

Abstract
One- or two-dimensional arrays of iron oxide nanoparticles were formed in colloidal assemblies of amphiphilic polymers. Electron tomography imaging revealed that nanoparticles are arranged into one-dimensional strings in magneto-micelles or two-dimensional sheets in magneto-core/shell assemblies. The distinct directional assembly behavior was attributed to the interparticle interaction relative to the nanoparticle–polymer interaction, which was modulated by varying the cosolvent used for the solution phase self-assembly. Magneto-core/shell assemblies with varying structural parameters were formed with a range of different sized as-synthesized nanoparticles. The transverse magnetic relaxivity rates (r2) of a series of different assemblies were determined to examine the effect of nanoparticle arrangement on the magnetic relaxivity for their potential applications in MRI. The results indicated that the assembly structure of nanoparticles in polymer micelles significantly affects the r2 of surrounding water, providing a way to control magnetic relaxivity.
Keywords: block copolymer, amphiphilic, nanoparticles, iron oxide, magnetic relaxivity
Introduction
The solution phase self-assembly of amphiphilic polymers and nanoparticles offers a way to form controllable clusters of nanoparticles embedded in colloidal polymer particles.(1, 2) Many different types of nanoparticles have been incorporated into diverse polymer assembly structures such as simple micelles, rod-like micelles, and vesicles for various applications ranging from biological imaging and therapeutic applications to painting and coating materials.(1, 3–5) For example, Moffitt et al. have fabricated such assemblies by synthesizing quantum dots using polymer assemblies as nanoreactors.(6) As a versatile alternative to the synthetic approach, Taton and co-workers have developed a protocol to prepare polymer micelles loaded with various types of nanoparticles by the simultaneous self-assembly of amphiphilic polymers and presynthesized nanoparticles.(5, 7, 8)
In many applications of nanoparticle/polymer hybrid materials, the ability to control the assembly structure (e.g., size of assemblies, particle arrangement) is important, as it can significantly affect their optical, magnetic, and mechanical properties.(9–11) For example, polymer assemblies encapsulated with multiple superparamagnetic nanoparticles have been actively studied as magnetic resonance imaging (MRI) contrast agents, as nanoparticle aggregates are more effective in shortening T2 relaxation time of surrounding water molecules than isolated particles, yielding a better contrast in MRI applications.(3, 4, 12–14) A number of recent theoretical and experimental studies showed that the transverse relaxation rate (r2) increases with the size of particle aggregates until it plateaus at a certain size range and then eventually decreases with a further increase of the aggregation number.(3, 15–19)
While the ability to control the arrangement of nanoparticles might provide an additional opportunity to further control the magnetic relaxivity, thus far, little research has been carried out on nonisotropic nanoparticle arrays. In most previous studies, nanoparticles were either segregated into closely packed isotropic aggregates in the polymer core (Figure 1a)(3, 19, 20) or dispersed throughout the polymer matrix (Figure 1d).(21) When the interactions between nanoparticles and polymers are unfavorable and/or there is a strong attractive interaction between particles, they tend to form closely packed three-dimensional aggregates in polymer micelles, as depicted in Figure 1a.(22) This phenomenon is often observed with gold nanoparticles functionalized with saturated alkylthiols due to the enthalphic gain by the packing of long alkyl chains.(23–26) The uniform dispersion of nanoparticles depicted in Figure 1d is often achieved in athermal nanoparticle–polymer mixtures where nanoparticles are grafted with the same polymer as the micelle core.(21, 22) The intermediate interaction between the two extreme cases is expected to result in anisotropic assemblies of nanoparticles into one- or two-dimensional arrays.(27, 28) We have previously reported that multiple polymer morphologies with various nanoparticle arrangements are accessible through the self-assembly of iron oxide nanoparticles and amphiphilic polymers of polystyrene and poly(acrylic acid) (PS-b-PAA) by simply changing the initial solvent composition used for self-assembly (Figure 2).(13) In particular, when N,N-dimethylformamide (DMF) was used as an initial solvent, spherical sheets of iron oxide nanoparticles were formed in unique core/shell-type polymer micelles, which are called “magneto-core/shell” assemblies (Figure 2b). On the other hand, when the common solvent tetrahydrofuran (THF) was used as the initial solvent, nanoparticles appeared to be distributed throughout the polymer matrix (Figure 2c), as it had previously reported by Taton and co-workers.(5) This structure was referred to as “magneto-micelles”.
Figure 1.
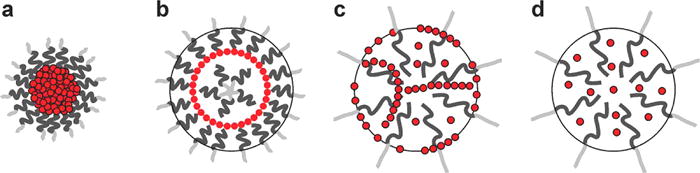
Simplified pictorial representations of the four different nanoparticle arrangements within polymeric micelles: (a) 3-D aggregates (b) 2-D sheets, (c) 1-D strings, and (d) well-dispersed particles.
Figure 2.

