

Abstract
Idiopathic thrombocytopenic purpura (ITP) is typically a diagnosis of exclusion, assigned by clinicians after ruling out other identifiable etiologies. Since a report by Gasbarrini et al. in 1998, an accumulating body of evidence has proposed a pathophysiological link between ITP and chronic Helicobacter pylori (H. pylori) infection. Clinical reports have described a spontaneous resolution of ITP symptoms in about 50% of chronic ITP patients following empirical treatment of H. pylori infection, but response appears to be geography dependent. Studies have also documented that ITP patients in East Asian countries are more likely to express positive antibody titers against H. pylori-specific cytotoxic-associated gene A (CagA), a virulence factor that is associated with an increased risk for gastric diseases including carcinoma. While a definitive mechanism by which H. pylori may induce thrombocytopenia remains elusive, proposed pathways include molecular mimicry of CagA by host autoantibodies against platelet surface glycoproteins, as well as perturbations in the phagocytic activity of monocytes. Traditional treatments of ITP have been largely empirical, involving the use of immunosuppressive agents and immunoglobulin therapy. However, based on the findings of clinical reports emerging over the past 20 years, health organizations around the world increasingly suggest the detection and eradication of H. pylori as a treatment for ITP. Elucidating the exact molecular mechanisms of platelet activation in H. pylori-positive ITP patients, while considering biogeographical differences in response rates, could offer insight into how best to use clinical H. pylori eradication to treat ITP, but will require well-designed studies to confirm the suggested causative relationship between bacterial infection and an autoimmune disease state.
Keywords: Helicobacter, thrombocytopenia, immunemediated, infectious, biogeography, CagA
Helicobacter pylori (H. pylori) is a gram-negative, spiral-shaped, flagellated, microaerophilic bacillus that colonizes the gastric mucosa and is likely transmitted via the fecal–oral, or oral–oral route during childhood [1–6]. Prevalent in more than half of the world’s population, H. pylori infection occurs more frequently in developing nations [1,3]. There are documented differences in virulence factors between Western and East Asian strains of H. pylori, with the Eastern Asian strains suspected to have higher pathogenicity in relation to gastritis and gastric carcinoma [7,8]. Helicobacter pylori is recognized as a causative agent for a variety of gastric diseases, including gastritis, peptic ulcer, and gastric atrophy, and is correlated with an increased risk for gastric cancer [9]. The bacterium has also been linked to a number of extragastric diseases including nutritional deficiencies, such as vitamin B12 deficiency, and hematological pathologies, such as idiopathic thrombocytopenic purpura (idiopathic immune-mediated thrombocytopenia, ITP) [10,11]. In this review, the “virulence” of a particular H. pylori strain will refer to the susceptibility of infected individuals to any of the above-mentioned diseases, as well as the severity of disease symptoms and associated pathophysiological changes.
ITP is an autoimmune disorder wherein platelets are opsonized by platelet-specific IgG autoantibodies (autoAbs), resulting in platelet destruction within the reticuloendothelial system [12,13]. Platelets are circulating anucelate cells derived from megakaryocytes within the bone marrow. In an adult human, there are approximately 100 billion new platelets produced each day with a life span of 8–10 days, which maintains a normal platelet count of 150–400 × 109 platelets per liter of blood [14,15]. Platelets are known to play a critical role in both hemostasis and inflammation [16]. The autodestruction of platelets can be a result of infectious, genetic, or environmental factors, with primary ITP having no identifiable underlying etiology and secondary ITP having a causative agent positively identified.
Previous reports have postulated various mechanisms for H. pylori’s role in ITP including molecular mimicry, increased plasmacytoid dendritic cell numbers, and variable host immune response to virulence factors, including vacuolating-associated cytotoxin gene A (VacA), and cytotoxin-associated gene A (CagA) – a virulence factor that H. pylori injects into host gastric epithelial cells (ECs) via type a IV secretion system [17–20]. The presence of anti-CagA antibodies (Abs) has been shown to be predictive of platelet recovery in ITP patients treated for the eradication of H. pylori infection [19], which suggests that anti-CagA Ab titers can be used as a marker to determine whether eradication of H. pylori may be indicated in certain ITP patients. Additionally, molecular mimicry between CagA and platelet- associated IgG (PAIgG) has been previously documented [21]. These findings suggest that the host Ab response to CagA exhibits cross-reactivity with platelet surface antigens (Ags), promoting both platelet aggregation via immune complex formation and increased platelet clearance rates, resulting in thrombocytopenia.
Validating the ostensible etiological link between H. pylori infection and ITP could have a major impact on the clinical diagnosis and treatment of ITP. Current guidelines in multiple countries – including North America, Canada, and a number of countries in Asia – recommend H. pylori eradication in ITP patients that fail to respond to traditional treatments, if they test positive for the bacterium or reside in highly endemic areas.
Immune Thrombocytopenic Purpura
ITP is an autoimmune-mediated hematological disorder characterized by the destruction of host platelets and the impairment of megakaryocyte platelet production within the bone marrow [22]. The diagnosis of ITP relies upon a platelet count of <100 × 109 platelets per liter of blood and can be categorized as acute (diagnosis to 3 months), persistent (3–12 months), or chronic (>12 months) [23]. ITP can be a primary disease or secondary to a variety of etiologies including infection, autoimmune, or neoplastic disease. A chronic inflammatory state following primary bacterial or viral infection can induce the host autoimmune response via multiple mechanisms, including the production of auto-Abs and immune complex formation. ITP has an incidence of about 2.7 per 100,000 persons in adults and 5 per 100,000 persons in children, with a female sex predilection of 1.7 (female: male ratio) [24]. While ITP is typically an incidental finding during routine hematological evaluation, the most common clinical presentation of ITP is mucocutaneous bleeding, with rare complications of life-threatening hemorrhages [24,25]. ITP has a more acute onset, is self-limited, and usually follows an infectious illness in children, while ITP is usually chronic with an insidious onset in adults [25–27].
ITP is a diagnosis of exclusion, where the underlying etiology of the thrombocytopenia is undetermined. Recent reviews of previously published clinical studies have shown that eradication of H. pylori infection in patients with chronic ITP (cITP) results in increased platelet counts in about half of the cases [24,28–31]. In the majority of these retrospective studies, this associative link is defined as a durable platelet response after H. pylori eradication. A 2009 systematic review and meta-analysis revealed a correlation between H. pylori infection and ITP with a positive effect of H. pylori eradication on platelet count [29]. A current review of the clinical literature confirms that eradication treatment in H. pylori-positive adult ITP patients resulted in about a 50% complete response rate (Table 1), although this is variable by geographic location and many of the studies did not differentiate between acute and chronic ITP. Despite mounting evidence suggesting that H. pylori infection may also play an etiological role in pediatric cITP, published studies are inconclusive and empirical treatment of H. pylori in children with ITP remains controversial [32,33]. Due to the scarcity of controlled clinical studies to date and some reports presenting conflicting evidence, further research must be performed to confirm H. pylori as a causative agent of cITP in both adults and children [24,30,34,35].
Table 1.
