

Abstract
TNFα and IL-17 secreted by proinflammatory T-cells (TEFF) promote bone erosion by activating osteoclasts. We previously demonstrated that in addition to bone resorption, osteoclasts act as antigen presenting cells to induce FoxP3 in CD8 T-cells (TcREG). The osteoclast-induced regulatory CD8 T-cells limit bone resorption in ovariectomized mice (a murine model of postmenopausal osteoporosis). Here we show that while low-dose RANKL maximally induces TcREG via Notch signaling pathway to limit bone resorption, high-dose RANKL promotes bone resorption. In vitro, both TNFα and IL-17, cytokines that are abundant in ovariectomized animals, suppress TcREG induction by osteoclasts by repressing Notch ligand expression in osteoclasts but this effect can be counteracted by addition of RANKL. Ovariectomized mice treated with low-dose RANKL induced TcREG that suppressed bone resorption, decreased TEFF levels and increased bone formation. High dose RANKL had the expected osteolytic effect. Low dose RANKL administration in ovariectomized mice lacking CD8 T-cells was also osteolytic, confirming that TcREG mediate this bone anabolic effect. Our results show that while RANKL directly stimulates osteoclasts to resorb bone, it also controls the osteoclasts’ ability to induce regulatory T-cells, engaging an important negative feedback loop. In addition to the conceivable clinical relevance to treatment of osteoporosis, these observations have potential relevance to induction of tolerance and autoimmune diseases.
Introduction
Osteoimmunology is an emerging study of the crosstalk between the immune and skeletal systems. Osteoimmunology arose from the recognition that cytokines produced by lymphocytes can affect bone homeostasis (1). While much is known about the cytokines and mechanisms that lead to bone erosion by the proinflammatory cytokines (2–5), much less is known about the mechanisms that maintain or restore homeostasis (i.e. the healthy state). It is anticipated that there must be feedback loops in both the immune and skeletal systems that maintain and restore homeostasis after perturbations or changes to the system that arise due to pathologic (e.g. infections) and normal physiological processes (e.g. pregnancy).
The current study builds on previous work where we have identified a novel regulatory feedback loop between CD8 T-cells and osteoclasts (6–10). Specifically, osteoclasts induced the transcription factor Forkhead box protein 3 (FoxP3) in CD8 T-cells. FoxP3 is a master regulator of regulatory T-cells (11). While regulatory CD8+ T-cells, called TcREG, have been documented in humans and mice (11–21) they have not been studied extensively, in part due to their low abundance (0.2 to 2% of CD8+ T-cells) in lymphoid organs. In comparison, the well-studied CD4+ regulatory T-cells, TREG, comprise 5–12% of CD4+ T-cell in the spleen. TcREG and TREG have overlapping and distinct functions. Both cells express CD25 and the transcription factor FoxP3, a marker of regulatory T-cells (22–24). TREG are critical for suppressing the aberrant activation of self-reactive T-cells (17) as depletion of TREG or genetic ablation of FoxP3 in CD4 T-cells leads to multi-organ autoimmune syndrome (25–27). TcREG, in contrast, ostensibly do not maintain global suppression of self-reactive T-cells (28), but have immune-suppressive activity (29). The two regulatory T-cells are activated differently: thymically and peripherally produced TREG require restimulation through their T-cell receptor (TCR) by MHC class II to express their suppressive effector functions (30). The maturation of antigen presenting cells (APC) that express MHC class II needed for restimulation is tightly regulated (31,32). In contrast, TcREG do not require restimulation (8). In any case, as all cells (except red blood cells) constitutively express MHC class I, any cell could potentially stimulate TcREG, and therefore an overabundance of TcREG may lead to immune suppression. A number of studies (33–35), including ours (8), have shown that TcREG are rapidly induced (24 to 48 hours) locally in tissue from naïve CD8 T-cells; hence their steady state abundance is low in lymphoid tissue.
In particular, we have shown in vitro that osteoclasts, in addition to having a bone resorbing activity, also have an antigen presenting activity. Osteoclasts crosspresent (extracellular) antigens and activate CD8 T-cells to induce FoxP3, CD25, interleukin (IL)-2, IL-6, IL-10 and interferon (IFN)-γ in the CD8 T-cells. The osteoclast-induced TcREG (OC-iTcREG) suppress bone resorption by osteoclasts to form a negative feedback loop. The suppression of bone resorption activity has been demonstrated in vitro (8) and in two different models of bone loss in vivo (10). Unlike the bone marrow TcREG, thymically produced TcREG do not efficiently suppress osteoclast activity (Buchwald and Aurora, unpublished observations). TcREG may also be induced in the tonsils (36,37) and elsewhere. Both the endogenous bone marrow TcREG and ex vivo generated OC-iTcREG suppressed bone resorption in mice in response to 1 mg/ml RANKL administration (10). Adoptively transferred OC-iTcREG also suppressed bone resorption by reducing the numbers of osteoclasts in ovariectomized mice (10). We have also shown that transfer of ex vivo generated OC-iTcREG are immunosuppressive in vivo because they decrease the levels of proinflammatory effector T-cells (TEFF) in the bone marrow, which increase in ovariectomized animals, to levels found in sham operated mice (10). These results established that OC-iTcREG negatively regulate osteoclast activity and the immune system.
Homeostasis, the ability to maintain a stable set point in response to physiologic or environmental changes, is achieved through a number of regulatory motifs (38–41). One of these motifs, referred to as the reactive negative regulator ensures that responses to stimuli are of the appropriate intensity, duration and are subsequently terminated or resolved (9,42). For example, acute inflammation is an appropriate and healthy response to an infection or trauma that clears or dilutes the offending agent and activates repair mechanisms. Acute inflammation is a healthy response, as long as it is brief and intense enough to clear the infection and then resolves with minimal collateral damage. The failure of activation of the reactive regulatory motif can often lead to pathology. As TcREG represent an example of a reactive negative regulator, we believe a better understanding of this system can provide insights into how such regulation is lost during pathogenesis and/or used to maintain or restore homeostasis.
Here, we initiated our study to test the ability of osteoclasts to induce TcREG in vivo. We found that activation of osteoclasts is needed to induce TcREG, consistent with our expectation that TcREG are reactive negative regulators. However, these results beg the question: what defect(s) in the osteoclast—TcREG feedback system allows excess bone resorption in ovariectomized mice (and by inference in postmenopausal osteoporosis)? Our studies provide a surprising result that could not have been anticipated in the absence of knowledge of the feedback loop: that under some conditions RANKL leads to an increase in bone density.
Materials and Methods
Mice
C57BL/6J mice (model 000664) were purchased from Jackson Laboratory or used from in-house breeding colonies. Breeders of FoxP3eGFP reporter (model 006772) and β2M−/− (model 002087) mice were purchased from Jackson Laboratory, and bred in-house for these experiments. OT-I Rag−/− mice were purchased from Taconic. Breeders of OT-I Thy1.1 Rag−/− mice were a gift of Dr. Ryan Teague (St. Louis University School of Medicine). All animals were maintained in the Department of Comparative Medicine, Saint Louis University School of Medicine in accordance with institutional and Public Health Service Guidelines. Saint Louis University School of Medicine Institutional Animal Care and Use Committee approved all procedures performed on mice (protocol numbers 2072 and 2184).
Ovariectomy
Bilateral ovariectomy was performed on 12–14 week old mice. Mice were anesthetized using 2.5% isoflurane to initiate anesthesia, and 1% for maintenance. The ovaries were accessed through a single incision in the skin, and exteriorized through muscle wall on each side. Each ovary was clamped using hemostat and removed by a single cut. Skin staples (3M) were used to close the skin incision. To minimize discomfort post-surgery, 0.025 mg/kg Buprenorphine was administered subcutaneously. Zoledronate (Selleck Chemicals) was administered at 30 μg/kg via tail vein.
Adoptive transfer of T-cells
All T-cells were transferred via tail vein. For injections mice were restrained and 20×106 T-cells, suspended in 100–150 μl PBS were injected into the lateral vein.
Generation of OC
OC precursors were isolated as previously described (7,8). Briefly, the mice were sacrificed by CO2 asphyxiation and the long bones harvested. One end cap of the bone was removed and the long bones were placed in a 0.7 ml microcentrifuge tube, pierced with a 22G needle at the bottom of the tube. The 0.7 ml tube was fitted inside a 1.5 ml microcentrifuge tube. The assembly was spun for 30 seconds at 16,000×g. The bone marrow cells were resuspended in α-minimum essential medium (αMEM, Invitrogen), and filtered through a 40-μ cell strainer. The cells were pelleted, resuspended and maintained in αMEM growth medium (αMEM supplemented with 10% heat-inactivated fetal bovine serum [Invitrogen]), penicillin-streptomycin-glutamine (Invitrogen) and recombinant murine M-CSF (Peprotech) at 20 ng/ml). OC were generated by addition of recombinant murine GST-RANKL (a gift of Prof. Steven Teitelbaum, Washington University in St. Louis) to a final concentration of 50 ng/ml. M-CSF and GST-RANKL were added every 48 to 72 h.
Isolation of T-cells
Single cell suspensions of spleens were prepared in PBS + 1% FBS by grinding with a sterile syringe plunger and dispersed by pipetting, then filtering through a 40 μ cell strainer. For co-culture experiments, splenic OT-I or WT CD8 T-cells were prepared by first enriching for T-cells using Pan-T-cell beads and further purified by negative selection using appropriate magnetic beads (Miltenyi). All bone marrow and splenic T-cells purified by positive selection were incubated for 30 m at 37° C to allow cells to allow dissociation or uptake of bound beads from cell surface. The resulting T-cells were routinely > 97% pure when stained with anti-CD3, anti-CD4 and anti-CD8 antibody.