(a) Schematic representation of self-assembly of nanoparticles and amphiphilic polymers into two distinct assemblies of magneto-core/shell assemblies (left) and magneto-micelles (right). (b) TEM image of a magneto-core/shell assembly prepared with 2.8 nm iron oxide particles and PAA38-b-PS247 using DMF as the common solvent. (c) TEM image of a magneto-micelle prepared with 2.8 nm iron oxide particles and PAA38-b-PS247 using THF as the common solvent.
Here, we investigated the three-dimensional (3-D) arrangement of iron oxide nanoparticles in the two distinct nanoparticle/polymer hybrid assembly structures (Figure 2b,c) by transmission electron microscopy (TEM) tomography. We confirmed that nanoparticles are indeed forming a spherical monolayer sheet in the core/shell structure formed in DMF, as shown in Figure 1b. For magneto-micelles prepared in THF, we found that nanoparticles are arranged into interesting one-dimensional strings in polymer micelles, as depicted in Figure 1c, in contrast to previous interpretations(5, 7, 8, 13, 29) that nanoparticles are homogeneously embedded in the polymer core. The results reported here demonstrate that the nanoparticle–nanoparticle interaction can be fine-tuned in the solution phase self-assembly by changing the solvent quality, and it can be used to form one- or two-dimensional arrays of nanoparticles in colloidal polymer particles. The effect of subtle differences in nanoparticle surface ligands in the binary self-assembly is discussed to explain the directional self-assembly of as-synthesized nanoparticles. Note that the distinct self-assembly behaviors of slightly different types of alkyl-terminated nanoparticles in polymer assemblies have not been explicitly compared and discussed previously. Furthermore, we investigated the magnetic relaxivity of the two distinct types of assemblies with varying structural parameters and found that the nanoparticle arrangement significantly affects the r2 relaxivity of nanoparticle-loaded polymer assemblies due to the changes in the proximity of water to nanoparticles.
Results and Discussion
The two distinct assembly structures shown in Figure 2 were prepared with oleic acid-stabilized iron oxide nanoparticles by following previously reported procedures.(13) Briefly, as-synthesized nanoparticles dissolved in 50 μL of THF and PAA-b-PS in 1500 μL of DMF or THF were mixed together. Then, water was slowly added to the solution to induce self-assembly of block copolymers and nanoparticles. Finally, initial organic solvents were removed by dialysis, and the assemblies were precipitated by centrifugation. Collected nanoparticle/polymer assemblies were then redispersed in water and analyzed by TEM and dynamic light scattering (DLS). As previously reported,(13, 30) TEM images of the assembly formed in DMF showed a dark ring of nanoparticles at a certain radial position inside a polymer assembly, indicating that nanoparticles preferentially accumulate at the radial position. The electron energy-loss spectroscopy (EELS) line scans of carbon-K and iron-L lines were consistent with the preferential radial arrangement of nanoparticles (Supporting Information). In the binary assembly formed in THF, on the other hand, nanoparticles appear to be distributed throughout the polymer matrix (Figure 2).(5, 13)
However, since TEM measurements are two-dimensional (2-D) projections, TEM images and EEELS analyses do not provide information on 3-D nanoparticle distributions. In order to map out the three-dimensional nanoparticle arrangement in colloidal polymer assemblies, the two distinct types of assemblies formed from DMF and THF solutions were characterized by TEM tomography (Figures 3 and 4). Figure 3 presents a series of 0.14 nm thick X–Y computational slices (Figure 3a.i–vii) of the 3-D tomographic volume containing a magneto-core/shell assembly. From the 3-D surface rendering of the tomographic volume of the micelle, it is clearly observed that a monolayer of particles decorates a polymer core as a 2-D array, and they are coated with a polymer shell layer. At roughly the halfway slicing point of the magneto-core/shell assembly (Figure 3a.iv), a “ring” of nanoparticles is observed. There were no nanoparticles embedded inside the polymer core, and nanoparticles were exclusively at a radial position, as previously hypothesized from TEM contrasts.(31) In addition, no contrast differences are observed inside the nanoparticle shell, which indicates that the core is filled with polymer. The 3-D surface rendering of the tomographic volume (Figure 3b, Movie 1) also demonstrates the radially arranged nanoparticles, confirming that the self-assembled magneto-core/shell structure is a layered structure composed of polymer core, polymer shell, and nanoparticle monolayer imbedded in between the polymer core and the shell.
Figure 3.
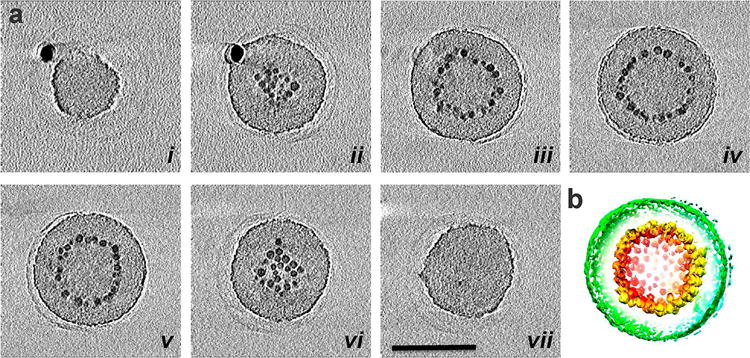
3-D structural analysis of a magneto-core/shell assembly by electron tomography. (a) X–Y computational slices (i–vii) of the 3-D volume containing a magneto-core/shell nanoparticle assembly, shown in every 40 slices (5.6 nm) through the volume. The scale bar is 100 nm. (b) 3-D surface rendering of the tomographic volume, colored red to green according to the radial position from the center. The density is contoured at a level that displays nanoparticles (red and yellow) and the outer surface of the polymer sphere (green). The imaged magneto-core/shell assembly was prepared with 2.8 nm iron oxide particles and PAA38-b-PS247 at 24 np wt % using DMF as the common solvent.