Platelet response to eradication treatment in Helicobacter pylori-positive adult ITP patients
| References | Agea | Disease durationb | Bacterial eradication (%) | Platelet count before eradicationc | Platelet count post eradicationc | Complete response rated (%) | Study location (primary patient ethnicity) |
|---|---|---|---|---|---|---|---|
| Gasbarrini et al. (1998) [104] | 43 ± 14 | NR | 8/11 (73) | 95 ± 39 | 140 ± 34 | 8/8 (100) | Italy (Caucasian) |
| Jarque et al. (2001) [105] | 54 (17–80) | NR | 23/32 (72) | 58 ± 24 | 65 ± 32 | 3/23 (13) | Spain (Caucasian) |
| Kohda et al. (2002) [106] | 54 ± 14 | 41.2 ± 38.2 | 19/19 (100) | 67 ± 54 | 120 ± 50 | 12/19 (63) | Japan (Japanese) |
| Hino et al. (2003) [107] | 55 ± 15 | 78 ± 42.4 | 18/21 (86) | 37 ± 21 | 67 ± 54 | 10/18 (56) | Japan (Japanese) |
| Hashino et al. (2003) [108] | 53.2 ± 12.9 | 125.5 ± 77.1 | 13/14 (93) | 58 ± 30 | 99 ± 56 | 5/13 (38) | Japan (Japanese) |
| Ando et al. (2003) [109] | 58 ± 11 | 78.1 ± 65.1 | 27/29 (93) | 56 ± 24 | 93 ± 49 | 16/27 (59) | Japan (Japanese) |
| Michel et al. (2004) [110] | 52.5 ± 15.9 | 10 (1–21) | 14/15 (93) | 32 ± 15 | 66 ± 98 | 0/14 (0) | USA (Caucasian) |
| Takahashi et al. (2004) [54] | 54 ± 13 | 52.5 ± 6.4 | 13/15 (87) | 40 ± 27 | 101 ± 86 | 7/13 (54) | Japan (Japanese) |
| Sato et al. (2004) [111] | 62 (37–87) | 59.4 (6–264) | 27/32 (84) | 54 ± 17 | 110 ± 21 | 10/27 (37) | Japan (Japanese) |
| Ando et al. (2004) [112] | 62 (38–83) | NR | 15/17 (88) | 49 ± 26 | 168 ± 43 | 10/15 (67) | Japan (Japanese) |
| Nomura et al. (2004) [113] | NR | NR | 12/28 (43) | 29 ± 6 | 78 ± 11 | 15/28 (54) | Japan (Japanese) |
| Veneri et al. (2005) [114] | 57 (24–72) | NR | 41/43 (95) | 57 ± 23 | 122 ± 33 | 20/41 (49) | Italy (Caucasian) |
| Inaba et al. (2005) [115] | 57 (25–82) | 48 (6–180) | 25/25 (100) | 52 ± 26 | NR | 11/25 (44) | Japan (Japanese) |
| Stasi et al. (2005) [116] | 58 ± 13 | 25 ± 19 | 52/64 (81) | 42 ± 25 | 129 ± 61 | 17/52 (33) | Italy/UK (Caucasian) |
| Fujimura et al. (2005) [117] | 59 ± 14 | 98.2 ± 81.6 | 161/207 (78) | NR | NR | 101/161 (63) | Japan (Japanese) |
| Suzuki et al. (2005) [118] | NR | NR | 11/13 (85) | 55 ± 27 | 114 ± 90 | 6/13 (46) | Japan (Japanese) |
| Suvajdzic et al. (2006) [119] | 54 ± 13 | NA | 23/30 (77) | 68 ± 33 | 84 ± 45 | 6/23 (26) | Serbia (Caucasian) |
| Ahn et al. (2006) [120] | 56.8 ± 18.5 | NA | 15/15 (100) | 72 ± 45 | 69 ± 65 | 2/15 (13) | USA (Caucasian) |
| Sayan et al. (2006) [121] | 50.8 ± 16.2 | NA | 18/20 (90) | 39 ± 16 | 100 ± 63 | NA | Turkey (Middle Eastern) |
| Asahi et al. (2006/08) [49,122] | NR | NR | 26/26 (100) | 35 ± 13 | 114 ± 61 | 16/26 (62) | Japan (Japanese) |
| Kodama et al. (2007) [19] | 57.9 ± 14.3 | 57.9 ± 14.3 | 44/52 (85) | 40 ± 29 | NR | 27/44 (62) | Japan (Japanese) |
| Campuzano-Maya (2007) [123] | NR | NR | 26/29 (90) | 57 ± 38 | 198 ± 98 | 20/26 (78) | Colombia (South American) |
| Estrada-Gomez et al. (2007) [124] 3 | NR | NR | 14/14 (100) | NR | NR | NR | Mexico (Mexican) |
| Satake et al. (2007) [125] | NR | 65 (1–272) | 23/25 (92) | NR | NR | 12/23 (52) | Japan (Japanese) |
| Emilia et al. (2007) [126] | 58 ± 19 | 11.5 ± 6.4 | 34/38 (89) | 41 ± 24 | 134 ± 96 | 23/34 (68) | Italy (Caucasian) |
| Rostami et al. (2008) [127] | 29.2 ± 7 | 64.5 ± 49.5 | 62/71 (87) | 60.2 ± 18.14 | 140.5 ± 34 | 28/62 (45) | Iran (Middle Eastern) |
| Jackson et al. (2008) [128] | 47.5 | NR | 2/4 (50) | NR | NR | 3/4 (75) | Canada (Caucasian) |
| Tag et al. (2010) [129] | 55 (35–76) | 2.5 (0–27.1) | 23/23 (100) | 78 (6–96) | 100 (46–172) | 11/23 (48) | Korea (Korean) |
| Sato et al. (2011) [130] | 62 ± 13 | 42 ± 41 | 31/31 (100) | <100 | >130 | 18/31 (58) | Japan (Japanese) |
| Veneri et al. (2011) [131] | 52.2 (15–87) | 52.7 ± 116.1 | 11/12 (92) | 9.6 ± 4 | >100 | 6/12 (50) | Italy (Caucasian) |
| Payandeh et al. (2012) [132] | 38.2 | NR | 26/29 (90) | 49.5 ± 15 | 69 ± 21.9 | 15/26 (58) | Iran (Middle Eastern) |
| Gan et al. (2013) [133] | 50 (19–71) | 109 (36–216) | NR | 47.5 ± 12 | 64.5 ± 33.2 | 2/11 (18) | Malaysia (Malay/Chinese/Indian) |
| Takezako et al. (2013) [134] | 55 ± 21 | 19 ± 13 | 17/19 (89) | 65 ± 48 | 200 ± 140 | 5/17 (29) | Japan (Japanese) |
Mean or median (range).
Months, (mean ± SD) (median, range).
×109/L.
≥6 months.
Proposed Mechanistic Pathways of ITP Induction
Different strains of H. pylori express a distinct range of virulence factors, resulting in increased pathogenesis (increased pathological changes in tissue histology and increased local inflammation). Notably, H. pylori strains that are positive (cagA+) for the cytotoxin-associated gene pathogenicity island (cag PAI) are more virulent than cag PAI-negative strains (cagA−) and have been strongly associated with gastric carcinoma [7]. The cag PAI encodes a type IV secretion mechanism, giving rise to inflammation via NF-kB activation [36,37] and interleukin 8 (IL-8) production [38] and mediates translocation of the 145-kDa cagA gene product, CagA, into gastric epithelial cells (ECs) (Fig. 1). Once injected, CagA undergoes intracellular tyrosine phosphorylation (pY) and perturbs host cell signaling, promoting disturbance of ECs and gastric carcinogenesis [7,39,40]. While incompletely understood, tyrosine phosphorylation of CagA by the Src family protein tyrosine kinase mediates a growth-factor-like morphological change in infected gastric ECs known as the “humming-bird” phenotype, which is characterized by needlelike cellular protrusions [41–43]. This change in EC morphology can lead to increased cellular proliferation and has been used by researchers to explain the carcinogenic effect of H. pylori infection [44]. The severity of the “hummingbird” morphological change depends on the virulence of a given H. pylori strain and is a plausible indication of a strain’s potential to induce ITP in infected subjects. CagA is noted for the diversity of its amino acid sequence among different H. pylori strains, and published reports have revealed differences in infection severity between Western and Eastern Asian strains of H. pylori, with the extent to which a given strain’s CagA undergoes intracellular pY serving as a strong indicator of virulence [41].
Figure 1.

Effects of Helicobacter pylori CagA virulence factor on host cell. Helicobacter pylori injects CagA (red) into a host gastric epithelial cell via a type IV secretion system (T4SS, tan). Dimerizatiom of primary receptors activated by T4SS core complex: α5β1 integrin and receptor tyrosine kinases (purple). Within ECs, CagA EPIYA motif is recognized by major host kinases, Src, and Abl, resulting in tyrosine phosphorylation (yellow). Phosphorylated CagA (red and yellow) induces host cell pathology. Both, intracellular CagA and phosphorylated CagA, induce major effects on host cell signaling, resulting in changes in cell morphology, cell cycle regulation, and pro-inflammatory cytokine transcription. Intercellular junctions (gray): AJ, adherens junctions; TJ, tight junctions. This figure is an adaptation of Figures 2, 3, and 4 from [103].
Various mechanisms have been proposed for the induction of ITP as a result of H. pylori infection, incorporating both the innate (T-cell lymphocytes, neutrophils, monocytes, and cytokines) and acquired (B-cell lymphocytes) immune system. These include (1) the production of Ag-specific Abs that cross-react with platelet surface glycoproteins [21], (2) genetic differences in HLA class II allele patterns [45], (3) enhanced platelet activation from the interaction of H. pylori-bound von Willebrand factor (VWF) interacting with platelet surface Ag (GPIb) with subsequent aggregation supported by IgG [46], (4) the induction of the B-cell-mediated expansion of platelet-reactive Abs [47,48], (5) enhanced platelet activation from the binding of vacuolating cytotoxin (VacA) virulence factor to multimerin 1 on platelets [20], and (6) the downregulation of FcγRIIB receptors on monocytes, resulting in increased phagocytic activity, by H. pylori infection [49]. Helicobacter pylori Ags are currently known to induce a T- and B-cell-mediated response, which results in host production of H. pylori-specific Abs [50,51]. A recent study evaluated cytokine levels in patients with chronic ITP before and 6 months after H. pylori eradication. Antibiotic treatment, in patients with a positive platelet response to H. pylori eradication, resulted in a reduction in the cytokine profile consisted with Th1 and Th17 cells; also, there was an enhancement of a Th2 cytokine profile, as well as an increased number of T regulatory cells [52]. Some of the Abs generated during this immune response demonstrate cross-reactivity with host cells, including gastric and duodenal ECs, renal tubular cells, and salivary gland cells [53]. Recent clinical studies have elucidated an association between cagA+ H. pylori and ITP [17–19,21]. Takahashi et al. [54] demonstrated that patients with ITP had PAIgGs that cross-reacted with H. pylori-specific CagA, and reported a decline in anti-CagA Abs, as well as an increase in total circulating platelet count, in patients who underwent successful H. pylori eradication treatment. These findings parallel those of Kodama et al. [19], who also reported that H. pylori eradication therapy improved platelet counts in H. pylori-positive ITP patients and led to a significant decrease in anti-CagA Abs. One explanation for this finding is the possibility of molecular mimicry between CagA Ags and platelet surface glycoproteins, resulting in the anti-CagA Ab targeting of host platelets (Fig. 2). Although no research published to date has definitively established CagA as a causative factor in ITP, retrospective clinical studies substantiate a correlation of H. pylori-specific CagA with clinical ITP [55].
