

Abstract
The BCR–ABL fusion oncoprotein accelerates differentiation and proliferation of myeloid cells during the chronic phase of chronic myeloid leukemia (CP-CML). Here, the role of CCAAT/enhancer binding protein β (C/EBPβ), a regulator for ‘emergency granulopoiesis,’ in the pathogenesis of CP-CML was examined. C/EBPβ expression was upregulated in Lineage− CD34+ CD38− hematopoietic stem cells (HSCs) and myeloid progenitors isolated from bone marrow of patients with CP-CML. In EML cells, a mouse HSC line, BCR–ABL upregulated C/EBPβ, at least in part, through the activation of STAT5. Myeloid differentiation and proliferation induced by BCR–ABL was significantly impaired in C/EBPβ-deficient bone marrow cells in vitro. Mice that were transplanted with BCR–ABL-transduced C/EBPβ knockout bone marrow cells survived longer than mice that received BCR–ABL-transduced wild-type (WT) bone marrow cells. Significantly higher levels of leukemic stem cells were maintained in BCR–ABL-transduced C/EBPβ-deficient cells than in BCR–ABL-transduced WT cells. These results suggest that C/EBPβ is involved in BCR–ABL-mediated myeloid expansion. Further elucidation of the molecular mechanisms underlying the C/EBPβ-mediated stem cell loss might reveal a novel therapeutic strategy for eradication of CML stem cells.
Keywords: C/EBPβ, BCR–ABL, chronic myeloid leukemia
INTRODUCTION
Chronic phase chronic myeloid leukemia (CP-CML) is characterized by massive proliferation and differentiation of myeloid cells.1,2 In sharp contrast to acute myeloid leukemia with leukemic hiatus, both myeloid progenitors and mature granulocytes accumulate in the bone marrow, peripheral blood and spleen in CP-CML.1 The myeloid expansion in CP-CML has been attributed to the BCR–ABL fusion protein, resulting from a translocation between chromosomes 9 and 22.2–4 Experiments using transgenic mouse models have shown that the BCR–ABL-mediated leukemic status is reversed by suppression of BCR–ABL,5–7 suggesting that BCR–ABL is the sole cause of the myeloid expansion in CP-CML. In practice, inhibition of the BCR–ABL tyrosine kinase activity effectively controls the disease during the chronic phase in most cases.8–12 Recent findings suggested a hierarchical organization of CML hematopoiesis, with CML stem cells giving rise to heterogeneous progeny.13–15 Accumulating clinical experiences, together with experimental data, have shown that leukemic stem cells in CP-CML are resistant to TKIs (tyrosine kinase inhibitors) and sometimes causes a relapse of the disease after discontinuation of TKI treatment.16–19 Progression of CML toward the accelerated phase and blast crisis is considered to be a consequence of further acquisition of genetic mutations, which makes the disease more resistant to TKIs and results in an extremely poor prognosis.20,21 Therefore, a better understanding of the characteristics of leukemic stem cells, as well as the pathogenesis of CP-CML, are essential for establishing a novel therapeutic strategy for CML.
Granulopoiesis is a process in which hematopoietic stem cells (HSCs) give rise to mature granulocytes throughout life. Our previous findings revealed that a member of the CCAAT/enhancer binding protein (C/EBP) family of transcription factors, C/EBPβ, is required for ‘emergency granulopoiesis,’ which is characterized by the accelerated differentiation and proliferation of granulocytic precursors in response to infections or cytokine stimulation.22 Myeloid expansion is a common feature of both emergency granulopoiesis and CP-CML. However, little is known regarding the role of C/EBPβ in the pathogenesis of CP-CML. In this study, the effects of BCR–ABL on the expression and function of C/EBPβ in BCR–ABL-induced myeloid expansion was determined and the therapeutic implications of these data are discussed.
MATERIALS AND METHODS
Primary human bone marrow cells and cell lines
Frozen samples of human bone marrow cells from healthy donors or untreated CP-CML patients were purchased from AllCells LLC (Emeryville, CA, USA). In all cases, written informed consent was obtained according to the Institutional Review Board or Human Subject Committee approved donor program. The characteristics of the patients are shown in Supplementary Table 1. EML cells23 (a kind gift from Dr Schickwann Tsai at the University of Utah, UT, USA) were maintained in IMDM (Iscove modified Dulbecco’s medium) supplemented with 20% heat-inactivated horse serum and 15% BHK/MKL cell-conditioned medium.
Mice
C57BL/6 mice were purchased from Clea Japan (Tokyo, Japan). C/EBPβ knockout (KO) mice24 were back crossed to C57BL/6 strain mice at least eight times. Whenever C/EBPβ KO mice were analyzed, wild-type (WT) littermates were used as control. All mice were maintained under specific pathogen-free conditions in the Institute of Laboratory Animals, Kyoto University. All experiments were performed according to the institutional guidelines of Kyoto University.
Reagents
A STAT5 inhibitor (N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide) and Ly294002, a PI3K inhibitor, were purchased from Merck (Darmstadt, Germany). U0126, a MEK inhibitor, was purchased from Cell Signaling Technology (Danvers, MA, USA). Stock solutions of the STAT5 inhibitor, Ly294002, and U0126 were made in DMSO (dimethyl sulfoxide).
Plasmids
The pMSCVneo vector and pMSCV-internal ribosome entry site-green fluorescent protein (GFP) vector (MIG),25 and their derivatives for the expression of BCR–ABL (p210), were kind gifts from Dr Keiko Okuda (Kyoto Prefectural University of Medicine, Kyoto, Japan). Retroviruses expressing the constitutively active STAT5 mutant (STAT51*6)26 and the dominant-negative STAT5 mutant (STAT5Δ749)27 were kind gifts from Dr Toshio Kitamura (University of Tokyo, Tokyo, Japan).
Retrovirus infection
Plat-E packaging cells28 were transfected with retrovirus vectors using FuGENE6 (Roche Diagnostics, Mannheim, Germany) as previously described.22,28 Bone marrow cells were harvested from mice (4–8 weeks of age) treated with 5-fluorouracil (5-FU) (150 mg/kg, intraperitoneally). The bone marrow cells were cultured at 37 °C for 48 h in IMDM containing 15% fetal bovine serum, 50 μM 2-mercaptoethanol, 50 ng/ml mouse stem cell factor, 50 ng/ml human thrombopoietin (a kind gift from Kyowa Hakko Kirin Co., Ltd), 50 ng/ml mouse fms-like tyrosine kinase 3 ligand (FL), and 10 ng/ml mouse interleukin-6 (IL-6). First round retroviral infection was carried out using RetroNectin (Takara Bio, Otsu, Japan) in the same medium. For the second round infection, polybrene was added with the retroviral supernatant.
