

Abstract
Emerging evidence suggests that ascorbate, the dominant form of vitamin C under physiological pH conditions, influences the genome activity via regulating epigenomic processes. Ascorbate serves as a cofactor for ten-eleven translocation (TET) dioxygenases that catalyze the oxidation of 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC), further to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which are ultimately replaced by unmodified cytosine. The JmjC domain-containing histone demethylases also require ascorbate as a cofactor for histone demethylation. Thus, by primarily participating in the demethylation of both DNA and histones, ascorbate appears to be a mediator of the interface between the genome and environment. Furthermore, redox status has a profound impact on the bioavailability of ascorbate in the nucleus. In order to bridge the gap between redox biology and genomics, we suggest an interdisciplinary research field that can be termed “Redox Genomics” to study dynamic redox processes in health and diseases. This review examines the evidence and potential molecular mechanism of ascorbate in demethylation of the genome, while highlighting potential epigenetic roles of ascorbate in various diseases.
Keywords: Vitamin C, iron and 2-oxoglutarate-dependent dioxygenase, ten-eleven translocation dioxygenase, JmjC domain-containing histone demethylase, DNA demethylation, histone demethylation
INTRODUCTION
Vitamin C (L-ascorbic acid) is an essential, water-soluble micronutrient that exists predominantly as the ascorbate anion under physiological pH conditions. It is well established that ascorbate is an antioxidant and free radical scavenger and further, an essential cofactor in numerous enzymatic reactions.
Most mammals, such as rodents, synthesize ascorbate de novo in the liver from glucose through a biosynthetic pathway. In contrast, humans as well as primates, guinea pigs and fruit bats no longer can synthesize ascorbate due to a mutant and nonfunctional enzyme, L-gulonolactone oxidase (Gulo), which catalyzes the last step of ascorbate biosynthesis (50). For these mammalian species, ascorbate is a vitamin that needs to be supplied through dietary sources and supplements.
Ascorbate, derived from either dietary sources or the liver, enters cells primarily through sodium-dependent vitamin C transporters (SVCTs). The high-capacity, low-affinity SVCT1 is primarily responsible for ascorbate absorption and re-absorption in intestinal and renal epithelial cells. The high-affinity, low-capacity SVCT2 distributes ascorbate to most tissues and is expressed more ubiquitously (91). The average concentration of ascorbate in the plasma of healthy humans or mice is ~50 μM. Currently, the recommended dietary allowances (RDA) by the Institute of Medicine (IOM) is 90 mg for adult males and 75 mg for adult females, although the tolerable upper intake level for adults is 2,000 mg per day. The daily adequate intakes (AI) for infants (0 – 12 months) is 40 ~ 50 mg. When the plasma ascorbate concentration drops to below 11.4 μM, there is a risk of developing scurvy, and is thus conventionally considered deficient. Once being transported across the plasma membrane, ascorbate accumulates within cells and its intracellular concentration can reach 1 ~ 10 mM (58). Thus, the majority of mammalian cells maintain highly elevated intracellular ascorbate concentrations compared to the extracellular milieu. For instance, neurons could have up to 10 mM of intracellular ascorbate, which is about 200 times higher than the extracellular ascorbate concentration (73).
Ascorbate is a relatively mild reducer and antioxidant. After a cascade of two-round oxidation and the loss of two electrons, the oxidized form of vitamin C, termed dehydroascorbic acid (DHA) is formed. Instead of utilizing SVCTs, DHA enters and leaves cells via facilitated glucose transporters (GLUTs). Once inside the cell, DHA can be rapidly reduced back to ascorbate. However, the reduced ascorbate is dominant and DHA is barely detectable in the plasma of healthy humans (43), suggesting that most cells take up and accumulate ascorbate primarily through SVCTs.
During evolution, primates and some other species have lost the ability to synthesize ascorbate due to accumulation of mutations in the Gulo gene. The antioxidant function of ascorbate generally appears to be compensated for by alternative reducing systems in these species (54). However, its role as a cofactor for iron and 2-oxoglutarate-dependent dioxygenases is irreplaceable, as these species develop scurvy, osteoporosis and other disease phenotypes unless provided with adequate dietary ascorbate. The iron and 2-oxoglutarate-dependent dioxygenases utilize Fe2+ as a cofactor and 2-oxoglutarate (2OG, also known as α-ketoglutarate) as a co-substrate; additionally, some of them require ascorbate as another cofactor for full catalytic activity (59). One classic member of this family is collagen prolyl 4-hydroxylase (P4H), which is well known for its involvement in scurvy. In the absence of ascorbate, the initial hydroxylation catalyzed by collagen P4H can proceed at a maximal rate. However, the catalytically inactive oxidized iron species (mostly Fe3+) soon inactivates collagen P4H, which leads to an incomplete hydroxylation of proline residues in collagen and ultimately the characteristic signs of scurvy (23). When available, ascorbate has the capacity to reduce oxidized iron species to catalytically active Fe2+. Ascorbate thus assists collagen P4H to complete the hydroxylation and prevent scurvy. Overall, it has been well established that ascorbate is required to maintain a number of Fe2+ and 2OG-dependent dioxygenases in their fully active forms (59).
In recent years, tremendous progress has been made in regards to the identification of a number of novel Fe2+ and 2OG-dependent dioxygenases that catalyze the hydroxylation of methylated nucleic acids (DNA and RNA) and methylated histones. DNA and histone methylation are the major epigenetic hallmarks in the mammalian genome. It has also been shown that some of these nuclear dioxygenases require ascorbate as a cofactor to start and complete both DNA demethylation and histone demethylation processes. These unexpected findings uncover a previously unknown function of ascorbate in regulating the epigenome, which calls for a re-evaluation of the role of ascorbate in human health and diseases.
VITAMIN C AND DNA DEMETHYLATION
TET dioxygenases are Fe2+ and 2OG-dependent dioxygenases
The epigenome reflects the interface of a dynamic environment and the genome. Known epigenetic events include covalent modifications on nucleotides and histones, chromatin remodeling, and non-coding RNAs, which collectively constitute the epigenome. Methylation at the C5 position of cytosine (5-methylcytosine, 5mC) is the major and best-characterized epigenetic mark of mammalian DNA. The transfer of a methyl group from the donor S-adenosylmethionine (SAM) to a cytosine is catalyzed by DNA methyltransferases (DNMTs), which are regarded as the writers of this epigenetic mark. After the methylation is completed, 5mC, especially in CpG dinucleotide context, can be recognized and then bound with a group of methyl-CpG binding proteins (MBPs). As readers, these MBPs then mediate multiple functions such as regulating transcription, initiating chromatin remodeling, maintaining genome stability and cellular identity. Although 5mC is a relatively stable epigenetic mark, it can be lost by passive dilution via a lack of maintenance by DNMT1 during DNA replication, which would result in passive demethylation. A few years ago, it remained largely unclear whether and how the methyl group in 5mC could be actively removed. The key question was whether or not there were DNA demethylases that existed as erasers to catalyze the removal of the methyl group from 5mC.
In 2009, the Heintz group reported the presence of an unusual DNA nucleotide, 5-hydroxymethylcytosine (5hmC), in the mouse brain (44). Although 5hmC constitutes less than 1% of total nucleotides, its percentage was higher in cerebellar Purkinje neurons than in granule cells. In contrast, there was no difference in 5mC content between these two distinct neuronal cell types. Interestingly, transcriptionally active euchromatin was enriched in Purkinje neurons while granule cells contained more of the transcriptionally inactive heterochromatin. Based on these results, Heintz et al suggested that 5hmC might play a role in the epigenetic regulation of neuronal functions (44). Interestingly, it appears that this is not the first time that 5hmC was identified in a genome. As early as 1952, 5hmC was detected in T-even bacteriophages (93). It was discovered that 5hmC could be incorporated into the viral genome to resist the attack of restriction enzymes from the bacteria. In 1972, 5hmC was also identified in mammalian DNA (70). However, 5hmC was generally regarded as oxidatively damaged cytosine in the mammalian genome and might eventually be replaced by DNA-repair mechanism. Since then, not much attention has been given to 5hmC in terms of elucidating its possible physiological functions in the genome until the Heintz report spurred new interests.
