

Abstract
Background
Despite a strong statistical correlation between dietary aflatoxin B1 (AFB1)-exposure and childhood stunting, the causal mechanism remains speculative. This issue is important because of emerging interest in reduction of human aflatoxin exposure to diminish the prevalence and complications of stunting. Pediatric liver diseases cause growth impairment, and AFB1 is hepatotoxic. Thus, liver injury might mediate AFB1-associated growth impairment. We have developed a rat model of dietary AFB1-induced stunting to investigate these questions.
Methods
Newly-weaned rats were given AFB1-supplemented- or control-diets from age 3-9 weeks, and then euthanized for serum- and tissue-collection. Food intake and weight were serially assessed, with tibial-length determined at the experimental endpoint. Serum AFB1-adducts, hepatic gene and protein expression, and liver injury markers were quantified using established methodologies.
Results
AFB1-albumin adducts correlated with dietary toxin contamination, but such contamination did not affect food consumption. AFB1-exposed animals exhibited dose-dependent wasting and stunting, liver pathology, and suppression of hepatic targets of growth hormone (GH) signaling, but did not display increased mortality.
Conclusion
These data establish toxin-dependent liver injury and hepatic GH-resistance as candidate mechanisms by which AFB1-exposure causes growth impairment in this mammalian model. Interrogation of modifiers of stunting using this model could guide interventions in at-risk and affected children.
Introduction
Stunting (height-for-age Z score <-2) and wasting (weight-for-height Z-score <-2) affect ∼200 million impoverished children worldwide.1, 2 These undergrown children are at increased risk for impaired cognition, metabolic syndrome, other morbidities, and mortality. Recognized causes of early stunting include intrauterine growth retardation (manifest as small for gestational age (SGA) infants), malnutrition, and infections; however, these factors account for only a subset of such impaired growth. Many observations link chronic aflatoxin ingestion with childhood stunting.3-5 Aflatoxins are toxic growth products (mycotoxins) produced by Aspergillus species6, 7, of which aflatoxin B1 (AFB1) is one of the most potent. Exposure typically occurs via ingestion of contaminated foods, with toxicity likely related to frequency and magnitude of exposure. Data supporting the association between aflatoxin exposure and growth impairment include: (i) the association between aflatoxin exposure and impaired weight gain in livestock; (ii) the high incidence of dietary aflatoxin exposure in sub-Saharan Africa and South Asia (regions with disproportionate prevalence of childhood stunting); and (iii) epidemiological and geographic correlation between the degree of stunting and the level of aflatoxinemia in African and Asian children.3-14 Aflatoxin exposure has also been implicated in kwashiorkor, and toxin-dependent intergenerational effects have been suggested by the association between maternal aflatoxinemia and SGA birth3-5.
Despite extensive epidemiological data correlating aflatoxin exposure and stunting, the causal role of dietary AFB1 exposure in childhood stunting has not been definitively established. Moreover, the physiological, cellular and molecular mechanisms that interconnect dietary AFB1 exposure and stunting remain unknown. Nevertheless, several indirect observations suggest that aflatoxin might hinder growth by injuring the liver and/or small intestine. For example, aflatoxins are metabolized by cytochrome P450s (CYPs) into reactive epoxides which form adducts with and disrupt the function of DNA, proteins, and other macromolecules6. Thus, abundant expression of CYPs 1A2 and 3A415 in human liver likely contributes to the known hepatotoxicity of aflatoxins7 and the established association between dietary aflatoxin exposure and risk of hepatocellular carcinoma (HCC) in hepatitis B-infected humans, livestock, and experimental models.6, 7 Because various pediatric liver diseases are associated with growth hormone (GH)-resistance and stunting16, aflatoxin-associated stunting might be mediated by hepatotoxic effects on GH-signaling. Enterocytes also express CYP3A4, and, thus, might be vulnerable to AFB1-induced toxicity.17, 18 Notably, stunted children in developing countries can exhibit an idiopathic form of enteropathy characterized by intestinal villous blunting, inflammation, and malabsorption.19 Furthermore, intestinal inflammation, like liver injury, is associated with GH resistance.20 Thus, aflatoxin-induced intestinal injury might also promote stunting via disruption of gut absorptive and/or barrier functions. These considerations implicate toxin-induced liver and intestinal injuries as mechanisms by which dietary AFB1 exposure might contribute to childhood stunting.
The experiments reported here were undertaken to develop an experimental animal model of dietary aflatoxin exposure with which to investigate the causal relationship between AFB1 toxin exposure and growth disturbance and to explore candidate mechanisms responsible for this association.
Results
Establishment of a Fischer Rat Model of Dietary Aflatoxin Induced Stunting
Rats are sensitive to AFB1-induced toxicity, and have been used for decades to investigate the biology of human aflatoxicosis.7, 8, 21 Therefore, we investigated the effects of dietary aflatoxin exposure on growth in newly-weaned rats. Inbred, male Fischer rats were initially studied so as to limit contributions of gender or genetics on phenotypic variation. The results demonstrated dose-dependent, significant reduction in the rate of weight gain in animals given ad libitum access to chow supplemented with 1-20 parts-per-million (ppm) of AFB1 compared to those provided with (vehicle-supplemented) control chow. Growth trajectory patterns showed deceleration in toxin-exposed vs. control animals within 1 week of diet initiation, and such divergence progressively increased throughout the 6 week duration of this experiment (Figure 1A). Tibial length, assessed at the experimental endpoint as a surrogate measure of linear growth22, was also diminished in a dose-dependent manner among treated rats (Figure 1B). Despite substantial effects on growth, no mortality was observed in association with toxin exposure.