Figure 4.
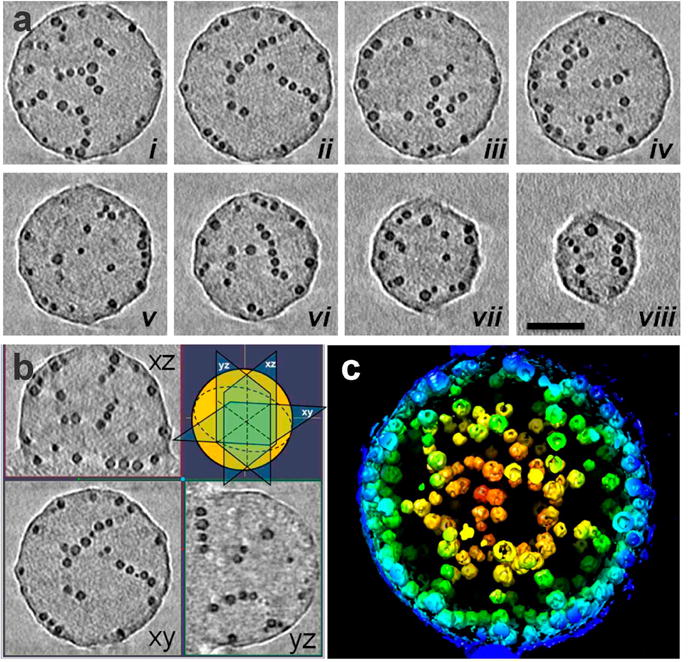
3-D structural analyses of a magneto-micelle by electron tomography. (a) X–Y computational slices (i–viii) of the 3-D volume containing a magneto-micelle, shown in every 40 slices (5.6 nm) through the volume. The scale bar is 50 nm. (b) Three orthogonal slices XY, YZ, and XZ through the tomogram with their orientations indicated in the cartoon (top right). (c) 3-D surface rendering of the tomographic volume, colored red to blue according to the radial position from the center. The magneto-micelle was prepared with 2.8 nm iron oxide particles and PAA38-b-PS247 at 62 np wt % using THF as the common solvent.
Magneto-micelles formed in THF were also characterized by TEM tomography for comparison. Interestingly, the tomography data of magneto-micelles revealed that nanoparticles are aligned into one-dimensional (1-D) “strings”, as seen in X–Y computational slices through the 3D volume (Figure 4a.i–viii) and three orthogonal slices of the tomographic volume (Figure 4b). Many of them are also seen along the outer edge of the magneto-micelle. These curved linear strings of 8–12 nanoparticles appear interconnected with each other, forming an intricate particle network, as shown in the surface rendering representation (Figure 4c, Movie 2). The linear particle assemblies reside within the spherical polymer matrix, which shows a slightly higher density (darker) compared to the surrounding background. This result is different from the assumption made in previous reports where nanoparticles were believed to be homogeneously distributed in the polymer core.(5) The 1-D assembly structure of nanoparticles in polymer micelles is pictorially described in Figure 1c.
The structures presented in Figure 1 represent four possible nanoparticle arrangements in spherical colloidal polymer assemblies: 3-D aggregates, 2-D sheets, 1-D strings, 0-D well-dispersed particles. As mentioned above, well-dispersed nanoparticles are often found when particles are functionalized with the same polymer as the polymer micelle core (Figure 1d).(21) At the other end, 3-D aggregates of nanoparticles are often found with gold nanoparticles coated with long alkylthiols such as dodecanethiol (Figure 1a).(26) We and others have found that these particles tend to segregate in the core of polymer micelles due to the relatively unfavorable particle–polymer interactions and the strong interparticle interaction.(3, 26) It is interesting that oleic acid-modified iron oxide nanoparticles behave differently from alkylthiol-modified gold nanoparticles, and they tend to self-assemble into low-dimensional arrays of strings and sheets rather than 3-D aggregates. Note that alkylthiols and oleic acids are the most frequently used surface ligands for noble metal particles and oxide particles, respectively. We attribute the distinct self-assembly behaviors of the two different as-synthesized nanoparticles to the relative interparticle interaction and to the nanoparticle–polymer interaction. Gold nanoparticles modified with long alkylthiols tend to aggregate easily due to the strong nanoparticle–nanoparticle interaction owing to the efficient packing of alkyl groups(24, 25) as well as the stronger van der Waals interaction of gold particles. The oleic acid layer on iron oxide nanoparticles, on the other hand, is oily and disordered due to the unsaturated bond and is not as well packed as alkylthiols on gold. Therefore, the nanoparticle–nanoparticle interaction of iron oxide particles should be weaker than that of alkylthiol-modified gold nanoparticles, leading to 2-D or 1-D nanoparticle arrays rather than 3-D aggregates. The 2-D arrays of nanoparticles were also found in quantum dots stabilized with trioctyl phosphine oxides (TOPO),(31) which are shorter and bulkier than typical alkylthiol ligands used for gold; thus they behave similarly to oleic acid-stabilized iron oxide particles. In short, the relatively weak interparticle interaction of oleic acid-modified iron oxide nanoparticles and TOPO-stabilized quantum dots results in the formation of anisotropic nanoparticle arrays in a polymer matrix as opposed to 3-D aggregates commonly found for alkylthiol-modified gold nanoparticles.
