

Abstract
Alcohol use and alcohol addiction represent dysfunctional brain circuits resulting from neuroadaptive changes during protracted alcohol exposure and its withdrawal. Alcohol exerts a potent effect on synaptic plasticity and dendritic spine formation in specific brain regions, providing a neuroanatomical substrate for the pathophysiology of alcoholism. Epigenetics has recently emerged as a critical regulator of gene expression and synaptic plasticity-related events in the brain. Alcohol exposure and withdrawal induce changes in crucial epigenetic processes in the emotional brain circuitry (amygdala) that may be relevant to the negative affective state defined as the “dark side” of addiction. Here, we review the literature concerning synaptic plasticity and epigenetics, with a particular focus on molecular events related to dendritic remodeling during alcohol abuse and alcoholism. Targeting epigenetic processes that modulate synaptic plasticity may yield novel treatments for alcoholism.
Keywords: alcoholism, epigenetics, synaptic plasticity, dendritic spines, histone deacetylases, histone acetylation, amygdala
1. Introduction
Alcohol abuse and alcoholism represent significant public health problems that impact both the individual and society as a whole [1]. Alcoholism is defined as compulsive drug seeking behavior that interferes with normal functioning and is related to various psychiatric states such as stress and anxiety [2-7]. Additionally, alcoholism and alcohol abuse are characterized by both positive and negative emotional states [4,5]. The behavioral switch between positive reinforcement (i.e., seeking a drug for its rewarding effects) and negative reinforcement (i.e., seeking a drug in order to remove the negative emotional state associated with withdrawal) is an important aspect of the cycle of alcohol addiction [4,5]. The negative affective states (e.g., anxiety, stress, and dysphoria) that drive the negative reinforcement of addiction, along with the physical manifestations of drug withdrawal, are referred to as the “dark side” of addiction [4-7].
The dark side of addiction represents a dysfunction of brain reward and emotion systems [5-7], and therefore, targeting aberrant molecular pathways in these circuitries may yield better therapeutic interventions for alcoholism and other addictive behaviors. In particular, the extended amygdala (consisting of the central nucleus of the amygdala [CeA], the bed nucleus of the stria terminalis [BNST] and a transitional zone of the nucleus accumbens [NAc] shell) [7,8] is posited to integrate stress signals in the brain with reward processing through dynamic molecular regulation leading to changes in synaptic plasticity and dendritic arborization [4,9,10]. Several molecular and cellular substrates in key brain regions play a crucial role in the development and maintenance of drug abuse and addictive behaviors [3,9-14].
Uncovering the precise molecular machinery responsible for abnormal dendritic branching and synaptic remodeling remains an important research area in the field of alcoholism. Recent evidence has implicated both genetic and epigenetic mechanisms in the control of synaptic plasticity, particularly as it relates to alcohol consumption and addiction [3,11]. In this review, we will summarize the current knowledge of molecular and epigenetic mechanisms underlying synaptic remodeling and maintenance of the “dark side” or negative affective state of alcohol addiction.
2. Synaptic plasticity and alcohol
2.1. Plasticity at the individual synapse
Drug and alcohol addiction are often characterized as a dysfunction of normal learning and memory circuits within the brain [4,11,12]. These complex processes are governed by the rapid forming and reforming of synaptic structures in particular brain regions that occur concomitantly with changes in signaling at the level of individual synapses, collectively known as synaptic plasticity [13]. At the single synapse level, synaptic transmission at a particular synapse enhances the efficacy of subsequent signaling at that same synapse in a process known as long-term potentiation (LTP) [14]. Conversely, the dampening of future synaptic transmission is called long-term depression (LTD) [15]. In addition to LTP and LTD, changes to the ultrastructure of synapses play a role in the pathogenesis of addiction through the action of several different neurotransmitter systems, particularly the mesolimbic dopamine system [16,17]. Glutamatergic and GABAergic systems also are implicated in synaptic remodeling and are affected by exposure to alcohol and drugs of abuse [17,18].
2.2. The effect of alcohol on dendritic spine formation
Modulation of signaling events at the single synapse are accompanied by the structural reorganization of neuronal dendritic spines [19], sometimes referred to as metaplasticity [16]. The observation of certain mRNA and protein transcripts, such as activity-regulated cytoskeleton-associated protein (Arc), found in recently activated synapses led to the hypothesis that specific molecular players were involved in structural synaptic remodeling [20,21]. Arc is transported to dendritic spines by actin-based motor proteins, where it modulates glutamatergic neurotransmission and structural organization [22–24]. By this process, dendrites form and re-form connections to other neurons in the surrounding area. Dendritic branching and arborization are robustly affected by many drugs of abuse, including cocaine, heroin, and alcohol [17,24], and several cellular mechanisms [1-4] have been implicated in long-lasting behavioral changes associated with drug addiction such as compulsive drug-seeking and negative affective states [4,7,12].
Alcohol exerts effects on the morphology of dendritic spines. In particular, acute alcohol administration is associated with an increase in dendritic spines in the CeA and medial nucleus of the amygdala (MeA) of rats [24]. These structural effects coincide with alcohol-induced anxiolysis and an increase in expression of brain-derived neurotrophic factor (BDNF) and Arc, gene products known to play a role in synaptic plasticity [24]. Interestingly, withdrawal from chronic alcohol exposure decreases dendritic spines and Arc and BDNF expression in the CeA and MeA, leading to anxiogenesis in rats [24,25]. Withdrawal from chronic alcohol exposure also decreases dendritic arborization in the hippocampus and the NAc [26,27]. This data mimics results from postmortem studies of human alcoholics showing decreased dendritic spine density in cortical pyramidal neurons [28]. These studies support the notion of a synaptic plasticity-driven model of alcoholism.