Generation of OC-iTcREG
Day 4 OC cultured in 20 ng/ml M-CSF and 50 ng/ml GST-RANKL, were seeded at 5×105 cells/well in the presence of 5 μM OVA (A-5503; Sigma-Aldrich) in 24-well tissue culture-treated plates (Corning). After 14–16 h of incubation, medium was removed and (adherent) cells were washed with pre-warmed medium. 2.5×105 freshly harvested splenic OT-I transgenic T cells purified by negative selection were added in 2 ml of complete T-cell media (RPMI, 10% ΔFBS, penicillin-streptomycin-glutamine, non-essential amino acids, sodium pyruvate, HEPES, and 55 μM β-mercaptoethanol). Following 48 h co-culture, T-cell aliquots were removed and intracellular staining was performed to assess FoxP3 expression. The TcREG were then further expanded, in the absence of OC, by splitting cells 1:2 and culturing in 100 U/ml IL-2 containing T-cell media for an additional 48 h. For polyclonal TcREG generation, T-cells were purified from spleens of C57BL/6 mice and incubated with day 4 OC in the presence of 1μg/ml anti-CD3. Control T-cells were activated with plate bound anti-CD3 (1 μg/ml) and anti-CD28 (2μg/ml; both from eBiosciences) for 48 hours; the activated T-cells were expanded further by splitting 1:2 and culturing for additional 48 hours in IL-2 (100 U/ml). 20×106 TcREG (in 200 μl) were then injected by tail vein into 8-week-old OT-I mice.
PCR assessment of expression of CD200 and Notch ligands
Osteoclasts seeded on day 4 at 1.5×106 cells/ml were used in all experiments. RNA was isolated at time point described in Figure legends. 10 to 50 ng of RNA was used for first-strand cDNA synthesis in 50μL reaction per kit instructions (Superscript III cDNA synthesis system; Invitrogen). In all cases ~10% of the cDNA product was used in a 50μL PCR reaction that contained 2 μM each forward and reverse primers. For quantitative PCR (qPCR) SYBR green system (Invitrogen) was used. Otherwise, cDNA was amplified (25 cycles) and the products resolved on 1.2 % agarose gel and visualized by ethidium bromide staining.
| Gene | Forward primer | Reverse primer | Annealing temp. (°C) | Product size (bp) |
|---|---|---|---|---|
| DLL1 | ACCTTCTTTCGCGTATGCCTCAAG | AGAGTCTGTATGGAGGGCTTC | 58 | 222 |
| DLL4 | CGAGAGCAGGGAAGCCATGA | CCTGCCTTATACCTCTGT | 58 | 379 |
| JAG1 | ATTCGATCTACATAGCCTGTGAG | CTATACGATGTATTCCATCCGGT | 50 | 671 |
| JAG2 | TGTCAGCCACGGAGCAGTCATT | TCTCACGTTCTTTCCTGCGCTT | 50 | 469 |
| CD200 | TGGTCGTCTCTTCCTCCACA | CTCCTGAATACCAGACTGCCC | 50 | 196 |
Antibodies and Fluorescence activated cells sorting (FACS)
Anti-mouse antibodies for FACS were: PE-conjugated anti-mouse CD8a (clone 53-6.7; BD Pharmingen), AF700-conjugated anti-mouse CD44 (IM7; BD Pharmingen), e450-conjugated anti-mouse FoxP3 (FJK-16s, eBioscience), anti-CD3e (500A2; Biolegend), anti-CD8a (5H10; Caltag), anti-CD4 (RM4-5; BD Pharmingen), V450-conjugated CD45.1 (A20; BD Biosciences), PE-Cy7 conjugated anti-CD45.2 (104; BD Biosciences) and anti-CD25 (Clone PC61; BD Pharmingen). Functional grade anti-CD3 (17A2) and anti-CD28 (37.51) were purchased from eBioscience. For FACS cells were blocked with anti-mouse FcgRIII/IIR (BD Pharmingen) for 10 m and then stained for 45 m on ice with fluorophore-conjugated antibody. Stained cells were washed, fixed with 3% paraformaldehyde and analyzed on LSRII instrument with CellQuest (BD Biosciences) software. Data analyses were performed with FlowJo software (version 8.73; Tree Star).
Serum CTX measurements
Food was withdrawn 6 to 10 h prior to bleeding. Peripheral blood (100 to 200 μl), obtained via sub-mandibular vein, was allowed to clot for one hour at room temperature and serum collected by spinning down the cell pellet (1000×g for 10 m). Serum was flash frozen and stored at −80°C. Serum C-terminal telopeptide of type 1 collagen (serum CTX) was measured using ELISA according to the manufacturer’s instructions (Immunodiagnostic Systems, Plc.)
Matrix dissolution assays
CD8 T-cells were isolated from the bone marrow cells (isolated as described in OC generation section above) using magnetic beads. The TcREG were further purified in some experiments by cell sorting and co-cultured with OC (5 × 105) that were previously seeded on 24-well hydroxyapatite coated plates (Corning). M-CSF and GST-RANKL were added every 48 h. On day 5, cells were removed with 10% bleach and pit area was photographed and quantified using NIH ImageJ.
μCT data collection and analysis
The bones were scanned in μCT40 (Scanco Medical) at 55 kV, 145 μA, and resolution of 16 μm. Gauss sigma of 1.2, Gauss support of 2, lower threshold of 237, and upper threshold of 1000 were used for all the analysis. Regions of interest were selected 50 slices below the growth plate of the proximal tibia to evaluate the trabecular compartment. Bone mineral density was obtained by quantitative μCT using Scanco Phantoms for calibration (43). All μCT data and bone histomorphometry data was collected and analyzed by C.Y. who was blinded to the treatment performed on the mouse samples.
Statistical Analysis
Statistical significance was assessed in all cases using paired two-tailed Mann-Whitney U test in GraphPad Prism 5.0f. One-way and two-way ANOVA was performed in GraphPad Prism 5.0f.
Results
Low dose of RANKL activates osteoclasts to induce TcREG
We first determined the dose-response of TcREG production by RANKL in the bone marrow. RANKL was administered into FoxP3eGFP reporter mice (44) at various doses for two consecutive days. Fifty hours after first dose, the mice were sacrificed and the numbers of CD8 T-cells that were GFP positive cells in the bone marrow were measured. This time was chosen because in vitro TcREG require 48 to 50 h for maximal induction (7). The lowest dose of RANKL (0.125 mg/kg) induced the largest proportion of FoxP3+ CD8 T cells (Fig. 1A). No change in TcREG levels was observed in the spleen (Fig. 1A), strongly suggesting that RANKL mediates on TcREG its effect via osteoclasts. To assess that RANKL activated osteoclasts, we also measured serum CTX levels. These results (Fig. 1B) are consistent with our previous study where we demonstrated in mice that CD8 T-cells were bone protective and adoptive transfer of CD8 T-cells from Scurfy mice (i.e. FoxP3−/−) did not protect against the osteolytic effects of RANKL (10). The increased levels of FoxP3 in response to RANKL in the bone marrow could either be due to recruitment of TcREG to the bone marrow or induction of FoxP3 expression in cells that were FoxP3 negative.
Figure 1. TcREG are induced by activated osteoclasts.

A. RANKL treatment increases TcREG levels in bone marrow in FoxP3eGFP reporter mice: The numbers of FoxP3+ CD8 T-cells (TcREG) that are found in the bone marrow and spleen are plotted (y-axis) as a function of RANKL dose (x-axis), 50 h after first dose. The cells were gated on the lymphocytes (by forward and side-scatter), then on double positive CD45 and CD3 T-cells and finally on CD8, GFP double positive cells for quantitation. B. RANKL treatment activates osteoclasts: Serum CTX levels increase in response to low-dose (0.125 mg/kg) and high-dose (1 mg/kg) RANKL. C. TcREG are induced in the bone marrow: To test for TcREG induction by RANKL activated osteoclasts, congenically marked (Thy 1.2) GFP negative CD8 T-cells (see panel D for FACS plots) were adoptively transferred into Thy 1.1 OT-I Rag−/− mice. In the absence of RANKL treatment very low level of conversion was observed after three days. Low-dose (0.125 mg/kg) RANKL robustly induced TcREG; TcREG induction was measured 50 h after RANKL treatment and 3 days after T-cell transfer. Cells were isolated from tibia and femurs (bone marrow) for FACS analysis. The induction required active osteoclasts as no induction of TcREG was observed in mice pre-treated (two-weeks prior to T-cell transfer) with Zoledronic acid (ZA). D. Representative FACS plots are shown for panel C. The results represent mean ± standard deviation (error bars) from 4 experiments (n = 4 mice/group/experiment). P values were calculated using Mann-Whitney test; * = P < 0.05; ** = P < 0.01; **** = P < 1×104; and NS = not statistically significant.
To distinguish between recruitment and induction we FACS sorted a naïve GFP negative population of CD8 T-cells (Thy1.2+ and CD44 negative) from the spleens and bone marrow of FoxP3eGFP reporter mice (44) to high purity (Fig. 1D first panel) and adoptively transferred them into congenically marked (Thy 1.1) OT-I Rag−/− mice. The OT-I Rag−/− mice were used as recipients because they are not lymphopenic and because they lack endogenous TcREG, which avoids issues with homeostatic proliferation and competition with endogenous TcREG, respectively, and hence increases the sensitivity of the assay. In the absence of RANKL administration very low levels GFP+ CD8 T-cell were detected, but RANKL administration (0.125 mg/kg) yielded ~1% GFP+ Thy1.2 T-cells (Fig. 1C and 1D third panel). The conversion from GFP− to GFP+ by low dose RANKL is a clear indication of TcREG induction. To determine whether osteoclasts were responsible for TcREG induction, we pretreated the OT-I Thy1.1 Rag−/− mice with the bisphosphonate, Zoledronic acid (ZA), two weeks prior to transferring GFP− cells. As the half life of ZA in plasma is ~20 minutes (45) but ~ 60 days in the bone (46), we expect no direct effect of ZA 14 days post-treatment on the transferred T-cells. In mice treated with ZA (Fig. 1C), no conversion of CD8 T-cells to GFP+ was observed indicating that actively resorbing osteoclasts are required for TcREG induction. These results show that RANKL leads to induction of endogenous TcREG in the reporter mice and in adoptive transfer T-cell model.