The breakthrough in understanding the presence of 5hmC in the genome came from studies on a gene family that is known as Ten-eleven translocation (TET). TET1 at chromosome 10q22 was originally named after the identification of this gene fused with the Mixed Lineage Leukemia (MLL) gene resulting from a chromosome translocation of 10q22 and 11q23 in acute myeloid leukemia (51). TET1 appears to be a member of a well-conserved gene family that contains two other members (TET2 at 4q24 and TET3 at 2p12) in the human genome. Soon after, somatic mutations in TET2 were identified in about 15% of patients affected by myeloid leukemia (12). However, the way in which the mutant TETs contributed to myeloid leukemia was unclear. As a matter of fact, the basic biological function of TETs was unknown at that time.
Through a series of elegant experiments, Rao’s lab demonstrated for the first time that TET1 enzymatic activity involved the oxidation of 5mC to 5hmC (85), providing the first bona-fide active demethylation of DNA. Via bioinformatics approaches, TET dioxygenases were identified as mammalian homologs of the proteins JBP1 and JBP2 in trypanosome, a unicellular protozoon. These two proteins were known to oxidize the 5-methyl group of thymine. Upon forced expression of wild type (WT) and mutant TET1 in cultured cells, a dramatic reduction of 5mC signal was observed in WT, but not the mutant, TET1 overexpressing cells. In vitro enzymatic analysis further demonstrated that the recombinant TET1 catalytic domain catalyzed 5mC to 5hmC in methylated DNA fragments (85). Both JBP1 and JBP2 belong to the Fe2+ and 2OG-dependent dioxygenases family, suggesting that the enzymatic activity of TET dioxygenases may also depend on Fe2+ and 2OG. Indeed, withdrawing 2OG from the in vitro reaction almost completely blocked the conversion of 5mC to 5hmC. Furthermore, removing Fe2+ dramatically inhibited, but did not abolish it completely. The explanation for this result was that a certain amount of Fe2+ might be contained in the recombinant TET1 catalytic domain during protein purification process. Thus, TET1 was identified as a Fe2+ and 2OG-dependent enzyme that converts 5mC to 5hmC. Soon after, mouse TETs, including TET1, TET2 and TET3, were also identified as being able to oxidize 5mC to 5hmC (34).
Subsequent experiments further confirmed that TETs are Fe2+ and 2OG-dependent dioxygenases. First, the binding motif of Fe2+ in TETs include at least two histidine residues and one asparagine residue, which are conserved across JBP1, JBP2, human TETs and mouse TETs (78). Mutations at the Fe2+-binding sites in TET dioxygenases diminished 5hmC signal in the cultured cells (34). Secondly, 2OG is a critical intermediate metabolite of the Krebs cycle. The oxidative decarboxylation of isocitrate to 2OG is catalyzed by enzymes that are termed isocitrate dehydrogenases (IDH). Mutations in IDH1 and IDH2 have been associated with certain cancers, in which 5hmC is depleted. Instead of 2OG, the mutant IDH produces 2-hydroxyglutarate (2HG), which is also correlated with the reduction of 5hmC in cancers (94). In cultured cells, supplementation of 2HG suppressed the conversion of 5mC to 5hmC, suggesting that 2HG is an inhibitor of 2OG competing for the binding site in TET dioxygenases (94). These lines of evidence have demonstrated that like P4H, the catalytic activity of TET dioxygenases is indeed dependent on Fe2+ and 2OG.
The Zhang group demonstrated that TETs further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (35). Both 5fC and 5caC could be excised by the DNA repair enzyme Thymine DNA glycosylase (TDG) to produce an abasic position, which is eventually replaced by an unmodified C as shown primarily by two groups (29, 53). Thus, 5hmC, 5fC and 5caC have been proposed as demethylation intermediates. So far, our knowledge of TETs-mediated DNA demethylation is the following: the Fe2+ and 2OG-dependent TETs consecutively oxide 5mC to 5hmC, then to 5fC and 5caC which can eventually be removed from the genome and substituted by unmodified C, thus completing the process of DNA demethylation. Although it involves multiple steps, the TET-mediated oxidation, combined with base excision repair (BER), constitutes the most important and consistent pathway responsible for the active demethylation of DNA.
Vitamin C – an additional cofactor for TET dioxygenases
The requirement for ascorbate as an additional cofactor for P4H and other dioxygenases suggested a potential role for this reducing cofactor in TET-mediated DNA demethylation. Interestingly, studies have shown that ascorbate has the capacity to modify the status of DNA methylation in mammalian cells. For instance, ascorbate causes the widespread DNA demethylation of nearly 2,000 genes in embryonic stem cells (11). Ascorbate also enhances the generation of induced pluripotent stem cells (iPSC) from terminally differentiated cells, which is usually accompanied by genome-wide DNA demethylation (18, 84). These results indicate that ascorbate could be involved in the DNA demethylation process. However, it is unclear whether ascorbate participates directly in DNA demethylation and whether the facilitated DNA demethylation is mediated by the enhanced catalytic activity of TETs by ascorbate.
Initial in vitro enzymatic analysis suggested that ascorbate was not essential for TET-mediated hydroxylation of 5mC. It was reported that in the absence of ascorbate, recombinant TET1 converted 5mC to 5hmC at a similar efficiency as when ascorbate was included or excluded in the reaction (85). It is now understood that Fe2+ and 2OG could solely satisfy the need of TET dioxygenases in the short term to initiate the hydroxylation of 5mC, as they do for P4H. A possible premature exhaustion of TET enzymatic activity in the absence of ascorbate was not fully explored. It was not clear how long this reaction could last without ascorbate, especially when the in vitro stoichiometry of the reaction was not completely controlled. In some in vitro experimental settings, Fe2+ was obviously overloaded (molar ration of Fe2+: TET1 > 20). In this case, Fe2+ might remain available for TET dioxygenases over short periods of time. Thus, under this condition, ascorbate is not required as a rejuvenator of TET dioxygenases via its reduction of the inactive oxidized iron species (Fe3+) to the active Fe2+ state after TET dioxygenases have been inactivated by coupled or uncoupled decarboxylation of 2OG, as occurs in collagen P4H. In contrast, free Fe2+ is limited and stringently controlled within the cell. Therefore, these in vitro enzymatic analyses do not reflect the whole scenario of TET dioxygenases in catalyzing 5mC hydroxylation in the cell. The role of ascorbate as a potential cofactor for TET dioxygenases to sustain and complete the hydroxylation of 5mC to 5hmC was therefore not excluded by these in vitro enzymatic analyses.
The Wang lab demonstrated for the first time that ascorbate enhanced 5hmC generation in vivo in cultured cells, most likely by acting as a cofactor for TET to hydroxylate 5mC (13, 62). This previously unknown function of ascorbate in DNA demethylation was subsequently validated in different cell types and modeled animals by other groups (2, 7, 96). 5hmC was originally thought to be detectable only in a few cell types such as stem cells and neurons, but not in other fully differentiated cells. For instance, 5hmC was previously reported as undetectable in cultured HEK-293 cells, and only detectable in the cells when TETs were forcibly overexpressed (35). Wang et al. found that mouse embryonic fibroblasts (MEF) expressed TETs at low but detectable levels as previously reported (14, 41), with TET3 at a higher level than TET1 and TET2. Thus, MEFs constituted an appropriate experimental system to analyze their enzymatic requirements in a cell-based experimental setting. Interestingly, standard cell culture media usually lack ascorbate in their formula. However, when ascorbate is available, it can effectively enter into cells via different transporters. The content of 5hmC was extremely low in MEFs cultured in ascorbate-free medium. Additions of ascorbate in a dose- and time-dependent manner enhanced the generation of 5hmC. The signal of 5hmC reached a plateau when MEFs were treated with 10 3M ascorbate. Possibly due to the low expression of TETs in MEFs, ascorbate at a relatively low level could fulfill the requirement of TET dioxygenases to reach their maximal enzymatic activity. Surprisingly, the effect of ascorbate on 5hmC was quite rapid. The generation of 5hmC in MEFs could be induced by ascorbate in as little as 1 hr. The rapid effect suggested that no protein synthesis was required, rather activation of existing TET dioxygenases was enough to generate 5hmC (62). Indeed, ascorbate treatment did not change the expression of either TETs or IDHs.