Figure 1. Dietary AFB1 Induced Stunting in Fischer Rats.

(A) AFB1 Dose Dependent Wasting: Weight gain over time in newly weaned Fischer rats exposed to dietary AFB1 (● 0 ppm, ▼ 1 ppm, ■ 5 ppm, ◆ 10ppm, ▲ 20 ppm; *p<0.02 for area-under-the-growth-curves vs. 0, 1, 5, and 10 ppm; **p<0.01 vs. 0, 1 ppm; †p=0.02 vs. 0 ppm). (B) AFB1 Dose Dependent Stunting: Linear growth, assessed by tibial length, at the experimental endpoint (*p<0.005 vs. 0, 1, 5, 10 ppm; **p<0.03 vs. 0, 1 ppm; †p=0.03 vs. 0 ppm). (C) Food consumption (grams intake/rat/day) in control and toxin- exposed rats (● 0 ppm, ▼ 1 ppm, ■ 5 ppm, ◆ 10ppm, ▲ 20 ppm). (D) Serum AFB-albumin burden vs. dietary exposure (ANOVA p=0.03; very low level of adduct in controls likely reflects the allowable threshold of AFB1 contamination in PicoLab Rodent Diet 20).
To determine if toxin-induced growth impairment in exposed rats was associated with decreased food consumption in animals exposed to higher toxin concentrations, we compared daily food intake between cages of cohabitating rats (3/cage) for each examined level of toxin exposure over the experimental time course. Food consumption was comparable among all groups (Figure 1C); thus, reduced nutrient intake does not explain dose-dependent wasting and stunting in this model. To further investigate this point, AFB1-albumin adducts, which reflect the magnitude of toxin exposure23 were quantified at the experimental endpoint. This analysis demonstrated strong correlation between level of dietary toxin contamination and detectable adduct in serum (Figure 1D). Together, these data establish a causal role of dietary AFB1 exposure in growth impairment in rats.
Evaluation of Liver Injury and Function in Dietary AFB1-Exposed Rats
Because AFB1 hepatotoxicity6, 7 might mediate toxin-induced stunting, we examined sera and liver from AFB1-exposed and control Fischer rats for evidence of liver injury and dysfunction. Liver histology showed toxin dose-dependent bile duct hyperplasia (Figure 2A, H&E), which has previously been reported in toxin-exposed rats24, and increased fibrosis, as assessed by Sirius Red staining (Figure 2A, Sirius Red). AFB1 also induced dose dependent elevation of serum alanine aminotransferase (ALT, Figure 2B, left panel). Together, these data reveal AFB1 dose-dependent liver injury. Consistent with that conclusion, hepatocellular proliferation, a measure of injury-induced liver regeneration25, also showed dose-dependent elevation in toxin-exposed rats (Figure 2A, BrdU and Figure 2B, middle panel). Serum glucose, albumin (Figure 2B, right panel), and bilirubin (below the limit of detection), which reflect liver function, were not significantly affected by toxin exposure. These data demonstrate dietary AFB1-induced liver injury, but not liver failure, in this model.
Figure 2. Dietary AFB1 Induced Liver Injury in Rats.

(A) AFB1-exposed rats demonstrate dose-dependent increases in bile duct hyperplasia (by H&E staining, image photographed at 50x magnification), fibrosis (by Sirius Red, 50x), and hepatocellular proliferation (by immunohistochemical detection of BrdU incorporation, 200x; 100 micron bar in lower left corner of left-most image in each series). (B) Quantification of serum ALT (left panel, ANOVA p=0.04); hepatocellular BrdU (middle panel, ANOVA p=0.02; *p<0.05 vs. control); serum glucose (black bars) and serum albumin (grey bars; right panel).
Hepatic Growth Hormone (GH) Signaling in Toxin-Exposed Animals
GH and its target insulin like growth factor 1 (IGF1) are important regulators of longitudinal growth26, and STAT5 activation mediates the growth-promoting effects of GH27. Hepatic GH-resistance likely contributes to the growth impairment associated with pediatric liver diseases16. Therefore, we next examined GH signaling in livers from AFB1-exposed and control rats. This analysis showed suppression of hepatic Igf1 mRNA expression at the experimental endpoint (i.e. 6 weeks after initiating dietary toxin exposure; Figure 3A). GH receptor (Ghr, Figure 3A) and Stat5b (but not Stat5a, Figure 3A) expression exhibited similar AFB1-induced suppression. To further examine the effects of AFB1 on hepatic GH signaling, we also assessed hepatic expression and activation (by phosphorylation) of STAT5 protein, which mediates GH effects on Igf1 expression28. The results demonstrated marked variation in levels of phospho-STAT5 across replicate livers from animals exposed to control or 10 ppm AFB1-supplemented chow (Figure 3B). Such variation is consistent with the pulsatile release of GH from pituitary that occurs every 3-4 hours in male rats29; nevertheless, there was also a downward trend in levels of phosphorylated:total STAT5 protein and significant reduction of total STAT5 protein in livers from toxin-exposed rats (Figure 3B). Together, these data suggest that AFB1 causes liver-based GH resistance.