We had concerns that the GST-RANKL fusion protein used in these experiments, which can form dimers or higher order oligomers through the GST moiety (47), could be activating osteoclasts to induce TcREG non-physiologically. To allay this concern, commercially purchased recombinant mouse RANKL (i.e. non-fused) was used for assessing changes in TcREG levels in the bone marrow and spleen. Murine-RANKL lacking the GST moiety gave identical results as shown in Fig. 1A (data not shown). Taken together with our in vitro results that osteoclasts induce TcREG, this experiment establishes that activation of resorption by osteoclasts by low-dose RANKL induces TcREG.
Equivalent levels of TcREG in ovariectomized and sham-operated mice
Based on the finding that activation of osteoclast induces TcREG we reasoned that TcREG should be induced by ovariectomy, as estrogen-depletion activates osteoclasts leading to osteoporosis. Therefore, we compared levels of TcREG in the bone marrow of sham-operated and ovariectomized mice. As shown in Fig. 2A, TcREG were present in ovariectomized mice, but no increase in levels of TcREG was observed between the sham-operated and ovariectomized mice (24 days post-ovariectomy). At this time point, we also observed no significant difference either in the number or percent (relative to CD3+ cells) of CD8 T-cells in the bone marrow between sham-operated and ovariectomized mice.
Figure 2. TcREG levels are not changed in ovariectomized mice but the TcREG can suppress osteoclast activity in vitro.
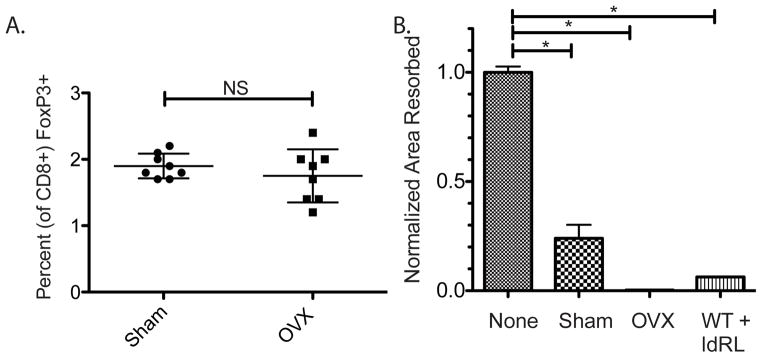
A. No change in TcREG levels despite increased osteoclast activity: The abundance of TcREG in sham-operated mice was similar to the levels found in ovariectomized (OVX) mice. B. TcREG from ovariectomized mice can suppress bone resorption: TcREG purified by magnetic beads from the bone marrow of sham-operated (Sham), ovariectomized (OVX) and WT (C57BL/6J) mice treated with low-dose RANKL (WT+ldRL; 0.125 mg/kg) were tested in a matrix dissolution assay. To assess the levels of repression, the results are expressed as normalized area resorbed relative to wells with osteoclasts with no TcREG added (None). All TcREG suppressed bone resorption by osteoclasts indicating that the TcREG are functional. The results represent mean ± standard deviation (error bars) from 2 experiments (n = 3 mice/group/experiment). P values were calculated using Mann-Whitney test; * = P < 0.05 and NS = not statistically significant.
We next assessed the endogenous TcREG for their ability to suppress osteoclasts from both sham-operated and ovariectomized mice. To this end we isolated the GFP+ CD8 T-cells from the bone marrow space of ovariectomized and sham operated mice by cell sorting. Equivalent numbers of T-cells (5×104 cells/well) and osteoclasts (1×105) were tested in a bone matrix dissolution assay. The results show that TcREG from both the ovariectomized and sham operated mice were effective in suppressing osteoclast bone resorbing activity in vitro (Fig. 2B). Osteoclast abundance increases in estrogen-depleted animal through a Fas ligand-dependent mechanism (48,49). The simplest interpretation of these results is that while the TcREG present in ovariectomized mice are functional, but their abundance relative to the number of osteoclasts is insufficient to limit bone resorption leading to osteoporosis. Further, increasing the number of TcREG in ovariectomized mice either by adoptive transfer or by induction should ameliorate osteoporosis. What remains unclear is why are TcREG not induced in estrogen-depleted animals despite increased osteoclast activity?
IL-17 and TNFα suppresses TcREG induction by osteoclasts
It has been reported that menopause leads to a low level persistent production of effector T-cells [TEFF; (50)]. We hypothesized that in the presence of pro-inflammatory cytokines, osteoclasts fail to induce TcREG. To test this hypothesis, we measured OC-iTcREG induction in the presence of IL-17 and TNFα. Antigen-pulsed osteoclasts were co-cultured with OT-I CD8 T-cells in the presence of RANKL and TNFα or IL-17. Indeed, in the presence of 10 ng/ml IL-17 (Fig. 3A) or increasing TNFα concentrations (Fig. 3B), the induction of TcREG by osteoclasts was greatly impaired. Equal levels of suppression in TcREG induction was observed at doses of 2 to 50 ng/ml IL-17A, therefore only the effect of only one dose is shown. Effect of similar dose of IL-17A has been observed in alveolar cells (51). We tested two different concentrations of RANKL to ascertain if the ratio of RANKL to TNFα acts as determinant of TcREG induction. Our data (Fig. 3B) did not support the notion that the ratio of TNFα to RANKL is important for TcREG induction. To understand the underlying mechanism of how TNFα affects osteoclasts’ ability to induce TcREG, we investigated the signals provided by osteoclasts to induce FoxP3 in the CD8 T-cells. Concurrently we assessed if these signals were affected by TNFα or IL-17.
Figure 3. The pro-inflammatory cytokines IL-17A and TNFα suppress TcREG induction by osteoclasts.
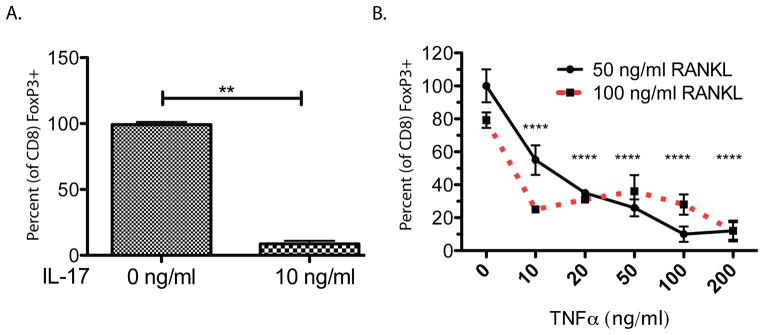
A. TcREG induction by osteoclasts in the presence of IL-17: TcREG were induced in vitro using OT-I CD8 T-cells co-cultured with bone marrow cells-derived osteoclasts, pulsed with SIINFEKL ovalbumin peptide. TcREG induction was observed in OT-I T-cells in the absence of (0 ng/ml) but significantly reduced in the presence of 10 ng/ml IL-17A. B. TcREG induction by osteoclasts in the presence of TNFα: TNFα also reduced TcREG induction in a dose dependent manner. Similar levels of suppression of TcREG induction was observed at 50 or 100 ng/ml RANKL each dose of TNFα indicating that the ratio of RANKL to TNFα is not critical. ANOVA analysis to determine the variation due to TNFα and RANKL concentration indicates that TNFα accounted for 85.5% of the total variance (P < 0.0001) and RANKL accounted for 0.65% of the total variance (P < 0.001). The interaction term accounted for remaining 9.3% of the total variance indicating that TNFα had a strong effect at each concentration of RANKL tested. The results represent mean ± standard deviation (error bars) from 4 experiments (n = 4 wells/group/experiment). P values were calculated using Mann-Whitney test; **** = P < 1×104 and ** = P < 0.01 relative to no (0 ng/ml of) TNFα.
Osteoclasts use CD200 as costimulatory signal to induce TcREG
Antigen presenting cells typically activate CD8 T-cells through three signals: antigen presented in the context of MHC class-I, a co-stimulatory signal, and finally a polarization signal that determines the effector phenotype of the T-cell. We have previously shown that osteoclasts cross-present antigens, and antigen presentation is required for TcREG induction (7,8). Here we wanted to identify the co-stimulatory signal provided by osteoclasts and to test if pro-inflammatory cytokines would regulate the expression of this molecule(s).