Utilizing a variety of cell-based experiments, it was then tested whether ascorbate works as a cofactor for TET to enhance the conversion of 5mC to 5hmC. (I) Treatment with other reducers such as glutathione (GSH) did not change the level of 5hmC, suggesting that the effect of ascorbate on 5hmC could not be attributed to its role as a general reducer. (II) Knocking down the expression of TETs by short interference RNAs (siRNA) largely abolished the effect of ascorbate on 5hmC, indicating that it is TET dioxygenases that mediate the action of ascorbate on 5hmC generation. (III) Ascorbate transporter inhibitors such as phloretin or sulfinpyrazone decreased the effect of ascorbate on 5hmC generation, suggesting that intracellular accumulation of ascorbate is necessary for activating the catalytic activity of TET dioxygenases. (IV) The presence of ascorbate at physiological concentration has been shown to enhance the uptake of iron by cells, raising the possibility that the effect of ascorbate on 5hmC might be indirect and mediated by an ascorbate-induced increase in the cellular uptake of iron (46–48). However, removing iron from the culture medium did not affect the induction of 5hmC by ascorbate, which suggests that the effect of ascorbate on 5hmC is independent of cellular uptake of iron. (V) Cells cultured with different concentrations of glucose, a major precursor of 2OG, exhibited the similar level of 5hmC in response to ascorbate treatment. These results indicate that the effect of ascorbate on 5hmC is not dependent upon iron uptake, the expression of TET and IDH, or the production of 2OG, which overall suggests that ascorbate may directly participate in the conversion of 5mC to 5hmC, most likely as a cofactor of TETs (13, 62). Embryonic stem (ES) cells are generally cultured in the medium without ascorbate. The Ramalho-Santos group observed that when added, ascorbate led to a rapid and global increase in 5hmC, which was followed by DNA demethylation of many gene promoters and upregulation of demethylated germline genes (2). Yin et al. reported that ascorbate directly enhanced the activity of purified C-terminal catalytic domain of TET2 to oxidize 5mC to 5hmC and 5fC, while other reducers such as spermidine, vitamin B1, vitamin E, glutathione, NADPH, and L-cysteine did not have this effect. After treatment with ascorbate, mouse ES cells displayed significant increases in 5mC oxidation products, particularly 5fC and 5caC. Furthermore, 5hmC was decreased in various tissues in Gulo knockout mice (96). These results suggest that ascorbate could be a cofactor for TET in the cascade oxidation of 5mC. Chen et al. reported that TET1, in an ascorbate-dependent manner, regulated 5hmC formation at loci critical for the reprogramming of iPSC (7).
From in vitro enzymatic activity analysis to cell and animal-based experiments, the available evidence strongly suggest that ascorbate is a cofactor for TET dioxygenases in the conversion of 5mC to 5hmC, and further 5fC and 5caC, thus modulating DNA demethylation (Fig. 1).
Figure 1. The role of ascorbate in DNA demethylation.

As a cofactor, ascorbate participates in the cascade oxidation of 5mC, to 5hmC, to 5fC and to 5caC catalyzed by TET dioxygenases. 5fC and 5caC are then replaced by unmodified 5C by BER machinery. Additionally, unable to maintain 5hmC in the newly synthesized DNA also leads to passive demethylation. The methylation of an unmodified 5C could be reestablished by DNMT1, thus complete a cycle of DNA methylation–demethylation.
Vitamin C, DNA demethylation and epigenetic reprogramming
The amount of ascorbate needed for TETs apparently depends on the protein level of TET dioxygenases. The expression level of TETs is low in most terminally differentiated cells such as fibroblasts but is relatively high in neuronal cells such as Purkinje cells (26, 44). However, TETs are expressed at a much higher level in embryonic tissues, especially at early developmental stages (86). It is known that epigenetic reprogramming occurs during the mammalian embryonic development, which involves DNA demethylation and re-methylation. For a long time, how the DNA is demethylated in pre-implanted embryos has been unclear. It is now known that both TET-mediated oxidation and passive dilution participate in the demethylation process. Immediately after fertilization, 5mC in the paternal chromatin is rapidly replaced by 5hmC via TET3-mediated oxidation, which cannot be maintained during the rapid DNA replication in pre-implanted embryos leading to passive demethylation and the erasing of paternal 5mC patterns (33). In contrast, the maternal DNA is only passively demethylated by dilution due to the inhibition of DNMT1; further TETs-catalyzed oxidization of 5mC is not involved (42). However, one recent report showed that both the paternal and maternal chromatin went through active demethylation from zygote to four-cell embryos (89). A second round of epigenetic reprogramming happens in primordial germ cells (PGC), which also involves in TETs-mediated active demethylation (42).
Currently available evidence suggests that TETs-mediated active demethylation is one of the major pathways for DNA demethylation during early embryonic development. As a result, it could be possible that varying levels of ascorbate during embryonic development affect epigenetic reprogramming of both germ cells and the developing embryo, which could lead to distinct consequences. To date, studies on the effects of ascorbate contents on DNA methylation-demethylation dynamics during embryonic development are still lacking. However, several studies suggest that deficiency of ascorbate is linked to certain types of developmental defects. For example, women at high risk for neural tube defect (NTD) recurrence tend to have lower leucocyte ascorbate levels compared with low-risk women (3, 77). Genetic variation in SVCT2, which might affect intracellular ascorbate levels, is also associated with the risk of preterm birth (16). However, due to the fact the ascorbate consumption from dietary sources and supplements is almost impossible to control quantitatively in human subjects, published results of ascorbate supplement on birth defects or preterm delivery are mixed. It is worth noting that ascorbate supplementation in pregnant women should be aimed at satisfying the needs of TETs and other Fe2+ and 2OG-dependent dioxygenases in both the mother and the embryo. In the case of sufficient ascorbate dietary intake, higher-doses of ascorbate may not necessarily exert additional benefits. Although ascorbate is most probably essential for TETs-mediated active demethylation in embryonic development, it is unclear how much ascorbate is needed from diet and/or supplements to completely satisfy the requirement of TETs in the prevention of embryonic defects caused by inappropriate DNA demethylation.
Vitamin C and the loss of 5hmC in cancer
In contrast to the relatively high level of 5hmC in embryos, especially in pre-implanted embryos and PGCs, cancer cells have very low or undetectable 5hmC. Studies have shown that loss of 5hmC is a novel epigenetic hallmark of most, if not all, types of human cancer (9, 36, 40, 45, 57, 65, 67, 71, 94, 95). The Rao lab was the first to provide convincing evidence that links the mutant TET1 and an impaired generation of 5hmC in leukemia (85). Since then, many groups have shown the loss of 5hmC in various cancers. In 2012, Shi et al published the inverse correlation of 5hmC with melanoma progression in humans, and a possible treatment in animal models. The content of 5hmC is relatively high in healthy melanocytes but is gradually lost during progression from benign nevi through advancing stages of primary and metastatic melanoma (49). The global loss of 5hmC disrupts the normal dynamics of DNA methylation-demethylation and affects genome-wide gene expression, which could eventually lead to malignant transformation. Besides mutations in TETs and IDHs (19), a decreased expression of TETs and IDHs is also attribute to the loss of 5hmC in cancer (49).