Figure 3. Effects of Dietary AFB1 on GH Signaling.
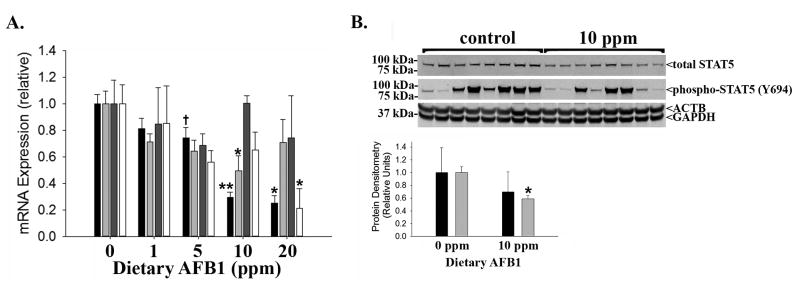
(A) Hepatic mRNA expression of Igf1 (black bars; *p<0.01 vs. 0, 1, 5 ppm; **p<0.02 vs. 0, 1, 5 ppm; †p=0.04 vs. 0 ppm), Ghr (light grey bars; *p=0.02 vs. 0 ppm), Stat5a (dark grey bars; no significant differences), and Stat5b (white bars; *p=0.03 vs. 0 ppm) in Fischer rats exposed to dietary AFB1. (B) Protein immunoblot (upper panel) and quantification (lower panel) of hepatic expression of phosphorylated (Y694) and total STAT5 (*p=0.001 vs. 0 ppm control; β-actin (ACTB) and GAPDH shown as loading controls; phospho:total STAT5, black bars; total STAT5:GAPDH, grey bars).
Gut Morphology in AFB1-Exposed Rats
Like hepatocytes, human enterocytes express CYP450s that convert AFB1 to its toxic reactive epoxide17, 18; however, the CYPs that activate AFB1 in rat intestine remain unknown. Therefore, we investigated the influence of dietary aflatoxin exposure on gut morphology. This analysis showed no significant difference in jejunal villous length in toxin-exposed vs. control rats; however, a downward trend was noted between 20 ppm AFB1-exposed vs. control rats (Figure 4, p=0.1). Thus, gut absorptive function might be compromised by AFB1 exposure.
Figure 4. Small Bowel Morphology in AFB1 Exposed Fischer Rats.
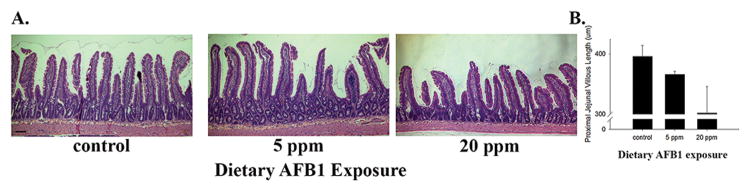
(A) Representative H&E stained sections of proximal jejunum from AFB1-exposed Fischer rats (40x; 100 micron bar in lower left corner of left-most image). (B) Quantification of jejunal villous length in toxin-exposed rats.
Evaluation of Dietary Aflatoxin Exposure-Induced Effects in Sprague-Dawley Rats
Finally, to begin to explore whether genetic modifiers impact dietary AFB1-induced stunting in this model, we investigated the growth of toxin-exposed, male, outbred Sprague-Dawley rats. Rats exposed to 10 ppm AFB1-containing chow over five weeks demonstrated relative growth impairment comparable to that seen in the Fischer animals (Figure 5A): relative weight gain from weaning (age 3 weeks) through age 7-8 weeks in toxin-exposed Fischer and Sprague-Dawley rats was 64-65% and 67-74%, respectively, of that seen in the corresponding controls. Serum AFB1-adduct levels were also similar between Fischer and Sprague-Dawley rats after 5 weeks of toxin exposure (Figure 5B). These data demonstrate that strain-related genetic differences do not significantly influence toxin-induced growth impairment, at least under these experimental conditions.
Figure 5. Dietary AFB1 Induced Stunting in Fischer vs. Sprague-Dawley Rats.
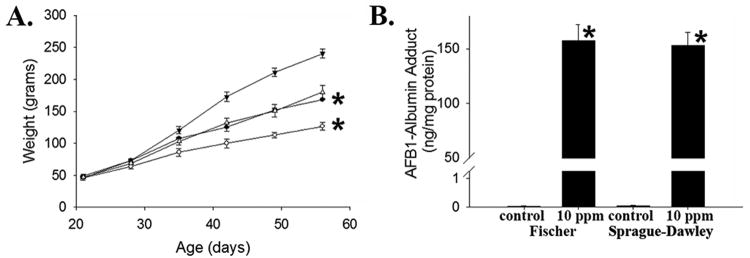
(A) Weight gain over time in newly weaned, male inbred Fischer and outbred Sprague-Dawley rats exposed to 10 ppm dietary AFB1 (● Fischer control, ○ Fischer 10 ppm, ▼ Sprague-Dawley control, ∆ Sprague-Dawley 10 ppm; *p<0.04 for area-under-the-growth-curves vs. corresponding control). (B) Serum AFB-albumin adduct levels in toxin-exposed Fischer vs. Sprague-Dawley rats (*p<0.01 vs. corresponding control).