Acute alcohol exposure provokes anxiolysis and increased dendritic spines in the amygdala, whereas continued exposure normalizes dendritic arborization and attenuates anxiolytic-like behavior. However, withdrawal from chronic or binge ethanol consumption, and especially repeated withdrawal, causes a stark decrease in the number of dendritic spines in the amygdala and other important brain regions, accompanied by increased anxiety-like behaviors. Relapse to drinking possibly normalizes dendritic spine density (Figure 1). Notably, changes in structural plasticity are accompanied by changes in BDNF and Arc gene expression in these studies, providing a molecular framework for the structural changes seen with altered synaptic plasticity. Several studies in the field have shown that Arc directly regulates dendritic spines both in the hippocampus and amygdala [24,29]. Interestingly, increased BDNF and Arc expression leads to increased dendritic spine density in the CeA and decreased anxiety-related and drinking behavior, while decreased BDNF and Arc expression in the CeA leads to decreased dendritic spine density and increased anxiety and drinking behavior (Figure 2) [24,25,30]. This cellular mechanism provides a possible explanation for many of the behavioral consequences of alcoholism, including compulsive drug-seeking and negative affective states seen during withdrawal.
Figure 1. Effect of alcohol exposure on dendritic spines.
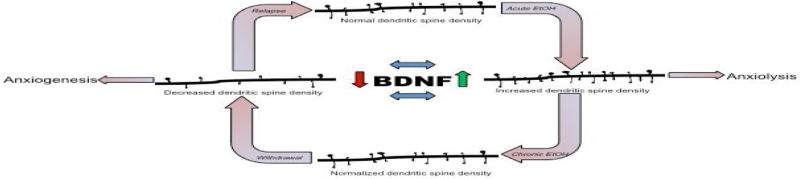
This diagram illustrates the modulation of dendritic spine density in the amygdala during various stages of alcohol (EtOH) exposure. Acute alcohol increases the density of dendritic spines, which is accompanied by marked anxiolysis. Continued alcohol exposure will normalize spine density, while withdrawal from alcohol intake causes decreased dendritic arborization and this is correlated with decreased BDNF system in the amygdala. This decrease in BDNF causes a subsequent increase in anxiety-like behaviors during alcohol withdrawal. Relapse to alcohol use possibly attenuates both the decreased spine density and anxiety-like behaviors. Diagram is based on published data from our laboratory [24,25,30].
Figure 2. Molecular players underlying alcohol-related alterations to dendritic spine density.

Brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton-associated protein (Arc) play a critical role in the induction and maintenance of structural synaptic plasticity in the amygdala. In particular, decreased BDNF and Arc expression are associated with decreased spine density, leading to an increase in anxiety and alcohol consumption. Conversely, increased expression of BDNF and Arc due to acute ethanol exposure induces spinogenesis in the amygdala and produces anxiolytic-like effects. Interestingly, decreases in dendritic spines in the central nucleus of amygdala (CeA) due to knock down of Arc is associated with increased consumption of alcohol and heightened anxiety states. Diagram derived from data published from our laboratory [24,25,30].
2.3. NMDA receptors and alcohol-dependent synaptic reorganization
The glutamatergic N-methyl-D-aspartate (NMDA) receptor is known to play an essential role in both short- and long-term activity-dependent synaptic plasticity [31–33]. These receptors act as modulators for the transmission of excitatory impulses to the postsynaptic nerve terminal. Interestingly, acute alcohol exposure potently inhibits NMDA-mediated currents, resulting in less depolarization in postsynaptic neurons [34,35]. Chronic ethanol exposure, on the other hand, increases the sensitivity of NMDA receptors to glutamate, potentiating neuronal depolarization and subsequent activation [35,36]. These effects are specific to NMDA receptors targeted to the synapse, rather than extrasynaptic receptors [37] and are localized to the amygdala in rodent models [35,36,38]. Withdrawal from alcohol produces robust anxiety-like behavior which is reversed upon administration of a glutamatergic antagonist in the amygdala [39]. This data suggests that the glutamatergic system and NMDA receptors are at least partly responsible for the anxiety-like behavior seen during alcohol withdrawal. Additionally, exposure to chronic ethanol caused a decrease in dendritic spine density in medium spiny neurons in the nucleus accumbens (NAc), which was associated with an increase in NMDA receptor NR1 subunit expression (discussed further below) [26]. Other studies showed the decrease in NAc spine density, particularly in “long thin” spines, was accompanied by a decrease in post-synaptic density-95 (PSD95)-positive and tyrosine hydroxylase (TH)-positive immunostaining in rats undergoing ethanol withdrawal [40]. This led to decreased NMDA flux and decreased LTD induction in NAc slices [40].