Figure 2.

Host production of anti-CagA Abs, resulting in ITP. (1) Helicobacter pylori injects CagA (red) into host gastric ECs via T4SS (tan). (2) CagA undergoes intracellular phosphorylation (red and yellow). (3) Two Ags are produced and presented on the cell surface of the infected host cell: one specific for CagA (light blue) and one that shows molecular mimicry to platelet surface glycoproteins (purple). (4) Host Abs recognizing either Ag undergo replication within the host lymph nodes. (5) These Abs are then released into the circulatory system, resulting in a secondary immune-mediated thrombocytopenia. (6) Increased platelet clearance is a result of Ab-Ag recognition in the reticuloendothelial system (R.E.S.), increased immune complex formation, and decreased platelet production in the bone marrow. (pink: platelet; orange: megakaryocyte; blue: mononuclear phagocyte; green: R.E.S.)
Injection of CagA into gastric epithelial cells results in a number of intracellular changes, morphological changes, and transcriptional activation (Fig. 1). A prospective clinical study performed by Asahi et al. [49], showed that H. pylori infection was correlated with an increase in monocyte phagocytic activity and a downregulation in FcγRIIB receptor expression on monocytes. Eradication treatment resulted in the resolution of these abnormalities as well as a decrease in circulating anti-CagA Abs. These findings suggest that the mechanism through which H. pylori induces an autoimmune disease state may very well be multifactorial: platelet clearance by macrophages in the reticular endothelial system, suppression of antigen presentation by macrophages, inhibition of lymphocyte responses to platelet surface antigens, direct increases in platelet activation, and cross-reactivity of autoplatelet Abs. A previous study exploring the pro-inflammatory activation of neutrophils and monocytes by H. pylori was not associated with the cagA or vacA genotype [56]. The lack of controlled scientific studies and the lack of establishment of Koch’s postulate require additional research to be performed to confirm a definitive mechanistic pathway linking H. pylori infection and ITP.
The Role of Biogeography
Computational phylogeography of H. pylori indicates a trend of spreading, alongside populations of its human host, from east Africa as early as 63,000 years ago [57]. The findings of Linz et al. demonstrate that the biogeographical evolution of modern H. pylori strains has been marked by high mutation rates and frequent interstrain recombination [58,59]. Helicobacter pylori strains present today subdivide into biogeographic populations that appear to reflect human migration patterns [60–62]. A comparison of 38 representative isolates of complete cag PAI genomes from known biogeographic islands was performed, showing that cagA gene content is highly conserved and its genetic diversity is reflective of geographic location [63]. The cag PAI is present in 95% of strains assigned to hpAfrica1, hpEastAsia, and hpAsia2 populations, while no hpAfrica2 strains possess the island, and cag PAI is variably found in hpEurope, hpNEAfrica, hpSahul, and the hspAmerind subpopulation of hpEastAsia (Fig. 3A) [63].
Figure 3.

Distribution of cag PAI in a collection of Helicobacter pylori strains from different world populations. Geographic sources of strains whose cag PAI sequences are now available. Each dot indicates the source of isolation of one of the 38 cag PAI sequences that were analyzed. The dots are color-coded by population or subpopulation as in (A). Representation of geographical distribution of platelet response after eradication treatment in H. pylori-positive adult patients (B). Data extrapolated from Table 1. Gray (0–25% response), yellow (25–50% response), green (50–75% response), and red (75–100% response) (modified with permission from [63]).
There is evidence for differences in infection severity between Western and Eastern Asian H. pylori strains. Eastern Asian H. pylori strains have been documented as more pathogenic, correlating with an increased occurrence of gastric carcinoma (among other GI pathologies) in H. pylori-positive patients in East Asia (including the countries of Japan, Korea, China, and Russia) [41,64]. This observed increase in pathogenicity correlates with an increase in CagA phosphosite abundance [7]. Furthermore, increased CagA pY can induce greater pathologic morphology changes in host gastric ECs [7]. CagA proteins of most Western H. pylori isolates have a 34-amino acid sequence that variably repeats among different strains [7]. This repeating sequence contains a conserved Glu-Pro-Ile-Tyr-Ala (EPIYA) sequence, which encodes the site for tyrosine phosphorylation (pY) [7]. CagA proteins having multiple EPIYA motifs undergo more extensive pY, exhibit increased specific binding to SHP-2 phosphatase, and induce greater morphological changes (“hummingbird phenotype”) in gastric ECs accompanied by sustained ERK activation, altered cellular migration (scattering), and induced apoptosis [65–67]. CagA proteins from Eastern Asian strains of H. pylori have distinct pY sequences at the region corresponding to the repeat sequence of Western CagA [7] (Fig. 4). These findings suggest that the CagA pY state underlies H. pylori’s pathologic effect in a range of diseases, including gastritis, gastric carcinoma, and ITP [41,68].
Figure 4.
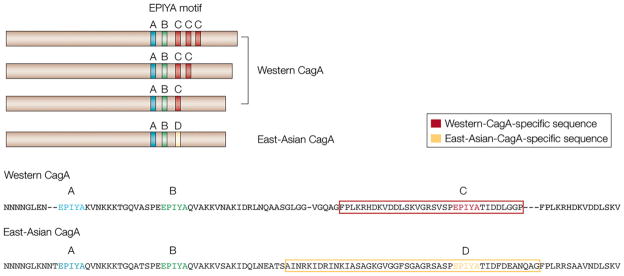
Diversity in CagA tyrosine phosphorylation (pY) sites. Tyrosine phosphorylation of cytotoxin-associated antigen A (CagA) by SRC kinase occurs at the EPIYA motif. There are four different EPIYA sites, called EPIYA-A, -B, -C, and -D, based on the sequence surrounding the EPIYA motif. Western strains of Helicobacter pylori express a form of CagA that contains the EPIYA-A and EPIYA-B sites, followed by 1–3 repeats of the 34-amino acid sequence that contains the EPIYA-C site (red boxes). East Asian strains of H. pylori express a form of CagA in which the EPIYA-C site is replaced with the EPIYA-D site (yellow box) (reprinted with permission from [41]).
In East Asia, CagA seropositivity is reported to be higher in patients diagnosed with gastric cancer than in those with gastritis alone [69], suggesting that anti-cagA Abs may be used as a biomarker for gastric cancer in East Asian countries [70]. Most H. pylori infections in Japan are positive for East Asian-type cagA [71,72]. However, in southwestern regions of Japan such as Okinawa, where the incidence of gastric cancer is lower, there are markedly lower rates of East Asian-type CagA. It has been postulated that the horizontal transmission of Western H. pylori strains to Japanese natives may have occurred during post-WWII occupation of Okinawa by US citizens [64,73]. Although the isoform of CagA found in southwestern Japan (“J-Western” CagA) differs from Western-type CagA via a 12-bp insertion in the cagA gene, the similarity between CagA multimerization sequence in Okinawan and Amerindal strains suggests they share ancestral lines [64,70,74]. The presence of both Western and East Asian-type CagA within Japan, along with correspondingly decreased incidence of gastric cancer among Japanese natives who test positive for only Western- or J-Western-type CagA, supports previous findings that variation in the virulence of CagA is important in the pathogenicity of H. pylori infection. Although few studies have probed the link between CagA virulence and ITP as comprehensively as gastric cancer, it is conceivable that similar trends underlie the increased prevalence of cITP in patients with cagA+ H. pylori infections.
Vacuolating cytotoxin (VacA) virulence factor has also been proposed as a factor in the development of ITP, via the binding to multimerin 1 on platelets leading to platelet activation [20]. Although not as thoroughly explored as that of CagA, the VacA genome may also be a variant that is based on bacterial and host migratory patterns. Lu et al. [75], performed a full genome comparative analysis of an H. pylori isolate in Australia, Sahul 64. This study suggested that Sahul 64, which was found to lack the cagPAI, have highly divergent cell envelope proteins, encode no transportable VacA protein, and might be better adapted to cause endogenous infections in Australians, despite the fact that these infections were less virulent compared to other cagPAI-positive strains. Evaluation of polymorphisms in virulence factors, CagA and VacA, in patients from different countries showed that the presence of the cagA gene correlated with vacA signal sequence s1/ m1 or s1/m2, and cagA-negative samples correlated with type s2/m2, suggesting that vacA genetic variation may be correlated to cagA biogeographical variation. Additionally, in the French study, cagA was positively correlated with the presence of the s1/m1 vacA genotype as well as with the induction of interleukin-8 (IL-8) [76–78]. Due to limited studies published on the genetic phylogeny of vacA, further investigation into the biogeography of this virulence factor in relationship to ITP may offer greater clinical insight.