Discussion
Childhood stunting is associated with increased morbidity and mortality1, 2. Indeed, more than one-third of pediatric deaths are attributable to under-nutrition, a common cause of stunting. Furthermore, under-grown children are at increased risk for chronic disease, cognitive impairment, and other morbidities1, 2. These observations have motivated efforts to reduce the prevalence (and consequences) of childhood growth impairment. Recent epidemiological links between dietary aflatoxin exposure and childhood stunting suggest that AFB1 exposure could be an important, inadequately addressed contributor to childhood stunting in resource-poor settings3-14. This possibility has led, in turn, to emerging interest in efforts to reduce aflatoxin exposure as a strategy to prevent complications of under-nutrition in children5. However, because of the heretofore lack of either intervention trials in at-risk human populations or a tractable experimental animal model, the specific causal role of dietary aflatoxin exposure in stunting has not yet been definitively established. Based on these considerations, we undertook the experiments here to develop such a model and investigate these questions. The results of our studies provide direct evidence supporting the causal role of AFB1 exposure in growth impairment in a mammalian model.
Rats have genetic and physiological similarity to humans; thus, the data reported here should have relevance to consideration of the mechanisms linking AFB1 exposure to impaired growth in children. Our results identify liver injury as one plausible intermediary between toxin exposure and stunting. GH resistance occurs in children with chronic liver injury16, and our data support GH-resistance as a candidate mechanism by which AFB1 might cause stunting, at least in some circumstances. These considerations are consistent with a recently published study showing that the magnitude of aflatoxin exposure is inversely associated with IGF1 expression in children and in transformed human hepatocytes in culture 30. Importantly, growth impairment occurred despite comparable food intake in rats exposed to a wide range of dietary toxin in this model. Thus, nutritional interventions alone might be insufficient to improve growth in toxin-exposed children. Because human intestinal epithelial cells, like hepatocytes, express CYPs capable of converting AFB1 into the reactive epoxide17, 18, AFB1 exposure might also promote stunting through enteropathic effects. With this in mind, it is intriguing that in areas where dietary AFB1 exposure is endemic, a malabsorptive condition known as environmental enteropathy is prevalent19. Such enteropathy is associated with histologic changes in the small intestine, particularly villous blunting. Although the studies reported here did not demonstrate significant toxin-induced blunting, we did observe a downward trend in intestinal villous length in animals exposed to the highest toxin concentration. Thus, future studies employing this model should investigate gut absorptive and barrier function in larger cohorts of animals.
The paradigm described here should also enable future efforts to discover genetic or environmental modifiers of aflatoxin-induced stunting in rats, which might suggest novel strategies to mitigate the growth-related consequences of childhood aflatoxin exposure. Although the studies here did not demonstrate strain-specific differences in AFB1-induced growth-impairment, genetic modifiers of aflatoxin-induced HCC have been reported in mice31. Environmental modifiers of toxin-induced liver cancer in mice have also been reported, with enteric microbiota affecting risk in that model.32 This observation is especially provocative in the context of emerging interest in understanding the enteric microbiome's role in human diseases. Enteric microbiotal contributions to AFB1-induced stunting could also be investigated with this model. For example, if commensal enteric microbial flora affect aflatoxin-epoxide formation or detoxification in rats, as suggested in other species7, altering such flora (e.g. by depletion with enteral antibiotics) might augment or diminish toxin-induced pathophysiology. Either possibility justifies further examination of the influence of the enteric microbiome on aflatoxin-related disease. For example, although speculative, comparative analyses of “germfree” rats and gnotobiotic rats reconstituted with enteric flora from aflatoxin-exposed stunted- or normally grown-children might identify specific microbes as environmental modifiers of aflatoxin-induced stunting. A homologous approach in mice was recently used to demonstrate microbiotal contributions to the development of kwashiorkor33. Such analyses should enlighten our current understanding of the scope of influence of the enteric microbiome on human health and disease more generally.
Finally, the relevance of the levels of dietary aflatoxin exposure of rats examined here to acute and chronic human exposures should be considered. The range of toxin exposure (from 1-20 ppm) in these studies was empirically selected based on pilot data showing reduced weight gain in newly weaned rats exposed to a 5 ppm AFB1-contaminated diet over ∼6 weeks (data not shown). Industrialized nations typically limit aflatoxin in human food to less than 20 parts per billion (ppb6). Nevertheless, a recent review of mycotoxin contamination of foods in African countries over the last 2 decades reported peak aflatoxin levels up to 355 ppb in maize and 500 ppb in other foods34. Furthermore, during outbreaks of human aflatoxicosis, levels of AFB1-contamination detected in the regional food supply fell within the range of those studied here. For example, analyses of locally-grown maize during the 2004-2005 outbreak in Kenya showed that 10% of analyzed samples exhibited AFB1 levels >1 ppm35 with peak levels from 4-46 ppm35. Nevertheless, the actual range and variability in acute and chronic human dietary aflatoxin exposure in resource-poor countries is difficult to estimate because of the lack of regular, standardized testing.