Furthermore, the weak interaction between oleic acid-stabilized nanoparticles allows for a fine-tuning of interparticle interactions relative to nanoparticle–polymer interactions by changing the solvent composition. Since THF is a good solvent for both nanoparticles and polymers, nanoparticles are distributed throughout the swollen PS matrix in the THF/water mixture. When the concentration of nanoparticles is sufficiently high, they can form strings in PS, as shown in Figure 4, presumably due to the weak dipole interaction between nanoparticles.(32) The dipole-induced one-dimensional association of nanoparticles is often observed in solution-dispersed nanoparticles.(33–35) In polymer matrices, the string formation can be promoted by the slightly unfavorable interaction between nanoparticles and polymers. On the other hand, in DMF, which is a marginal solvent for PS and a bad solvent for nanoparticles, nanoparticles are not well-dispersed in a compact PS matrix, and the relative interparticle interaction should be stronger. This fine balance between the interparticle interaction and nanoparticle–polymer interaction leads to the formation of 2-D sheets in the DMF/water mixture instead of 1-D strings or 3-D aggregates (Figure 1b).
These low-dimensional nanoparticle arrangements within colloidal polymer assemblies are reminiscent of the work by Kumar and co-workers on controlled aggregation of polymer-grafted nanoparticles in polymer melts.(27) In the study, the nanoparticle–nanoparticle interaction was modulated by varying the polymer grafting density, and the aggregation behavior was explained by the balance between the nanoparticle–nanoparticle interaction and the steric repulsion of grafted polymers.(27) In the solution phase assembly reported here, the interparticle interaction is effectively controlled by varying the solvent quality, allowing the fabrication of different types of nanoparticle arrays from the same batch of nanoparticles. The telebridging mechanism reported by Schweizer and co-workers(22, 36, 37) might also play a role, where the adsorption of nanoparticles on PAA through ligand exchange contributes to the formation of nanoparticle strings.
As mentioned above, the clusters of magnetic nanoparticles embedded in polymer micelles are of great interest due to their potential as MRI contrast agents, hyperthermia agents, drug carriers, magnetic relaxation switching assays, and separation techniques.(38–44) We carried out a series of r2 measurements of 2-D shells and 1-D strings of magnetic nanoparticles fabricated here to understand how various structural parameters of anisotropic assemblies affect the magnetic relaxivity of surrounding water for their potential applications in MRI. To this end, magneto-core/shell assemblies with 2-D nanoparticle arrays were prepared with a series of different sized iron oxide nanoparticles (Figure 5a–c). The radius of synthesized nanoparticles (rNP) determined by TEM ranged from 2.3 ± 0.3 to 7.5 ± 0.5 nm (Supporting Information).(45) Here, all reported nanoparticle sizes are in radius and do not include the oleic acid layer unless otherwise specified. The nanoparticle weight percent (np wt %), which is defined here by the weight of nanoparticles over the combined weight of nanoparticles and polymers, was kept constant for a given series of experiments in comparison. For example, iron oxide particles with radii of 2.3, 2.8, 3.4, 5.3, 5.4, and 7.5 nm were self-assembled with PAA38-b-PS154 at 16.0 np wt %. For a shorter polymer of PAA38-b-PS108, it was necessary to use a lower nanoparticle weight percent (10.0 np wt %) to accommodate a range of different sized particles. The largest nanoparticles that can be incorporated into the core/shell-type assemblies varied with the length of polymers as well as the nanoparticle weight percent. For PAA38-b-PS108, the critical nanoparticle radius that allowed for the formation of well-defined discrete core–shell assemblies was found between 3.3Rg and 4.0Rg, where Rg is the radius of gyration of the PS chain calculated from Rg = (bN1/2)/61/2 with the polymerization number (N) of 108 and the statistical segment length (b) of 0.68 nm for PS.(46–48) It is worth noting that, in previous bulk and thin film studies, a filler with an rNP larger than Rg was shown to macroscopically segregate from polymers.(49) In the present study of solution phase assemblies, the largest rNP/Rg ratio was found to be 3.4 for an rNP of 7.5 nm and PAA38-b-PS154, showing that the solution phase binary assembly can accommodate larger fillers than predicted for polymer melt studies.
Figure 5.