In addition to bacterial biogeography, a patient’s biogeography may also play a role in the etiology of H. pylori-induced ITP. For example, monocyte FcγR receptor expression can be influenced by both environmental factors, such as infection, and genetic factors, such as polymorphisms [49]. An alteration in the expression of the FcγR gene can result in a change in a monocyte’s affinity for binding IgG, which can cause a change in the clearance rate of immune complexes [79]. Changes in receptor expression, such as the presence of single nuclear polymorphisms in the FcγRIIB gene or its promoter region, can change transcriptional activity and has been shown to be predictive of the development of chronic disease in children presenting acute ITP [80–83]. Studies evaluating these genetic changes in receptors have not evaluated cases of H. pylori-positive, eradication-responsive ITP. We hypothesize that the migratory patterns, genetic profiles, and biogeography of both patient and pathogen, H. pylori, are likely to play a role in the induction of an autoimmune disease state. The scarcity of published literature to date warrants further exploration on the genetic makeup of both the patient and the infecting H. pylori strains in relation to ITP.
Current Policies/Recommendations for Helicobacter pylori Eradication
Although the molecular basis for the association between ITP and H. pylori infection has not been conclusively established, various health organizations around the world currently advocate for the detection and eradication of H. pylori infection in the face of ITP. In 1987, the European Helicobacter Study Group (EHSG) was founded to promote multidisciplinary research centered on H. pylori-associated pathologies. The EHSG convenes consensus meetings on a regular basis to recommend when and how to treat patients infected with H. pylori. In 2010, 44 experts from 24 countries participated in the Maastricht IV/Florence Consensus Report and concluded that ITP is one of the extragastric diseases for which the detection and eradication of H. pylori infection is indicated [84]. While more conservative, current guidelines (2011) from the American Society of Hematology acknowledge H. pylori as a potential etiology for ITP and recommend that more extensive testing for infection be reserved for patients who either reside in highly endemic regions, or exhibit symptoms beyond thrombocytopenia (+/− anemia with concurrent bleeding) [26].
In 2009, the Asia-Pacific Conference convened to review the most current information on H. pylori management to synthesize new guidelines for the indication of its treatment and eradication and concluded that H. pylori eradication in infected patients with ITP was indicated [85]. The Japanese Society for Helicobacter Research considers H. pylori infection to be an indication for eradication therapy for four diseases: peptic ulcer, gastric MALT lymphoma, early gastric cancer, and ITP [85,86]. Because of the high prevalence of H. pylori infection in Japan, in addition to the increased pathogenicity of the endemic strains and subsequent risk of gastric cancer with long-term infection, eradication treatment has been approved by the Japanese health insurance system as a result of the recommendations from the Japanese Society for Helicobacter Research [70]. Other East Asian countries, including Korea, have also adopted H. pylori eradication guidelines for the treatment of both gastric and extragastric diseases (e.g., cITP) [87]. The findings of some reports suggest that ITP disease duration, along with the pathogenicity of infecting H. pylori strain (primarily the likelihood that a patient will be seropositive for CagA-specific autoAbs), is the chief predictor of patient platelet response following H. pylori eradication treatment, but more controlled clinical studies are required to confirm the biogeographical link between H. pylori strain and the likelihood of a positive platelet response (Fig. 3B, Table 1).
Clinical Relevance
A principle concern for individuals with ITP is life-threatening hemorrhage. A study of 117 adult cases of cITP revealed that 33% of treated adults achieved stable remission, with the incidence of hemorrhagic complications being more common in patients greater than 60 years of age (10.4%) than in patients younger than 40 years (0.4%) [88]. A case series review including 1817 patients with ITP revealed that the risk of fatal hemorrhage, before age adjustment, was 0.0162–0.0389 cases per patient-year [89]. Intracranial hemorrhage is the most life-threatening complication in patients with severe thrombocytopenia, with a mortality rate up to 5% in adults with cITP [90,91]. Thirty percent of pediatric patients will have a prolonged course and 5–10% will develop chronic severe refractory disease [92,93], with a 0.1–0.5% risk of intracranial hemorrhage, usually with platelet counts lower than 10 × 109/L [94]. Treatment for ITP is recommended for patients with a platelet count below 50 × 109/L, bleeding due to platelet dysfunction, the presence of other hemostatic defects, surgery, trauma, or other identified comorbidities [55].
Current treatment for ITP in the United States involves the use of immunosuppressive agents, such as corticosteroids, intravenous immunoglobulin therapy (IVIg), anti-D immunoglobulin (anti-D), Rituximab, or salvage splenectomy, all of which can be expensive and have associated risks and adverse effects [26]. About 20% of patients do not sustain a normal platelet count after medical therapy or splenectomy, and 10–20% of responders to splenectomy will relapse [55]. In a 2009 meta-analysis conducted by Stasi et al., positive platelet responses were found in roughly 50% of adult ITP patients after H. pylori eradication treatment, with more significant platelet count increases observed in less severe cases of ITP. As most of the studies included in the meta-analysis by Stasi et al. [29] considered H. pylori eradication in primarily severe cases of ITP, absolute patient response rates to bacterial eradication treatment may exceed 50% efficacy. In patients that respond to H. pylori eradication therapy, autoplatelet Ab responses are completely resolved, without relapse for over 7 years [95]. However, platelet response to H. pylori eradication for the treatment of ITP appears to correlate with geographic location: higher response rates are observed in Japan and Italy (ranging from 28 to 100%) than in the United States and other European countries (<13%) [95]. A study from Iran reported that H. pylori eradication led to increased platelet count in those with mild ITP (platelet count >50 × 103 μL), but was less successful in patients with severe ITP (platelet count <50 × 103 μL) [96]. Meanwhile, eradication of infection in H. pylori-positive children and adolescent ITP patients in Brazil had a response rate of 60% compared to untreated, infected patients, 18.2% [97]. Taken in aggregate, these clinical observations underscore the importance of patient biogeography as a predictor of response to H. pylori treatment and further implicate regional H. pylori strain variation.
Active H. pylori infections are diagnosed using the 13C-urea breath test, detecting bacterial Ag in the stool, with bacterial culture or PCR via gastric biopsy, or silver staining of gastric tissue for visualization of H. pylori. Additionally, ELISA-based serum Ab testing may be performed, but may be insensitive, not necessarily indicative of active bacterial infection, and may be falsely positive after IVIg therapy. The currently recommended treatment regimen for H. pylori infection consists of a combination of antibiotic therapies, which usually poses significantly less adverse effects and less overall costs than most ITP treatments. Historically, recommended eradication treatment has been a triple therapy cocktail, consisting of a proton-pump inhibitor, amoxicillin, and clarithromycin or metronidazole for up to 2 weeks. The addition of various probiotic compound supplements, containing Lactobacillus, Bifidobacterium, Saccharomyces, and other bacteria, have also been implicated in both reducing the adverse effects of eradication treatment and improving eradication rates, although studies supporting this are contradictory [98]. The antibiotics chosen may vary by region due to the emergence of H. pylori antibiotic resistance in highly endemic areas [99,100]. This rise in antimicrobial resistance, particularly against clarithromycin and metronidazole, combined with poor patient compliance, has resulted in a <70% eradiation rate in most countries [101,102]. Resistant H. pylori strains necessitate sequential or quadruple therapy (either bismuth-based or nonbismuth-based), and the use of rifabutin and levofloxacin as salvage treatments when initial therapy fails [100,102]. Although new diagnostic tools are being developed for defining susceptibility profiles for H. pylori infections, there is no current “tailored” eradication therapy protocol in effect [102]. Taking into account the literature supporting the link between H. pylori infection and ITP, H. pylori should be considered as a potential etiology for ITP in both children and adults; however, eradication therapy should be reserved for those who test positive for active infection.
Conclusions
Literature over the past 20 years has elucidated the potential link between chronic H. pylori infection and the development of ITP in both adults and children. Most publications are retrospective clinical studies with variable treatment response rates, potentially due to biogeography and subsequent genetic variation of the bacteria and/or patient. Many studies have proposed that H. pylori virulence factor, CagA, stimulates the development of anti-CagA antibodies that are cross-reactive with platelet surface antigens, resulting in thrombocytopenia. Future studies exploring the potential molecular link between chronic H. pylori infection and ITP are justified by the fact that (1) upper gastrointestinal (GI) infection by H. pylori is prevalent in over 50% of the world’s human population [1], and because (2) clinical studies of ITP to date have remained largely observational in nature, with few having substantiated the proposed molecular mechanisms of disease pathogenesis by experimental means. Further studies exploring the potential pathophysiological mechanism between infectious agents (e.g., H. pylori) and ITP, via the role of genetic variation and biogeography, could open a new avenue with regard to the diagnosis and treatment of patients with idiopathic hematological disorders. Lack of robust animal models of ITP associated with H. pylori infections has hampered mechanistic studies underlying its potential relationship with cITP. However, with the recognition that controlled experiments are necessary in establishing a causative link, current published literature is substantial enough to suggest an associative link between H. pylori and ITP and to warrant empirical eradication treatment in patients that test positive for active H. pylori infection or those in highly endemic areas, whose populations are infected with CagA-virulent strains.