Serum AFB1-albumin adduct levels in response to specific AFB1 exposures across species with differing susceptibility to AFB1-induced carcinogenicity have previously been reported, offering additional opportunity to consider the relevance of the exposures here to observed human intoxications. For example, one study reported a ratio of AFB1 adduct-to-toxin exposure of ∼1 pg adduct/μg AFB1/day/kg body weight in rats exposed to 20 μg AFB1/kg/day for 14 days36. That study also estimated that human aflatoxin-adduct formation more closely resembles that of AFB1-sensitive (e.g. rat) versus resistant (e.g. mice) species. Another study demonstrated that humans and rats exposed to a single, comparable, lower (15 ng/kg) dose of AFB1 exhibit similar adduct levels37. In this case, a ratio of ∼40 pg adduct/μg AFB1/kg was reported. Thus, AFB1-albumin adduct formation varies substantially with respect to magnitude and chronicity of toxin exposure, with higher adduct-to-exposure ratios levels of reported in rats and humans exposed to less toxin. These findings are consistent with other analyses suggesting that adduct formation as a function of toxin exposure is saturable38, 39. By comparison, the rats examined in the studies reported here were exposed to a higher range of daily and cumulative AFB1 (calculated based on measured food intake) and also exhibited greater adduct-to-toxin exposure ratios (assessed at the experimental endpoint; Table 1). Nevertheless, as in the studies above, these ratios also declined with increasing levels of exposure. For example, rats exposed to 1 ppm AFB1 toxin-containing diet ingested ∼54-126 μg AFB1/kg/day over the experimental course and exhibited a serum adduct-to-toxin exposure ratio at the experimental endpoint of ∼1 ng adduct/μg AFB1/kg/day; in contrast, those given 20 ppm-containing diet took ∼1.1-4.2 mg/kg/day and exhibited a ratio of ∼100 pg adduct/μg AFB1/kg/day (Table 1). Notably, the levels of AFB1-albumin adduct in the experiments reported here (68-181 ng/mg, Figure 1) exceed those reported in human cases of acute aflatoxicosis (including fatal cases) associated with the 2004 outbreak in Kenya40. Together with the lack of mortality or apparent liver synthetic dysfunction in the rats studied here, these observations suggest that humans are relatively more susceptible than rats to toxic effects of dietary AFB1. One possible explanation for this observation relates to species-specific differences in hepatic aflatoxin-epoxide detoxification, specifically with respect to the glutathione-S-transferases (GSTs) which detoxify the unstable CYP-derived aflatoxin-epoxide preventing its toxic reaction with DNA, albumin, and other cellular components. Rats express an inducible, α class GST isoform highly efficient in AFB1 detoxification, whereas humans express a μ class GST with lower activity. Thus, differences in hepatic GST expression likely affect susceptibility to AFB1-induced toxicity. Whether the AFB1 exposure regimen employed here affected hepatic α GST expression is unknown. Nevertheless, these data raise the possibility that humans are even more susceptible than rats to toxin-induced stunting, and justify further use of this animal model to investigate strategies with which to mitigate such effects.
Table 1. AFB1 Adduct and Exposure Range in Fischer Rats.
AFB1 adduct and exposure levels are summarized for each level of dietary contamination studied. Cumulative exposure and range of daily exposure were calculated from measured daily food intake and animal weights. Adduct:Exposure ratio was calculated over the last 3 days of the experiment based on the ∼3 day half-life of serum albumin in rat 39.
| Dietary AFB1 (ppm) | Cumulative exposure (μg/animal) | Range of daily exposure μg/kg/day/animal (median) | Adduct:Exposure ratio ng AFB-albumin/μg AFB1/kg/day |
|---|---|---|---|
| 1 | 484 | 54-126 (108) | 1.16 |
| 5 | 2547 | 482-1254 (661) | 0.47 |
| 10 | 3735 | 261-1541 (1011) | 0.19 |
| 20 | 8266 | 1118-4168 (2324) | 0.08 |
Experimental Methods
Chemicals and other reagents
AFB1 (Sigma-Aldrich, St. Louis, MO) was suspended in glycerol trioctanoate (Sigma-Aldrich) to create a stock solution (0.5 mg/mL), which was added to standard rodent chow (PicoLab Rodent Diet 20; LabDiet, St. Louis, MO) to create diets containing 0, 1, 5, 10, or 20 ppm of AFB1. Food, with and without AFB1, was stored at 4 ° C until placed in animal cages.
Animal husbandry and experimental design
Three-week old (newly weaned) male inbred F344 Fischer or outbred Sprague-Dawley rats (Charles River Laboratory, Portage, MI) were housed in groups of three animals per cage for each level of toxin exposure under controlled light/dark cycles, and allowed ad libitum access to water and toxin-supplemented or vehicle-treated (i.e. control) chow throughout the experiment. Dietary consumption (per cage) was quantified daily and rats were weighed once or twice weekly. At 8-9 am on the day of the experimental endpoint, rats were injected with 100 mg/kg BrdU (Sigma-Aldrich) 1 hour prior to bleeding and sacrifice (from 9-10 am) for collection of sera and tissues. Liver tissue was immediately frozen in liquid nitrogen and stored at -80°C or fixed in formalin for histological and immunohistochemical analyses (described below).