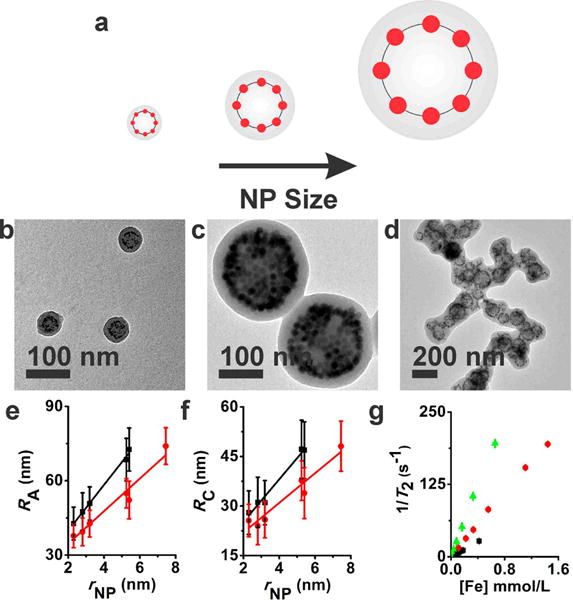
Structural parameters and relaxivity measurements of magneto-core/shell assemblies formed with different sized nanoparticles. (a) Pictorial description showing that the incorporation of larger nanoparticles results in larger sized assemblies. (b–d) TEM images of magneto-core/shell assemblies prepared with PAA38-b-PS247 and two different sized iron oxide nanoparticles, 2.3 nm (b) and 7.5 nm (c) at 24.0 np wt %. A tomography image of the core–shell assembly formed at 24.0 np wt % (c) is presented in Figure S6. (d) One-dimensional aggregates of magneto-core/shell assemblies formed with 7.5 nm particles and a shorter polymer, PAA38-b-PS108, at 10.0 np wt %. (e, f) RA (e) and RC (f) dependence on rNP for PAA38-b-PS154 (red circle) and PAA38-b-PS108 (black square). The weight percentages of nanoparticles were 10.0% and 16.0% for PAA38-b-PS108 and PAA38-b-PS154, respectively. All assemblies were prepared using DMF as the common solvent. (g) Inverse transverse relaxation time (1/T2) versus the iron molar concentration [Fe] for magneto-core/shell assemblies prepared with three different sized particles: 2.3 nm (black squares), 3.2 nm (red circles), and 7.5 nm (green triangles). The error bars are standard deviations from three different measurements.
The structure of 2-D assemblies (i.e., radius of assembly (RA), radius of core (RC), and shell thickness (LS)) can be exactly determined and described by simple TEM imaging. The structural parameters of a series of magneto-core/shell assemblies were measured by TEM and are plotted in Figure 5e,f. Consistent with our previous report on the self-assembly of quantum dots,(50) the size of core/shell assemblies and the radial position of nanoparticles gradually increased with increasing the size of the magnetic particles. This behavior has been attributed to the stretching energy of the shell polymer due to the bumpy surface created by nanoparticle sheets.(50) Additional experiments with several different length polymers (PAA15-b-PS107 and PAA38-b-PS189) showed the same trend (Supporting Information). When the nanoparticle size increased further beyond the critical radius described above, the core/shell assemblies coalesced into chains (Figure 5d) to relieve the excess polymer stretching. The transverse magnetic relaxivity was measured for three different sets of magneto-core/shell assemblies prepared with different sized magnetic particles (2.3, 3.2, and 7.5 nm) and PAA38-b-PS247. Figure 5g presents inverse transverse relaxation time (1/T2) as a function of iron molar concentration. The transverse relaxivity rates, r2, calculated from the data were 63 ± 1, 144 ± 1, and 301 ± 2 mM−1 s−1 for 2.3, 3.2, and 7.5 nm particles, respectively. The increase of r2 with increasing nanoparticle sizes should be the result of both the size of nanoparticles and the size of assemblies.
In order to examine the effect of the aggregate size alone on the magnetic relaxivity, a set of magneto-core/shell assemblies were prepared using the same batch of nanoparticles with radii of 3.2 nm and PAA38-b-PS247. The size of the core/shell assemblies was effectively controlled by changing the concentration of polymers and nanoparticles, while keeping other conditions the same including the nanoparticle weight percent (Figure 6). The size of assemblies increased with increasing the concentration, as the aggregation number is a function of concentration and the critical micelle concentration.(51) When the polymer concentration in DMF was increased from 0.01 to 0.08 wt % at 24 np wt %, RA gradually increased from 62 ± 2 nm to 110 ± 6 nm (Figure 6). A further increase of polymer concentration beyond 0.08 wt % did not increase the size of assemblies further presumably due to the excessive polymer stretching. As the polymer concentration increases even further beyond 0.12 wt %, the core/shell assemblies coalesced into large chains of aggregates (Supporting Information) similar to the structure formed with large nanoparticles (Figure 5d).
Figure 6.