NMDA receptors are heteromeric molecules composed of different combinations of the subunits NR1, NR2, and NR3 [33]. During early development, immature synapses are enriched with NMDA receptors composed of NR1-NR2B heteromers. Conversely, mature synapses undergo a developmental switch to express higher levels of NR1-NR2A heteromer receptors [41–44]. NR2B-containing receptors are thought to be important for the formation of strong synaptic connections during a critical developmental period based on the observation that overexpression of this subunit increases the density of dendritic spines [45]. Activation of NMDA receptors causes downstream activation of calcium (Ca2+)/calmodulin-dependent protein kinase II (CaMKII) signaling pathways, and NR2B-containing receptors show a higher affinity for CaMKII than NR2A-containing receptors [46]. Activation of CaMKII regulates activity-dependent synaptic changes, thus providing a signaling mechanism for the developmental switch underlying synaptic maturation [47]. Hippocampal slices exposed to alcohol show selective internalization of NR2A subunits, changing the composition of NMDA receptors at the synapse to essentially pure NR2B-containing heteromers [48]. Enhanced NR1 and NR2B expression and increased mushroom-type dendritic spines were observed in vivo in postsynaptic density fractions taken from the mouse prefrontal cortex in animals exposed to ethanol chronically [49]. Chronic intermittent ethanol exposure increases NR2B without increasing NR2B phosphorylation or PSD95 [50]. Interestingly, the prefrontal cortex has been implicated in long-term learning mechanisms that lead to relapse even a longer period of abstinence [7,51]. Similar increases in NR2B-containing receptors were observed in the hippocampus of ethanol-exposed rat brain slices [52]. These receptors were associated with clusters of F-actin and PSD95, indicating an increase in dendritic spine density [52]. The increased expression of NR2B subunits also is observed in response to other drugs of abuse, such as cocaine, heroin, and nicotine [53–55].
NR2B subunits likely mediate the enhanced LTP seen in animals chronically exposed to drugs of abuse due to the higher affinity for downstream activating mechanisms. The increased responsivity of these receptors, when compared to NR2A subunits, forms a critical period for learning and synaptic plasticity in the brain, and the loss of NR2B receptor subunits may play an important role in the decreased plasticity seen in the aging brain [56,57]. The re-enrichment of synapses with NR2B subunits may hijack normal dendritic spine formation to reopen a critical period-like state. This process is hypothesized to contribute to the aberrant learning mechanisms responsible for the compulsive drug seeking and negative affective states associated with the dark side of addiction [7,12,18]. Understanding the molecular processes that cause these effects will likely lead to new approaches to treat alcohol and drug addiction.
3. Epigenetic basis of the dark side of addiction
3.1. Epigenetic modifications
Recently, epigenetics has emerged as a novel mechanism underlying the processes of synapse formation and maintenance. The term “epigenetics” defines modifications of the genome that modulate transcription and/or translation without affecting the underlying DNA sequence [58,59]. Epigenetic mechanisms include the covalent modifications of histone proteins around which DNA is wrapped (i.e., the chromatin complex) [59] and the addition of methyl groups directly to DNA, known as DNA methylation [60]. These chemical modifications to chromatin can alter the accessibility of DNA to transcriptional machinery, leading to changes in gene expression without any change to the DNA sequence [61,62].
Several histone chemical modifications [59,63] can either promote or inhibit transcription of the underlying genetic information, depending on the type of epigenetic mark and the specific residue on which it is located. For example, acetylation, mainly occurring at lysine residues, is a marker of active gene transcription [59]. Methylation of histone proteins, particularly methylation of lysine and arginine, can either promote or repress transcription, depending on the context of the modified residue. Namely, methylation of H3K4 (histone H3 lysine residue K4), H3K36, and H3K79 are associated with active transcription, while methylation of H3K9, H3K27, and H4K20 are associated with decreased transcriptional states [59,63]. These covalent modifications are regulated by a group of enzymes that function to add and/or remove specific epigenetic markers from specific histone residues. This group includes, for example, histone acetyltransferases (HATs; responsible for adding acetyl groups to histone lysine residues) and histone deacetylases (HDACs; responsible for removing acetyl groups from histone lysine residues). The language created by the dynamic interaction of these enzymes and their respective chromatin marks is enormously complex, but understanding these interactions is crucial to determining the precise regulation of gene transcription [64]. In the context of this review, it is important to note that covalent histone modifications are important for ongoing synaptic plasticity and are involved in various neurological and psychiatric disease processes [65,66].
In addition to covalent histone modifications, methyl groups can be added directly to cytosine residues within DNA (i.e., DNA methylation), usually in genomic regions rich in CpG (cytosine-phosphate-guanine) residues known as CpG islands [67,68]. DNA methylation most often causes transcriptional repression of the underlying DNA sequence via steric hindrance of the binding of transcriptional machinery and, additionally, via the recruitment of methyl-CpG-binding proteins (MeCPs) and HDACs [69,70]. DNA methylation plays a vital role in brain development, neuronal differentiation, synaptogenesis and ongoing synaptic plasticity in differentiated neurons [71–73]. Much like HATs and HDACs, the DNA methylation process is catalyzed by particular enzymes that add or remove methyl groups to DNA [74]. The DNA methyltransferase (DNMT) family is responsible for the catalytic addition of methyl groups. After this process occurs, newly methylated CpG islands are bound by members of the MeCP family of proteins which influence transcription [75,76]. The MeCP2 protein is particularly important for the regulation of synaptic plasticity and dendritic spine maturation [72,77,78].
Methylated DNA in the form of methylcytosine can be further converted to hydroxymethylcytosine by a family of enzymes known as the ten eleven translocation (TET) group [79]. The presence of hydroxymethylcytosine is enriched in the brain and appears to be associated with actively transcribed genes [80,81]. Another family of enzymes, known as growth arrest and DNA damage repair (Gadd45), participate in active demethylation of DNA and are responsible for epigenetic activation [82,83]. Notably, TET1 was recently shown to directly influence active DNA demethylation of essential synaptic plasticity genes in two different memory extinction studies [84,85]. Genetic knockout of Gadd45b in a rodent model causes lasting changes in memory formation and synaptic plasticity [86]. Taken together, these studies implicate DNA methylation and demethylation as critically important regulators of neuronal function and dendritic spine formation. In the subsequent sections, we will outline the epigenetic contribution to synaptic regulation in response to alcohol abuse and addiction.