Small bowel longitudinal length was measured and the proximal 25% isolated and processed for histological analysis of villous height as previously described41. Briefly, the intestinal lumen was flushed with phosphate-buffered saline followed by 10% neutral-buffered-formalin, with the mucosal surface subsequently exposed by longitudinally incising the bowel; the proximal jejunum was pinned mucosal surface up on a wax tray and fixed in 10% formalin at 4°C overnight; the tissue was subsequently covered with 2% agar, then cut into uniform sections, layered, and placed into a tissue cassette and in formalin for subsequent processing. Tibias were recovered and cleaned in 1 M NaOH prior to determination of tibial length, performed using a stadiometer.
Laboratory safety and precautions
AFB1 is mutagenic, and it and its derivatives should be handled using appropriate precautions. Aflatoxin-containing chow was prepared in an approved fume hood using PicoLab Rodent Diet 20 (LabDiet) and commercially available AFB1. Toxin was first suspended in glyceryl trioctanoate prior to mixing with powdered chow. Chow was prepared biweekly with strict adherence to use of personal protective equipment including lab coat, face mask, eye protection and gloves. Work areas were thoroughly cleansed with 10% bleach after chow preparation. Toxin exposed and control animals were housed in marked cages in an approved, dedicated, marked animal room in which no other experimental animals were housed. Animal cages were changed weekly, with soiled bedding, residual chow, and disposable equipment saturated with 10% bleach prior to disposal in a HEPA-filtered dump station. At the experimental endpoint, animals were moved to chow- and toxin-free clean cages prior to transfer to the lab where serum and tissue harvest occurred. Non-harvested animal tissues were disposed of as biohazardous waste.
Histological and immunohistochemical analyses
Liver and gut tissues were stained with hematoxylin and eosin- (H&E) and Sirius Red (using standard methodology) and for hepatocellular BrdU incorporation (as previously described25). Villous length was quantified in H&E stained sections of gut using AxioVision microscope software (Zeiss, Thornwood, NY), with 200-300 villi measured from each animal in each toxin-exposed cohort as described41.
Gene and Protein Expression Analyses
Hepatic expression of specific genes was characterized using qRT-PCR as previously described42. Data were standardized to the expression of β2-microglobulin (B2M) to calculate fold-differences in gene expression. Specificity was verified for each gene using melt-curves and by simultaneous analysis of a reaction mixture containing all components except reverse transcriptase. Gene specific qRT-PCR primers included: B2M forward-GCTCGGTGACCGTGATCTTT, reverse-TTGAGGAAGTTGGGCTTCCC; Igf1 forward-TGGTGGACGCTCTTCAGTTC, reverse-TCCGGAAGCAACACTCATCC; Ghr forward-ATGTTTCCTGGAAGTGGGGC, reverse-TTCAGGGGAACGACACTTGG; Stat5a forward-CCCTCAGGCTCACTACAACA, reverse-CGGCGTAAAAGTTCCTCCAC; Stat5b forward-CCTTGTACGGCCAGCATTTC, reverse-AGGAGCTGGGTGGCCTTAAT. Liver tissue lysates were prepared and protein immunoblotting conducted as previously described42 using anti-STAT5 and anti-phospho-STAT5 (Y694) antibodies (Cell Signaling, Boston, MA).
Serum analyses
Sera were submitted to the St. Louis Children's Hospital Clinical laboratory for analyses. AFB1-Lysine adduct was determined by isotope dilution mass spectrometry as described23.
Statistical analyses
Data were analyzed using PASW Statistics version 22 (SPSS, Chicago, IL). Numerical data comparisons between groups were conducted using the unpaired Student's t-test for pair-wise comparisons and ANOVA for multiple groups with Tukey's test used for post-hoc comparisons. Rates and proportions were compared between groups using chi-square analysis. Significance (α) was set at 0.05. Data are reported as mean ± standard error.
Acknowledgments
We thank Phillip Tarr for provocative discussions that inspired these studies and his advice about study design, data interpretation, and manuscript preparation. We are also grateful to Terrence Riehl and William Stenson for guidance regarding intestinal tissue preparation for morphometry and to the Washington University School of Medicine Digestive Disease Research Core Center (DDRCC) for histology and immunohistochemistry.
Financial Support: These studies were supported by funding from the Washington University Digestive Disease Research Core Center (NIH-NIDDK P30-DK052574) and the Washington University Department of Pediatrics (to DAR).
Footnotes
Disclosure Statement: The authors have no financial ties to products in the study and no potential or perceived conflicts of interest to report.