Bone marrow transplantation
Recipient C57BL/6 mice (8–10 weeks of age) were lethally irradiated (10 Gy). MIG or MIG-BCR–ABL-transduced bone marrow cells (0.5–1×105 GFP-positive cells per mouse) were injected into the tail vein of primary recipient mice. For radioprotection, 2×105 cells of freshly harvested whole bone marrow were co-transplanted. For secondary transplantation, bone marrow cells were harvested from mice that received the primary transplants and 0.2–2×106 GFP-positive cells were intravenously injected into sublethally irradiated secondary recipient mice. The frequencies of leukemic stem cells were calculated using the L-Calc software (StemCell Technologies, Vancouver, Canada).
Methylcellulose colony-forming assay
MIG-BCR–ABL-transduced mouse bone marrow cells from C/EBPβ KO or WT mice were subjected to a methylcellulose colony-forming assay using the cytokine-free medium Methocult 3231 (StemCell Technologies). The fluorescence images of the colonies were obtained using AxioCam MRm digital camera and Axio Vision software in combination with SteREO Lumar V12 microscope and Neolumar S objective lens (×0.8) (Carl Zeiss, Oberkochen, Germany).
Wright Giemsa staining
Smears of mice peripheral blood or bone marrow cells and cytospin slides of EML cells or colony-forming cells were stained using a Diff-Quik kit (Sysmex, Kobe, Japan), a modified Wright Giemsa staining system. Images were obtained using DP71 digital camera and DP Controller software in combination with CX41 microscope and PlanCN objective lens (×40/0.65 numerical aperture) (Olympus, Tokyo, Japan).
Flow cytometric analysis
Flow cytometric analysis and cell sorting were performed with a FACSCalibur, FACSCanto II, or FACSAria instrument (BD Biosciences, San Jose, CA, USA). As lineage markers for human bone marrow mononuclear cells, phycoerythrin (PE)-Cy5-conjugated anti-CD235, biotin-conjugated anti-CD3, CD4, CD8, CD11b, CD14, CD19, CD20 (eBioscience, San Diego, CA, USA) and anti-CD56 antibodies (Bio Legend, San Diego, CA, USA) were used and followed by staining with streptavidin-PE-Cy5. Cells were further stained with APC (allophycocyanin)-conjugated anti-CD34 (8G12), PE-conjugated anti-CD38 (HIT2), fluorescein isothiocyanate-conjugated anti-CD45RA (HI100) (all from BD Pharmingen, San Diego, CA, USA) and PE-Cy7-conjugated anti-CD123 antibodies (eBioscience) for definition of HSCs and myeloid progenitors. For the staining of mouse cells, PerCP-Cy5.5-conjugated anti-CD3, CD4, CD11b, B220 and Ter119 antibodies, and PE-Cy5.5-conjugated anti-CD8 and Gr-1 antibodies (all from eBioscience) were used as lineage markers. PE-Cy7-conjugated anti-Sca-1, PE-conjugated anti-CD11b and APC-conjugated anti-c-kit antibodies (all from eBioscience) were used for definition of HSCs and myeloid cells. Data were analyzed with the FlowJo software (Tree Star, Ashland, OR, USA).
Real-time reverse transcriptase PCR
Total RNA was extracted with an RNeasy Micro Kit (Qiagen, Valencia, CA, USA) and converted to cDNA using random primers. The cDNA was amplified using an Applied Biosystems Step One Plus thermal cycler (Applied Biosystems, Carlsbad, CA, USA). The following parameters were used: 95 °C for 20 s, followed by 45 cycles at 95 °C for 1 s and 60 °C for 20 s. The following primers, and probes from the Universal Probe Library (Roche Applied Science, Mannheim, Germany) were used: for mouse C/EBPβ, probe #55, and primers 5′-ATCGACTTCAGCCCCTACCT-3′ and 5′-TA GTCGTCGGCGAAGAGG-3′; for mouse GAPDH (glyceraldehyde 3-phosphate dehydrogenase), probe #80, and primers 5′-TGTCCGTCGTGGATCTGAC-3′ and 5′-CCTGCTTCACCACCTTCTTG-3′; for human C/EBPβ, probe #74, and primers 5′-CGCTTACCTCGGCTACCA-3′ and 5′-ACGAGGAGGACGTGGAGAG-3′; and for human GAPDH, probe #60, and primers 5′-AGCCACATCGCTCAGA CAC-3′ and 5′-GCCCAATACGACCAAATCC-3′. Results were normalized by the level of GAPDH mRNA.
Western blot analysis
Cells were diluted with equal amounts of Laemmli sample buffer and boiled at 100 °C for 10 min. Samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. A ‘Can Get’ Signal immunoreactions enhancer kit (Toyobo, Osaka, Japan) was used to dilute the primary and secondary antibodies. Antibodies specific for C/EBPβ (sc-150, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and GAPDH (sc-25778, Santa Cruz Biotechnology) were used as primary antibodies. Immunoreactive proteins were detected using horseradish peroxidase-conjugated anti-rabbit IgG (NA934V, GE Healthcare, Little Chalfont, UK) and visualized using enhanced chemiluminescence (GE Healthcare).
Statistics
Statistical analyses were performed using Student’s t-test. Survival of mice was analyzed using the log-rank test. P-values <0.05 were considered statistically significant.
RESULTS
C/EBPβ is upregulated in bone marrow HSCs and myeloid progenitors from patients with CP-CML
C/EBPβ expression is upregulated or maintained in normal myeloid progenitors during emergency granulopoiesis, while the expression of all other C/EBP family members was downregulated.22 Therefore, the expression of C/EBPβ in bone marrow cells of patients with CP-CML was examined. Among the lineage marker negative (Lin−) bone marrow cells, the frequency of CD34+ CD38− HSCs was lower and the frequency of CD34+ CD38+ myeloid progenitors was higher in CP-CML bone marrow than in bone marrow from healthy donors (Figures 1a and b; Supplementary Figure S1). The CD34+ CD38+ population was further subdivided into common myeloid progenitors (CMPs), granulocyte–macrophage progenitors (GMPs) and megakaryocyte–erythrocyte progenitors (MEPs) based on the expression levels of CD123 and CD45RA29 (Figure 1a). The frequency of GMPs was significantly lower and that of MEPs was significantly higher (approximately twofold) in CP-CML bone marrow than in bone marrow from healthy donors (Figure 1c). The differences between normal and CP-CML bone marrow in the frequency of CD34+ CD38− HSCs and of myeloid progenitors were consistent with previous findings.13,15,30 The expression of C/EBPβ in the purified progenitors was measured. The levels of C/EBPβ mRNA in HSCs and all the myeloid progenitors from CP-CML bone marrow were significantly higher than the levels in bone marrow from healthy donors (Figure 1d, healthy donors, n = 6; CP-CML, n = 5; GMPs, P<0.01; HSCs, CMPs and MEPs, P<0.05). These results suggest that C/EBPβ is upregulated in HSCs and myeloid progenitors in patients with CP-CML.
Figure 1.
