology. 1998;75:192–197. doi: 10.1016/S0171-9335(98)80061-7. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science (New York, NY) 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nature reviews Molecular cell biology. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Rethinking the regulation of cellular metabolism. Cold Spring Harbor symposia on quantitative biology. 2011;76:23–29. doi: 10.1101/sqb.2012.76.010496. [DOI] [PubMed] [Google Scholar]
- West MA, Bretscher MS, Watts C. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. The Journal of cell biology. 1989;109:2731–2739. doi: 10.1083/jcb.109.6.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science (New York, NY) 2011;332:1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science (New York, NY) 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.