Acknowledgments
We would like to acknowledge Dr. Jacquin C. Niles, MD, PhD, and Dr. Ram Sasisekharan, PhD, in the Department of Biological Engineering at Massachusetts Institute of Technology for their guidance and support in the development of this review. We would like to thank Alyssa Terestre for her assistance in manuscript preparation. This review was supported by the following grants: NIH T320D010978-26(JGF), P01CA028842-23 (JGF), P30ES002109(JGF).
Footnotes
Competing interests: the authors of this paper have no conflict of interest to disclose.
References
- 1.Rothenbacher D, Brenner H. Burden of Helicobacter pylori and H. pylori-related diseases in developed countries: recent developments and future implications. Microbes Infect. 2003;5:693–703. doi: 10.1016/s1286-4579(03)00111-4. [DOI] [PubMed] [Google Scholar]
- 2.Rothenbacher D, Winkler M, Gonser T, Adler G, Brenner H. Role of infected parents in transmission of Helicobacter pylori to their children. Pediatr Infect Dis J. 2002;21:674–9. doi: 10.1097/00006454-200207000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Frenck RW, Jr, Clemens J. Helicobacter in the developing world. Microbes Infect. 2003;5:705–13. doi: 10.1016/s1286-4579(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 4.Malaty HM, El-Kasabany A, Graham DY, Miller CC, Reddy SG, Srinivasan SR, Yamaoka Y, Berenson GS. Age at acquisition of Helicobacter pylori infection: a follow-up study from infancy to adulthood. Lancet. 2002;359:931–5. doi: 10.1016/S0140-6736(02)08025-X. [DOI] [PubMed] [Google Scholar]
- 5.Parsonnet J, Shmuely H, Haggerty T. Fecal and oral shedding of Helicobacter pylori from healthy infected adults. JAMA. 1999;282:2240–5. doi: 10.1001/jama.282.23.2240. [DOI] [PubMed] [Google Scholar]
- 6.Zou QH, Li RQ. Helicobacter pylori in the oral cavity and gastric mucosa: a meta-analysis. J Oral Pathol Med. 2011;40:317–24. doi: 10.1111/j.1600-0714.2011.01006.x. [DOI] [PubMed] [Google Scholar]
- 7.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002;99:14428–33. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiota S, Matsunari O, Yamaoka Y. Relationship between J-Western CagA subtype and the vacA m2 region of Helicobacter pylori. J Clin Microbiol. 2010;48:3033–4. doi: 10.1128/JCM.00983-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–9. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franceschi F, Zuccala G, Roccarina D, Gasbarrini A. Clinical effects of Helicobacter pylori outside the stomach. Nat Rev Gastroenterol Hepatol. 2014;11:234–42. doi: 10.1038/nrgastro.2013.243. [DOI] [PubMed] [Google Scholar]
- 11.Roubaud BC, Franceschi F, Salles N, Gasbarrini A. Extragastric diseases and Helicobacter pylori. Helicobacter. 2013;18(Suppl 1):44–51. doi: 10.1111/hel.12077. [DOI] [PubMed] [Google Scholar]
- 12.Semple JW. Infections, antigen-presenting cells, T cells, and immune tolerance: their role in the pathogenesis of immune thrombocytopenia. Hematol Oncol Clin North Am. 2009;23:1177– 92. doi: 10.1016/j.hoc.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol. 2010;17:590–5. doi: 10.1097/MOH.0b013e32833eaef3. [DOI] [PubMed] [Google Scholar]
- 14.Kaushansky K. Lineage-specific hematopoietic growth factors. N Engl J Med. 2006;354:2034–45. doi: 10.1056/NEJMra052706. [DOI] [PubMed] [Google Scholar]
- 15.Italiano JE, Jr, Hartwig JE. Chapter 2 - Megakaryocyte development and platelet formation. In: Michelson AD, editor. Platelets. 2. Burlington: Academic Press; 2007. pp. 23–44. [Google Scholar]
- 16.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–74. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 17.Franceschi F, Christodoulides N, Kroll MH, Genta RM. Helicobacter pylori and idiopathic thrombocytopenic purpura. Ann Intern Med. 2004;140:766–7. doi: 10.7326/0003-4819-140-9-200405040-00028. [DOI] [PubMed] [Google Scholar]
- 18.Saito A, Yokohama A, Osaki Y, et al. Circulating plasmacytoid dendritic cells in patients with primary and Helicobacter pylori-associated immune thrombocytopenia. Eur J Haematol. 2012;88:340–9. doi: 10.1111/j.1600-0609.2011.01745.x. [DOI] [PubMed] [Google Scholar]
- 19.Kodama M, Kitadai Y, Ito M, Kai H, Masuda H, Tanaka S, Yoshihara M, Fujimura K, Chayama K. Immune response to CagA protein is associated with improved platelet count after Helicobacter pylori eradication in patients with idiopathic thrombocytopenic purpura. Helicobacter. 2007;12:36–42. doi: 10.1111/j.1523-5378.2007.00477.x. [DOI] [PubMed] [Google Scholar]
- 20.Satoh K, Hirayama T, Takano K, Suzuki-Inoue K, Sato T, Ohta M, Nakagomi J, Ozaki Y. VacA, the vacuolating cytotoxin of Helicobacter pylori, binds to multimerin 1 on human platelets. Thromb J. 2013;11:23. doi: 10.1186/1477-9560-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol. 2004;124:91–6. doi: 10.1046/j.1365-2141.2003.04735.x. [DOI] [PubMed] [Google Scholar]
- 22.Neunert CE. Current management of immune thrombocytopenia. Hematology Am Soc Hematol Educ Program. 2013;2013:276–82. doi: 10.1182/asheducation-2013.1.276. [DOI] [PubMed] [Google Scholar]
- 23.Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–93. doi: 10.1182/blood-2008-07-162503. [DOI] [PubMed] [Google Scholar]
- 24.Franchini M, Veneri D. Helicobacter pylori-associated immune thrombocytopenia. Platelets. 2006;17:71–7. doi: 10.1080/09537100500438057. [DOI] [PubMed] [Google Scholar]
- 25.British Committee for Standards in Haematology General Haematology Task Force Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol. 2003;120:574–96. doi: 10.1046/j.1365-2141.2003.04131.x. [DOI] [PubMed] [Google Scholar]
- 26.Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Jr, Crowther MA. The American Society of Hematology 2011 evidence- based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190–207. doi: 10.1182/blood-2010-08-302984. [DOI] [PubMed] [Google Scholar]
- 27.Provan D, Newland A. Fifty years of idiopathic thrombocytopenic purpura (ITP): management of refractory itp in adults. Br J Haematol. 2002;118:933–44. doi: 10.1046/j.1365-2141.2002.03669.x. [DOI] [PubMed] [Google Scholar]
- 28.Teawtrakul N, Sawadpanich K, Sirijerachai C, Chansung K, Wanitpongpun C. Clinical characteristics and treatment outcomes in patients with Helicobacter pylori-positive chronic immune thrombocytopenic purpura. Platelets. 2014;25(7):548–51. doi: 10.3109/09537104.2013.841883. [DOI] [PubMed] [Google Scholar]
- 29.Stasi R, Sarpatwari A, Segal JB, Osborn J, Evangelista ML, Cooper N, Provan D, Newland A, Amadori S, Bussel JB. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood. 2009;113:1231–40. doi: 10.1182/blood-2008-07-167155. [DOI] [PubMed] [Google Scholar]
- 30.Franchini M, Cruciani M, Mengoli C, Pizzolo G, Veneri D. Effect of Helicobacter pylori eradication on platelet count in idiopathic thrombocytopenic purpura: a systematic review and meta-analysis. J Antimicrob Chemother. 2007;60:237–46. doi: 10.1093/jac/dkm195. [DOI] [PubMed] [Google Scholar]
- 31.Franchini M, Veneri D. Helicobacter pylori infection and immune thrombocytopenic purpura: an update. Helicobacter. 2004;9:342–6. doi: 10.1111/j.1083-4389.2004.00238.x. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara M, Capozzi L, Russo R. Effect of Helicobacter pylori eradication on platelet count in children with chronic idiopathic thrombocytopenic purpura. Hematology. 2009;14:282–5. doi: 10.1179/102453309X12473408860181. [DOI] [PubMed] [Google Scholar]
- 33.Kuhne T, Imbach P. Management of children and adolescents with primary immune thrombocytopenia: controversies and solutions. Vox Sang. 2013;104:55–66. doi: 10.1111/j.1423-0410.2012.01636.x. [DOI] [PubMed] [Google Scholar]
- 34.Samson AD, Schipperus MR, Langers AM, Dekkers OM. Helicobacter pylori infection is not correlated with subclinical thrombocytopenia: a cross-sectional study. Platelets. 2014;25:221–3. doi: 10.3109/09537104.2013.803063. [DOI] [PubMed] [Google Scholar]