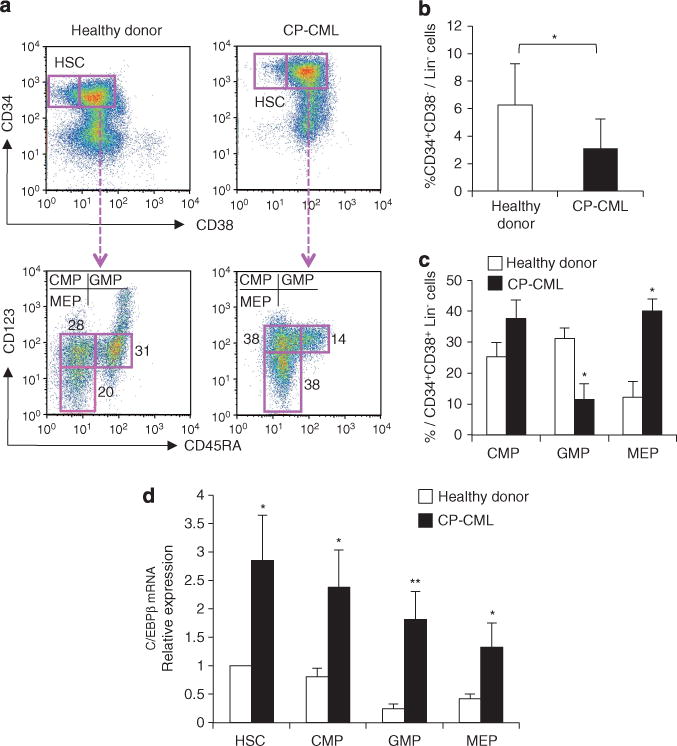
C/EBPβ expression in hematopoietic stem and progenitor cells from the bone marrow of CP-CML patients. (a) Flow cytometric analysis of lineage marker negative (Lin−) bone marrow cells from healthy donors and CP-CML patients identified CD34+ CD38− HSCs and CD34+ CD38+ myeloid progenitors (upper panels). Myeloid progenitors were further subdivided into CMPs, GMPs and MEPs based on the expression of CD45RA and CD123 (lower panels). The data shown are representative of six independent experiments. (b) Frequency of CD34+ CD38− HSCs within the Lin− fraction of bone marrow cells. Error bars indicate s.d. (healthy donors, n=6; CP-CML, n=5; *P<0.05). (c) Percentage of CMPs, GMPs and MEPs in the Lin− CD34+ CD38+ fraction. Error bars indicate s.d. (healthy donors, n=6; CP-CML, n=5; *P<0.001). (d) C/EBPβ mRNA levels in purified HSCs, CMPs, GMPs and MEPs. Results were normalized to the expression level of HSCs from healthy donors (healthy donors, n = 6; CP-CML, n = 5; *P<0.05; **P<0.01).
BCR–ABL upregulates C/EBPβ in EML cells, a mouse HSC line
The presence of the BCR–ABL fusion protein is thought to be the only difference between normal hematopoiesis and BCR–ABL-mediated myeloid expansion in CP-CML.2,4 To assess whether BCR–ABL could upregulate C/EBPβ, the BCR–ABL gene was retrovirally introduced into a factor-dependent mouse HSC line, EML cells, and the expression of C/EBPβ was compared with EML cells transduced with a control vector (EML-control). After transduction with BCR–ABL, EML cells (EML-BCR–ABL) became factor-independent and could be cultured long term in the absence of stem cell factor-containing conditioned medium. The morphologies of the EML-control and EML-BCR–ABL cells were indistinguishable by Giemsa staining (Figure 2a, day 0). The parent EML cells expressed c-kit but not CD11b. Flow cytometric analysis of the transduced cells revealed that a small subset of EML-BCR–ABL expressed c-kit at a slightly lower intensity and expressed CD11b weakly (Figure 2b, day 0). Myeloid differentiation of EML cells can be induced by the addition of IL-3, retinoic acid and GM-CSF (granulocyte–macrophage colony stimulating factor).23 As shown in Figure 2a (day 5), myeloid differentiation of both EML-control and EML-BCR–ABL cells was effectively induced. Lower c-kit and higher CD11b expression by EML-control cells was observed 5 days after the induction of differentiation and the extent of the changes in the expression of c-kit and CD11b was more evident in EML-BCR–ABL cells (Figure 2b). These results suggest that BCR–ABL enhanced myeloid differentiation of immature cells such as EML cells.
Figure 2.
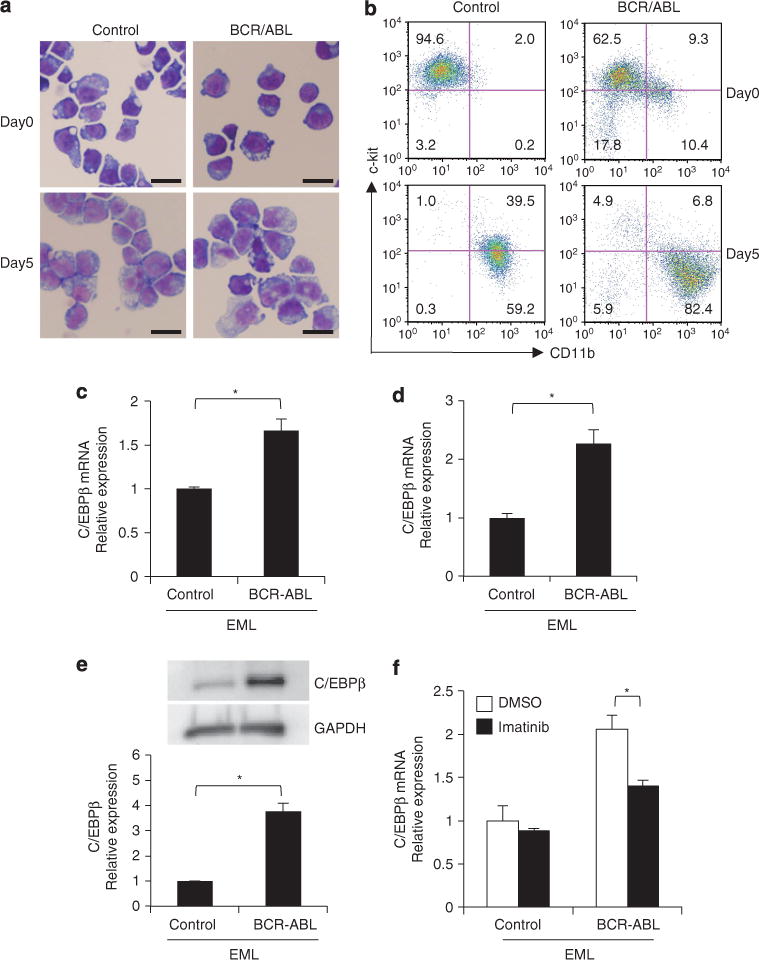
Effects of BCR–ABL on the expression of C/EBPβ in EML cells. (a) Wright Giemsa staining of pMSCVneo vector-transduced EML cells (EML-control) and BCR–ABL-containing pMSCVneo vector-transduced EML cells (EML-BCR–ABL) before (day 0) and after (day 5) the induction of myeloid differentiation (scale bar, 20 μm; original magnification, ×400). (b) Flow cytometric analysis of c-kit and CD11b expression in EML-control and EML-BCR–ABL cells (day 0 and day 5 after myeloid differentiation). Numbers in each quadrant indicate the percentage of live cells. (c) C/EBPβ mRNA levels in EML-control and EML-BCR–ABL cells. Results were normalized to the expression level of control. Error bars indicate s.d. from duplicate samples. Results are representative of three independent experiments. *P=0.011. (d) C/EBPβ mRNA levels in c-kit+ CD11b− fraction of the EML-control cells and EML-BCR–ABL cells. Results were normalized to the expression level of control. Error bars indicate s.d. from duplicate samples. *P<0.01. (e) C/EBPβ protein levels in EML-control and EML-BCR–ABL cells. Results were normalized to the expression level of control. Error bars indicate s.d. from duplicate samples. Results are representative of three independent experiments. *P<0.01. (f) C/EBPβ mRNA levels in EML-control and EML-BCR–ABL cells with or without a 48-h treatment with imatinib mesylate (100 nM). Dimethyl sulfoxide (DMSO) was used as the control. Results were normalized to the expression level of control. Error bars indicate s.d. from duplicate samples. Results are representative of three independent experiments. *P=0.016.