The most common and well-studied costimulatory signals on antigen-presenting cells are CD80 and CD86. As CD80/CD86 double knockout mice are commercially available, we generated osteoclasts from these mice. The CD80/CD86 null osteoclasts (as well as wild-type controls) were then used to prime OT-I T-cells in the presence and absence of antigen. We found that the osteoclasts derived from CD80/CD86−/− mice were able to induce TcREG as effectively as wild-type controls (Fig. 4A). OT-I CD8 T-cells activated by CD80/CD86 null osteoclasts produced IL-6, IL-2 and IFN-γ to levels indistinguishable from wild-type mice (Fig. 4B). Having ruled out CD80/CD86 as the costimulatory molecules, we queried our osteoclast microarray dataset (6) for other costimulatory molecules expressed by osteoclasts. We found that osteoclasts express CD200 (up regulated 8.2 ± 1.2 fold relative to pre-osteoclasts; P ≤ 10−4) and c-Mer (upregulated 4.4 ± 0.9 fold; P ≤ 0.008). We chose to test the role of CD200 based on the phenotype of CD200−/− mice and the reported role for CD200 in regulating TREG (52–54). Treatment of OT-I CD8 T-cells with (soluble) CD200-Fc prior to co-culturing with osteoclasts impaired FoxP3 induction (Fig. 4C). In contrast, treatment of the OT-I CD8 T-cells with a control IgG1-Fc had no effect on FoxP3 induction (Fig. 4C). Consistent with a previous study (55), quantitative real-time PCR (qPCR) shows that CD200 is not expressed in osteoclast precursors but is expressed in mature osteoclasts (i.e. was induced by RANKL in the precursors; Fig. 4D). CD200 mRNA expression was not affected by treatment of osteoclasts with TNFα or IL-17 (Fig. 4D). These results show that osteoclasts use CD200, which is expressed in mature osteoclasts, as a costimulation signal for FoxP3 induction.
Figure 4. CD200 but not CD80/CD86 are used by osteoclasts as costimulatory signal to T-cells.

A. Osteoclasts from CD80/CD86−/− can induce TcREG: Osteoclasts derived from bone marrow cells of CD80/CD86 double knockout mice were capable of inducing TcREG to a similar extent to wild-type (WT) derived osteoclasts. B. TcREG produce similar levels of cytokines when primed by WT or double-knockout osteoclasts: The levels of cytokines produced by TcREG induced by WT osteoclast and CD80/CD86 double knockouts were indistinguishable. Taken together, these results indicate that CD80/CD86 are not used for costimulation of CD8 T-cells by osteoclasts. C. Blockade by CD200 decreases TcREG induction: Soluble CD200-Fc added to OT-I CD8 T-cells prior to coculturing with osteoclasts blocked FoxP3 induction. D. CD200 expression is not altered by TNFα or IL-17: CD200 expression is not detected in osteoclast precursors but is expressed in mature osteoclast. Recombinant murine TNFα (20 ng/ml) or IL-17A (10 ng/ml) had no effect on CD200 expression in mature osteoclasts. The results represent mean ± standard deviation (error bars) from 4 experiments (n = 4 mice/group/experiment). P values were calculated using Mann-Whitney test; **** = P < 1×104 and ** P < 0.01.
Osteoclasts induce TcREG using the Notch ligand DLL4
The most well characterized inducer (i.e. polarizing signal) of FoxP3 in T-cells is TGFβ. We have previously shown that neutralization or addition of TGFβ has no effect on the induction of TcREG (8). We therefore sought to identify other pathways that can regulate the FoxP3 promoter in T-cells. A number of previous studies have identified that Notch signaling contributes to FoxP3 induction (56–59). To test if Notch signaling is important for TcREG induction by osteoclasts, we initially used the γ-Secretase inhibitor DAPT. Ligation of the Notch receptor by its ligand leads to cleavage of Notch by γ-secretase to release Notch intra-cellular domain (60). Inclusion of 10 μM DAPT in co-cultures of osteoclasts and OT-I CD8 T-cells completely abrogated FoxP3 induction (Fig. 5A). Next, we identified the Notch ligands expressed in osteoclasts using reverse-transcription followed by PCR. We found that of the five Notch ligands encoded in the mouse genome, mature osteoclasts express: Jagged (Jag)1, Jag2, Delta-like (DLL)1 and DLL4 (Fig. 5B). Of the four, the osteoclast-precursors (bone marrow cells treated with M-CSF but not RANKL) express all of the ligands except DLL4 (data not shown and Fig. 5C). Surprisingly, DLL4 transcription was significantly repressed after culturing mature osteoclasts overnight in 20 ng/ml TNFα or 10 ng/ml IL-17 (Fig. 5C). To assess the role of DLL4 in TcREG induction we added (soluble) DLL4-Fc to OT-I CD8 T-cells prior to co-culturing with osteoclasts. DLL4-Fc effectively abrogated TcREG induction by osteoclasts (data not shown). To assess the role of DLL4 in vivo we performed the in vivo induction experiment as described in Fig. 1: (polyclonal) GFP− CD8 T-cells from FoxP3eGFP reporter mice (44) were purified by cell sorting and transferred into OT-II Rag−/− mice. Two hours prior to RANKL treatment, DLL4-Fc (30 mg/kg given intravenously) or a control IgG1-Fc, were injected intravenously into the recipient mice. Low-dose RANKL (0.125 mg/kg) was then administered on two consecutive days. No induction of TcREG was observed in mice receiving DLL4-Fc (Fig 5D). These results demonstrate that osteoclasts use Notch ligand DLL4 to signal into CD8 T-cells to induce the expression of FoxP3.
Figure 5. Notch signaling by ligand DLL4, expressed on osteoclasts, induces TcREG in vitro.

A. Inhibition of γ-secretase blocks TcREG induction by osteoclasts: DAPT, a γ-secretase inhibitor, dissolved in DMSO was used to test the role of Notch signaling in the induction of TcREG. 10 μM DAPT completely inhibited TcREG induction. Data represents four independent experiments with four wells/group/experiment. B. Notch ligands expressed in mature osteoclasts: RT-PCR was used to determine which Notch ligands are expressed on osteoclasts and CD8 T-cells. Of the six Notch ligands encoded in the mouse genome, mature osteoclasts express Jagged (Jag)-1, Jag-2, Delta-like (DLL)-1 and DLL4. The CD8 T-cells also express Jag2 and DLL-1. C. RANKL induces DLL4 expression while IL-17 and TNFα represses it: Of these four Notch ligands expressed in mature osteoclasts, only DLL4 was absent in the osteoclast precursors by qPCR. DLL4 expression was repressed in mature osteoclasts in the presence of recombinant murine 20 ng/ml TNFα and 10 ng/ml IL-17A. D. Blockade of DLL4 inhibits TcREG induction in vivo: Soluble DLL4-Fc, but not a control IgG1-Fc, administered 2 hours prior to RANKL administration blocked TcREG induction in vivo. The design of this in vivo induction experiment was identical to that used in Fig. 1B and 1C. E. DLL4 acts as switch in response to RANKL levels: Addition of RANKL increased the expression of DLL4 even in the presence of 20 ng/ml TNFα. F. RANKL addition in presence of TNFα restores TcREG induction by osteoclasts: Concomitant with the expression of DLL4, addition of RANKL also restores induction of TcREG in the presence of 10 (solid line) or 20 ng/ml (dashed line) TNFα. Statistical comparisons (P values) are relative to 25 ng/ml RANKL and 20 ng/ml TNFα treatment. ANOVA indicates that 90.7% of the variance was accounted for by RANKL treatment (P < 0.0001), and 1.83% of the variance by TNFα treatment (P = 0.0002). The interaction term accounted for 1.6% of the variance (P = 0.0147) indicating that the effect of RANKL was dominant at both concentrations of TNFα tested. The results represent mean ± standard deviation (error bars) from three independent experiments with 4 wells/group/experiment. P values were calculated using Mann-Whitney test; **** = P < 1×104; *** = P < 0.001; ** P < 0.01 and NS = not statistically significant.
DLL-4 expression is repressed in the presence of TNFα and IL-17 but can be de-repressed by RANKL
Having identified that DLL4 was required for osteoclasts to induce Foxp3 expression in CD8+ T cells we next tested if DLL-4 expression was affected by the pro-inflammatory cytokines. As RANKL induced DLL4 in osteoclast precursors, we hypothesized that adding additional RANKL may overcome the repression of DLL4 by TNFα. To assess the reversibility of DLL4 expression, mature osteoclasts were cultured overnight in 50 ng/ml RANKL and 20 ng/ml TNFα, and then additional RANKL was added to the culture media (while maintaining 20 ng/ml TNFα). Indeed, addition of RANKL overcomes (Fig. 5E; within 24 hours) the repression of DLL4 by TNFα as assessed by qPCR. Based on this result, we tested the ability of RANKL to restore induction of TcREG by osteoclasts. The experiment was performed as above, but in this case FoxP3 induction in OT-I CD8 T-cells in the presence of OVA was measured at either 10 ng/ml or 20 ng/ml TNFα (Fig. 5F). Consistent with restored expression of DLL4 on osteoclasts by RANKL the induction of TcREG was also restored in the presence of TNFα. Our results indicate that TNFα repressed DLL4 expression and that addition of RANKL could de-repress the effect of TNFα leading to increased DLL4 expression and to increased TcREG induction by osteoclasts.
Low dose RANKL induces functional TcREG in ovariectomized mice
Next we tested the ability of RANKL to induce TcREG in ovariectomized mice as proof of principle for de-repression by increased levels of RANKL, in pro-inflammatory environment. Here again, the dose of RANKL that induced the highest levels of TcREG in vivo was used (Fig. 1A). Low dose RANKL (ldRL = 0.125 mg/kg) was administered two weeks after ovariectomy. After 10 days of RANKL administration TcREG levels increased by an average of 3.6-fold relative to untreated mice (Fig. 6A). We administered low-dose RANKL two weeks post-ovariectomy because peak bone loss occurs in the tibia in rodents at this time (61,62). Additionally, effector T-cell levels also peak two-weeks post-ovariectomy (63,64). To determine if the induced TcREG are functional, bone resorption and bone formation rates in these (low-dose) RANKL-treated mice was assessed. As we have previously established that ex vivo generated OC-iTcREG limit bone resorption, increase bone mass, decrease activated effector T-cells, and increase bone formation and mineral apposition rates (10), we used this treatment as a benchmark in these experiments. Obtaining the same effect by low-dose RANKL and TcREG also indicates that the effect on bone parameters is, in fact, due to the effect of TcREG.