- 35.Bisogno G, Errigo G, Rossetti F, Sainati L, Pusiol A, Da Dalt L, Colleselli P, Grotto P, Carli M. The role of Helicobacter pylori in children with chronic idiopathic thrombocytopenic purpura. J Pediatr Hematol Oncol. 2008;30:53–7. doi: 10.1097/MPH.0b013e3181615613. [DOI] [PubMed] [Google Scholar]
- 36.Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–5. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology. 1997;113:1099–109. doi: 10.1053/gast.1997.v113.pm9322504. [DOI] [PubMed] [Google Scholar]
- 38.Tummuru MK, Sharma SA, Blaser MJ. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol Microbiol. 1995;18:867–76. doi: 10.1111/j.1365-2958.1995.18050867.x. [DOI] [PubMed] [Google Scholar]
- 39.Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol. 2002;43:971–80. doi: 10.1046/j.1365-2958.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- 40.Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775–8. doi: 10.1074/jbc.C100754200. [DOI] [PubMed] [Google Scholar]
- 41.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–94. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 42.Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999;96:14559–64. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segal ED, Falkow S, Tompkins LS. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc Natl Acad Sci USA. 1996;93:1259–64. doi: 10.1073/pnas.93.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Argent RH, Thomas RJ, Letley DP, Rittig MG, Hardie KR, Atherton JC. Functional association between the Helicobacter pylori virulence factors VacA and CagA. J Med Microbiol. 2008;57(Pt 2):145–50. doi: 10.1099/jmm.0.47465-0. [DOI] [PubMed] [Google Scholar]
- 45.Veneri D, De Matteis G, Solero P, et al. Analysis of B- and T-cell clonality and HLA class II alleles in patients with idiopathic thrombocytopenic purpura: correlation with Helicobacter pylori infection and response to eradication treatment. Platelets. 2005;16:307–11. doi: 10.1080/09537100400028685. [DOI] [PubMed] [Google Scholar]
- 46.Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, Cox DM. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology. 2003;124:1846–54. doi: 10.1016/s0016-5085(03)00397-4. [DOI] [PubMed] [Google Scholar]
- 47.Roark JH, Bussel JB, Cines DB, Siegel DL. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood. 2002;100:1388–98. [PubMed] [Google Scholar]
- 48.Emilia G, Luppi M, Torelli G. Infectious agents and human immune diseases: lessons from Helicobacter pylori. Am J Med. 2005;118:420–1. doi: 10.1016/j.amjmed.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 49.Asahi A, Nishimoto T, Okazaki Y, Suzuki H, Masaoka T, Kawakami Y, Ikeda Y, Kuwana M. Helicobacter pylori eradication shifts monocyte Fcgamma receptor balance toward inhibitory FcgammaRIIB in immune thrombocytopenic purpura patients. J Clin Invest. 2008;118:2939–49. doi: 10.1172/JCI34496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nurgalieva ZZ, Conner ME, Opekun AR, Zheng CQ, Elliott SN, Ernst PB, Osato M, Estes MK, Graham DY. B-cell and T-cell immune responses to experimental Helicobacter pylori infection in humans. Infect Immun. 2005;73:2999–3006. doi: 10.1128/IAI.73.5.2999-3006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Futagami S, Takahashi H, Norose Y, Kobayashi M. Systemic and local immune responses against Helicobacter pylori urease in patients with chronic gastritis: distinct IgA and IgG productive sites. Gut. 1998;43:168–75. doi: 10.1136/gut.43.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rocha AM, Souza C, Melo FF, Clementino NC, Marino MC, Rocha GA, Queiroz DM. Cytokine profile of patients with chronic immune thrombocytopenia affects platelet count recovery after Helicobacter pylori eradication. Br J Haematol. 2014 doi: 10.1111/bjh.13141. [DOI] [PubMed] [Google Scholar]
- 53.Ko GH, Park HB, Shin MK, Park CK, Lee JH, Youn HS, Cho MJ, Lee WK, Rhee KH. Monoclonal antibodies against Helicobacter pylori cross-react with human tissue. Helicobacter. 1997;2:210–5. doi: 10.1111/j.1523-5378.1997.tb00090.x. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi T, Yujiri T, Tanizawa Y. Helicobacter pylori and chronic ITP: the discrepancy in the clinical responses to eradication therapy might be due to differences in the bacterial strains. Blood. 2004;104:594. doi: 10.1182/blood-2004-02-0702. [DOI] [PubMed] [Google Scholar]
- 55.Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168–86. doi: 10.1182/blood-2009-06-225565. [DOI] [PubMed] [Google Scholar]
- 56.Hansen PS, Go MF, Varming K, Andersen LP, Graham DY, Nielsen H. Proinflammatory activation of neutrophils and monocytes by Helicobacter pylori is not associated with cagA, vacA or picB genotypes. APMIS. 1999;107:1117–23. [PubMed] [Google Scholar]
- 57.Linz B, Balloux F, Moodley Y, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–8. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA. 2005;102:15942–7. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prugnolle F, Manica A, Balloux F. Geography predicts neutral genetic diversity of human populations. Curr Biol. 2005;15:R159–60. doi: 10.1016/j.cub.2005.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu H, Prugnolle F, Manica A, Balloux F. A geographically explicit genetic model of worldwide human-settlement history. Am J Hum Genet. 2006;79:230–7. doi: 10.1086/505436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, NJ: Princeton University Press; 1994. [Google Scholar]
- 62.Rosenberg NA, Mahajan S, Ramachandran S, Zhao C, Pritchard JK, Feldman MW. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 2005;1:e70. doi: 10.1371/journal.pgen.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olbermann P, Josenhans C, Moodley Y, Uhr M, Stamer C, Vauterin M, Suerbaum S, Achtman M, Linz B. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 2010;6:e1001069. doi: 10.1371/journal.pgen.1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsunari O, Shiota S, Suzuki R, Watada M, Kinjo N, Murakami K, Fujioka T, Kinjo F, Yamaoka Y. Association between Helicobacter pylori virulence factors and gastroduodenal diseases in Okinawa, Japan. J Clin Microbiol. 2012;50:876–83. doi: 10.1128/JCM.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Higashi H, Nakaya A, Tsutsumi R, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–16. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 66.Saberi S, Douraghi M, Azadmanesh K, Shokrgozar MA, Zeraati H, Hosseini ME, Mohagheghi MA, Parsaeian M, Mohammadi M. A potential association between Helicobacter pylori CagA EPIYA and multimerization motifs with cytokeratin 18 cleavage rate during early apoptosis. Helicobacter. 2012;17:350– 7. doi: 10.1111/j.1523-5378.2012.00954.x. [DOI] [PubMed] [Google Scholar]
- 67.Lee IO, Kim JH, Choi YJ, Pillinger MH, Kim SY, Blaser MJ, Lee YC. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem. 2010;285:16042–50. doi: 10.1074/jbc.M110.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst. 1995;87:1777–80. doi: 10.1093/jnci/87.23.1777. [DOI] [PubMed] [Google Scholar]
- 69.Shiota S, Matsunari O, Watada M, Yamaoka Y. Serum Helicobacter pylori CagA antibody as a biomarker for gastric cancer in east-Asian countries. Future Microbiol. 2010;5:1885–93. doi: 10.2217/fmb.10.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shiota S, Murakawi K, Suzuki R, Fujioka T, Yamaoka Y. Helicobacter pylori infection in Japan. Expert Rev Gastroenterol Hepatol. 2013;7:35–40. doi: 10.1586/egh.12.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. Variants of the 30 region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J Clin Microbiol. 1998;36:2258–63. doi: 10.1128/jcm.36.8.2258-2263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamaoka Y, Orito E, Mizokami M, et al. Helicobacter pylori in North and South America before Columbus. FEBS Lett. 2002;517:180–4. doi: 10.1016/s0014-5793(02)02617-0. [DOI] [PubMed] [Google Scholar]