The amount of C/EBPβ mRNA in undifferentiated EML-BCR–ABL cells was 1.87-fold higher than in undifferentiated EML-control cells (Figure 2c). When the c-kit+ CD11b− fraction of the EML-control cells and EML-BCR–ABL cells was analyzed, a significant difference was still observed 2.26-fold higher in EML-BCR–ABL cells (Figure 2d), suggesting that the upregulation of C/EBPβ was not the result of contamination of differentiated cells. The level of C/EBPβ protein was 3.76-fold higher in EML-BCR–ABL cells relative to EML-control cells (Figure 2e). When EML-BCR–ABL cells were treated with imatinib mesylate, the upregulation of C/EBPβ by BCR–ABL was reduced (Figure 2f), while the level of C/EBPβ in EML-control cells was not affected. These results suggest that C/EBPβ is upregulated directly in response to signaling downstream of BCR–ABL.
STAT5 is involved in the BCR–ABL-mediated upregulation of C/EBPβ
Various signaling pathways are activated by BCR–ABL, including the JAK/STAT, Raf/MEK/ERK and PI3K/AKT pathways.31–36 To elucidate the signaling pathways responsible for the upregulation of C/EBPβ, each of the known downstream signaling pathways was inhibited. When EML-BCR–ABL cells were treated with the MEK inhibitor U0126 or the PI3K inhibitor Ly294002, C/EBPβ expression was not affected (Figure 3a). In contrast, treatment with the STAT5 inhibitor (N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide) significantly reduced C/EBPβ expression in EML-BCR–ABL cells (Figure 3b). A dominant-negative STAT5 mutant, STAT5Δ749, was introduced into the EML-derived cell lines to inhibit STAT5. STAT5Δ749 significantly repressed the expression of C/EBPβ in EML-BCR–ABL cells but had no effect in EML-control cells (Figure 3c). Conversely, when a constitutively active STAT5 mutant, STAT51*6, was retrovirally transduced into the parental EML cells (EML-CA-STAT5), C/EBPβ mRNA levels were significantly greater compared with the level in EML cells transduced with a control vector (Figure 3d). These results suggest that STAT5 is involved in the BCR–ABL-mediated upregulation of C/EBPβ.
Figure 3.
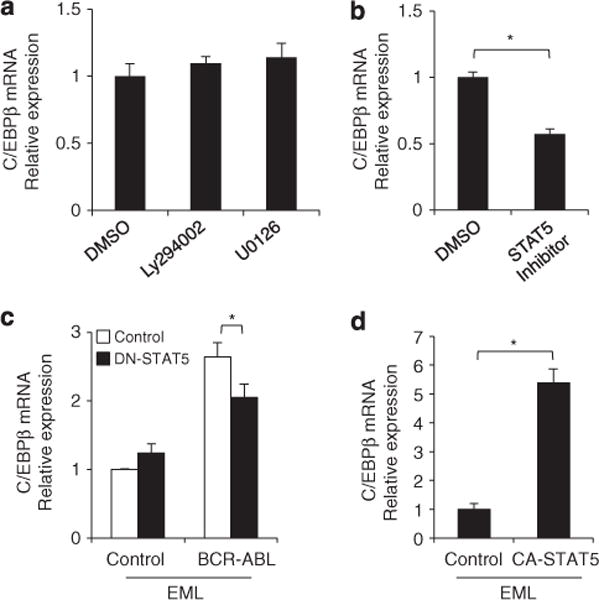
Involvement of BCR–ABL downstream signaling pathways in the upregulation of C/EBPβ. Changes in C/EBPβ mRNA in EML-BCR–ABL cells 24 h after treatment with the PI3K inhibitor Ly294002 (2.5 μM), the MEK inhibitor U0126 (25 μM) (a), or a STAT5 inhibitor (40 μM) (b). DMSO was used as the control. Results were normalized to the expression level of control. Error bars indicate s.d. from duplicate samples. Results are representative of two independent experiments. *P<0.01. (c) C/EBPβ mRNA in EML-control and EML-BCR–ABL cells transduced with a dominant-negative STAT5 mutant, STAT5Δ749. Results were normalized to the expression level of control. Error bars indicate s.d. from duplicate samples. Results are representative of two independent experiments. *P<0.05. (d) C/EBPβ mRNA levels in EML cells transduced with a constitutively active STAT5 mutant (CA-STAT5: STAT51*6) or empty-vector. Results were normalized to the expression level of control. Error bars indicate the s.d. from duplicate samples. Results are representative of two independent experiments. *P<0.01.
C/EBPβ regulates BCR–ABL-mediated proliferation and differentiation of myeloid cells in vitro
The upregulation of C/EBPβ in the presence of BCR–ABL was confirmed using primary bone marrow cells. BCR–ABL was retrovirally introduced into bone marrow cells obtained from 5-FU-treated WT mice and RNA was extracted from purified c-kit+ Sca-1+ Lin− (KSL) cells after the transduced cells were cultured for 2 days. The RNA was analyzed by quantitative reverse transcriptase PCR. The expression of C/EBPβ in BCR–ABL-transduced KSL cells was significantly greater than in control vector-transduced KSL cells (n = 3 each, P<0.01) (Figure 4a).
Figure 4.