3.2. Epigenetic regulation of the κ-opioid system by alcohol
The action of drugs of abuse is likely mediated through multiple signaling molecules and molecular cascades, many of which involve epigenetic processes. The endogenous dynorphin system is of particular importance, as binding of dynorphin to κ-opioid receptors likely mediates negative affective states seen in alcohol withdrawal [87–89]. Prodynorphin mRNA, the molecular precursor to dynorphin, is induced in the NAc following dopamine release (and, consequently, drug use) [90]. This increase is postulated to mediate feedback inhibition of dopamine release, contributing to the dysphoria and anxiety seen in the latter stages of addiction and other psychiatric disorders [91,92]. Acute ethanol administration in rats causes an increase in H3K9 acetylation and a decrease in repressive H3K27 methylation at the prodynorphin gene promoter in the amygdala, providing an epigenetic mechanism for the acute increase of prodynorphin expression [93]. Antagonism of κ-opioid receptors, the major postsynaptic target of dynorphin, curbs ethanol self-administration in alcohol-dependent rodents [94]. Interestingly, a study of human alcoholics showed an increase in DNA methylation in the prefrontal cortex at the prodynorphin gene associated with a single nucleotide polymorphism (SNP) [95].
Dynorphin is known to activate corticotrophin releasing factor (CRF), an important mediator of stress responses in the brain and the rest of the body [96,97]. Of note, CRF is a well-known modulator of synaptic plasticity and dendritic branching in the amygdala and other brain regions such as the hippocampus and the ventral tegmental area (VTA) [97,98]. CRF antagonists injected into the brain reverse the anxiogenic effect of ethanol withdrawal [99,100], and this reversal is mainly mediated by CRF neurons in the CeA [100]. CRF receptor antagonism suppresses the response to stress which is normally increased during ethanol withdrawal [101].
3.3. Epigenetic regulation of synaptic plasticity by alcohol
Recently, chromatin remodeling has emerged as a novel mechanism which could explain the effects of alcohol on anxiety [102,103]. Alcohol exposure exerts potent effects on cyclic AMP response-element binding protein (CREB), a transcription factor implicated in neuronal plasticity and cognition [2,104,105]. CREB is crucial for synaptogenesis, axonal growth, dendritic spine density, and the fine-tuning of synaptic connectivity [106–108]. Rats exposed to acute ethanol displayed anxiolysis that paralleled increases in phosphorylated CREB, CREB binding protein (CBP), acetylated histone H3 and H4, neuropeptide Y (NPY) expression in the CeA and MeA, and, perhaps most importantly, inhibition of HDAC activity in the amygdala [102]. These results indicate an open chromatin conformation in response to acute alcohol exposure. It is important to note that CBP itself acts as a histone acetyltransferase, or HAT [109]. Ethanol withdrawal after chronic ethanol treatment produces a significant decrease in phosphorylated CREB, CBP, acetylated H3 and H4, and NPY expression in the amygdala, with a parallel increase in HDAC activity and development of anxiety-like behaviors. Opposite to the acute alcohol effects, these results indicate a condensed chromatin conformation upon repeated alcohol exposure. These effects were rescued by treatment with trichostatin A (TSA), a pan HDAC inhibitor, which prevented the alcohol withdrawal related anxiety [102].
As mentioned earlier, BDNF and Arc are gene products important for synaptic plasticity, and activation of these genes lies downstream of CREB phosphorylation [110,111]. Both BDNF and Arc are induced upon acute ethanol exposure, but these signaling molecules show decreased amygdaloid function during chronic ethanol exposure and/or withdrawal [24,30]. Decreased BDNF and Arc expression are linked to increased HDAC activity, as well as decreased histone H3 acetylation and dendritic spine density in the CeA and MeA. TSA treatment returned BDNF and Arc expression to normal levels and restored dendritic spine density to control amounts [25,102]. In a recent study, TSA treatment was effective in reversing deficits in NPY expression and deficits in global and NPY-specific H3K9 acetylation in the amygdala of alcohol-preferring (P) as compared to non-preferring (NP) rats [112]. Additional research by our group found the reversal of rapid tolerance to the anxiolytic effects of ethanol, including NPY expression in the CeA and MeA, upon treatment with TSA [113]. In an ethanol-induced behavioral sensitization paradigm, treatment with sodium butyrate (NaB), an HDAC inhibitor, increased BDNF expression in the striatum [114]. These studies cleary indicate that histone modifications regulate important synaptic plasticity-associated genes in the amygdala and other brain regions, thereby modulating alcohol-related behaviors.