- 73.Truong BX, Mai VT, Tanaka H, et al. Diverse characteristics of the CagA gene of Helicobacter pylori strains collected from patients from southern vietnam with gastric cancer and peptic ulcer. J Clin Microbiol. 2009;47:4021–8. doi: 10.1128/JCM.00504-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suzuki M, Kiga K, Kersulyte D, et al. Attenuated CagA onco-protein in Helicobacter pylori from Amerindians in Peruvian Amazon. J Biol Chem. 2011;286:29964–72. doi: 10.1074/jbc.M111.263715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu W, Wise MJ, Tay CY, Windsor HM, Marshall BJ, Peacock C, Perkins T. Comparative analysis of the full genome of Helicobacter pylori isolate Sahul64 identifies genes of high divergence. J Bacteriol. 2014;196:1073–83. doi: 10.1128/JB.01021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salehi Z, Abadi AS, Ismail PB, Kqueen CY, Jelodar MH, Kamalidehghan B. Evaluation of Helicobacter pylori vacA genotypes in Iranian patients with peptic ulcer disease. Dig Dis Sci. 2009;54:2399–403. doi: 10.1007/s10620-008-0633-z. [DOI] [PubMed] [Google Scholar]
- 77.Audibert C, Janvier B, Grignon B, Salaun L, Burucoa C, Lecron JC, Fauchere JL. Correlation between IL-8 induction, cagA status and vacA genotypes in 153 French Helicobacter pylori isolates. Res Microbiol. 2000;151:191–200. doi: 10.1016/s0923-2508(00)00139-x. [DOI] [PubMed] [Google Scholar]
- 78.Rudi J, Kuck D, Rudy A, Sieg A, Maiwald M, Stremmel W. Helicobacter pylori vacA genotypes and cagA gene in a series of 383 H. pylori-positive patients. Z Gastroenterol. 2000;38:559–64. doi: 10.1055/s-2000-7449. [DOI] [PubMed] [Google Scholar]
- 79.van der Pol W, van de Winkel JG. IgG receptor polymorphisms: risk factors for disease. Immunogenetics. 1998;48:222– 32. doi: 10.1007/s002510050426. [DOI] [PubMed] [Google Scholar]
- 80.Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, Kimberly RP. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. 2003;48:3242–52. doi: 10.1002/art.11313. [DOI] [PubMed] [Google Scholar]
- 81.Bruin M, Bierings M, Uiterwaal C, Revesz T, Bode L, Wiesman ME, Kuijpers T, Tamminga R, de Haas M. Platelet count, previous infection and FCGR2B genotype predict development of chronic disease in newly diagnosed idiopathic thrombocytopenia in childhood: results of a prospective study. Br J Haematol. 2004;127:561–7. doi: 10.1111/j.1365-2141.2004.05235.x. [DOI] [PubMed] [Google Scholar]
- 82.Floto RA, Clatworthy MR, Heilbronn KR, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. 2005;11:1056–8. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 83.Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. II. Differential binding of GATA4 and Yin-Yang1 transcription factors and correlated receptor expression and function. J Immunol. 2004;172:7192–9. doi: 10.4049/jimmunol.172.11.7192. [DOI] [PubMed] [Google Scholar]
- 84.Malfertheiner P, Megraud F, O’Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/Florence Consensus Report. Gut. 2012;61:646–64. doi: 10.1136/gutjnl-2012-302084. [DOI] [PubMed] [Google Scholar]
- 85.Fock KM, Katelaris P, Sugano K, et al. Second Asia-Pacific Consensus Guidelines for Helicobacter pylori infection. J Gastroenterol Hepatol. 2009;24:1587–600. doi: 10.1111/j.1440-1746.2009.05982.x. [DOI] [PubMed] [Google Scholar]
- 86.Asaka M, Kato M, Takahashi S, Fukuda Y, Sugiyama T, Ota H, Uemura N, Murakami K, Satoh K, Sugano K. Guidelines for the management of Helicobacter pylori infection in Japan: 2009 revised edition. Helicobacter. 2010;15:1–20. doi: 10.1111/j.1523-5378.2009.00738.x. [DOI] [PubMed] [Google Scholar]
- 87.Kim N, Kim JJ, Choe YH, Kim HS, Kim JI, Chung IS. Diagnosis and treatment guidelines for Helicobacter pylori infection in Korea. Korean J Gastroenterol. 2009;54:269–78. doi: 10.4166/kjg.2009.54.5.269. [DOI] [PubMed] [Google Scholar]
- 88.Cortelazzo S, Finazzi G, Buelli M, Molteni A, Viero P, Barbui T. High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood. 1991;77:31–3. [PubMed] [Google Scholar]
- 89.Cohen YC, Djulbegovic B, Shamai-Lubovitz O, Mozes B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch Intern Med. 2000;160:1630–8. doi: 10.1001/archinte.160.11.1630. [DOI] [PubMed] [Google Scholar]
- 90.Mueller-Eckhardt C. Idiopathic thrombocytopenic purpura (ITP): clinical and immunologic considerations. Semin Thromb Hemost. 1977;3:125–59. doi: 10.1055/s-0028-1086134. [DOI] [PubMed] [Google Scholar]
- 91.Berchtold P, McMillan R. Therapy of chronic idiopathic thrombocytopenic purpura in adults. Blood. 1989;74:2309–17. [PubMed] [Google Scholar]
- 92.Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program. 2012;2012:444–9. doi: 10.1182/asheducation-2012.1.444. [DOI] [PubMed] [Google Scholar]
- 93.Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. Am J Hematol. 2010;85:174–80. doi: 10.1002/ajh.21616. [DOI] [PubMed] [Google Scholar]
- 94.Butros LJ, Bussel JB. Intracranial hemorrhage in immune thrombocytopenic purpura: a retrospective analysis. J Pediatr Hematol Oncol. 2003;25:660–4. doi: 10.1097/00043426-200308000-00017. [DOI] [PubMed] [Google Scholar]
- 95.Kuwana M. Helicobacter pylori-associated immune thrombocytopenia: clinical features and pathogenic mechanisms. World J Gastroenterol. 2014;20:714–23. doi: 10.3748/wjg.v20.i3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Payandeh M, Raeisi D, Sohrabi N, Zare ME, Kansestani AN, Keshavarz N, Gholami S, Hashemian AH. Poor platelet Count Response to Helicobacter Pylori Eradication in Patients with Severe Idiopathic Thrombocytopenic Purpura. Int J Hematol Oncol Stem Cell Res. 2013;7:9–14. [PMC free article] [PubMed] [Google Scholar]
- 97.Brito HS, Braga JA, Loggetto SR, Machado RS, Granato CF, Kawakami E. Helicobacter pylori infection & immune thrombocytopenic purpura in children and adolescents: a randomized controlled trial. Platelets. 2014:1–6. doi: 10.3109/09537104.2014.911836. [DOI] [PubMed] [Google Scholar]
- 98.Medeiros JA, Pereira M-I. The Use of Probiotics in Helicobacter pylori Eradication Therapy. J Clin Gastroenterol. 2013;47:1–5. doi: 10.1097/MCG.0b013e3182702dbc. [DOI] [PubMed] [Google Scholar]
- 99.Seo JH, Woo HO, Youn HS, Rhee KH. Antibiotics resistance of Helicobacter pylori and treatment modalities in children with H. pylori infection. Korean J Pediatr. 2014;57:67–71. doi: 10.3345/kjp.2014.57.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vakil N, Vaira D. Treatment for H. pylori infection: new challenges with antimicrobial resistance. J Clin Gastroenterol. 2013;47:383–8. doi: 10.1097/MCG.0b013e318277577b. [DOI] [PubMed] [Google Scholar]
- 101.Georgopoulos SD, Papastergiou V, Karatapanis S. Current options for the treatment of Helicobacter pylori. Expert Opin Pharmacother. 2013;14:211–23. doi: 10.1517/14656566.2013.763926. [DOI] [PubMed] [Google Scholar]
- 102.Kanizaj TF, Kunac N. Helicobacter pylori: future perspectives in therapy reflecting three decades of experience. World J Gastroenterol. 2014;20:699–705. doi: 10.3748/wjg.v20.i3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hagymasi K, Tulassay Z. Helicobacter pylori infection: new pathogenetic and clinical aspects. World J Gastroenterol. 2014;20:6386–99. doi: 10.3748/wjg.v20.i21.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gasbarrini A, Franceschi F, Tartaglione R, Landolfi R, Pola P, Gasbarrini G. Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori. Lancet. 1998;352:878. doi: 10.1016/S0140-6736(05)60004-9. [DOI] [PubMed] [Google Scholar]
- 105.Jarque I, Andreu R, Llopis I, et al. Absence of platelet response after eradication of Helicobacter pylori infection in patients with chronic idiopathic thrombocytopenic purpura. Br J Haematol. 2001;115:1002–3. doi: 10.1046/j.1365-2141.2001.03194.x. [DOI] [PubMed] [Google Scholar]
- 106.Kohda K, Kuga T, Kogawa K, Kanisawa Y, Koike K, Kuroiwa G, Hirayama Y, Sato Y, Niitsu Y. Effect of Helicobacter pylori eradication on platelet recovery in Japanese patients with chronic idiopathic thrombocytopenic purpura and secondary autoimmune thrombocytopenic purpura. Br J Haematol. 2002;118:584–8. doi: 10.1046/j.1365-2141.2002.03612.x. [DOI] [PubMed] [Google Scholar]