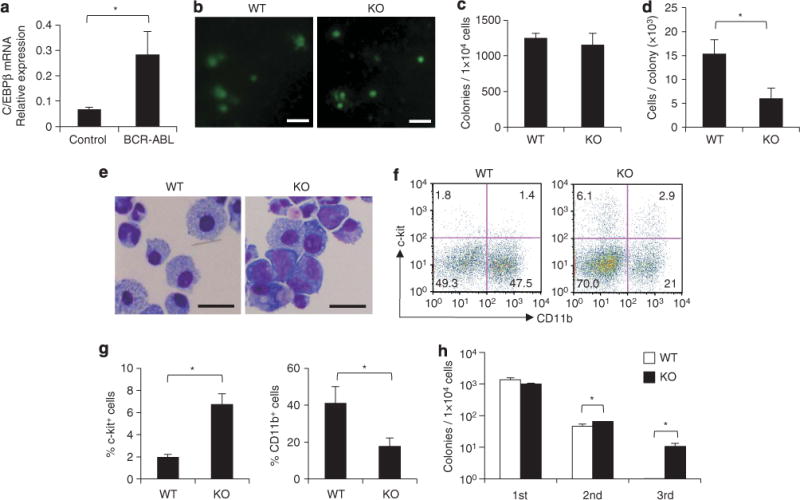
BCR–ABL-mediated in vitro colony formation in the absence of C/EBPβ. (a) C/EBPβ mRNA levels in c-kit+ Sca-1+ Lin− cells from WT bone marrow cells transduced with a control MIG vector or a MIG-BCR–ABL vector (n=3 each, P<0.01). (b) Colonies formed by BCR–ABL-transduced C/EBPβ KO bone marrow cells and WT bone marrow cells after culture for 7 days in cytokine-free methylcellulose (scale bar, 1 mm). Colony numbers (c), cell numbers per colony (d), and Wright Giemsa staining (scale bar, 20 mm; original magnification, ×400) (e) of the colony-forming cells are shown. Error bars indicate s.d. from triplicate cultures. Results are representative of three independent experiments. *P<0.01. (f) Flow cytometric analysis of the cells forming primary colonies by day 7. Numbers in each quadrant indicate the percentage of live cells. (g) Frequency of c-kit+ cells and CD11b+ cells in the cells forming primary colonies by day 7. Results are representative of two independent experiments. *P<0.05. (h) Serial colony-replating of 1×104 BCR–ABL-transduced C/EBPβ WT and KO bone marrow cells. The colonies were counted and collected on day 10 (second) or 14 (third), respectively. Error bars indicate s.d. from triplicate cultures. Results are representative of three independent experiments. *P<0.01.
To understand the role of C/EBPβ in BCR–ABL-mediated myeloid expansion, BCR–ABL and GFP were retrovirally introduced into bone marrow cells obtained from 5-FU-treated C/EBPβ KO mice and their properties were compared with BCR–ABL-transduced bone marrow cells from WT mice. The transduced cells were first subjected to cytokine-free semisolid methylcellulose culture. All the colonies formed on day 7 were GFP-positive and the numbers of colonies were equivalent between transduced bone marrow cells from WT and KO mice (Figures 4b and c), suggesting that the frequencies of clonogenic progenitors were similar. The majority of C/EBPβ KO cell-derived colonies were smaller in size (Figure 4b) and the cell number per C/EBPβ KO cell colony was significantly lower than WT cell colonies (P<0.01) (Figure 4d). These findings suggest that the loss of C/EBPβ impaired BCR–ABL-mediated cell proliferation. Having observed a difference between WT and C/EBPβ KO cells in proliferative capacity in the presence of the BCR–ABL fusion protein, the morphologies of the colony-forming cells were assessed by Giemsa staining (Figure 4e). The BCR–ABL-transduced C/EBPβ KO cell colonies contained more immature myeloid cells with larger nuclei and basophilic cytoplasm, whereas the BCR–ABL-transduced WT cell colonies contained more mature neutrophilic granulocytes and macrophages (Figure 4e). Flow cytometric analysis revealed that the BCR–ABL-transduced C/EBPβ KO cells gave rise to a significantly higher proportion of c-kit+ cells and a lower proportion of CD11b+ cells, relative to BCR–ABL-transduced WT cells (%c-kit+ cells = 6.7±1.0% vs 2.0±0.2%, P=0.01; %CD11b+ cells=17.9±4.4% vs 41.3±8.8%, P=0.039) (Figures 4f and g). In addition, BCR–ABL-transduced C/EBPβ KO cells-derived colonies could be replated more than three times in cytokine-free medium, whereas BCR–ABL-transduced WT cells stopped growing after being replated only twice (Figure 4h; Supplementary Figure S2). These results suggest that the C/EBPβ deficiency abolished the BCR–ABL-mediated proliferation and differentiation of myeloid cells.
Loss of C/EBPβ delays BCR–ABL-mediated myeloid expansion in vivo
BCR–ABL-transduced bone marrow cells from C/EBPβ KO mice or WT mice were transplanted into lethally irradiated recipient mice to clarify the role of C/EBPβ in the in vivo myeloproliferation induced by BCR–ABL. After transplantation of transduced cells, increases in neutrophilic granulocytes were observed in the peripheral blood of mice having received either WT cells or KO cells (Figure 5a). Flow cytometric analysis of peripheral blood also revealed that over 70% of nucleated cells were CD11b+ in both WT and KO transplant recipients (Figure 5b). In mice transplanted with BCR–ABL-transduced C/EBPβ KO cells, the increase in white blood cell count (Figure 5c) and the development of splenomegaly (Figure 5d) were delayed compared with mice receiving transplants of WT BCR–ABL-transduced cells. The median survival of mice transplanted with BCR–ABL-transduced WT cells was 19 days. In contrast, the median survival of mice transplanted with BCR–ABL-transduced C/EBPβ KO cells was 31 days, significantly longer than mice receiving transplants of BCR–ABL-transduced WT cells (P=0.0005) (Figure 5e). These results suggest that C/EBPβ is involved in BCR–ABL-induced enhanced myelopoiesis in vivo.
Figure 5.
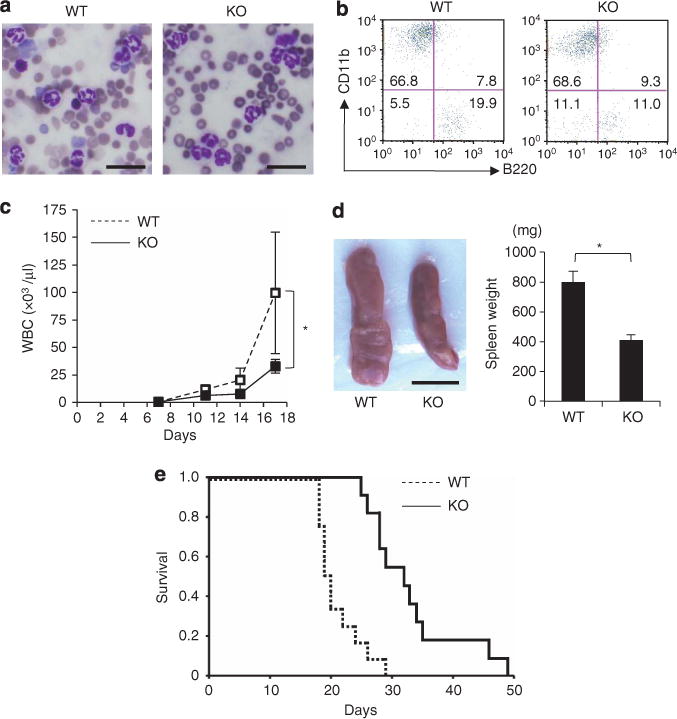
C/EBPβ deficiency alters the BCR–ABL-dependent myeloproliferative status in vivo. Wright Giemsa staining (scale bar, 20 μm; original magnification, ×400) (a), 20 days post-transplantation, and flow cytometric analysis (b), 28 days post-transplantation, of peripheral blood from recipient mice transplanted with BCR–ABL-transduced WT cells or BCR–ABL-transduced C/EBPβ KO cells. Numbers in quadrants indicate the percentages within peripheral blood nucleated cells. (c) Peripheral white blood cell count of recipients after transplantation. Data are expressed as mean±s.d. (n = 4 each; *P<0.05). (d) Splenomegaly observed in recipients at day 19 after transplantation (scale bar, 10 mm). Spleen weights are presented as mean±s.d. (WT, n = 9; KO, n = 6; *P<0.01). (e) Survival of recipients transplanted with BCR–ABL-transduced WT cells or BCR–ABL-transduced C/EBPβ KO cells (WT, n=12; KO, n = 11; *P=0.0005).