3.4. Direct HDAC2 regulation of dendritic spines in alcoholism and cognition
In recent studies conducted in our lab, increased innate HDAC2 isoform expression and decreased H3K9 acetylation were observed in the CeA and MeA of P versus NP rats, a well-established model used to study the genetic predisposition to alcoholism [103]. P rats displayed decreased BDNF and Arc expression in the CeA and MeA and markedly decreased dendritic spine density in those same brain regions as compared to NP rats [30,103]. BDNF and Arc expression were normalized by an infusion of small interfering RNA (siRNA) against HDAC2 into the CeA. HDAC2 siRNA infusion subsequently increased dendritic spine density which were innately lower compared to NP rats [30,103]. HDAC2 siRNA infusion into the CeA also corrected the deficits in histone H3 acetylation of BDNF and Arc genes and expression of these genes. HDAC2 siRNA infusion into the CeA attenuated the anxiety-like and alcohol-drinking behaviors of P rats. These data firmly established the regulation of synaptic plasticity and dendritic arborization in alcoholism by direct HDAC2 regulation of BDNF and Arc [30,103].
Notably, HDAC2 is crucial for other neurocognitive domains, such as learning and memory [115,116]. A recent study showed that increases in HDAC2 were responsible for decreased gene expression seen in Alzheimer's disease-like neurological deficits. These deficits were reversed by injection of a short hairpin RNA (shRNA) targeted knockdown of HDAC2, which restored normal levels of synaptic plasticity [117]. HDAC2 overexpression in neurons decreases dendritic spine density, synapse number and memory formation. Conversely, HDAC2-deficient mice show increased dendritic spine density and synapse number, which is paralleled by increased memory formation [115]. This study was the first to show the direct regulation of dendritic spine density by the HDAC2 isoform and its involvement in cognitive processes.
As demonstrated above, HDACs, and in particular the HDAC2 isoform, have emerged as significant players in synaptic plasticity-related chromatin remodeling and subsequent dendritic spine alteration. Increased HDAC2 expression decreases the expression of genes important for the maintenance of dendritic spine density such as BDNF, Arc, and NPY, leading to increased anxiety and alcohol-seeking behavior. Decreasing HDAC2 reverses both the molecular and behavioral consequences of alcohol addiction, thus implicating this enzyme as a potential treatment target (Figure 3). HDAC2 is also crucial for the induction and maintenance of structural synaptic plasticity in other neurological domains such as memory formation [115]. Taken together, these findings underscore the potential usefulness of HDAC inhibition in treating alcohol use disorders and other neurocognitive ailments.
Figure 3. Reversal of the dark side of addiction by inhibition of HDAC2.

Increased expression of histone deacetylase isoform 2 (HDAC2) and/or higher HDAC activity can be caused either by a genetic predisposition (exemplified by alcohol-preferring P rats) or by prior alcohol abuse. Higher HDAC activity and subsequent deacetylation of histone H3 induces a condensed chromatin conformation around crucial synaptic plasticity genes such as BDNF, Arc and neuropeptide Y (NPY), leading to decreased expression in the amygdala. The resultant decrease in dendritic spine density causes the expression of negative affective states (anxiety) and compulsive drug-seeking behavior, collectively referred to as the “dark side” of addiction [5,6]. This process can be reversed by the inhibition of HDAC activity by HDAC2 siRNA infusion into central nucleus of amygdala (CeA) or by direct pharmacological inhibition (HDAC inhibitors), thereby increasing gene expression and dendritic spine density. Diagram is derived from published data from our laboratory [25,102,103,112].
4. HDAC inhibitors as novel treatments for addiction
Given the ability of HDAC inhibitors to potently modulate the synaptic plasticity of learning and memory [118], these drugs hold potential as treatment for substance abuse-related disorders. The HDAC2 isoform has emerged as the principal effector of epigenetic remodeling in both cognitive decline and alcohol addiction (Figure 3) [103,112,117,119]. All aforementioned HDAC inhibitors may be acting via inhibition of HDAC2 in the brain. The lack of specificity often leads to concerns about side effects and unintended consequences of HDAC inhibitor administration [120,121]. These drugs have been utilized as cancer therapy agents [121,122]. The clinical side effects observed during HDAC inhibitor cancer therapy include benign problems, such as gastrointestinal disturbances and fatigue, and more serious issues including thrombocytopenia, anemia, hypotension, inflammation, infection, electrolyte disturbances, and cardiovascular complications [123–125]. This evidence, combined with the knowledge that HDAC inhibitors are toxic to neurons in culture at high concentrations [126,127], has tempered optimism regarding the use of these drugs to treat brain diseases. Nonetheless, it must be noted that HDAC inhibitors are exceedingly well tolerated in most cases [118,128]. Additionally, it is possible that the dose needed to modify brain chromatin conformation in addictive and other neurological disorders may be significantly lower than the doses used as adjuvants in cancer chemotherapy.
HDAC inhibitors have shown promising results in the treatment of other neurological disorders, particularly with regard to rescuing aberrant synaptic plasticity. For example, HDAC inhibitors have successfully ameliorated the symptoms of neurocognitive decline and the decreased dendritic arborization seen in a variety of animal models of Alzheimer disease (AD) [129–131]. This data is further corroborated by lower BDNF expression and higher levels of repressive H3K9 methylation (a chromatin marker which prohibits H3K9 acetylation) in neuronal cultures obtained from a mouse model of AD [132].
Additionally, older mice with lower levels of acetylated H4K12 displayed increased associative memory formation, gene expression and histone acetylation when administered HDAC inhibitors [133]. HDAC inhibitors also have been effective in reducing the motor symptoms of Huntington disease (HD) in animal models of the disease [134,135]. In particular, genes involved in HD pathology show hypo-acetylation in the promoter region that is corrected with HDAC inhibitor treatment [136]. Hypo-acetylation also was observed in dying motor neurons in a rodent model of amyotrophic lateral sclerosis (ALS) [137]. The HDAC inhibitor sodium valproate provided neuroprotective effects in this same model in an effect mediated through CBP [138]. HDAC inhibitors have been effective in a rare neurodevelopmental disease known as Rubinstein-Taybi syndrome (RTS), which is caused by mutations in the CBP gene [139]. CBP activates gene promoters by acting as a HAT at critical synaptic plasticity genes [140]. The histone acetylation deficits and the observed neurodevelopmental defects of RTS are reversed by the administration of the HDAC inhibitor TSA [140,141]. These drugs have also proven effective in improving symptoms of depression, schizophrenia, and cocaine addiction in animal models [142–144].