- 107.Hino M, Yamane T, Park K, Takubo T, Ohta K, Kitagawa S, Higuchi K, Arakawa T. Platelet recovery after eradication of Helicobacter pylori in patients with idiopathic thrombocytopenic purpura. Ann Hematol. 2003;82:30–2. doi: 10.1007/s00277-002-0579-8. [DOI] [PubMed] [Google Scholar]
- 108.Hashino S, Mori A, Suzuki S, et al. Platelet recovery in patients with idiopathic thrombocytopenic purpura after eradication of Helicobacter pylori. Int J Hematol. 2003;77:188–91. doi: 10.1007/BF02983220. [DOI] [PubMed] [Google Scholar]
- 109.Ando K, Shimamoto T, Tauchi T, Ito Y, Kuriyama Y, Gotoh A, Miyazawa K, Kimura Y, Kawai T, Ohyashiki K. Can eradication therapy for Helicobacter pylori really improve the thrombocytopenia in idiopathic thrombocytopenic purpura? Our experience and a literature review. Int J Hematol. 2003;77:239–44. doi: 10.1007/BF02983780. [DOI] [PubMed] [Google Scholar]
- 110.Michel M, Cooper N, Jean C, Frissora C, Bussel JB. Does Helicobater pylori initiate or perpetuate immune thrombocytopenic purpura? Blood. 2004;103:890–6. doi: 10.1182/blood-2003-03-0900. [DOI] [PubMed] [Google Scholar]
- 111.Sato R, Murakami K, Watanabe K, et al. Effect of Helicobacter pylori eradication on platelet recovery in patients with chronic idiopathic thrombocytopenic purpura. Arch Intern Med. 2004;164:1904–7. doi: 10.1001/archinte.164.17.1904. [DOI] [PubMed] [Google Scholar]
- 112.Ando T, Tsuzuki T, Mizuno T, et al. Characteristics of Helicobacter pylori-induced gastritis and the effect of H. pylori eradication in patients with chronic idiopathic thrombocytopenic purpura. Helicobacter. 2004;9:443–52. doi: 10.1111/j.1083-4389.2004.00261.x. [DOI] [PubMed] [Google Scholar]
- 113.Nomura S, Inami N, Kanazawa S. The effects of Helicobacter pylori eradication on chemokine production in patients with immune thrombocytopenic purpura. Eur J Haematol. 2004;72:304–5. doi: 10.1111/j.1600-0609.2004.00220.x. [DOI] [PubMed] [Google Scholar]
- 114.Veneri D, Krampera M, Franchini M. High prevalence of sustained remission of idiopathic thrombocytopenic purpura after Helicobacter pylori eradication: a long-term follow-up study. Platelets. 2005;16:117–9. doi: 10.1080/09537100400015153. [DOI] [PubMed] [Google Scholar]
- 115.Inaba T, Mizuno M, Take S, et al. Eradication of Helicobacter pylori increases platelet count in patients with idiopathic thrombocytopenic purpura in Japan. Eur J Clin Invest. 2005;35:214–9. doi: 10.1111/j.1365-2362.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- 116.Stasi R, Rossi Z, Stipa E, Amadori S, Newland AC, Provan D. Helicobacter pylori eradication in the management of patients with idiopathic thrombocytopenic purpura. Am J Med. 2005;118:414–9. doi: 10.1016/j.amjmed.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 117.Fujimura K, Kuwana M, Kurata Y, et al. Is eradication therapy useful as the first line of treatment in Helicobacter pyloripositive idiopathic thrombocytopenic purpura? Analysis of 207 eradicated chronic ITP cases in Japan. Int J Hematol. 2005;81:162–8. doi: 10.1532/ijh97.04146. [DOI] [PubMed] [Google Scholar]
- 118.Suzuki T, Matsushima M, Masui A, Watanabe K, Takagi A, Ogawa Y, Shirai T, Mine T. Effect of Helicobacter pylori eradication in patients with chronic idiopathic thrombocytopenic purpura-a randomized controlled trial. Am J Gastroenterol. 2005;100:1265–70. doi: 10.1111/j.1572-0241.2005.41641.x. [DOI] [PubMed] [Google Scholar]
- 119.Suvajdzic N, Stankovic B, Artiko V, Cvejic T, Bulat V, Bakrac M, Colovic M, Obradovic V, Atkinson HD. Helicobacter pylori eradication can induce platelet recovery in chronic idiopathic thrombocytopenic purpura. Platelets. 2006;17:227–30. doi: 10.1080/09537100500462487. [DOI] [PubMed] [Google Scholar]
- 120.Ahn ER, Tiede MP, Jy W, Bidot CJ, Fontana V, Ahn YS. Platelet activation in Helicobacter pylori-associated idiopathic thrombocytopenic purpura: eradication reduces platelet activation but seldom improves platelet counts. Acta Haematol. 2006;116:19–24. doi: 10.1159/000092343. [DOI] [PubMed] [Google Scholar]
- 121.Sayan O, Akyol Erikci A, Ozturk A. The Efficacy of Helicobacter pylori eradication in the treatment of idiopathic thrombocytopenic purpura–the first study in Turkey. Acta Haematol. 2006;116:146–9. doi: 10.1159/000093648. [DOI] [PubMed] [Google Scholar]
- 122.Asahi A, Kuwana M, Suzuki H, Hibi T, Kawakami Y, Ikeda Y. Effects of a Helicobacter pylori eradication regimen on anti-platelet autoantibody response in infected and uninfected patients with idiopathic thrombocytopenic purpura. Haematologica. 2006;91:1436–7. [PubMed] [Google Scholar]
- 123.Campuzano-Maya G. Proof of an association between Helicobacter pylori and idiopathic thrombocytopenic purpura in Latin America. Helicobacter. 2007;12:265–73. doi: 10.1111/j.1523-5378.2007.00502.x. [DOI] [PubMed] [Google Scholar]
- 124.Estrada-Gomez RA, Parra-Ortega I, Martinez-Barreda C, Ruiz-Arguelles GJ. Helicobacter pylori infection and thrombocytopenia: a single-institution experience in Mexico. Rev Invest Clin. 2007;59:112–5. [PubMed] [Google Scholar]
- 125.Satake M, Nishikawa J, Fukagawa Y, Akashi K, Okamoto T, Yoshida T, Hirano A, Maetani N, Iida Y, Sakaida I. The longterm efficacy of Helicobacter pylori eradication therapy in patients with idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol. 2007;22:2233–7. doi: 10.1111/j.1440-1746.2007.04845.x. [DOI] [PubMed] [Google Scholar]
- 126.Emilia G, Luppi M, Zucchini P, et al. Helicobacter pylori infection and chronic immune thrombocytopenic purpura: long-term results of bacterium eradication and association with bacterium virulence profiles. Blood. 2007;110:3833–41. doi: 10.1182/blood-2006-12-063222. [DOI] [PubMed] [Google Scholar]
- 127.Rostami N, Keshtkar-Jahromi M, Rahnavardi M, Esfahani FS. Effect of eradication of Helicobacter pylori on platelet recovery in patients with chronic idiopathic thrombocytopenic purpura: a controlled trial. Am J Hematol. 2008;83:376–81. doi: 10.1002/ajh.21125. [DOI] [PubMed] [Google Scholar]
- 128.Jackson SC, Beck P, Buret AG, O’Connor PM, Meddings J, Pineo G, Poon MC. Long term platelet responses to Helicobacter pylori eradication in Canadian patients with immune thrombocytopenic purpura. Int J Hematol. 2008;88:212–8. doi: 10.1007/s12185-008-0138-8. [DOI] [PubMed] [Google Scholar]
- 129.Tag HS, Lee HS, Jung SH, Kim BK, Kim SB, Lee A, Lee JS, Shin SH, Kim YS. Effects of Helicobacter pylori eradication in patients with immune thrombocytopenic purpura. Korean J Hematol. 2010;45:127–32. doi: 10.5045/kjh.2010.45.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sato R, Murakami K, Okimoto T, Watanabe K, Kodama M, Fujioka T. Development of corpus atrophic gastritis may be associated with Helicobacter pylori-related idiopathic thrombocytopenic purpura. J Gastroenterol. 2011;46:991–7. doi: 10.1007/s00535-011-0416-8. [DOI] [PubMed] [Google Scholar]
- 131.Veneri D, Bonani A, Franchini M, Fedrizzi A, Pizzolo G. Idiopathic thrombocytopenia and Helicobacter pylori infection: platelet count increase and early eradication therapy. Blood Transfus. 2011;9:340–2. doi: 10.2450/2011.0014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Payandeh M, Sohrabi N, Zare ME, Kansestani AN, Hashemian AH. Platelet Count Response to Helicobacter pylori Eradication in Iranian Patients with Idiopathic Thrombocytopenic Purpura. Mediterr J Hematol Infect Dis. 2012;4:e2012056. doi: 10.4084/MJHID.2012.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gan GG, Norfaizal AL, Bee PC, Chin EF, Habibah AH, Goh KL. Helicobacter pylori infection in chronic immune thrombocytopenic purpura patients in Malaysia. Med J Malaysia. 2013;68:231–3. [PubMed] [Google Scholar]
- 134.Takezako N, Sekiguchi N, Tanimura A, Homma C, Shikai T, Takezako Y, Yamagata N, Miwa A. Lymphocytosis in Idiopathic Thrombocytopenic Purpura Patients Infected by Helicobacter pylori. Open J Blood Dis. 2013;3:32–5. [Google Scholar]