BCR–ABL-induced loss of self-renewing hematopoietic/leukemic stem cells was attenuated in the absence of C/EBPβ
In vitro experiments revealed that C/EBPβ KO bone marrow cells retained more c-kit+ immature cells and could be replated more times after transduction with BCR–ABL (Figures 4f–h; Supplementary Figure S2), suggesting that C/EBPβ is involved in the BCR–ABL-mediated loss of self-renewing hematopoietic/leukemic stem cells. Consistent with these observations, BCR–ABL-transduced C/EBPβ KO cells had given rise to a higher proportion of c-kit+ cells than BCR–ABL-transduced WT cells on day 19 after transplantation (16.0±2.6% vs 5.5±4.6%, P=0.01) (Figures 6a and b).
Figure 6.
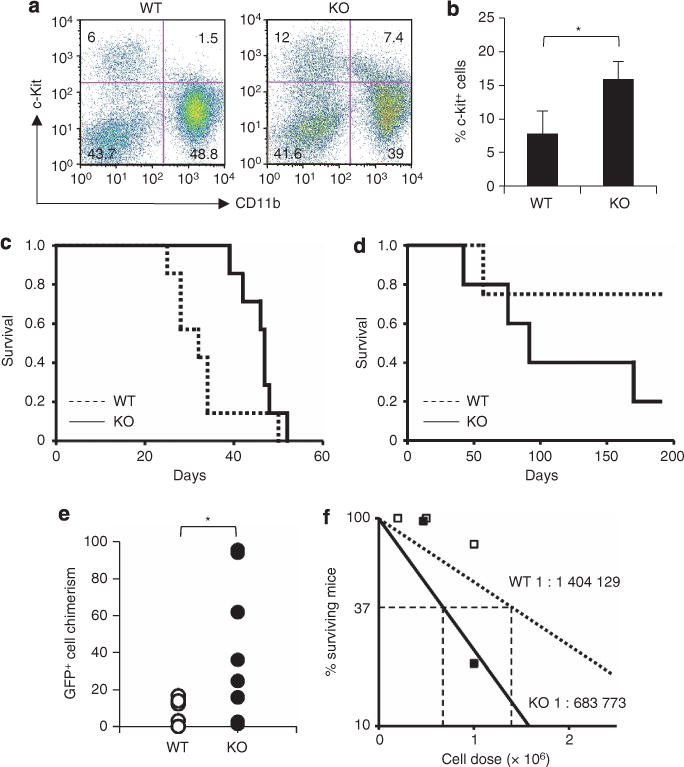
Involvement of C/EBPβ in BCR–ABL-mediated loss of immature hematopoietic cells. Flow cytometric analysis of GFP+ bone marrow cells from recipients 19 days after transplantation of BCR–ABL-transduced WT cells or BCR–ABL-transduced C/EBPβ KO cells (a, b). Numbers in quadrants indicate the percentages within GFP+ bone marrow cells (WT, n=2; KO, n=3; *P=0.01). (c, d) Survival of secondary transplantation recipients. 2×106 (c; WT, n=7; KO, n=7) or 1×106 BCR–ABL-transduced GFP+ cells (d; WT, n=4; KO, n=5) from the first recipients were transplanted. (e) GFP+ cell chimerism on day 38 after the secondary transplantation of BCR–ABL-transduced WT cells or BCR–ABL-transduced C/EBPβ KO cells (n=8 each; *P=0.013). (f) The frequencies of leukemia-initiating cells in bone marrow of primary recipient mice transplanted with BCR–ABL-transduced WT cells or BCR–ABL-transduced C/EBPβ KO cells.
Serial transplantation experiments were carried out to determine the role of C/EBPβ in the loss of self-renewal capacity. When 2×106 GFP+ bone marrow cells from the primary recipients were transplanted into sublethally irradiated secondary recipients, all the mice having received either BCR–ABL-transduced WT cells or BCR–ABL-transduced KO cells developed a myeloproliferative status, reminiscent of the first transplantation (Figure 6c). After transplantation of 1×106 GFP+ bone marrow cells from the primary transplant recipients, one of the four recipient mice that received a secondary transplant of BCR–ABL-transduced WT cells developed a myeloproliferative status and four out of five recipient mice that received a secondary transplant of BCR–ABL-transduced C/EBPβ KO cells developed a myeloproliferative disorder (Figure 6d). On day 38 after the secondary transplantation of 1×106 GFP+ bone marrow cells, BCR–ABL-transduced C/EBPβ KO cells achieved a significantly higher degree of chimerism in the peripheral blood of the secondary recipients than BCR–ABL-transduced WT cells (n=8 each, P=0.013) (Figure 6e). None of the mice that received <0.5×106 GFP+ bone marrow cells from the primary recipients developed a myeloproliferative status. Based on these transplantation experiments, the estimated frequencies of leukemia-initiating cells in bone marrow of primary recipient mice transplanted with BCR–ABL-transduced WT cells and BCR–ABL-transduced C/EBPβ KO cells were 1 in 1 404 129 and 1 in 683 773, respectively (Figure 6f). These results suggest that enhanced C/EBPβ expression induced by BCR–ABL was involved in the loss of the self-renewal potential of leukemic stem cells.
DISCUSSION
Proliferation and differentiation of myeloid cells are unique to CP-CML. This is the first report demonstrating that the BCR–ABL-mediated myeloid expansion in CP-CML is promoted by C/EBPβ, a regulator of ‘emergency granulopoiesis.’