Genetic variation in SVCT1 and SVCT2 has been associated with the risk of certain types of cancer including advanced colorectal adenoma (17), muscle-invasive bladder cancer (25), gastric cancer (92) and non-Hodgkin lymphoma (82). It should be noted that these genetic associations have not yet been independently verified in other cohorts. Also worth important consideration are the potential functional consequences of these associated variants on ascorbate transportation, which remain largely unclear. A search of Cosmics, a cancer somatic mutation database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), reveals that mutations in SVCTs have been identified in many types of cancer such as breast cancer, colorectal adenoma, brain tumors and others. Recurrent mutations in the splicing factor SF3B1 have been identified in chronic lymphocytic leukemia, uveal melanoma as well as other cancers (27, 72). In at least one of these types of cancers, chronic lymphocytic leukemia, the mutant SF3B1 causes a truncated, most likely nonfunctional, SVCT2 that can result in intracellular deficiency of ascorbate in cancer cells (72). Furthermore, the uptake rate of ascorbate, the dominant form of ascorbate in the plasma, by a melanoma cell line is only ~50% of the uptake rate of the healthy melanocytes (83). These studies suggest that in addition to the genetic alterations in TETs and IDHs, variations in SVCTs might also contribute to the loss of 5hmC in cancer cells. Further studies are required to determine whether there is a local ascorbate deficiency in cancer cells.
In 1976, Linus Pauling proposed the treatment of cancer patients with intravenous ascorbate, followed by oral maintenance. This proposal was met with some skepticism, in part due to controversial experimental support, but also due to the lack of a complete picture of the effect of ascorbate on cancer cells; the newly recognized role of ascorbate in the epigenetic modulation of gene activity might shed a new light on this issue. It has been shown that overexpressing TET1 in breast cancer and overexpressing TET2 in melanoma could partially reestablish a normal 5hmC profile in the cancer cells and decreases malignancy especially invasiveness (32, 49). These findings suggest that the means of rebuilding the 5hmC content could offer a potential treatment for these cancers. However, it might not be feasible to clinically overexpress TET or IDH in patients. Our unpublished data show that treatment with a physiological concentration of ascorbate can decrease the malignant phenotype of melanoma cells in vitro by partially reestablishing the global content of 5hmC. As a cofactor for TETs, ascorbate enhances and possibly maximizes the catalytic activity of the existing TETs in cancer cells. The content of 5hmC induced by ascorbate in a melanoma cell line is comparable to the effect of overexpressing TET2 in the same cell line (49).
Ascorbate is a safe and well-tolerated dietary supplement that is readily available. Thus, it is feasible that ascorbate could be conveniently utilized in patient care. It is notable that ascorbate especially at high pharmacological concentrations has a long and widely discussed history in treating cancer. Further, possibly due to the fact that it is nearly impossible to quantitatively control dietary ascorbate consumption in human subjects, a solid epidemiological link between ascorbate deficiency and the incidence of cancer has not been established. However, a recent meta-analysis shows that higher intake of ascorbate might have a protective effect against the risk of lung cancer (52). Another meta-analysis supports the notion that post-diagnosis ascorbate supplementation may be associated with a reduced risk of mortality; dietary ascorbate intake is associated with a reduced risk of total mortality and breast cancer-specific mortality (28). It will not be surprising if some of the beneficial effects of ascorbate on cancer were found to be mediated by the induction of 5hmC generation in cancer cells.
Vitamin C and peripheral neuropathy
In the late 1960s, it was found that peripheral neuropathy developed in volunteers after ascorbate had been withdrawn from diet and dietary supplement for a longer period (~70 days). Reported complaints of the volunteers included numbness of the calves, hypoesthesia to light touches and pain (30, 31). These complaints, along with other signs of peripheral neuropathy gradually disappeared after ascorbate was resupplied to the volunteers. Since the potentially damaged peripheral nerves are nearly impossible to obtain from volunteers suffering ascorbate deficiency, a detailed histopathology of this induced peripheral neuropathy in humans is still unclear. Further, it also remains unknown how deficiency in ascorbate damages the peripheral nerve. Recent in vitro studies and animal modeling have provided convincing evidence linking deficient ascorbate and peripheral neuropathy.
In peripheral nerves, Schwann cells form myelin sheaths, which encircles axons to provide metabolic support and allow for fast nerve conduction. In the 1980s, ascorbate was identified as an essential factor to initiate and promote myelin formation by Schwann cells in vitro (5). In the defined medium used for co-culture of neurons and Schwann cells, myelin is not formed if ascorbate is absent in the medium (66). In contrast, myelin formation by olfactory ensheathing cells is not dependent on ascorbate (1), suggesting the specificity of the requirement of ascorbate for the myelin formation by Schwann cells. Previous studies focused on the known regulatory role of ascorbate in the formation of collagen- and laminin-containing extracellular matrix (ECM) (60, 15). However, recent animal-based studies suggest that the effect of ascorbate on ECM such as collagen cross-linking may not play a key role in promoting axonal myelination.
It has been shown that ascorbate enters and accumulates in Schwann cells primarily via SVCT2 (20). Loss of one allele in the gene SVCT2 (SVCT2+/−) causes a lower protein level of SVCT2, which would result in deficient ascorbate within Schwann cells, but not in extracellular milieu. Hypomyelination develops in peripheral nerves of SVCT2+/− mice (21), suggesting that the intracellular ascorbate is the key to myelination. Thus, collagen cross-linking induced by ascorbate in the extracellular matrix might not play an important role in the myelination formed by Schwann cells as previously thought. G. Wang’s unpublished observation indicates that ascorbate significantly increases 5hmC content and shifts the transcriptome pattern in cultured Schwann cells. It is now believed that ascorbate may have much broader impacts on the critical stages of myelination including Schwann cell activation, proliferation, differentiation and myelination by shifting their epigenome and transcriptome.
Charcot-Marie-Tooth disease (CMT) is a hereditary peripheral neuropathy. Type 1A of CMT (CMT1A), which is characterized by abnormalities in myelin, is mainly caused by duplication of a region on chromosome 17p12-p11.2 and mutations in the gene peripheral myelin protein 22 (PMP22), which reside in the same region (74). Treatment with large doses of ascorbate ameliorates the neuropathy of a CMT1A mouse model, in which PMP22 is overexpressed (69). Unfortunately, subsequent clinical trials on CMT1A patients presented somewhat disappointing results – no significant benefits were observed after treatment with ascorbate (1.5 g/day) for 1~2 years (6, 68). The reasons for this disappointing clinical trial are still under debate. Perhaps, 1~2 years of treatment are still too short for ascorbate to exert a measurable clinical outcome improvements in this slowly progressive disease (22). Alternatively, the CMT1A mouse model may have certain fundamental differences in the phenotype manifestation. For example, the mouse model synthesizes endogenous ascorbate whereas patients do not have this biosynthetic pathway. Additionally, one study showed that ascorbate combined with vitamin B2 and a synthetic reducer idebenone accelerates the recovery of vision in patients affected by Leber hereditary optic neuropathy (LHON) (56). Overall, the mechanistic role of ascorbate in myelin formation requires further examinations, which will aid in the design of future clinical trials aiming at the delay or prevention, rather than the cure, of peripheral neuropathy in patients carrying a CMT1A mutation.
VITAMIN C AND HISTONE DEMETHYLATION
Histone demethylation
The basic unit of eukaryotic chromatin is the nucleosome, which is composed of a short length of DNA wrapped around an octamer that consists of 2 copies of each histone (H2A, H2B, H3 and H4). Although not a component of the core histones, H1 binds the nucleosome “beads” and could be involved in the higher-order assembly of chromatin. Like other proteins, histones are also substrates for post-translational modifications (PTM). The known PTMs on histones include methylation, acetylation, phosphorylation, ubiquitination, sumyolation and others (75). These PTMs, which are catalyzed by different enzymes can target, yet sometimes compete for, amino acid residues in the histones. The dynamic PTMs in the histone modulate chromatin structure, genome stability and gene transcription.