Figure 6. Low-dose RANKL induces functional TcREG in ovariectomized mice.

A. Induction of TcREG in the bone marrow of ovariectomized mice in response to low-dose RANKL: Intraperitoneal administration of low dose RANKL (RL; 0.125 mg/kg) induced TcREG in the bone marrow of ovariectomized mice. Representative FACS contour plots are shown on the first and second panel and the distribution (N= 8 mice per group) is shown in the third panel. B. TcREG induction in ovariectomized mice suppresses bone resorption by lowering osteoclast numbers in ovariectomized mice: Relative to sham-operated (S) mice the serum CTX levels increased in ovariectomized mice, but decreased in groups of ovariectomized mice treated with low-dose RANKL (RL), Zoledronate (ZA) or treated by adoptive-transfer of ex vivo generated TcREG (TcREG). The decrease in serum CTX by TcREG is mediated by decreasing numbers of osteoclasts per area of bone surface (N.OC/BS; center panel) and is reflected in the decrease of bone surface occupied by osteoclast surface (OC.S/BS; right panel). Representative images are shown below. C. TcREG induction in ovariectomized mice increases bone volume and bone density: The tibias of groups of mice were evaluated by μCT to determine ratio of bone volume to total volume (BV/TV; left panel) and bone mineral density (BMD; right panel). Representative images from proximal tibia are shown. D. TcREG induction in ovariectomized mice increases bone formation rate: Low-dose RANKL treatment also increased mineral apposition rate (left panel) and bone formation rate (right panel) to a greater extent than Zoledronate and ex vivo generated TcREG. Representative images from the double-labeled femur (calcein green and alizarin red) from each group are shown. Arrows are shown to emphasize the distance between dyes. E. TcREG induced by low-dose RANKL treated mice have immunosuppressive function: Low-dose RANKL and adoptive transfer of ex vivo generated TcREG decreased proinflammatory TEFF (gated on lymphocyte side-scatter/forward-scatter, then CD3 and CD45 double positive, and CD3 and CD44 double positive) cells in the bone marrow. The results represent mean ± standard deviation (error bars) from 4 experiments (n = 4 mice/group/experiment). P values were calculated using Mann-Whitney test; **** = P < 1×10−4; *** = P < 1×10−3, ** = P < 0.01 * = P < 0.05 and NS = not statistically significant.
Ovariectomized mice treated with low-dose RANKL had lower levels of serum CTX compared to mice treated with OC-iTcREG (Fig. 6B left panel). We also observed fewer osteoclasts per unit area of bone surface (Fig. 6B center panel) and less bone surface was occupied by osteoclasts (Fig. 6B right panel). The low-dose RANKL treated mice also had increased bone volume (BV/TV), increased bone mineral density (Fig. 6C) and improved trabecular bone parameters (Fig. S1) relative to the adoptively transferred OC-iTcREG and ZA treated mice. Finally, the low-dose RANKL treated mice also had increased bone formation and mineral apposition rates over the adoptively transferred OC-iTcREG treated mice (Fig. 6D). Ovariectomy increases proinflammatory effector T-cells (TEFF; CD3+ CD44+) in the bone marrow (Fig. 6E). In contrast to ZA treatment, low-dose RANKL decreased the levels of TEFF in the bone marrow (Fig 6E). The suppression of TEFF production is consistent with the regulatory T-cell activity of osteoclast-induced TcREG (7). Together, these results indicate that TcREG induced by low-dose RANKL was a more effective treatment than adoptive transfer of OC-iTcREG and than the bisphosphonate ZA.
The effect of low-dose RANKL is mediated through TcREG
As adoptive transfer of TcREG and low-dose RANKL give the same phenotype viz. decrease in bone resorption, increase bone formation, and decrease in TEFF in the bone marrow, these results support our hypothesis that ovariectomy leads to a deficit in TcREG numbers. Nonetheless, to directly confirm that low-dose RANKL mediates its effect in ovariectomized mice through TcREG we used mice that lack CD8 T-cells (β2M−/−). In the absence of CD8 T-cells, TcREG cannot be generated and therefore we anticipated that low-dose RANKL would have no effect on bone resorption, (CD44+) effector T-cells, or bone formation as observed in wild-type mice. Our results show that relative to sham-operated mice ovariectomized β2M−/− mice treated with low-dose RANKL administration had increased levels of effector T-cells (Fig. 7A) and bone resorption (Fig. 7B). The increased bone loss in ovariectomized mice and mice treated with low-dose RANKL were corroborated by μCT (Fig. 7C and D). Furthermore, as β2M−/− mice have an intact CD4 T-cell compartment, our results show that (FoxP3+ CD4) TREG are not able to substitute for the loss of TcREG, indicating a unique role for TcREG.
Figure 7. The anabolic effect of low-dose RANKL treatment in ovariectomized mice is mediated through CD8 T-cells.

To confirm that decrease in bone erosion and effector T-cell levels is mediated via TcREG, β2M−/− mice lacking CD8 T-cells, were sham-operated (S; n=6), ovariectomized (OVX; n=6) and ovariectomized then treated with low-dose RANKL (ldRL = 0.125 mg/kg; n=6). In some experiments mice are also treated with high-dose RANKL (hdRL = 1 mg/kg). A. Low-dose RANKL treatment of ovariectomized mice lacking CD8 T-cells does not decrease TEFF: Ovariectomy of β2M−/− mice increased effector T-cells (CD3+ and CD44+) relative to sham-operated (indicated by S). However, unlike treatment of CD8 T-cell-replete mice, no decrease in levels of effector T-cells was observed due to low-dose RANKL (ldRL) treatment in β2M−/− mice. B. Low-dose RANKL treatment of ovariectomized mice lacking CD8 T-cells does not decrease bone resorption: Bone resorption levels, as assayed by serum CTX, increase in CD8 T-cell (β2M−/−) deficient ovariectomized mice relative to sham-operated mice. A modest increase in serum CTX is observed in ovariectomized β2M−/− mice treated with low-dose RANKL. C. Low-dose RANKL treatment of ovariectomized mice lacking CD8 T-cells does not increase bone volume or bone density: Ovariectomy leads to significant levels of bone loss relative in β2M−/− mice. Low-dose RANKL leads to further decrease in bone volume (BV/TV) and bone mineral density (BMD) in β2M−/− mice, indicating that CD8 T-cells in WT mice protect against bone loss. D. Representative μCT renderings for panels C and E. E. Low-dose RANKL treatment of ovariectomized mice is bone anabolic but not in mice lacking CD8 T-cells: In WT mice low-dose RANKL is anti-resorptive in ovariectomized mice but promotes bone loss in estrogen-replete mice. Treatment with high-dose RANKL (1mg/kg; n= 6 mice per group) leads to bone resorption in both estrogen-deficient and replete mice. In CD8 T-cell-deficient mice low dose RANKL leads to bone loss in both estrogen-replete (Sham) and ovariectomized (OVX) mice. Percentage of bone volume lost (or gained; BV/TV) relative to reference state (untreated sham surgery or ovariectomy - indicated by horizontal dashed line) is also given. The results represent mean ± standard deviation (error bars). P values were calculated using Mann-Whitney test; *** = P < 0.001 and * = P < 0.05. F. A model of the negative feedback loop between osteoclast-induced TcREG: In ovariectomized mice increased levels of proinflammatory cytokines TNFα and IL-17 suppress DLL4 expression needed for TcREG induction (left panel). In the absence of TcREG and in the presence of the proinflammatory cytokine the activated osteoclasts have increased bone resorption leading to osteoporosis. Treatment with low-dose RANKL (right panel) reverses the repression of DLL4 in ovariectomized mice leading to induction of TcREG by osteoclasts to engage the negative feedback loop. Induction of TcREG not only limits osteoclastogenesis but also reduced the proinflammatory effector T-cells. The reduction in TEFF leads to increased bone formation rate.
Finally, as all doses of RANKL induce TcREG (Fig. 1A), we tested if high dose of RANKL (e.g. 1 mg/kg) provides a bone anabolic effect in ovariectomized mice. Our data shows that high doses of RANKL promote bone resorption in both estrogen replete and deficient mice and in CD8 deficient mice relative to low-dose RANKL (Fig. 7E). In this comparison, it is noteworthy that β2M−/− mice lost significant levels of bone by ovariectomy alone (75% in β2M−/− mice versus 28% in WT mice), indicating that endogenous TcREG (i.e. not induced by low-dose RANKL treatment) are partially successful in limiting bone loss. This result is consistent with our hypothesis that the abundance of endogenous TcREG is insufficient relative to the abundance of osteoclasts in estrogen-depleted animals. Low-dose RANKL was only effective in limiting bone loss through TcREG because this effect is only observed in CD8 T-cell-sufficient mice. High-dose RANKL activates osteoclasts and induces TcREG in wild-type mice, but the TcREG induction is apparently insufficient to limit bone loss and counter the effect of RANKL on the osteoclasts. These results show that RANKL has a biphasic effect: it activates osteoclasts to induce TcREG at low doses, but at high doses RANKL overrides the limiting effect of the TcREG. Our results indicate that TcREG act as homeostatic buffering system to “smooth out” variations in RANKL levels to limit bone loss. Increasing the levels of TcREG by RANKL pulse can compensate and restore balance from bone loss to bone formation.