Reference List
- 1.Black RE, Allen LH, Bhutta ZA, et al. Maternal and child undernutrition: global and regional exposures and health consequences. Lancet. 2008;371(9608):243–260. doi: 10.1016/S0140-6736(07)61690-0. [DOI] [PubMed] [Google Scholar]
- 2.Black RE, Victora CG, Walker SP, et al. Maternal and child undernutrition and overweight in low-income and middle-income countries. Lancet. 2013;382(9890):427–451. doi: 10.1016/S0140-6736(13)60937-X. [DOI] [PubMed] [Google Scholar]
- 3.Khlangwiset P, Shephard GS, Wu F. Aflatoxins and growth impairment: a review. Crit Rev Toxicol. 2011;41(9):740–755. doi: 10.3109/10408444.2011.575766. [DOI] [PubMed] [Google Scholar]
- 4.Wu F, Groopman JD, Pestka JJ. Public health impacts of foodborne mycotoxins. Annu Rev Food Sci Technol. 2014;5:351–372. doi: 10.1146/annurev-food-030713-092431. [DOI] [PubMed] [Google Scholar]
- 5.Etzel RA. Reducing malnutrition: time to consider potential links between stunting and mycotoxin exposure? Pediatrics. 2014;134(1):4–6. doi: 10.1542/peds.2014-0827. [DOI] [PubMed] [Google Scholar]
- 6.Kensler TW, Roebuck BD, Wogan GN, Groopman JD. Aflatoxin: a 50-year odyssey of mechanistic and translational toxicology. Toxicol Sci. 2011;120(Suppl 1):S28–S48. doi: 10.1093/toxsci/kfq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newberne PM. Mycotoxins: toxicity, carcinogenicity, and the influence of various nutritional conditions. Environ Health Perspect. 1974;9:1–32. doi: 10.1289/ehp.9-1475399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussein HS, Brasel JM. Toxicity, metabolism, and impact of mycotoxins on humans and animals. Toxicology. 2001;167(2):101–134. doi: 10.1016/s0300-483x(01)00471-1. [DOI] [PubMed] [Google Scholar]
- 9.Gong YY, Cardwell K, Hounsa A, et al. Dietary aflatoxin exposure and impaired growth in young children from Benin and Togo: cross sectional study. BMJ. 2002;325(7354):20–21. doi: 10.1136/bmj.325.7354.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong YY, Egal S, Hounsa A, et al. Determinants of aflatoxin exposure in young children from Benin and Togo, West Africa: the critical role of weaning. Int J Epidemiol. 2003;32(4):556–562. doi: 10.1093/ije/dyg109. [DOI] [PubMed] [Google Scholar]
- 11.Egal S, Hounsa A, Gong YY, et al. Dietary exposure to aflatoxin from maize and groundnut in young children from Benin and Togo, West Africa. Int J Food Microbiol. 2005;104(2):215–224. doi: 10.1016/j.ijfoodmicro.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Shirima CP, Kimanya ME, Kinabo JL, et al. Dietary exposure to aflatoxin and fumonisin among Tanzanian children as determined using biomarkers of exposure. Mol Nutr Food Res. 2013;57(10):1874–1881. doi: 10.1002/mnfr.201300116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asiki G, Seeley J, Srey C, et al. A pilot study to evaluate aflatoxin exposure in a rural Ugandan population. Trop Med Int Health. 2014;19(5):592–599. doi: 10.1111/tmi.12283. [DOI] [PubMed] [Google Scholar]
- 14.Routledge MN, Kimanya ME, Shirima CP, Wild CP, Gong YY. Quantitative correlation of aflatoxin biomarker with dietary intake of aflatoxin in Tanzanian children. Biomarkers. 2014;19(5):430–435. doi: 10.3109/1354750X.2014.924998. [DOI] [PubMed] [Google Scholar]
- 15.Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58(5-6):737–747. doi: 10.1007/PL00000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucuvalas JC, Horn JA, Chernausek SD. Resistance to growth hormone in children with chronic liver disease. Pediatr Transplant. 1997;1(1):73–79. [PubMed] [Google Scholar]
- 17.Zhang QY, Raner G, Ding X, Dunbar D, Coon MJ, Kaminsky LS. Characterization of the cytochrome P450 CYP2J4: expression in rat small intestine and role in retinoic acid biotransformation from retinal. Arch Biochem Biophys. 1998;353(2):257–264. doi: 10.1006/abbi.1998.0654. [DOI] [PubMed] [Google Scholar]
- 18.Zhang QY, Dunbar D, Ostrowska A, Zeisloft S, Yang J, Kaminsky LS. Characterization of human small intestinal cytochromes P-450. Drug Metab Dispos. 1999;27(7):804–809. [PubMed] [Google Scholar]
- 19.Korpe PS, Petri WA., Jr Environmental enteropathy: critical implications of a poorly understood condition. Trends Mol Med. 2012;18(6):328–336. doi: 10.1016/j.molmed.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Pascalis B, Bianchi A, Satta MA, et al. Growth hormone in inflammatory bowel disease. Eur Rev Med Pharmacol Sci. 2006;10(1):13–16. [PubMed] [Google Scholar]
- 21.Steyn M, Pitout MJ, Purchase IF. A comparative study on aflatoxin B 1 metabolism in mice and rats. Br J Cancer. 1971;25(2):291–297. doi: 10.1038/bjc.1971.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abrahamyan DO, Gazarian A, Braillon PM. Estimation of stature and length of limb segments in children and adolescents from whole-body dual-energy X-ray absorptiometry scans. Pediatr Radiol. 2008;38(3):311–315. doi: 10.1007/s00247-007-0720-x. [DOI] [PubMed] [Google Scholar]