Methylation at lysine and arginine residues is the major epigenetic modification in histones. Along with DNA methylation, histone methylation is a key component in the epigenome. The most extensively studied methylation occurs on histone H3 at lysine (K) 4 (H3K4), H3K9, H3K27, H3K36, H3K79 and H4K20 as well as on histone H3 at arginine (R) 2 (H3R2), H3R8, H3R17, H3R26 and H4R3. The methyl donor in histone methylation is S-adenosylmethionine (SAM), the same as the donor for DNA methylation. The addition of methyl groups to either lysine residues or arginine residues is catalyzed by histone methyltransferases (HMT) as writers of this epigenetic mark. There are many HMTs that belong to three families of enzymes; SET-domain-containing protein family, DOT1-like protein family methylate lysines and protein arginine N-methyltransferase (PRMT) family methylates arginines (24). In DNA methylation, only one methyl group is added to the cytosine. In histone methylation, there could be one, two or three methyl groups added to the single nitrogen of lysines resulting in monomethylated, dimethylated or trimethylated lysines respectively. Similarly, arginines could also be monomethylated and dimethylated (either symmetric at two nitrogens or asymmetric at a single nitrogen of the arginine), but not trimethylated. A large number of histone methylation binding proteins, as readers, bind methylated histones and further influence the recruitment of chromatin-modifying effectors, local chromatin structure and exert distinct impacts on genome functions depending on specific lysine or arginine residues. For example, methylation of H3K4 is associated with actively transcribed genes while methylation at H3K27 is a hallmark of silenced chromatin (37).
Histone methylation was once thought to be irreversible, thus, an everlasting PTM on histones. In 2004, the Shi group successfully identified the first lysine-specific histone demethylase 1 (LSD1 also known as KDM1A) (79) to remove methyl groups from monomethylated or dimethylated H3K4 and H3K9, thus resolving the debate of whether or not the histone methylation is reversible. LSD2, the other member of the LSD family, was later isolated to have similar enzymatic activity of histone demethylation like LSD1 (38). Via a flavin adenine dinucleotide (FAD)-dependent amine oxidase reaction, LSD family demethylases cleave the α-carbon bond of the methylated lysine to form an imine intermediate, which is hydrolyzed to form formaldehyde, releasing one molecule of H2O2 and the demethylated lysine (80). It appears that the LSD family demethylases can only demethylate monomethylated and dimethylated lysines, but not the trimethylated (me3) lysines in histone, suggesting the existence of other types of histone demethylase (64).
Vitamin C – an additional cofactor for JmjC domain-containing histone demethylases
In 2006, the Zhang group purified a JmjC domain-containing histone demethylase 1 (JHDM1), which specifically demethylates H3K36 in the presence of Fe2+ and 2OG. (88). Shortly after, the Zhang group also identified that the transcriptional repressor JHDM3A demethylates trimethylated H3K9 and H3K36 (39). So far, about 20 proteins that belong to the JmjC domain-containing histone demethylase family have been discovered to have the catalytic capacity to demethylate histones (63). It is now known that the JmjC domain-containing demethylases can demethylate mono-, di-, and trimethylated histone lysine residues. JmjC domain-containing histone demethylases, like TETs, also belong to the Fe2+ and 2OG dioxygenase superfamily. The Zhang group for the first time reported that ascorbate is required for optimal catalytic activity of JHDM1 and the demethylation mediated by JHDM3A is halted when ascorbate is withdrawn from the in vitro assay (39, 88). Although the role of ascorbate in histone demethylation was only examined in in vitro assays in these studies and the effect of ascorbate in other members in the JmjC domain-containing histone demethylases has not yet been reported, it is reasonable to deduce that ascorbate could be a cofactor for the JmjC domain-containing histone demethylase family, thus modulating histone demethylation in a similar way as it does on DNA demethylation (Fig. 2).
Figure 2. The role of ascorbate in histone demethylation.
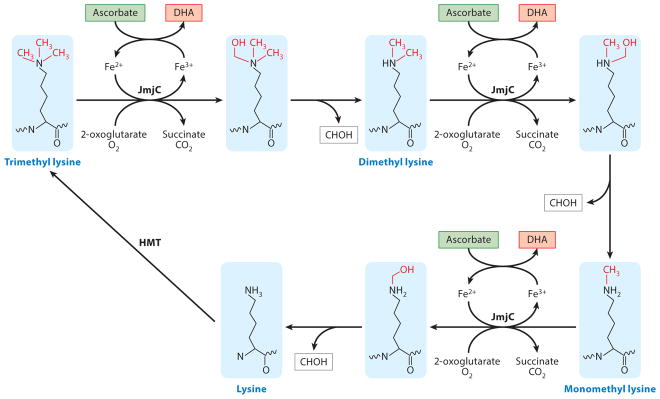
As a cofactor for Jmjc domain-containing histone demethylases (JmjC), ascorbate participates in the oxidation of tri-methylated, di-methylated and mono-methylated lysine in histones, which is followed by a spontaneous removal of the hydroxymethyl group. The methylation of an unmodified lysine in histones could be reestablished by HMT, thus complete a cycle of histone methylation–demethylation.
Vitamin C, histone demethylation and epigenetic reprogramming
Ascorbate appears to be important in the late phase of reprogramming from terminally differentiated cells (10). The key role of ascorbate in the transition from pre-iPSC phase to fully reprogrammed iPSCs was first uncovered by the Pei group (8). It appears that H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Ascorbate helps histone demethylases switch the pre-iPSC fate on or off by regulating the H3K9 methylation status of the core pluripotency loci; in addition to modulating DNA demethylation (11, 18, 84). Furthermore, ascorbate stimulates proliferation of bone marrow mesenchymal stem cells (MSC) and improves the efficiency of iPSC generation from the MSC (97). The impact on DNA and histone demethylation may underlie the gene expression profiles altered by ascorbate treatment in MEFs during iPSC induction (90) (Table 1).
Table 1.
Gene expression changes induced by ascorbate administration in MEFs.
| Chr | Gene | Gene description | Affymetrix ID | Effect |
|---|---|---|---|---|
| 11 | Ccl8 | Chemokine (C-C motif) ligand 8 | 10379535 | increased |
| 19 | As3mt | Arsenic (+3 oxidation state) methyltransferase | 10463704 | increased |
| 19 | Ifit1 | Interferon-induced protein with tetratricopeptide repeats 1 | 10462623 | increased |
| 5 | Oasl2 | 2′-5′ oligoadenylate synthetase-like 2 | 10524621 | increased |
| 6 | Usp18 | Ubiquitin specific peptidase 18 | 10541307 | increased |
| 16 | Rtp4 | Receptor transporter protein 4 | 10434778 | increased |
| 8 | Ddx60 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 | 10571984 | increased |
| 10 | Aldh1l2 | Aldehyde dehydrogenase 1 family, member L2 | 10371332 | decreased |
| 11 | Tgtp1 | T cell specific GTPase 1 | 10385533 | increased |
| 11 | Xaf1 | XIAP associated factor 1 | 10378068 | increased |
| 3 | Gbp3 | Guanylate binding protein 3 | 10496580 | increased |
| 1 | Csprs | Component of Sp100-rs | 10347925 | increased |
| 13 | Akr1c18 | Aldo-keto reductase family 1, member C18 | 10407435 | increased |
| 6 | Strip2 | Striatin interacting protein 2 | 10536949 | increased |
| 7 | Irf7 | Interferon regulatory factor 7 | 10569102 | increased |
| 10 | Ddit3 | DNA-damage inducible transcript 3 | 10366881 | decreased |
| 2 | Zbp1 | Z-DNA binding protein 1 | 10490150 | increased |
| 2 | Trib3 | Tribbles homolog 3 (Drosophila) | 10488608 | decreased |
| 16 | Mx2 | MX dynamin-like GTPase 2 | 10437224 | increased |
| 11 | Dhx58 | DEXH (Asp-Glu-X-His) box polypeptide 58 | 10391207 | increased |
Ascorbate is also involved in cell differentiation. For example, ascorbate enhances T cell maturation, an effect that is intrinsic to lymphoid cells and independent of T cell receptor (TCR) rearrangement. Inhibition of DNA and histone methylation further promotes the effect of ascorbate on T-cell differentiation, which suggests a role of ascorbate participated active demethylation of DNA and histone in T cell maturation (55).
CELLULAR AVAILABILITY OF VITAMIN C AND REDOX GENOMICS
The available data provide evidence for the role of ascorbate as a critical mediator of the interface between the genome and environment, principally by participating in epigenomic demethylation. Thus, any genetic and environmental factors that influence the synthesis, absorption, transportation and metabolism of ascorbate could have significant consequences for genome integrity, development, pluripotency, and ultimately health and disease by modulating the epigenetic control of genome activity.