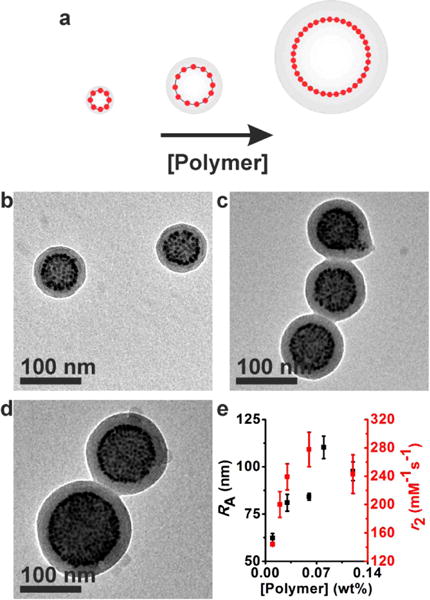
Structural parameters and relaxivity measurements of different sized magneto-core/shell assemblies formed with 3.2 nm (rNP) particles and PAA38-b-PS247 using DMF as the common solvent. The size of assemblies was controlled by changing the concentration of polymers and nanoparticles while keeping the nanoparticle weight percent constant (24 np wt %). (a) Pictorial description showing the concentration dependence of the assembly structure. (b–d) TEM images of magneto-core/shell assemblies formed at polymer concentrations of 0.01 wt % (b), 0.05 wt % (c), and 0.11 wt % (d). (e) The hydrodynamic radius determined by DLS (black) and r2 (red) of magneto-core/shell assemblies formed at different polymer concentrations.
The r2 values of a series of different sized magneto-core/shell assemblies were measured and are presented in Figure 6 along with their size data. In general, the size dependency of r2 relaxivity can be explained by the diffusion length of water molecules relative to the size of nanoparticles.(3, 15, 17–19) Theoretical studies showed that there are three different regimes showing distinct size dependencies: the motional averaging regime (MAR), the static dephasing regime (SDR), and the echo-limiting regime (ELR).(15, 19) In MAR, where the sizes of nanoparticles are sufficiently small, water molecules experience changing magnetic fields as they diffuse; thus the r2 value increases with nanoparticle size. At a certain particle size, a water molecule feels a constant magnetic field during transverse relaxation, and the r2 value no longer increases with the nanoparticle size. The maximum relaxivity is achieved in this regime called the SDR. As the nanoparticle size increases further in the ELR, water molecules very close to nanoparticles are completely dephased due to the strong magnetic field, leading to a reduced r2 value.(19) This behavior has been experimentally demonstrated with isotropic aggregates of nanoparticles embedded in silica or polymer matrix.(3, 17, 19) The r2 of 2-D nanoparticle shells studied here initially increased with concentration until the micelle size peaks at 0.06 wt %, which is characteristic of MAR, and then it plateaued as the size of the assembly did not increase further (Figure 6e). Compared to the literature values for isotropic assemblies, our 2-D shells showed an increased maximum r2 at a larger micelle size.(3) For example, Weller and co-workers observed a maximum r2 of about 220 mM−1 s−1 at the micelle diameter of about 80 nm (hydrodynamic diameter) for iron oxide nanoparticles with a diameter of 7.5 nm.(3) Here, our 2-D assemblies showed a maximum r2 of 278 mM−1 s−1 at the micelle diameter of 160 nm (hydrodynamic diameter) using a similar sized particle (diameter: 6.4 nm). A noticeable ELR behavior was not observed in the 2-D assemblies studied here, implying that the magnetic field generated by the 2-D nanoparticle arrays is not too strong to completely dephase nearby water over a large size range.
Since the motion of water molecules is restricted around nanoparticles in polymer assemblies due to the hydrophobic polymer layer encasing nanoparticles, the distance between nanoparticles and the hydrophilic corona should be an important factor in determining the magnetic relaxivity. In the core/shell assemblies studied here, the separation between the external water molecules and magnetic particles can be precisely controlled by varying the shell thickness. A series of assemblies with different shell thickness was prepared by changing the nanoparticle weight percent (24, 49, and 62 np wt %) using 2.9 nm particles to examine the effect of shell thickness on the magnetic relaxivity (Figure 7). TEM and DLS analyses showed that the radial position of nanoparticles increases with increasing nanoparticle weight percent to accommodate a larger number of nanoparticles (Figure 7a–f). The size of the assemblies on the other hand stayed roughly the same (Figure 7e,f), leading to reduced shell thicknesses with increasing nanoparticle weight percent. The r2 values of the magneto-core/shell assemblies were determined to be 119 ± 5, 155 ± 10, and 190 ± 20 mM−1 s−1 for 24, 49, and 62 np wt%, respectively (Figure 7g), showing that the magnetic relaxivity increased with decreasing the shell thickness due to the better water accessibility with a thinner shell.
Figure 7.