One of the major findings of this study is the upregulation of C/EBPβ by BCR–ABL in CP-CML. In EML cells or in immature mouse hematopoietic cells, BCR–ABL upregulated C/EBPβ and accelerated the differentiation of these cells (Figures 2b–e and Figure 4a). In contrast, previous reports showed that, in 32Dcl3 cells transduced with BCR–ABL, C/EBPβ was downregulated and granulocytic differentiation was blocked.37,38 One explanation for the discrepancy between the observations in EML cells and 32Dcl3 cells may be due to the differentiation status. 32Dcl3 cells give rise only to neutrophilic granulocytes in the presence of G-CSF,39 while EML cells are multipotent and can give rise to monocytic, erythroid and lymphoid lineages, in addition to the granulocytic lineage,23 suggesting that EML cells represent more immature hematopoietic cells than 32Dcl3 cells. The higher C/EBPβ expression in 32Dcl3 than in EML cells (Supplementary Figure S3) also supports this idea, as C/EBPβ is upregulated during myeloid differentiation.40,41 Taking into account the previous findings that HSCs are the target cell population for BCR–ABL during chronic phase,4 the data from EML cells are most likely to reflect conditions of CP-CML. Consistent with our observations, Minami et al.42 found that C/EBPβ is one of the markedly upregulated genes in a pluripotent hematopoietic cell line transduced with BCR–ABL. Most importantly, C/EBPβ is upregulated in purified HSCs and myeloid progenitors obtained from patients with CP-CML, as shown in Figure 1d. The differences in BCR–ABL-mediated regulation of C/EBPβ in EML cells and in 32Dcl3 cells also suggested the involvement of cell context-dependent machinery. Guerzoni et al.37 described a downregulation of C/EBPβ associated with the progression of CML toward a blast crisis, whereas our study focused on the upregulation of C/EBPβ in the chronic phase. The changes in the BCR–ABL-mediated regulation of C/EBPβ during the progression of CML may be a consequence of genetic or epigenetic changes, which result in a cell context similar to 32Dcl3 cells, in terms of the regulation of C/EBPβ.
BCR–ABL activates a number of signaling cascades through its tyrosine kinase activity. STAT5 is a well-known target of BCR–ABL. STAT5 is phosphorylated by JAK2(ref. 43) and is thought to activate target genes to induce or maintain CP-CML.33,44 Our current data strongly suggest that C/EBPβ resides downstream of STAT5. Transplantation of BCR–ABL-transduced STAT5 KO bone marrow cells resulted in delayed progression of a myeloproliferative disorder in mice,33 a phenotype highly similar to our CP-CML model using C/EBPβ KO cells. The similarities between the behavior of BCR–ABL-transduced STAT5 KO cells and BCR–ABL-transduced C/EBPβ KO cells strongly suggested that these two molecules act sequentially in the same pathway. STAT5 and C/EBPβ are both essential for cytokine-induced granulopoiesis,22,45–47 suggesting that STAT5 and C/EBPβ may also interact when emergency granulopoiesis is stimulated. There has been no evidence, thus far, to demonstrate that STAT5 directly regulates C/EBPβ. The consensus binding site for STAT5 (TTCN3GAA) is not found in the proximal promoter region of C/EBPβ. Recently, Zhang et al.48 showed that STAT3 regulates C/EBPβ transcription through binding to the IL-6 response element II (CTGGGA) located at −1180 base pairs in the C/EBPβ promoter. However, our preliminary analysis of the 2000 base pairs proximal promoter region of C/EBPβ by reporter assay failed to identify the positive regulatory elements which respond to STAT5 (data not shown). In addition, specific binding of STAT5 to the particular regulatory element was not observed in published chromatin immunoprecipitation sequencing data analyzing T cells stimulated with IL-2 (GSE12346(ref. 49) and GSE26553(ref. 50)). These data suggest that STAT5 might upregulate C/EBPβ through binding to regulatory elements outside this IL-6 response element II or through other indirect mechanisms. Elucidation of the direct or indirect interactions between STAT5 and C/EBPβ is necessary for further understanding of BCR–ABL-mediated myeloid expansion and emergency granulopoiesis.
Our present data showed that myeloid expansion was delayed when BCR–ABL was transduced into C/EBPβ KO bone marrow cells, clearly suggesting the involvement of C/EBPβ in the enhanced myelopoiesis observed in patients with CP-CML. The effects of BCR–ABL on the self-renewing potential of HSCs are still controversial. Schemionek et al.51 recently reported that BCR–ABL partially impaired long-term HSCs. In the present study, a phenotypically and functionally immature status was maintained in a greater fraction of BCR–ABL-transduced C/EBPβ KO cells than in BCR–ABL-transduced WT cells both in vitro and in vivo. These data suggested that C/EBPβ is involved in the BCR–ABL-mediated loss of self-renewing potential of the HSCs. From the therapeutic point of view, repression of C/EBPβ in CP-CML patients would delay the progression of the disease, although leaving leukemic stem cells relatively intact. This strategy might be effective for patients with BCR–ABL mutations that are resistant to TKIs, as the role of C/EBPβ in the pathogenesis of CP-CML should be common to all BCR–ABL mutants. Alternatively, upregulation of C/EBPβ in leukemic stem cells might induce exhaustion of the leukemic stem cells, leading to a complete cure for the disease. Actually, Guerzoni et al.37,52 showed that transduction of C/EBPβ promotes differentiation of BCR–ABL-expressing cells. The effects of upregulation of endogenous C/EBPβ on leukemic stem cells should be determined in the future. Further understanding of the regulation of the self-renewal and differentiation of leukemic stem cells in CP-CML may lead to identification of novel therapies for CML based on the regulation of C/EBPβ.
The leukocytosis observed during infections (=emergency granulopoiesis) is sometimes called a ‘leukemoid’ reaction because of the great increase in the number of myeloid cells with a ‘left shift’ in the shape of the nucleus.53 Here, a molecular link between leukemoid reactions and CP-CML was identified. C/EBPβ fine tunes the proliferation and differentiation of HSCs and CML leukemic stem cells in response to their respective upstream signals. Further elucidation of the molecular mechanisms that regulate the self-renewal and differentiation of stem cells in CP-CML and emergency granulopoiesis will facilitate the development of novel strategies for the treatment of CML.