Insufficient dietary intake is the major cause for systemic ascorbate deficiency. Diet and lifestyle are known to dramatically influence the level of ascorbate in the human body. Even in a developed country like the USA, more than 7% of the population (>20 million individuals) is estimated to be deficient in ascorbate (concentrations <11.4 μM in plasma), according to a recent survey (76). Cigarette smokers, low-income families and individuals carrying certain genetic variants in SVCTs have higher risk of ascorbate deficiency (87). Further, the turnover rate of vitamin C appears to be quite rapid, suggesting that the number of people with short-term ascorbate deficiency could be even higher (81). Ascorbate deficiency also can happen locally in certain types of cells. For example, intracellular ascorbate concentration in hepatocytes is decreased in aged mice due to a lower expression of SVCT1, as compared to young controls (61). Certain environmental exposures such as arsenic compounds also can diminish ascorbate levels and alter the epigenome of peripheral blood mononuclear cells (4).
Both systemic and local deficiency affects the availability of ascorbate to the cell. Furthermore, the transport of ascorbate to the nucleus is the key for ascorbate to reach to the chromosomes, which then influences the epigenome by participating in the demethylation of DNA and histones. More importantly, ascorbate is merely one component of the redox network in the cell. The degradation and recycling of ascorbate is largely controlled by the redox status. For example, the status of redox couples including GSH/GSSG, NADH/NAD and FADH2/FAD are capable of impacting the ascorbate/DHA redox couple, and thus, the availability of ascorbate to the DNA and histone demethylation machinery. Consequently, redox status in the cell, especially in the nucleus, will have a profound impact on the epigenome via ascorbate. It is well known that redox status changes along with development, aging, health and diseases. Bridging the gap between redox biology and genomics, we suggest an interdisciplinary research field that can be termed “Redox Genomics” to study the dynamic role of redox in health and diseases. Embryonic development, aging and many diseases such as chronic inflammation, cancer, diabetes, and neurodegenerative disorders, could be reassessed from the perspective of Redox Genomics.
Furthermore, many pre-clinical studies utilized cell- and animal (especially rodents) based models. It appears that ascorbate is not even contained in the formulation of most culture media. Thus, the critical regulatory role of ascorbate in the epigenome might have been overlooked in many cell-based studies. In addition, rodents can synthesize ascorbate de novo (unlike humans), therefore, these previous rodent studies were not equipped to detect how variation in ascorbate availability might affect the epigenome. We suggest that the availability of ascorbate should be addressed in future pre-clinical studies.
SUMMARY POINTS
Ascorbate regulates DNA demethylation as an essential cofactor for TET dioxygenases.
Ascorbate regulates histone demethylation as an essential cofactor for Jmjc domain-containing histone demethylases.
Ascorbate is a key mediator of the interface between the genome and environment.
Ascorbate is critical in maintaining the epigenome, especially at early embryonic stages.
Failure to maintain the catalytic activity of TET dioxygenases and Jmjc domain-containing histone demethylases by deficient ascorbate contributes to different diseases.
Ascorbate is an important micronutrient with functions far beyond scurvy prevention.
FUTURE ISSUES
Considering its cofactor role for the epigenetic enzymes, ascorbate, if deficient temporally and spatially, will impair the epigenome and contribute to certain phenotypic changes, and eventually lead to diseases. However, it is also understood that excessive ascorbate may not necessarily be beneficial for the epigenome. From an epigenomics perspective, the future goal should be to address ascorbate deficiency rather than elevating ascorbate to a very high level by large doses supplementation or injection. This novel epigenetic function of ascorbate needs to become known to the general public. Individuals with genetic variations affecting SVCTs should be alerted to the need for proper supplementation. RDA of ascorbate for women at early pregnancy may be reevaluated when the role of ascorbate in epigenetic reprogramming is taken into consideration. Deficient ascorbate may be involved in many diseases such as birth defects, cancer and diabetic complications. These need to be studied in patients as well as cell- and animal-based models. As previously noted, ascorbate is largely absent in the formulation of most media used for culturing cells. This suggests that the critical regulatory role of ascorbate in the epigenome has been completely overlooked in numerous cell-based studies. On the other hand, rodents have been overwhelmingly used in various disease modeling and drug screening. However, unlike humans, rodents synthesize ascorbate de novo in the liver. This implicates that the rodents-based studies have completely ignored the effect of variation in ascorbate availability (as in humans) on the epigenome. These issues need to be addressed sooner rather than later in the biomedical research field.
CONCLUSIONS
A novel function of ascorbate in epigenomic regulation has been uncovered by recently published studies. Ascorbate directly participates in epigenomic demethylation by serving as a cofactor for the demethylases. The bioavailability of ascorbate thus has a profound impact on the epigenome. Based on this knowledge, a novel interdisciplinary research field termed Redox Genomics is emerging to meet the needs to study the epigenomic role of redox in health and diseases.
Acknowledgments
We thank Ana Lorenzo, Kevin Dickson and Joshua Chen for their assistance. We apologize to our colleagues whose work we were not able to cite in this review due to the space limitation. The work on the epigenomic regulation by vitamin C in the Wang lab is supported by a grant from the National Institutes of Health (1R01NS089525) and a James and Esther King Biomedical Research Award (3KN08).
LITERATURE CITED
- 1.Babiarz J, Kane-Goldsmith N, Basak S, Liu K, Young W, Grumet M. Juvenile and adult olfactory ensheathing cells bundle and myelinate dorsal root ganglion axons in culture. Exp Neurol. 2011;229:72–9. doi: 10.1016/j.expneurol.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 2.Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, Lorincz MC, Ramalho-Santos M. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–6. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brender JD, Werler MM, Kelley KE, Vuong AM, Shinde MU, Zheng Q, Huber JC, Jr, Sharkey JR, Griesenbeck JS, Romitti PA, Langlois PH, Suarez L, Canfield MA The National Birth Defects Prevention Study . Nitrosatable drug exposure during early pregnancy and neural tube defects in offspring: National Birth Defects Prevention Study. Am J Epidemiol. 2011;174:1286–95. doi: 10.1093/aje/kwr254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brocato J, Costa M. 10th NTES Conference: Nickel and arsenic compounds alter the epigenome of peripheral blood mononuclear cells. J Trace Elem Med Biol. 2014 doi: 10.1016/j.jtemb.2014.04.001. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunge RP, Bunge MB, Eldridge CF. Linkage between axonal ensheathment and basal lamina production by Schwann cells. Annu Rev Neurosci. 1986;9:305–28. doi: 10.1146/annurev.ne.09.030186.001513. [DOI] [PubMed] [Google Scholar]
- 6.Burns J, Ouvrier RA, Yiu EM, Joseph PD, Kornberg AJ, Fahey MC, Ryan MM. Ascorbic acid for Charcot-Marie-Tooth disease type 1A in children: a randomised, double-blind, placebo-controlled, safety and efficacy trial. Lancet Neurol. 2009;8:537–44. doi: 10.1016/S1474-4422(09)70108-5. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H, Wang X, Hu X, Gu T, Zhou Z, Liu J, Liu J, Wu H, Mao SQ, Mo K, Li Y, Lai K, Qi J, Yao H, Pan G, Xu GL, Pei D. Vitamin C modulates Tet1 function during somatic cell reprogramming. Nat Genet. 2013;45:1504–9. doi: 10.1038/ng.2807. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, Liang H, Chen Y, Chen J, Wu Y, Guo L, Zhu J, Zhao X, Peng T, Zhang Y, Chen S, Li X, Li D, Wang T, Pei D. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 9.Chen ML, Shen F, Huang W, Qi JH, Wang Y, Feng YQ, Liu SM, Yuan BF. Quantification of 5-Methylcytosine and 5-Hydroxymethylcytosine in Genomic DNA from Hepatocellular Carcinoma Tissues by Capillary Hydrophilic-Interaction Liquid Chromatography/Quadrupole Time-of-Flight Mass Spectrometry. Clin Chem. 2013;59:824–32. doi: 10.1373/clinchem.2012.193938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Gao S, He W, Kou X, Zhao Y, Wang H, Gao S. Xist repression shows time-dependent effects on the reprogramming of female somatic cells to induced pluripotent stem cells. Stem Cells. 2014;32:2642–56. doi: 10.1002/stem.1775. [DOI] [PubMed] [Google Scholar]