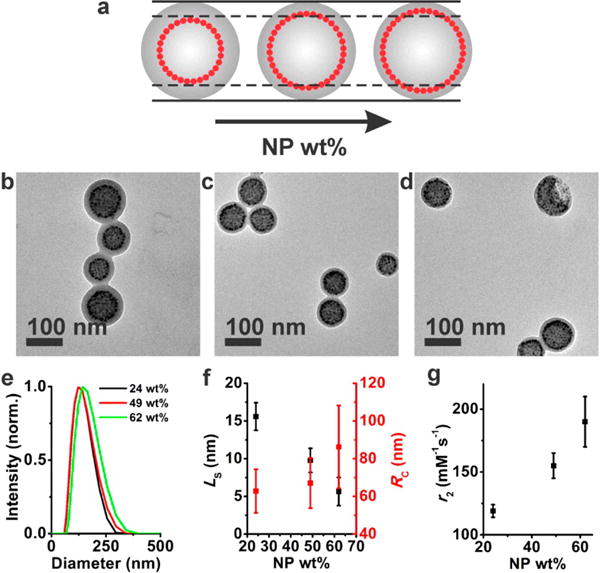
Structural characterization and r2 data of magneto-core/shell assemblies formed at varying nanoparticle weight percent. In this set of experiments, the nanoparticle weight percent was controlled by changing the nanoparticle concentration while keeping the polymer concentration constant (0.01 wt %). (a) Pictorial description showing that the increased nanoparticle weight percent results in a reduced shell thickness. (b–d) TEM images of magneto-core/shell assemblies formed at a nanoparticle weight percent of 24 (b), 49 (c), and 62 (d). The assemblies were prepared with 2.9 nm particles and PAA38-b-PS247 using DMF as the common solvent. Tomography data of the core–shell assemblies formed at 24 np wt % (b) and 62 np wt % (d) are presented in Figure S7. (e) Normalized DLS intensity plot of the magneto-core/shell assemblies shown in b–d. (f) LS (black) and RC (red) dependence on nanoparticle weight percent for the corresponding assemblies in b–d. (g) r2 dependency on nanoparticle weight percent for the corresponding assemblies in b–d.
The r2 dependency on the shell thickness is consistent with our previous result that the magneto-core/shell assemblies formed in DMF were more efficient in shortening the transverse relaxation time of water than the magneto-micelle structure formed in THF.(13) Here, we prepared the magneto-micelles at two different nanoparticle weight percents (24 and 62 np wt %) (Figure 8a,b) and compared their r2 values with those of magneto-core/shell assemblies prepared at the same nanoparticle weight percent (Figure 8d). At 24 np wt %, the hydrodynamic diameters of the two structures were similar to 136 and 127 nm for DMF and THF samples, respectively. However, the r2 of magneto-core/shell assemblies (119 ± 5 mM−1 s−1) was significantly larger than that of magneto-micelles (84 ± 5 mM−1 s−1), consistent with our previous report.(13) Since the two assemblies were similar in size at 24 np wt %, the different r2 relaxivity values found in the two structures can be attributed to the nanoparticle arrangement effect. This result is in contrast to theoretical predictions assuming a free diffusion of water molecules(18) and illustrates the importance of nanoparticle arrangement in a polymer matrix in real systems. Because magnetic nanoparticles are arranged close to the hydrophilic PAA layer in the core/shell assemblies, water molecules are more accessible to nanoparticles in the core/shell structure, leading to higher r2 relaxivity values.
Figure 8.
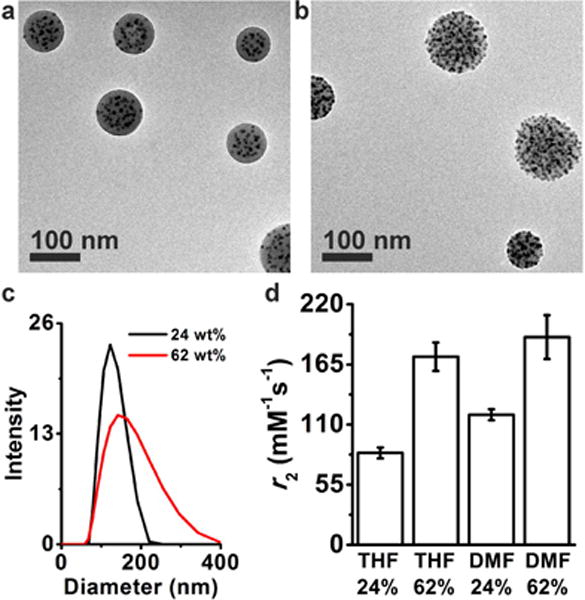
(a, b) TEM images of magneto-micelles formed at a nanoparticle wt % of 24 (a) and 62 (b). The assemblies were prepared with 2.9 nm particles and PAA38-b-PS247 using THF as the common solvent. The nanoparticle weight percent was adjusted by varying the nanoparticle concentration while keeping the polymer concentration constant (0.01 wt %). Tomography data of magneto-micelles formed at 24 np wt % (a) and 62 np wt % (b) are presented in Figure S8. The internal nanoparticle arrangements are more clearly seen in Movie 3 and Movie 4 (Supporting Information), which present z-slices of reconstructed tomography images of 24 and 62 np wt % samples, respectively. (c) DLS intensity plots of magneto-micelles prepared at 24 np wt % (black) and 62 np wt % (red). (d) r2 rate dependence on the micelle morphology and nanoparticle weight percent.
As expected, the r2 of both magneto-core/shell and magneto-micelles increased with increasing nanoparticle weight percent (Figure 8d). Interestingly, however, the difference in the r2 values between magneto-core/shell assemblies and magneto-micelles became smaller at the high nanoparticle content of 62 np wt% (Figure 8d). The steeper r2 change of magneto-micelles with the nanoparticle weight percent should be the result of both nanoparticle arrangement and the aggregate size. From the TEM tomography data (Figure 4, Figure S8d, Movie 4) and conventional TEM images (Figure 8b), it is apparent that a number of nanoparticles form strings and they are located close to the surface of magneto-micelles at 62 np wt %, while a majority of nanoparticles are embedded inside the polymer core at 24 np wt % (Figure 8a, Figure S8b, Movie 3). Therefore, a larger fraction of nanoparticles in magneto-micelles are in close proximity with water at 62 np wt %, which explains the significantly increased r2 value of magneto-micelles at the high nanoparticle weight percent. In addition, the size of magneto-micelles increased with nanoparticle weight percent (Figure 8c) as previously reported,(5) while the size of magneto-core/shell assemblies stays roughly constant (Figure 7). This should also contribute to the larger r2 change of magneto-micelles with nanoparticle weight percent.