Our lab and others have published extensively on the ability of HDAC inhibitors to reverse the gene expression deficits caused by multiple models of alcoholism and alcohol abuse, the results of which were discussed above [25,112,113]. This data supports further examination of histone modifying agents as potential therapeutic drugs in the treatment of alcohol addiction. Treatment with TSA and HDAC2 siRNA was able to normalize the deficits in dendritic spines in the CeA under various conditions of alcoholism, suggesting that HDACs are important targets within the epigenome regulating synaptic remodeling.
5. Conclusions
Here, we have summarized the current knowledge regarding alcohol-induced modulations of dendritic branching and the epigenetic and molecular mechanisms underlying these processes (Figs 1-3). As discussed above, alcohol is an important regulator of synaptic organization and function, and that these processes are controlled, at least in part, by specific epigenetic regulators. In particular, alcohol exposure induces near-immediate changes to chromatin and synaptic plasticity-associated gene expression. These alterations could induce a change in neuronal phenotype marked by re-enrichment of immature synapses, aberrant synaptic plasticity and dysfunctional dendritic spine formation. These structural changes are posited to underlie the compulsive drug seeking, negative affective states, and behavioral changes seen in alcohol addiction (Figure 3). We also have highlighted the potential usefulness of HDAC inhibitors as a novel treatment for alcohol use disorders (25,102,103,112). Future studies should continue to elucidate the specific epigenetic mechanisms underlying compulsive alcohol use and alcoholism, as this is likely to provide new molecular targets for clinical intervention.
Acknowledgements
This work was supported by National Institute on Alcohol Abuse and Alcoholism Grants AA-019971 (NADIA project), AA-010005, and AA-013341, and by the Department of Veterans Affairs (Merit Review Grant, I01BX000143; Research Career Scientist award) to S.C.P. SCP reports that a US patent application entitled “Histone acetyl transferase activators and histone deacetylase inhibitors in the treatment of alcoholism” (serial number 60/848237 filed on September 29th, 2006) is currently pending.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Other author reports no potential conflicts of interest.
References
- 1.Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol. Rev. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- 2.Pandey SC. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol. Sci. 2003;24:456–460. doi: 10.1016/S0165-6147(03)00226-8. [DOI] [PubMed] [Google Scholar]
- 3.Moonat S, Pandey SC. Stress, epigenetics, and alcoholism. Alcohol Res. 2012;34:495–505. [PMC free article] [PubMed] [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koob GF. Addiction is a reward deficit and stress surfeit disorder. Front. Psychiatry. 2013;4:72. doi: 10.3389/fpsyt.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koob GF, Le Moal M. Plasticity of reward neurocircuitry and ‘dark side’ of drug addiction. Nat. Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- 7.Koob GF. Alcoholism: allostasis and beyond. Alcohol. Clin. Exp. Res. 2003;27:232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- 8.Alheid GF. Extended amygdala and basal forebrain. Ann. N. Y. Acad. Sci. 2003;985:185–205. doi: 10.1111/j.1749-6632.2003.tb07082.x. [DOI] [PubMed] [Google Scholar]
- 9.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 10.Spiga S, Mulas G, Piras F, Diana M. The “addicted” spine. Front. Neuroanat. 2014;8:110. doi: 10.3389/fnana.2014.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maze I, Nestler EJ. The epigenetic landscape of addiction. Ann. N. Y. Acad. Sci. 2011;1216:99–113. doi: 10.1111/j.1749-6632.2010.05893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Y, Nestler EJ. The neural rejuvenation hypothesis of cocaine addiction. Trends Pharmacol. Sci. 2014;35:374–383. doi: 10.1016/j.tips.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat. Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- 14.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 15.Yoon B-J, Smith GB, Heynen AJ, Neve RL, Bear MF. Essential role for a long-term depression mechanism in ocular dominance plasticity. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9860–9865. doi: 10.1073/pnas.0901305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creed MC, Lüscher C. Drug-evoked synaptic plasticity: beyond metaplasticity. Curr. Opin. Neurobiol. 2013;23:553–558. doi: 10.1016/j.conb.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCool BA. Ethanol modulation of synaptic plasticity. Neuropharmacology. 2011;61:1097–1108. doi: 10.1016/j.neuropharm.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 20.Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the lEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- 21.Steward O, Worley PF. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron. 2001;30:227–240. doi: 10.1016/s0896-6273(01)00275-6. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura A, Fujii R, Watanabe Y, Okabe S, Fukui K, Takumi T. Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr. Biol. 2006;16:2345–2351. doi: 10.1016/j.cub.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 23.Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K. Effector immediate-early gene arc in the amygdala plays a critical role in alcoholism. J. Neurosci. 2008;28:2589–2600. doi: 10.1523/JNEUROSCI.4752-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC. Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int. J. Neuropsychopharmacol. 2014;17:313–22. doi: 10.1017/S1461145713001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou FC, Anthony B, Dunn KW, Lindquist WB, Xu ZC, Deng P. Chronic alcohol drinking alters neuronal dendritic spines in the brain reward center nucleus accumbens. Brain Res. 2007;1134:148–161. doi: 10.1016/j.brainres.2006.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riley JN, Walker DW. Morphological alterations in hippocampus after long-term alcohol consumption in mice. Science. 1978;201:646–648. doi: 10.1126/science.566953. [DOI] [PubMed] [Google Scholar]
- 28.Ferrer I, Fabregues I, Rairiz J, Galofre E. Decreased numbers of dendritic spines on cortical pyramidal neurons in human chronic alcoholism. Neurosci Lett. 1986;69:115–9. doi: 10.1016/0304-3940(86)90425-8. [DOI] [PubMed] [Google Scholar]