Supplementary Material
Acknowledgments
We thank Dr Toshio Kitamura (University of Tokyo, Tokyo, Japan) and Dr Keiko Okuda (Kyoto Prefectural University of Medicine, Kyoto, Japan) for providing us with STAT5 mutant vectors and BCR–ABL-expressing vectors, respectively. We are grateful to Mikiko Katakami, Yoko Nakagawa and Yoshiko Manabe for their excellent technical assistance. This work was partly supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science and the Global COE Program ‘Center for Frontier Medicine’ from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, Takeda Science Foundation, the Kobayashi Foundation for Cancer Research, and Senshin Medical Research Foundation.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
References
- 1.Cotta CV, Bueso-Ramos CE. New insights into the pathobiology and treatment of chronic myelogenous leukemia. Ann Diagn Pathol. 2007;11:68–78. doi: 10.1016/j.anndiagpath.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 3.Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340:1330–1340. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- 4.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 5.Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 6.Huettner CS, Koschmieder S, Iwasaki H, Iwasaki-Arai J, Radomska HS, Akashi K, et al. Inducible expression of BCR/ABL using human CD34 regulatory elements results in a megakaryocytic myeloproliferative syndrome. Blood. 2003;102:3363–3370. doi: 10.1182/blood-2003-03-0768. [DOI] [PubMed] [Google Scholar]
- 7.Koschmieder S, Gottgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood. 2005;105:324–334. doi: 10.1182/blood-2003-12-4369. [DOI] [PubMed] [Google Scholar]
- 8.Goldman JM, Melo JV. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 9.Kimura S, Naito H, Segawa H, Kuroda J, Yuasa T, Sato K, et al. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood. 2005;106:3948–3954. doi: 10.1182/blood-2005-06-2209. [DOI] [PubMed] [Google Scholar]
- 10.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 11.Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelo-genous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110:3540–3546. doi: 10.1182/blood-2007-03-080689. [DOI] [PubMed] [Google Scholar]
- 12.Hochhaus A, Baccarani M, Deininger M, Apperley JF, Lipton JH, Goldberg SL, et al. Dasatinib induces durable cytogenetic responses in patients with chronic mye-logenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia. 2008;22:1200–1206. doi: 10.1038/leu.2008.84. [DOI] [PubMed] [Google Scholar]
- 13.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granu-locyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 14.Passegue E, Weisman IL. Leukemic stem cells: where do they come from? Stem Cell Rev. 2005;1:181–188. doi: 10.1385/SCR:1:3:181. [DOI] [PubMed] [Google Scholar]
- 15.Bruns I, Czibere A, Fischer JC, Roels F, Cadeddu RP, Buest S, et al. The hemato-poietic stem cell in chronic phase CML is characterized by a transcriptional profile resembling normal myeloid progenitor cells and reflecting loss of quiescence. Leukemia. 2009;23:892–899. doi: 10.1038/leu.2008.392. [DOI] [PubMed] [Google Scholar]
- 16.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 17.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107:4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 18.Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 19.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121:396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 21.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 22.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPβeta is required for ‘emergency’ granulopoiesis. Nat Immunol. 2006;7:732–739. doi: 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 23.Tsai S, Bartelmez S, Sitnicka E, Collins S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 1994;8:2831–2841. doi: 10.1101/gad.8.23.2831. [DOI] [PubMed] [Google Scholar]
- 24.Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, et al. Lympho-proliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. EMBO J. 1995;14:1932–1941. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1:136–138. [PubMed] [Google Scholar]
- 26.Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18:3871–3879. doi: 10.1128/mcb.18.7.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moriggl R, Gouilleux-Gruart V, Jahne R, Berchtold S, Gartmann C, Liu X, et al. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996;16:5691–5700. doi: 10.1128/mcb.16.10.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 29.Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99:11872–11877. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz-Blanco E, Bruns I, Neumann F, Fischer JC, Graef T, Rosskopf M, et al. Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia. 2007;21:494–504. doi: 10.1038/sj.leu.2404549. [DOI] [PubMed] [Google Scholar]
- 31.Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–254. [PubMed] [Google Scholar]
- 32.Ilaria RL, Jr, Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271:31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 33.Ye D, Wolff N, Li L, Zhang S, Ilaria RL., Jr STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood. 2006;107:4917–4925. doi: 10.1182/blood-2005-10-4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 35.Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005;65:2047–2053. doi: 10.1158/0008-5472.CAN-04-3888. [DOI] [PubMed] [Google Scholar]
- 36.Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619–1630. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerzoni C, Bardini M, Mariani SA, Ferrari-Amorotti G, Neviani P, Panno ML, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood. 2006;107:4080–4089. doi: 10.1182/blood-2005-08-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuster C, Forster K, Dierks H, Elsasser A, Behre G, Simon N, et al. The effects of Bcr-Abl on C/EBP transcription-factor regulation and neutrophilic differentiation are reversed by the Abl kinase inhibitor imatinib mesylate. Blood. 2003;101:655–663. doi: 10.1182/blood-2002-01-0043. [DOI] [PubMed] [Google Scholar]
- 39.Steinman RA, Tweardy DJ. Granulocyte colony-stimulating factor receptor mRNA upregulation is an immediate early marker of myeloid differentiation and exhibits dysfunctional regulation in leukemic cells. Blood. 1994;83:119–127. [PubMed] [Google Scholar]
- 40.Du Y, Campbell JL, Nalbant D, Youn H, Bass AC, Cobos E, et al. Mapping gene expression patterns during myeloid differentiation using the EML hematopoietic progenitor cell line. Exp Hematol. 2002;30:649–658. doi: 10.1016/s0301-472x(02)00817-2. [DOI] [PubMed] [Google Scholar]
- 41.Bjerregaard MD, Jurlander J, Klausen P, Borregaard N, Cowland JB. The in vivo profile of transcription factors during neutrophil differentiation in human bone marrow. Blood. 2003;101:4322–4332. doi: 10.1182/blood-2002-03-0835. [DOI] [PubMed] [Google Scholar]
- 42.Minami Y, Stuart SA, Ikawa T, Jiang Y, Banno A, Hunton IC, et al. BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc Natl Acad Sci USA. 2008;105:17967–17972. doi: 10.1073/pnas.0808303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie S, Wang Y, Liu J, Sun T, Wilson MB, Smithgall TE, et al. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001;20:6188–6195. doi: 10.1038/sj.onc.1204834. [DOI] [PubMed] [Google Scholar]
- 44.Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2:98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feldman GM, Rosenthal LA, Liu X, Hayes MP, Wynshaw-Boris A, Leonard WJ, et al. STAT5A-deficient mice demonstrate a defect in granulocyte-macrophage colony-stimulating factor-induced proliferation and gene expression. Blood. 1997;90:1768–1776. [PubMed] [Google Scholar]
- 46.Kimura A, Rieger MA, Simone JM, Chen W, Wickre MC, Zhu BM, et al. The transcription factors STAT5A/B regulate GM-CSF-mediated granulopoiesis. Blood. 2009;114:4721–4728. doi: 10.1182/blood-2009-04-216390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akagi T, Saitoh T, O’Kelly J, Akira S, Gombart AF, Koeffler HP. Impaired response to GM-CSF and G-CSF, and enhanced apoptosis in C/EBPβeta-deficient hematopoietic cells. Blood. 2008;111:2999–3004. doi: 10.1182/blood-2007-04-087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol. 2008;9:1288–1296. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schemionek M, Elling C, Steidl U, Baumer N, Hamilton A, Spieker T, et al. BCR-ABL enhances differentiation of long-term repopulating hematopoietic stem cells. Blood. 2010;115:3185–3195. doi: 10.1182/blood-2009-04-215376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guerzoni C, Ferrari-Amorotti G, Bardini M, Mariani SA, Calabretta B. Effects of C/EBPalpha and C/EBPβeta in BCR/ABL-expressing cells: differences and similarities. Cell Cycle. 2006;5:1254–1257. doi: 10.4161/cc.5.12.2808. [DOI] [PubMed] [Google Scholar]
- 53.Sakka V, Tsiodras S, Giamarellos-Bourboulis EJ, Giamarellou H. An update on the etiology and diagnostic evaluation of a leukemoid reaction. Eur J Intern Med. 2006;17:394–398. doi: 10.1016/j.ejim.2006.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.