Discussion
All organisms need to maintain physiological stability to survive changes in their environment. A number of mechanisms have evolved to achieve this physiological stability, chiefly positive and negative feedback loops. Our studies have revealed such a negative feedback loop between osteoclasts and CD8 T-cells that appears to be important for bone and immune homeostasis (9). Our study demonstrates that TcREG induction, while not critical for globally regulating the aberrant activation of self-reactive T-cells, are important in local immune suppression.
Here we used a low-dose of RANKL, which induces the highest proportion of TcREG in bone marrow (Fig. 1) to induce TcREG in ovariectomized mice. We show that the low-dose RANKL-induced TcREG are functional: they limit bone loss in the estrogen-depleted mice. As ex vivo generated TcREG (produced under a non-inflammatory environment) also limit bone loss, are immunosuppressive and increase bone formation, our results indicate that the overall loss of TcREG induction contributes to the observed osteoporosis. Furthermore, as TcREG adoptive transfer recapitulates the same effect as the induced TcREG on bone and the immune system indicates that other cell types contribute minimally, if at all.
To understand the reason for the lack of TcREG induction, we examined the effect of TNFα and IL-17 on TcREG induction by osteoclasts in vitro. IL-17, expressed by TH17 has been previously shown to increase bone loss (65). Inhibition of IL-17 signaling has also been shown to ameliorate bone loss in ovariectomized mice (66,67). Similarly, TNFα increases in ovariectomized mice (63,64,68) and disruption of TNFα signaling protects against bone loss post-ovariectomy(69). Consistent with lack of increase of TcREG in ovariectomized mice, TcREG induction by osteoclasts was inhibited in the presence of TNFα or IL-17 in culture. These results indicate that the proinflammatory cytokines increase bone resorption, but repress TcREG induction by osteoclasts. To understand the mechanism for the loss of TcREG induction, we examined the signals that osteoclasts provide to CD8 T-cells. We have previously demonstrated that antigen presentation is required for TcREG induction (7,8). Here we identified CD200 as a costimulatory molecule used by osteoclasts to induce TcREG (Fig. 4). While CD200 is induced by RANKL, the expression levels of CD200 were not affected by TNFα or IL-17. CD200 has been shown to decrease bone mass via its effect on osteoclasts (55). Moreover, and consistent with our results with TcREG induction, CD200 knockout mice are also more susceptible to bone loss in the collagen-induced arthritis model (70).
Notch signaling has been previously been identified in impacting FoxP3 expression in T-cells (57,58,71). Therefore, we tested for the role of Notch signaling in TcREG induction. We identified that DLL4, expressed on mature osteoclasts but not in osteoclast precursors, was required for FoxP3 expression in CD8 T-cells. As Notch signaling plays an important role in osteoclast (72,73) and osteoblast differentiation (74) these results will be important for correctly interpreting the role of Notch ligand-receptor interactions in bone remodeling (75). Blockade of DLL4 abrogated TcREG induction in vivo, and DLL4 expression was repressed in the presence of IL-17 and TNFα, which concomitantly suppressed TcREG induction by osteoclasts. Further, adding back RANKL reversed DLL-4 repression and restored TcREG induction by osteoclasts (Fig 7F). Thus, TH17 could limit the ability of osteoclasts to induce TcREG by reducing the expression of DLL4, an effect moderated by the addition of RANKL. A genetic test for the requirement of DLL4 in inducing TcREG will require generation of osteoclast-specific DLL4 deletion.
It is remarkable that administering low-dose RANKL leads to reduced bone loss and increased bone formation because RANKL is exclusively believed to be a pro-resorptive. This result could not be predicted or understood in the absence of the knowledge of the osteoclast-TcREG feedback loop elucidated here. Low-dose RANKL is anabolic because it maximally induces TcREG and engages the feedback loop, but also because TcREG reduce the proinflammatory TEFF. Thus, low-dose RANKL induction of TcREG not only decreases the number of osteoclasts but also reduces, or eliminates, the production of the cytokines that drive osteoclast activity (Fig. 7F).
RANKL is produced by osteoblasts and osteocytes in both a membrane-bound and soluble form (76,77). To regulate osteoclast activity and numbers these cells also secrete osteoprotegrin (OPG), which binds to RANKL and competes to prevent RANK (receptor) activation. Assessing the levels of biologically active RANKL in vivo therefore is challenging because it is neither accurate nor precise. The situation in osteoporosis is further complicated by presence of pro-resorptive cytokines that directly affect both the osteoclasts activities: bone resorption and activation of CD8 T-cells. Nonetheless, it has been shown that reducing RANKL levels is beneficial by anti-RANKL therapies: Prolia (Denosumab) (78), and by OPG (79) for postmenopausal osteoporosis. However, this raises the question: if there are significant level of endogenous RANKL already produced postmenopause, how is a small change in RANKL level sensed by osteoclasts to induce DLL4 expression? One possible explanation is that the system is finely balanced in osteoporosis, such that low-dose RANKL can mediate its affect on osteoclasts. Alternatively, delivering RANKL as a pulse is important. For example, parathyroid hormone (PTH) demonstrates differences in how it is delivered: whereas, intermittent doses of PTH are anabolic, continuous exposure lead to bone loss (80). Additional studies are needed to understand this phenomenon.
While RANKL, TNFα and IL-17 all promote bone resorption, RANKL induces TcREG induction to limit bone loss (Fig. 7F). In contrast, TNFα and IL-17 suppress (at the transcriptional level) the expression of DLL4 in osteoclasts, one of the signals required for TcREG induction. Therefore, DLL4 unexpectedly acts as a switch for TcREG induction in osteoclasts in response to TNFα or RANKL. Consistent with the in vitro observations we observed an increase in TcREG levels in ovariectomized mice treated with low dose RANKL. Nonetheless, at doses above 0.5 mg/kg (Fig. 1A and 7E) or prolonged (> 5 days) dosing RANKL promotes bone resorption in estrogen-replete (81) and ovariectomized mice (Fig. 7E). Indeed, whereas low dose RANKL may be in the physiological range, high doses (i.e. 1 mg/kg) used to promote bone resorption are super-physiological (in the sense that this dose exceeds RANKL levels experienced by an animal during life). This work is the first demonstration of RANKL having a biphasic response. There are a number of demonstrated examples in biology (82) of mediators that produce a biphasic response.
Regulatory T-cells (TREG) are a subset of CD4+ T cells that play a critical role in negatively regulating self-reactive T-cells and in resolving inflammatory responses. It is well documented that a reduction in the number and/or function of TREG causes the breakdown of immunological tolerance leading to lymphoproliferation and autoimmune diseases (11,17,26). However, it is not clear why TREG fail to control inflammation in some individuals that develop autoimmune diseases. One reason suggested for this loss of tolerance is due to TREG instability (or more explicitly conversion of ex-TREG to TH17 cells (83)) when exposed to an inflammatory environment (84). Our inquiry of why TcREG fail to suppress osteoclast activity and allow osteoporosis to develop followed a similar line of investigation. Our results indicate that inflammatory cytokines do not lead to defects in TcREG, but suppress induction of TcREG by osteoclasts (i.e. the antigen-presenting cells) through regulating DLL4 expression. By extension, our results are consistent with the emerging paradigm that inflammatory cytokines (i.e. induced by adjuvant or PAMPS) affect the antigen-presenting cells, and not the TREG, to tip the balance from induction of tolerance towards immunity (85,86). Further studies are needed to determine the mechanisms underlying the effect on antigen-presenting cells for TREG activation.
Our laboratory was first to establish that murine osteoclasts have an antigen presentation activity and prime CD8 T-cells (7). Li et al. have shown that human peripheral blood-derived osteoclasts can also prime T-cells (87). This study is the first in vivo demonstration that osteoclasts can induce FoxP3+ CD8 T-cells and to verify these TcREG, although induced under inflammatory conditions are functional in that they are immunosuppressive and anti-resorptive. These studies also, therefore establish a proof-of-concept that induction of TcREG can be used for the treatment of osteoporosis in a well-accepted murine model of human postmenopausal osteoporosis (88).
Summarizing, we have shown that low doses of RANKL activate osteoclasts to induce TcREG. We show that in the presence of TNFα and IL-17, TcREG induction is suppressed. We have identified CD200 and DLL4 as costimulatory and differentiation signals respectively, used by osteoclasts to induce TcREG. Furthermore, we demonstrate that TNFα and IL-17 suppress DLL4 expression, and thus providing a plausible mechanism for why despite increased activity of the osteoclasts in ovariectomized mice, TcREG levels are not increased. Thus, in vitro DLL4 acts as a switch that responds to TNFα and IL-17 to turn off TcREG induction. RANKL turns on DLL4 expression and concomitantly TcREG induction. Finally, we demonstrate that low dose RANKL induces functional TcREG in ovariectomized mice leading to lower bone resorption, increased bone mass and density and increased formation of new bone. Thus, low dose RANKL has the potential to be a novel therapy to treat postmenopausal osteoporosis and perhaps other inflammatory bone erosion diseases. Induction of TcREG offers an advantage over anti-RANKL and bisphosphonate therapy because it not only inhibits bone resorption and promotes new bone formation, but because as we show TcREG also decrease the levels of pro-inflammatory effector T-cells in ovariectomized mice [this study and (10)] and have the potential to restore immune homeostasis as well.
Supplementary Material
Acknowledgments
We are grateful for assistance provided by Department of Comparative Medicine, in particular by Erin Touchette and Dr. Cheri West. We thank Dr. Steve Teitelbaum for providing the GST-RANKL bacterial expression system, as well as for discussion and advice. Crystal Idleburg is acknowledged for preparing slides for histomorphometry. Washington University Musculoskeletal Research Core (NIH P30 AR057235) provided partial support for these studies. Research reported in this study was partially supported by National Institute of Arthritis and Musculoskeletal and Skin Disease of the NIH under Award Number RO1AR064821 to R.A.