- 11.Chung TL, Brena RM, Kolle G, Grimmond SM, Berman BP, Laird PW, Pera MF, Wolvetang EJ. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells. 2010;28:1848–55. doi: 10.1002/stem.493. [DOI] [PubMed] [Google Scholar]
- 12.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F, Fontenay M, Vainchenker W, Bernard OA. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 13.Dickson KM, Gustafson CB, Young JI, Züchner S, Wang G. Ascorbate-induced generation of 5-hydroxymethylcytosine is unaffected by varying levels of iron and 2-oxoglutarate. Biochem Biophys Res Commun. 2013;439:522–7. doi: 10.1016/j.bbrc.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, Fujita R, Guarnieri P, Bhagat G, Vanti WB, Shih A, Levine RL, Nik S, Chen EI, Abeliovich A. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–5. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eldridge CF, Bunge MB, Bunge RP, Wood PM. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J Cell Biol. 1987;105:1023–34. doi: 10.1083/jcb.105.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erichsen HC, Engel SA, Eck PK, Welch R, Yeager M, Levine M, Siega-Riz AM, Olshan AF, Chanock SJ. Genetic variation in the sodium-dependent vitamin C transporters, SLC23A1, and SLC23A2 and risk for preterm delivery. Am J Epidemiol. 2006;163:245–54. doi: 10.1093/aje/kwj035. [DOI] [PubMed] [Google Scholar]
- 17.Erichsen HC, Peters U, Eck P, Welch R, Schoen RE, Yeager M, Levine M, Hayes RB, Chanock S. Genetic variation in sodium-dependent vitamin C transporters SLC23A1 and SLC23A2 and risk of advanced colorectal adenoma. Nutr Cancer. 2008;60:652–9. doi: 10.1080/01635580802033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esteban MA, Wang T, Qin B, Yang J, Qin D, Cai J, Li W, Weng Z, Chen J, Ni S, Chen K, Li Y, Liu X, Xu J, Zhang S, Li F, He W, Labuda K, Song Y, Peterbauer A, Wolbank S, Redl H, Zhong M, Cai D, Zeng L, Pei D. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 2010;6:71–79. doi: 10.1016/j.stem.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gess B, Lohmann C, Halfter H, Young P. Sodium-dependent vitamin C transporter 2 (SVCT2) is necessary for the uptake of L-ascorbic acid into Schwann cells. Glia. 2010;58:287–99. doi: 10.1002/glia.20923. [DOI] [PubMed] [Google Scholar]
- 21.Gess B, Röhr D, Fledrich R, Sereda MW, Kleffner I, Humberg A, Nowitzki J, Strecker JK, Halfter H, Young P. Sodium-dependent vitamin C transporter 2 deficiency causes hypomyelination and extracellular matrix defects in the peripheral nervous system. J Neurosci. 2011;31:17180–92. doi: 10.1523/JNEUROSCI.3457-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gess B, Röhr D, Young P. Ascorbic acid and sodium-dependent vitamin C transporters in the peripheral nervous system: from basic science to clinical trials. Antioxid Redox Signal. 2013;19:2105–14. doi: 10.1089/ars.2013.5380. [DOI] [PubMed] [Google Scholar]
- 23.Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–24. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guey LT, García-Closas M, Murta-Nascimento C, Lloreta J, Palencia L, Kogevinas M, Rothman N, Vellalta G, Calle ML, Marenne G, Tardón A, Carrato A, García-Closas R, Serra C, Silverman DT, Chanock S, Real FX, Malats N EPICURO/Spanish Bladder Cancer Study investigators . Genetic susceptibility to distinct bladder cancer subphenotypes. Eur Urol. 2010;57:283–92. doi: 10.1016/j.eururo.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45:133–5. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris HR, Orsini N, Wolk A. Vitamin C and survival among women with breast cancer: a meta-analysis. Eur J Cancer. 2014;50:1223–31. doi: 10.1016/j.ejca.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 29.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges RE, Baker EM, Hood J, Sauberlich HE, March SC. Experimental scurvy in man. Am J Clin Nutr. 1969;22:535–48. doi: 10.1093/ajcn/22.5.535. [DOI] [PubMed] [Google Scholar]
- 31.Hood J. Femoral neuropathy in scurvy. N Engl J Med. 1969;281:1292–3. doi: 10.1056/NEJM196912042812309. [DOI] [PubMed] [Google Scholar]
- 32.Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, Chu CS, Jeng YM, Chen YT, Lin FM, Huang HD, Lu YY, Teng YC, Lin ST, Lin RK, Tang FM, Lee SB, Hsu HM, Yu JC, Hsiao PW, Juan LJ. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568–79. doi: 10.1016/j.celrep.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 33.Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of TET dioxygenases in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. TET dioxygenases can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin SG, Jiang Y, Qiu R, Rauch TA, Wang Y, Schackert G, Krex D, Lu Q, Pfeifer GP. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71:7360–5. doi: 10.1158/0008-5472.CAN-11-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Justin N, De Marco V, Aasland R, Gamblin SJ. Reading, writing and editing methylated lysines on histone tails: new insights from recent structural studies. Curr Opin Struct Biol. 2010;20:730–8. doi: 10.1016/j.sbi.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 38.Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284:17775–82. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–6. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 40.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koshiishi I, Mamura Y, Liu J, Imanari T. Evaluation of an acidic deproteinization for the measurement of ascorbate and dehydroascorbate in plasma samples. Clinical Chemistry. 1998;44:863–8. [PubMed] [Google Scholar]
- 44.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kudo Y, Tateishi K, Yamamoto K, Yamamoto S, Asaoka Y, Ijichi H, Nagae G, Yoshida H, Aburatani H, Koike K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103:670–6. doi: 10.1111/j.1349-7006.2012.02213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lane DJ, Chikhani S, Richardson V, Richardson DR. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochim Biophys Acta. 2013;1833:1527–41. doi: 10.1016/j.bbamcr.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 47.Lane DJ, Lawen A. Non-transferrin iron reduction and uptake are regulated by transmembrane ascorbate cycling in K562 cells. J Biol Chem. 2008;283:12701–8. doi: 10.1074/jbc.M800713200. [DOI] [PubMed] [Google Scholar]
- 48.Lane DJ, Robinson SR, Czerwinska H, Bishop GM, Lawen A. Two routes of iron accumulation in astrocytes: ascorbate-dependent ferrous ironuptake via the divalent metal transporter (DMT1) plus an independent route for ferric iron. Biochem J. 2010;432:123–132. doi: 10.1042/BJ20101317. [DOI] [PubMed] [Google Scholar]
- 49.Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee CW, Hu D, Lian BQ, Kleffel S, Yang Y, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, Scolyer RA, Thompson JF, Kakavand H, Houvras Y, Zon LI, Mihm MC, Jr, Kaiser UB, Schatton T, Woda BA, Murphy GF, Shi YG. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–46. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linster CL, Van Schaftingen E, Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 51.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t (10;11)(q22;q23) Leukemia. 2003;17:637–41. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 52.Luo J, Shen L, Zheng D. Association between vitamin C intake and lung cancer: a dose-response meta-analysis. Sci Rep. 2014;4:6161. doi: 10.1038/srep06161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–8. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mandl J, Szarka A, Bánhegyi G. Vitamin C: update on physiology and pharmacology. Br J Pharmacol. 2009;157:1097–110. doi: 10.1111/j.1476-5381.2009.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manning J, Mitchell B, Appadurai DA, Shakya A, Pierce LJ, Wang H, Nganga V, Swanson PC, May JM, Tantin D, Spangrude GJ. Vitamin C promotes maturation of T-cells. Antioxid Redox Signal. 2013;19:2054–67. doi: 10.1089/ars.2012.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mashima Y, Kigasawa K, Wakakura M, Oguchi Y. Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy? J Neuroophthalmol. 2000;20:166–70. doi: 10.1097/00041327-200020030-00006. [DOI] [PubMed] [Google Scholar]