- 29.Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J. Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moonat S, Sakharkar AJ, Zhang H, Pandey SC. The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addict. Biol. 2011;16:238–250. doi: 10.1111/j.1369-1600.2010.00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: Multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 32.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 33.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 34.Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- 35.Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J. Neurosci. 2004;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–996. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- 37.Carpenter-Hyland EP, Woodward JJ, Chandler LJ. Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J. Neurosci. 2004;24:7859–7868. doi: 10.1523/JNEUROSCI.1902-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu W, Bie B, Pan ZZ. Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J. Neurosci. 2007;27:289–298. doi: 10.1523/JNEUROSCI.3912-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Läck AK, Diaz MR, Chappell A, DuBois DW, McCool BA. Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J. Neurophysiol. 2007;98:3185–3196. doi: 10.1152/jn.00189.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiga S, Talani G, Mulas G, Licheri V, Fois GR, Muggironi G, et al. Hampered long-term depression and thin spine loss in the nucleus accumbens of ethanol-dependent rats. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E3745–E3754. doi: 10.1073/pnas.1406768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-k. [DOI] [PubMed] [Google Scholar]
- 44.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 45.Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–53. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- 46.Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 47.Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, et al. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suvarna N, Borgland SL, Wang J, Phamluong K, Auberson YP, Bonci A, et al. Ethanol alters trafficking and functional N-methyl-D-aspartate receptor NR2 subunit ratio via H-Ras. J. Biol. Chem. 2005;280:31450–31459. doi: 10.1074/jbc.M504120200. [DOI] [PubMed] [Google Scholar]
- 49.Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One. 2012;7:e37541. doi: 10.1371/journal.pone.0037541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim A, Zamora-Martinez ER, Edwards S, Mandyam CD. Structural reorganization of pyramidal neurons in the medial prefrontal cortex of alcohol dependent rats is associated with altered glial plasticity. Brain Struct. Funct. 2014 Mar 26; doi: 10.1007/s00429-014-0755-3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gass JT, Chandler LJ. The Plasticity of Extinction: Contribution of the prefrontal cortex in treating addiction through inhibitory learning. Front. Psychiatry. 2013;4:46. doi: 10.3389/fpsyt.2013.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carpenter-Hyland EP, Chandler LJ. Homeostatic plasticity during alcohol exposure promotes enlargement of dendritic spines. Eur. J. Neurosci. 2006;24:3496–506. doi: 10.1111/j.1460-9568.2006.05247.x. [DOI] [PubMed] [Google Scholar]
- 53.Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gipson CD, Reissner KJ, Kupchik YM, Smith ACW, Stankeviciute N, Hensley-Simon ME, et al. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc. Natl. Acad. Sci. U. S. A. 2013;110:9124–9. doi: 10.1073/pnas.1220591110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc. Natl. Acad. Sci. 2011;108:19407–19412. doi: 10.1073/pnas.1112052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- 57.Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- 58.Murrell A, Rakyan VK, Beck S. From genome to epigenome. Hum. Mol. Genet. 2005;14:R3–R10. doi: 10.1093/hmg/ddi110. [DOI] [PubMed] [Google Scholar]
- 59.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 61.Starkman BG, Sakharkar AJ, Pandey SC. Epigenetics-beyond the genome in alcoholism. Alcohol Res. 2012;34:293–305. [PMC free article] [PubMed] [Google Scholar]
- 62.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 64.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 65.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 66.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007;8:355–67. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 67.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 68.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 69.Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64:1123–1134. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- 70.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 73.Gavin DP, Chase KA, Sharma RP. Active DNA demethylation in post-mitotic neurons: a reason for optimism. Neuropharmacology. 2013;75:233–245. doi: 10.1016/j.neuropharm.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 75.Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell. 1989;58:499–507. doi: 10.1016/0092-8674(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 76.Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Z, Hong EJ, Cohen S, ning Zhao W, yi H, Ho H, Schmidt L, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan SK, Handa V, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 83.Ma DK, Jang M-H, Guo JU, Kitabatake Y, Chang M-L, Pow-Anpongkul N, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kaas G, Zhong C, Eason D, Ross D, Vachhani R, li Ming G, et al. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086–1093. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rudenko A, Dawlaty M, Seo J, Cheng A, Meng J, Le T, et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109–1122. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sultan FA, Wang J, Tront J, Liebermann DA, Sweatt JD. Genetic deletion of gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity. J. Neurosci. 2012;32:17059–17066. doi: 10.1523/JNEUROSCI.1747-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koob GF, Le Moal M. Addiction and the brain antireward system. Annu. Rev. Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- 89.Tejeda HA, Shippenberg TS, Henriksson R. The dynorphin/κ-opioid receptor system and its role in psychiatric disorders. Cell. Mol. Life Sci. 2012;69:857–896. doi: 10.1007/s00018-011-0844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Knoll AT, Carlezon WA. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nestler EJ. Historical review: Molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol. Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 93.D'Addario C, Caputi FF, Ekström TJ, Di Benedetto M, Maccarrone M, Romualdi P, et al. Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J. Mol. Neurosci. 2013;49:312–319. doi: 10.1007/s12031-012-9829-y. [DOI] [PubMed] [Google Scholar]