Footnotes
Conflict Statement: The authors have declared that no conflict of interest exists.
Author Contributions: R.A. and Z.B. designed the experiments with advice from D.V.N. and R.D.P. Experiments were carried out by Z.B., S.N., J.K., E.V.S., A.C., and R.A. Readouts of bone histomorphometry and μCT was collected and analyzed by C.Y. or J.L.D. who were blinded to treatments. Z.B., D.V.N., R.D.P. and R.A. interpreted the experiments. R.A. wrote the manuscript with input from Z.B, D.V.N and R.D.P.
References cited
- 1.Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408(6812):535–536. doi: 10.1038/35046196. [DOI] [PubMed] [Google Scholar]
- 2.Rauner M, Sipos W, Thiele S, Pietschmann P. Advances in osteoimmunology: pathophysiologic concepts and treatment opportunities. Int Arch Allergy Immunol. 2013;160(2):114–125. doi: 10.1159/000342426. [DOI] [PubMed] [Google Scholar]
- 3.Schett G. Osteoimmunology in rheumatic diseases. Arthritis Res Ther. 2009;11(1):210. doi: 10.1186/ar2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takayanagi H. Inflammatory bone destruction and osteoimmunology. J Periodontal Res. 2005;40(4):287–293. doi: 10.1111/j.1600-0765.2005.00814.x. [DOI] [PubMed] [Google Scholar]
- 5.Takayanagi H. New developments in osteoimmunology. Nat Rev Rheumatol. 2012;8(11):684–689. doi: 10.1038/nrrheum.2012.167. [DOI] [PubMed] [Google Scholar]
- 6.Kiesel J, Miller C, Abu-Amer Y, Aurora R. Systems level analysis of osteoclastogenesis reveals intrinsic and extrinsic regulatory interactions. Dev Dyn. 2007;236(8):2181–2197. doi: 10.1002/dvdy.21206. [DOI] [PubMed] [Google Scholar]
- 7.Kiesel JR, Buchwald ZS, Aurora R. Cross-presentation by osteoclasts induces FoxP3 in CD8+ T cells. J Immunol. 2009;182(9):5477–5487. doi: 10.4049/jimmunol.0803897. [DOI] [PubMed] [Google Scholar]
- 8.Buchwald ZS, Kiesel JR, DiPaolo R, Pagadala MS, Aurora R. Osteoclast Activated FoxP3(+) CD8(+) T-Cells Suppress Bone Resorption in vitro. PLoS ONE. 2012;7(6):e38199. doi: 10.1371/journal.pone.0038199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchwald ZS, Aurora R. Osteoclasts and CD8 T Cells Form a Negative Feedback Loop That Contributes to Homeostasis of Both the Skeletal and Immune Systems. Clin Dev Immunol. 2013;2013:Article ID 429373. doi: 10.1155/2013/429373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchwald ZS, Kiesel J, Yang C, DiPaolo R, Novack D, Aurora R. Osteoclast-induced Foxp3+ CD8 T-Cells Limit Bone Loss in Mice. Bone. 2013;56:163–173. doi: 10.1016/j.bone.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4(4):337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 12.Allez M, Brimnes J, Dotan I, Mayer L. Expansion of CD8+ T cells with regulatory function after interaction with intestinal epithelial cells. Gastroenterology. 2002;123(5):1516–1526. doi: 10.1053/gast.2002.36588. [DOI] [PubMed] [Google Scholar]
- 13.Banham AH, Powrie FM, Suri-Payer E. FOXP3+ regulatory T cells: Current controversies and future perspectives. Eur J Immunol. 2006;36(11):2832–2836. doi: 10.1002/eji.200636459. [DOI] [PubMed] [Google Scholar]
- 14.Brimnes J, Allez M, Dotan I, Shao L, Nakazawa A, Mayer L. Defects in CD8+ regulatory T cells in the lamina propria of patients with inflammatory bowel disease. J Immunol. 2005;174(9):5814–5822. doi: 10.4049/jimmunol.174.9.5814. [DOI] [PubMed] [Google Scholar]
- 15.Colovai AI, Mirza M, Vlad G, Wang S, Ho E, Cortesini R, Suciu-Foca N. Regulatory CD8+CD28− T cells in heart transplant recipients. Hum Immunol. 2003;64(1):31–37. doi: 10.1016/s0198-8859(02)00742-5. [DOI] [PubMed] [Google Scholar]
- 16.Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. 2010;67(5):625–638. doi: 10.1002/ana.21944. [DOI] [PubMed] [Google Scholar]
- 17.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 18.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445(7129):771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 19.Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, Thompson TC, Old LJ, Wang RF. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13(23):6947–6958. doi: 10.1158/1078-0432.CCR-07-0842. [DOI] [PubMed] [Google Scholar]
- 20.Meloni F, Morosini M, Solari N, Passadore I, Nascimbene C, Novo M, Ferrari M, Cosentino M, Marino F, Pozzi E, Fietta AM. Foxp3 expressing CD4+ CD25+ and CD8+CD28− T regulatory cells in the peripheral blood of patients with lung cancer and pleural mesothelioma. Hum Immunol. 2006;67(1–2):1–12. doi: 10.1016/j.humimm.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Niederkorn JY. Emerging concepts in CD8(+) T regulatory cells. Curr Opin Immunol. 2008;20(3):327–331. doi: 10.1016/j.coi.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shevach EM. From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity. 2006;25(2):195–201. doi: 10.1016/j.immuni.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 24.Stephens GL, Andersson J, Shevach EM. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. J Immunol. 2007;178(11):6901–6911. doi: 10.4049/jimmunol.178.11.6901. [DOI] [PubMed] [Google Scholar]
- 25.Rudensky A. Foxp3 and dominant tolerance. Philos Trans R Soc Lond B Biol Sci. 2005;360(1461):1645–1646. doi: 10.1098/rstb.2005.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 27.Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, Nomura T, Toda M, Takahashi T. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 28.Blair PJ, Bultman SJ, Haas JC, Rouse BT, Wilkinson JE, Godfrey VL. CD4+CD8− T cells are the effector cells in disease pathogenesis in the scurfy (sf) mouse. J Immunol. 1994;153(8):3764–3774. [PubMed] [Google Scholar]
- 29.Mayer CT, Floess S, Baru AM, Lahl K, Huehn J, Sparwasser T. CD8+ Foxp3+ T cells share developmental and phenotypic features with classical CD4+ Foxp3+ regulatory T cells but lack potent suppressive activity. Eur J Immunol. 2011;41(3):716–725. doi: 10.1002/eji.201040913. [DOI] [PubMed] [Google Scholar]
- 30.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blander JM. Signalling and phagocytosis in the orchestration of host defence. Cell Microbiol. 2007;9(2):290–299. doi: 10.1111/j.1462-5822.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- 32.Villadangos JA, Schnorrer P, Wilson NS. Control of MHC class II antigen presentation in dendritic cells: a balance between creative and destructive forces. Immunol Rev. 2005;207:191–205. doi: 10.1111/j.0105-2896.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki M, Jagger AL, Konya C, Shimojima Y, Pryshchep S, Goronzy JJ, Weyand CM. CD8+CD45RA+CCR7+FOXP3+ T Cells with Immunosuppressive Properties: A Novel Subset of Inducible Human Regulatory T Cells. J Immunol. 2012;189(5):2118–2130. doi: 10.4049/jimmunol.1200122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerret NM, Houlihan JL, Kheradmand T, Pothoven KL, Zhang ZJ, Luo X. Donor-Specific CD8(+) Foxp3(+) T Cells Protect Skin Allografts and Facilitate Induction of Conventional CD4(+) Foxp3(+) Regulatory T Cells. Am J Transplant. 2012;12(9):2335–2347. doi: 10.1111/j.1600-6143.2012.04120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robb RJ, Lineburg KE, Kuns RD, Wilson YA, Raffelt NC, Olver SD, Varelias A, Alexander KA, Teal BE, Sparwasser T, Hammerling GJ, Markey KA, Koyama M, Clouston AD, Engwerda CR, Hill GR, MacDonald KP. Identification and expansion of highly suppressive CD8(+)FoxP3(+) regulatory T cells after experimental allogeneic bone marrow transplantation. Blood. 2012;119(24):5898–5908. doi: 10.1182/blood-2011-12-396119. [DOI] [PubMed] [Google Scholar]
- 36.Anderson ME, Jr, Buchwald ZS, Ko J, Aurora R, Sanford T. Patients with Pediatric Obstructive Sleep Apnea Show Altered T-Cell Populations with a Dominant TH17 Profile. Otolaryngol Head Neck Surg. 2014 doi: 10.1177/0194599814521780. [DOI] [PubMed] [Google Scholar]
- 37.Siegmund K, Ruckert B, Ouaked N, Burgler S, Speiser A, Akdis CA, Schmidt-Weber CB. Unique phenotype of human tonsillar and in vitro-induced FOXP3+CD8+ T cells. J Immunol. 2009;182(4):2124–2130. doi: 10.4049/jimmunol.0802271. [DOI] [PubMed] [Google Scholar]
- 38.Savageau MA, Coelho PM, Fasani RA, Tolla DA, Salvador A. Phenotypes and tolerances in the design space of biochemical systems. Proc Natl Acad Sci U S A. 2009;106(16):6435–6440. doi: 10.1073/pnas.0809869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savageau MA. Design principles for elementary gene circuits: Elements, methods, and examples. Chaos. 2001;11(1):142–159. doi: 10.1063/1.1349892. [DOI] [PubMed] [Google Scholar]
- 40.Savageau MA. Optimal design of feedback control by inhibition. Journal of molecular evolution. 1974;4(2):139–156. doi: 10.1007/BF01732019. [DOI] [PubMed] [Google Scholar]
- 41.Sneppen K, Krishna S, Semsey S. Simplified models of biological networks. Annual review of biophysics. 2010;39:43–59. doi: 10.1146/annurev.biophys.093008.131241. [DOI] [PubMed] [Google Scholar]