- 57.Mason EF, Hornick JL. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol. 2013;26:1492–7. doi: 10.1038/modpathol.2013.86. [DOI] [PubMed] [Google Scholar]
- 58.May JM. The SLC23 family of ascorbate transporters: ensuring that you get and keep your daily dose of vitamin C. Br J Pharmacol. 2011;164:1793–801. doi: 10.1111/j.1476-5381.2011.01350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–72. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 60.McGarvey ML, Baron-Van Evercooren A, Kleinman HK, Dubois-Dalcq M. Synthesis and effects of basement membrane components in cultured rat Schwann cells. Dev Biol. 1984;105:18–28. doi: 10.1016/0012-1606(84)90257-4. [DOI] [PubMed] [Google Scholar]
- 61.Michels AJ, Joisher N, Hagen TM. Age-related decline of sodium-dependent ascorbic acid transport in isolated rat hepatocytes. Arch Biochem Biophys. 2003;410:112–20. doi: 10.1016/s0003-9861(02)00678-1. [DOI] [PubMed] [Google Scholar]
- 62.Minor EM, Court BL, Young JI, Wang G. Ascorbate induces Ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288:13669–74. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monfort A, Wutz A. Breathing-in epigenetic change with vitamin C. EMBO Rep. 2013;14:337–46. doi: 10.1038/embor.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–79. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 65.Müller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D, Freihoff J, Becker A, Simon M, Hammes J, Denkhaus D, zur Mühlen A, Pietsch T, Waha A. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am J Pathol. 2012;181:675–83. doi: 10.1016/j.ajpath.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 66.Olsen CL, Bunge RP. Requisites for growth and myelination of urodele sensory neurons in tissue culture. J Exp Zool. 1986;238:373–84. doi: 10.1002/jez.1402380310. [DOI] [PubMed] [Google Scholar]
- 67.Orr BA, Haffner MC, Nelson WG, Yegnasubramanian S, Eberhart CG. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS One. 2012;7:e41036. doi: 10.1371/journal.pone.0041036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pareyson D, Reilly MM, Schenone A, Fabrizi GM, Cavallaro T, Santoro L, Vita G, Quattrone A, Padua L, Gemignani F, Visioli F, Laurà M, Radice D, Calabrese D, Hughes RA, Solari A CMT-TRIAAL; CMT-TRAUK groups . Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol. 2011;10:320–8. doi: 10.1016/S1474-4422(11)70025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontés M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- 70.Penn NW, Suwalski R, O’Riley C, Bojanowski K, Yura R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem J. 1972;126:781–90. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pérez C, Martínez-Calle N, Martín-Subero JI, Segura V, Delabesse E, Fernandez-Mercado M, Garate L, Alvarez S, Rifon J, Varea S, Boultwood J, Wainscoat JS, Cruz Cigudosa J, Calasanz MJ, Cross NC, Prósper F, Agirre X. TET2 mutations are associated with specific 5-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PLoS One. 2012;7:e31605. doi: 10.1371/journal.pone.0031605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Giné E, Hernández JM, González-Díaz M, Puente DA, Velasco G, Freije JM, Tubío JM, Royo R, Gelpí JL, Orozco M, Pisano DG, Zamora J, Vázquez M, Valencia A, Himmelbauer H, Bayés M, Heath S, Gut M, Gut I, Estivill X, López-Guillermo A, Puente XS, Campo E, López-Otín C. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1gene in chronic lymphocytic leukemia. Nat Genet. 2011;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 73.Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–23. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- 74.Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013;9:562–71. doi: 10.1038/nrneurol.2013.179. [DOI] [PubMed] [Google Scholar]
- 75.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839:627–43. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schleicher RL, Carroll MD, Ford ES, Lacher DA. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES) Am J Clin Nutr. 2009;90:1252–63. doi: 10.3945/ajcn.2008.27016. [DOI] [PubMed] [Google Scholar]
- 77.Schorah CJ, Wild J, Hartley R, Sheppard S, Smithells RW. The effect of periconceptional supplementation on blood vitamin concentrations in women at recurrence risk for neural tube defect. Br J Nutr. 1983;49:203–11. doi: 10.1079/bjn19830026. [DOI] [PubMed] [Google Scholar]
- 78.Shen L, Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr Opin Cell Biol. 2013;25:289–96. doi: 10.1016/j.ceb.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 80.Shi YG, Tsukada Y. The discovery of histone demethylases. Cold Spring Harb Perspect Biol. 2013;5:a017947. doi: 10.1101/cshperspect.a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simpson GL, Ortwerth BJ. The non-oxidative degradation of ascorbic acid at physiological conditions. Biochim Biophys Acta. 2000;1501:12–24. doi: 10.1016/s0925-4439(00)00009-0. [DOI] [PubMed] [Google Scholar]
- 82.Skibola CF, Bracci PM, Halperin E, Nieters A, Hubbard A, Paynter RA, Skibola DR, Agana L, Becker N, Tressler P, Forrest MS, Sankararaman S, Conde L, Holly EA, Smith MT. Polymorphisms in the estrogen receptor 1 and vitamin C and matrix metalloproteinase gene families are associated with susceptibility to lymphoma. PLoS One. 2008;3:e2816. doi: 10.1371/journal.pone.0002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Spielholz C, Golde DW, Houghton AN, Nualart F, Vera JC. Increased facilitated transport of dehydroascorbic acid without changes in sodium-dependent ascorbate transport in human melanoma cells. Cancer Res. 1997;57:2529–37. [PubMed] [Google Scholar]
- 84.Stadtfeld M, Apostolou E, Ferrari F, Choi J, Walsh RM, Chen T, Ooi SS, Kim SY, Bestor TH, Shioda T, Park PJ, Hochedlinger K. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat Genet. 2012;44:398–405. doi: 10.1038/ng.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139:1895–902. doi: 10.1242/dev.070771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Timpson NJ, Forouhi NG, Brion MJ, Harbord RM, Cook DG, Johnson P, McConnachie A, Morris RW, Rodriguez S, Luan J, Ebrahim S, Padmanabhan S, Watt G, Bruckdorfer KR, Wareham NJ, Whincup PH, Chanock S, Sattar N, Lawlor DA, Davey Smith G. Genetic variation at the SLC23A1 locus is associated with circulating concentrations of L-ascorbic acid (vitamin C): evidence from 5 independent studies with >15,000 participants. The American journal of clinical nutrition. 2010;92:375–82. doi: 10.3945/ajcn.2010.29438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–6. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 89.Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W, Li W, Zhou Q, Aluru N, Tang F, He C, Huang X, Liu J. Programming and inheritance of parental DNA methylomes in mammals. Cell. 2014;157:979–91. doi: 10.1016/j.cell.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi X, Qin B, Zeng L, Esteban MA, Pan G, Pei D. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell. 2011;9:575–87. doi: 10.1016/j.stem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 91.Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr. 2005;25:105–25. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- 92.Wright ME, Andreotti G, Lissowska J, Yeager M, Zatonski W, Chanock SJ, Chow WH, Hou L. Genetic variation in sodium-dependent ascorbic acid transporters and risk of gastric cancer in Poland. Eur J Cancer. 2009;45:1824–30. doi: 10.1016/j.ejca.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wyatt GR, Cohen SS. A new pyrimidine base from bacteriophage nucleic acids. Nature. 1952;170:1072–3. doi: 10.1038/1701072a0. [DOI] [PubMed] [Google Scholar]
- 94.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of a-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL, Xiong Y. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663–9. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, Zhang D, Huang H, Gao J, Li Z, Jiao Y, Li C, Liu S, Wu D, Gu W, Yang YG, Xu GL, Wang H. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135:10396–403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- 97.Yulin X, Lizhen L, Lifei Z, Shan F, Ru L, Kaimin H, Huang H. Efficient generation of induced pluripotent stem cells from human bone marrow mesenchymal stem cells. Folia Biol (Praha) 2012;58:221–3. [PubMed] [Google Scholar]