- 94.Walker BM, Zorrilla EP, Koob GF. Systemic κ-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict. Biol. 2011;16:116–119. doi: 10.1111/j.1369-1600.2010.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Taqi MM, Bazov I, Watanabe H, Sheedy D, Harper C, Alkass K, et al. Prodynorphin CpG-SNPs associated with alcohol dependence: elevated methylation in the brain of human alcoholics. Addict. Biol. 2011;16:499–509. doi: 10.1111/j.1369-1600.2011.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valdez GR, Platt DM, Rowlett JK, Rüedi-Bettschen D, Spealman RD. Kappa agonist-induced reinstatement of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J. Pharmacol. Exp. Ther. 2007;323:525–533. doi: 10.1124/jpet.107.125484. [DOI] [PubMed] [Google Scholar]
- 97.Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J. Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schierloh A, Deussing J, Wurst W, Zieglgänsberger W, Rammes G. Corticotropin-releasing factor (CRF) receptor type 1-dependent modulation of synaptic plasticity. Neurosci. Lett. 2007;416:82–86. doi: 10.1016/j.neulet.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 99.Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT. CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl) 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- 100.Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- 101.Valdez GR, Zorrilla EP, Roberts AJ, Koob GF. Antagonism of corticotropin-releasing factor attenuates the enhanced responsiveness to stress observed during protracted ethanol abstinence. Alcohol. 2003;29:55–60. doi: 10.1016/s0741-8329(03)00020-x. [DOI] [PubMed] [Google Scholar]
- 102.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J. Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC. Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol. Psychiatry. 2013;73:763–773. doi: 10.1016/j.biopsych.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 105.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 106.Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- 108.Aguado F, Díaz-Ruiz C, Parlato R, Martínez A, Carmona MA, Bleckmann S, et al. The CREB/CREM transcription factors negatively regulate early synaptogenesis and spontaneous network activity. J. Neurosci. 2009;29:328–333. doi: 10.1523/JNEUROSCI.5252-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 110.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 111.Poo MM. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 112.Sakharkar AJ, Zhang H, Tang L, Baxstrom K, Shi G, Moonat S, et al. Effects of histone deacetylase inhibitors on amygdaloid histone acetylation and neuropeptide Y expression: a role in anxiety-like and alcohol-drinking behaviours. Int. J. Neuropsychopharmacol. 2014;17:1207–20. doi: 10.1017/S1461145714000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol. Clin. Exp. Res. 2012;36:61–71. doi: 10.1111/j.1530-0277.2011.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Legastelois R, Botia B, Naassila M. Blockade of ethanol-induced behavioral sensitization by sodium butyrate: descriptive analysis of gene regulations in the striatum. Alcohol. Clin. Exp. Res. 2013;37:1143–1153. doi: 10.1111/acer.12088. [DOI] [PubMed] [Google Scholar]
- 115.Guan J-S, Haggarty SJ, Giacometti E, Dannenberg J-H, Joseph N, Gao J, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gräff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell. 2014;156:261–276. doi: 10.1016/j.cell.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gräff J, Rei D, Guan J-S, Wang W-Y, Seo J, Hennig KM, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gräff J, Tsai L-H. Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013;14:97–111. doi: 10.1038/nrn3427. [DOI] [PubMed] [Google Scholar]
- 119.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer's disease: decrements in DNA methylation. Neurobiol. Aging. 2010;31:2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int. J. Dev. Biol. 2009;53:275–289. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- 121.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 122.Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenetics. 2010;1:117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim H-J, Bae S-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 125.Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010;3:5. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim HS, Kim EM, Kim NJ, Chang KA, Choi Y, Ahn KW, et al. Inhibition of histone deacetylation enhances the neurotoxicity induced by the C-terminal fragments of amyloid precursor protein. J. Neurosci. Res. 2004;75:117–124. doi: 10.1002/jnr.10845. [DOI] [PubMed] [Google Scholar]
- 127.Salminen A, Tapiola T, Korhonen P, Suuronen T. Neuronal apoptosis induced by histone deacetylase inhibitors. Mol. Brain Res. 1998;61:203–206. doi: 10.1016/s0169-328x(98)00210-1. [DOI] [PubMed] [Google Scholar]
- 128.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 129.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai L-H. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 130.Ricobaraza A, Cuadrado-Tejedor M, Pérez-Mediavilla A, Frechilla D, Del Río J, García-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 131.Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. 2009;18:131–139. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Walker MP, Laferla FM, Oddo SS, Brewer GJ. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer's disease. Age (Omaha) 2013;35:519–531. doi: 10.1007/s11357-011-9375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 134.Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, et al. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum. Mol. Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- 137.Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rouaux C, Panteleeva I, René F, Gonzalez de Aguilar J-L, Echaniz-Laguna A, Dupuis L, et al. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 140.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 142.Covington HE, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kurita M, Holloway T, García-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat. Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Malvaez M, Sanchis-Segura C, Vo D, Lattal KM, Wood MA. Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol. Psychiatry. 2010;67:36–43. doi: 10.1016/j.biopsych.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]