- 42.Germain RN. Maintaining system homeostasis: the third law of Newtonian immunology. Nat Immunol. 2012;13(10):902–906. doi: 10.1038/ni.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nazarian A, Snyder BD, Zurakowski D, Muller R. Quantitative micro-computed tomography: a non-invasive method to assess equivalent bone mineral density. Bone. 2008;43(2):302–311. doi: 10.1016/j.bone.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 44.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178(5):2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 45.Chen T, Berenson J, Vescio R, Swift R, Gilchick A, Goodin S, LoRusso P, Ma P, Ravera C, Deckert F, Schran H, Seaman J, Skerjanec A. Pharmacokinetics and pharmacodynamics of zoledronic acid in cancer patients with bone metastases. J Clin Pharmacol. 2002;42(11):1228–1236. doi: 10.1177/009127002762491316. [DOI] [PubMed] [Google Scholar]
- 46.Weiss HM, Pfaar U, Schweitzer A, Wiegand H, Skerjanec A, Schran H. Biodistribution and plasma protein binding of zoledronic acid. Drug Metab Dispos. 2008;36(10):2043–2049. doi: 10.1124/dmd.108.021071. [DOI] [PubMed] [Google Scholar]
- 47.Maru Y, Afar DE, Witte ON, Shibuya M. The dimerization property of glutathione S-transferase partially reactivates Bcr-Abl lacking the oligomerization domain. J Biol Chem. 1996;271(26):15353–15357. doi: 10.1074/jbc.271.26.15353. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 49.Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27(3):535–545. doi: 10.1038/sj.emboj.7601984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pacifici R. T cells: Critical bone regulators in health and disease. Bone. 2010 doi: 10.1016/j.bone.2010.04.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Mei J, Gonzales L, Yang G, Dai N, Wang P, Zhang P, Favara M, Malcolm KC, Guttentag S, Worthen GS. IL-17A and TNF-alpha exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J Immunol. 2011;186(5):3197–3205. doi: 10.4049/jimmunol.1002016. [DOI] [PubMed] [Google Scholar]
- 52.Gorczynski RM, Chen Z, Hu J, Kai Y, Lei J. Evidence of a role for CD200 in regulation of immune rejection of leukaemic tumour cells in C57BL/6 mice. Clin Exp Immunol. 2001;126(2):220–229. doi: 10.1046/j.1365-2249.2001.01689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mukhopadhyay S, Pluddemann A, Hoe JC, Williams KJ, Varin A, Makepeace K, Aknin ML, Bowdish DM, Smale ST, Barclay AN, Gordon S. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host Microbe. 2010;8(3):236–247. doi: 10.1016/j.chom.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. 2008;9(9):1074–1083. doi: 10.1038/ni.1637. [DOI] [PubMed] [Google Scholar]
- 55.Cui W, Cuartas E, Ke J, Zhang Q, Einarsson HB, Sedgwick JD, Li J, Vignery A. CD200 and its receptor, CD200R, modulate bone mass via the differentiation of osteoclasts. Proc Natl Acad Sci U S A. 2007;104(36):14436–14441. doi: 10.1073/pnas.0702811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bassil R, Zhu B, Lahoud Y, Riella LV, Yagita H, Elyaman W, Khoury SJ. Notch ligand delta-like 4 blockade alleviates experimental autoimmune encephalomyelitis by promoting regulatory T cell development. J Immunol. 2011;187(5):2322–2328. doi: 10.4049/jimmunol.1100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ou-Yang HF, Zhang HW, Wu CG, Zhang P, Zhang J, Li JC, Hou LH, He F, Ti XY, Song LQ, Zhang SZ, Feng L, Qi HW, Han H. Notch signaling regulates the FOXP3 promoter through RBP-J- and Hes1-dependent mechanisms. Mol Cell Biochem. 2009;320(1–2):109–114. doi: 10.1007/s11010-008-9912-4. [DOI] [PubMed] [Google Scholar]
- 58.Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, Das P, Golde TE, Osborne BA. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. 2008;112(5):1813–1821. doi: 10.1182/blood-2008-03-144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen Z, Chen L, Hao F, Wu J. Transcriptional regulation of Foxp3 gene: multiple signal pathways on the road. Med Res Rev. 2009;29(5):742–766. doi: 10.1002/med.20152. [DOI] [PubMed] [Google Scholar]
- 60.Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66(10):1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jee WS, Yao W. Overview: animal models of osteopenia and osteoporosis. J Musculoskelet Neuronal Interact. 2001;1(3):193–207. [PubMed] [Google Scholar]
- 62.Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boyce B, Broxmeyer H, Manolagas SC. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257(5066):88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- 63.Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, Sierra O, Pacifici R. Estrogen deficiency induces bone loss by increasing T cell proliferation and lifespan through IFN-gamma-induced class II transactivator. Proc Natl Acad Sci U S A. 2003;100(18):10405–10410. doi: 10.1073/pnas.1533207100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, Pacifici R. Up-regulation of TNF-producing T cells in the bone marrow: a key mechanism by which estrogen deficiency induces bone loss in vivo. Proc Natl Acad Sci U S A. 2001;98(24):13960–13965. doi: 10.1073/pnas.251534698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deselm CJ, Takahata Y, Warren J, Chappel JC, Khan T, Li X, Liu C, Choi Y, Kim YF, Zou W, Teitelbaum SL. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem. 2012;113(9):2895–2902. doi: 10.1002/jcb.24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deselm CJ, Zou W, Teitelbaum SL. Halofuginone prevents estrogen-deficient osteoporosis in mice. J Cell Biochem. 2012 doi: 10.1002/jcb.24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, Pacifici R. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest. 2000;106(10):1229–1237. doi: 10.1172/JCI11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ammann P, Rizzoli R, Bonjour JP, Bourrin S, Meyer JM, Vassalli P, Garcia I. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J Clin Invest. 1997;99(7):1699–1703. doi: 10.1172/JCI119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290(5497):1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 71.Barbarulo A, Grazioli P, Campese AF, Bellavia D, Di Mario G, Pelullo M, Ciuffetta A, Colantoni S, Vacca A, Frati L, Gulino A, Felli MP, Screpanti I. Notch3 and canonical NF-kappaB signaling pathways cooperatively regulate Foxp3 transcription. J Immunol. 2011;186(11):6199–6206. doi: 10.4049/jimmunol.1002136. [DOI] [PubMed] [Google Scholar]
- 72.Sekine C, Koyanagi A, Koyama N, Hozumi K, Chiba S, Yagita H. Differential regulation of osteoclastogenesis by Notch2/Delta-like 1 and Notch1/Jagged1 axes. Arthritis Res Ther. 2012;14(2):R45. doi: 10.1186/ar3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283(10):6509–6518. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- 74.Regan J, Long F. Notch signaling and bone remodeling. Curr Osteoporos Rep. 2013;11(2):126–129. doi: 10.1007/s11914-013-0145-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zanotti S, Canalis E. Notch signaling in skeletal health and disease. Eur J Endocrinol. 2013;168(6):R95–103. doi: 10.1530/EJE-13-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Honma M, Ikebuchi Y, Kariya Y, Suzuki H. Regulatory mechanisms of RANKL presentation to osteoclast precursors. Curr Osteoporos Rep. 2014;12(1):115–120. doi: 10.1007/s11914-014-0189-0. [DOI] [PubMed] [Google Scholar]
- 77.Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, San Martin J, Dansey R. Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov. 2012;11(5):401–419. doi: 10.1038/nrd3705. [DOI] [PubMed] [Google Scholar]
- 78.Sutton EE, Riche DM. Denosumab, a RANK ligand inhibitor, for postmenopausal women with osteoporosis. Ann Pharmacother. 2012;46(7–8):1000–1009. doi: 10.1345/aph.1Q543. [DOI] [PubMed] [Google Scholar]
- 79.Shimizu-Ishiura M, Kawana F, Sasaki T. Osteoprotegerin administration reduces femural bone loss in ovariectomized mice via impairment of osteoclast structure and function. J Electron Microsc (Tokyo) 2002;51(5):315–325. doi: 10.1093/jmicro/51.5.315. [DOI] [PubMed] [Google Scholar]
- 80.Greenfield EM. Anabolic effects of intermittent PTH on osteoblasts. Current molecular pharmacology. 2012;5(2):127–134. [PubMed] [Google Scholar]
- 81.Tomimori Y, Mori K, Koide M, Nakamichi Y, Ninomiya T, Udagawa N, Yasuda H. Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J Bone Miner Res. 2009;24(7):1194–1205. doi: 10.1359/jbmr.090217. [DOI] [PubMed] [Google Scholar]
- 82.Kaiser J. Hormesis. Sipping from a poisoned chalice. Science. 2003;302(5644):376–379. doi: 10.1126/science.302.5644.376. [DOI] [PubMed] [Google Scholar]
- 83.Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3 T cells into TH17 cells in autoimmune arthritis. Nat Med. 2013;20(1):62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 84.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10(9):1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, Bluestone JA. Self-antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity. 2013;39(5):949–962. doi: 10.1016/j.immuni.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mayer CT, Berod L, Sparwasser T. Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front Immunol. 2012;3:183. doi: 10.3389/fimmu.2012.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood. 2010;116(2):210–217. doi: 10.1182/blood-2009-11-255026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kimmel DB. Animal models for in vivo experimentation in osteoporosis research. Osteoporosis. 1996;2:29–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.