- 23.Egner PA, Gange SJ, Dolan PM, Groopman JD, Munoz A, Kensler TW. Levels of aflatoxin-albumin biomarkers in rat plasma are modulated by both long-term and transient interventions with oltipraz. Carcinogenesis. 1995;16(8):1769–1773. doi: 10.1093/carcin/16.8.1769. [DOI] [PubMed] [Google Scholar]
- 24.Seawright AA, Snowden RT, Olubuyide IO, Riley J, Judah DJ, Neal GE. A comparison of the effects of aflatoxin B1 on the livers of rats and duck hepatitis B virus-infected and noninfected ducks. Hepatology. 1993;18(1):188–197. [PubMed] [Google Scholar]
- 25.Rudnick DA. Liver Regeneration: The Developmental Biologists Approach. In: Orlando G, Lerut JP, Soker S, Stratta RJ, editors. editors Regenerative Medicine Applications in Organ Transplantation. 1st. Waltham, MA: Elsevier/Academic Press; 2014. pp. 353–374. [Google Scholar]
- 26.Ahmed SF, Farquharson C. The effect of GH and IGF1 on linear growth and skeletal development and their modulation by SOCS proteins. J Endocrinol. 2010;206(3):249–259. doi: 10.1677/JOE-10-0045. [DOI] [PubMed] [Google Scholar]
- 27.Akira S. Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells. 1999;17(3):138–146. doi: 10.1002/stem.170138. [DOI] [PubMed] [Google Scholar]
- 28.Chia DJ, Varco-Merth B, Rotwein P. Dispersed Chromosomal Stat5b-binding elements mediate growth hormone-activated insulin-like growth factor-I gene transcription. J Biol Chem. 2010;285(23):17636–17647. doi: 10.1074/jbc.M110.117697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi HK, Waxman DJ. Pulsatility of growth hormone (GH) signalling in liver cells: role of the JAK-STAT5b pathway in GH action. Growth Horm IGF Res. 2000;10(Suppl B):S1–S8. doi: 10.1016/s1096-6374(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 30.Castelino JM, Routledge MN, Wilson S, et al. Aflatoxin exposure is inversely associated with IGF1 and IGFBP3 levels in vitro and in Kenyan schoolchildren. Mol Nutr Food Res. 2014 doi: 10.1002/mnfr.201300619. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGlynn KA, Hunter K, LeVoyer T, et al. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Res. 2003;63(15):4594–4601. [PubMed] [Google Scholar]
- 32.Fox JG, Feng Y, Theve EJ, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59(1):88–97. doi: 10.1136/gut.2009.183749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith MI, Yatsunenko T, Manary MJ, et al. Gut Microbiomes of Malawian Twin Pairs Discordant for Kwashiorkor. Science. 2013;339(6119):548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darwish WS, Ikenaka Y, Nakayama SM, Ishizuka M. An overview on mycotoxin contamination of foods in Africa. J Vet Med Sci. 2014;76(6):789–797. doi: 10.1292/jvms.13-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mwihia JT, Straetmans M, Ibrahim A, et al. Aflatoxin levels in locally grown maize from Makueni District, Kenya. East Afr Med J. 2008;85(7):311–317. doi: 10.4314/eamj.v85i7.9648. [DOI] [PubMed] [Google Scholar]
- 36.Wild CP, Hasegawa R, Barraud L, et al. Aflatoxin-albumin adducts: a basis for comparative carcinogenesis between animals and humans. Cancer Epidemiol Biomarkers Prev. 1996;5(3):179–189. [PubMed] [Google Scholar]
- 37.Cupid BC, Lightfoot TJ, Russell D, et al. The formation of AFB(1)-macromolecular adducts in rats and humans at dietary levels of exposure. Food Chem Toxicol. 2004;42(4):559–569. doi: 10.1016/j.fct.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 38.Scholl PF, McCoy L, Kensler TW, Groopman JD. Quantitative analysis and chronic dosimetry of the aflatoxin B1 plasma albumin adduct Lys-AFB1 in rats by isotope dilution mass spectrometry. Chem Res Toxicol. 2006;19(1):44–49. doi: 10.1021/tx050251r. [DOI] [PubMed] [Google Scholar]
- 39.Qian G, Tang L, Wang F, et al. Physiologically based toxicokinetics of serum aflatoxin B1-lysine adduct in F344 rats. Toxicology. 2013;303:147–151. doi: 10.1016/j.tox.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azziz-Baumgartner E, Lindblade K, Gieseker K, et al. Case-control study of an acute aflatoxicosis outbreak, Kenya, 2004. Environ Health Perspect. 2005;113(12):1779–1783. doi: 10.1289/ehp.8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riehl TE, Ee X, Stenson WF. Hyaluronic acid regulates normal intestinal and colonic growth in mice. Am J Physiol Gastrointest Liver Physiol. 2012;303(3):G377–G388. doi: 10.1152/ajpgi.00034.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang J, Barr E, Rudnick DA. Characterization of the regulation and function of zinc-dependent histone deacetylases during rodent liver regeneration. Hepatology. 2013;57(5):1742–1751. doi: 10.1002/hep.26206. [DOI] [PMC free article] [PubMed] [Google Scholar]