Conclusions
Here, we demonstrated that low-dimensional arrays of nanoparticle strings and sheets can be formed in colloidal polymer particles by controlling the nanoparticle–nanoparticle interaction relative to the nanoparticle–polymer interaction in the binary self-assembly of nanoparticles and amphiphilic polymers. While nanoparticles with strong interparticle interactions tend to form 3-D aggregates in polymer micelles, oleic acid-stabilized iron oxide nanoparticles with intermediate interparticle interaction can be assembled into 1-D strings in magneto-micelles or 2-D sheets in magneto-core/shell assemblies. The interparticle interaction relative to nanoparticle–polymer interaction could be fine-tuned by varying the cosolvent used for self-assembly. The self-assembly from a marginal solvent such as DMF led to the formation of 2-D nanoparticle sheets, while a good solvent such as THF resulted in 1-D strings. A range of different sized iron oxide particles was assembled into magneto-core/shell structures, and their transverse magnetic relaxivity rates were measured with varying structural parameters (i.e., size of nanoparticles, size of assemblies, radial position of nanoparticles, and shell thickness) to examine the effect of nanoparticle arrangement on the r2 of surrounding water. The size of assemblies increased with the size of nanoparticles and the concentration of polymer and nanoparticles. The r2 increased with increasing the size of assemblies, similarly to the previous observations in 3-D aggregates. The radial position of nanoparticles and the shell thickness were controlled by changing the nanoparticle weight percent while keeping the polymer concentration constant. The assemblies with reduced shell thickness showed increased r2 due to the better water accessibility. Magneto-micelles showed a steeper dependency of r2 on nanoparticle weight percent than magneto-core/shell structure due to the formation of nanoparticle strings residing close to the surface of micelles at high nanoparticle contents.
Experimental Section
Materials and Instrumentation
All polymers except for PAA14-b-PS250 (PAA(1000)-b-PS(26 000), Polymer Source Inc.) were synthesized by reversible addition–fragmentation chain transfer polymerization by a previously reported procedure.(31) All TEM measurements were taken using a JEOL 1400. All STEM images and the EDS mapping were acquired using a JEOL 2010F. DLS measurements were taken with a Malvern Zetasizer Nano Series. Transverse (T2) relaxivity times of the various micelles were determined using a Bruker mq60 MR relaxometer operating at 1.41 T (60 MHz). The iron concentration was determined using inductively coupled plasma–atomic emission spectroscopy (ICP-AES) (Spectro Genesis). TEM samples were prepared by placing a few droplets of aqueous micelle solution on TEM grids (Carbon Film Only, 200 mesh, Cat#: CF200-Cu, Electron Microscopy Sciences) and wicking away water from the other side of the TEM grid. For 3-D electron tomographic analysis, a series of projection images were recorded by tilting the specimen from −78° to 78° in an increment of 1° using a Gatan 916 ultrahigh tilt tomography holder (Gatan Inc., Pleasanton, CA, USA), with a Gatan 4K × 4K charge-coupled device (CCD) camera mounted on a Tecnai F20 electron microscope (FEI Corporation, Hillsboro, OR, USA). Images were recorded at a nominal magnification of 50 000 to 80 000 (effective pixel size is 0.14 to 0.37 nm) and an underfocus value of 0.5–2.0 μm along the tilt axis. A weighted back-projection algorithm, as implemented in the IMOD reconstruction package,(52) was used to convert the information present in the series of tilted projection images into three-dimensional density maps. The surface rendering was generated using the Chimera software.(53)
Synthesis of Iron Oxide Nanoparticles
Iron oxide nanoparticles were synthesized using oleic acid as a stabilizing agent following a modified literature method.(45) The iron oleate was synthesized in a 100 mL flask by dissolving 1.5 g of iron chloride (FeCl3·6H2O, 5.5 mmol, Aldrich, 97%) and 5.2 g of sodium oleate (17 mmol, TCI, 95%) in 20 mL of hexane, 11.5 mL of ethanol, and 8.8 mL of distilled water. The solution was heated to reflux (∼70 °C) and kept at that temperature for 4 h. After the 4 h, the upper organic layer was washed three times with 30 mL of water and separated by centrifugation. After the final washing, hexane was evaporated from the dark brown organic layer in a rotary evaporator and kept under vacuum overnight (∼12 h). For the synthesis of rNP = 2.3 nm iron oxide nanoparticles, 5.5 g of iron-oleate and 1.5 g of oleic acid (5.3 mmol, Aldrich, 90%) were reacted in 31 g of 1-octadecene (Aldrich, 90%) in a 100 mL round-bottom flask. For larger nanoparticles of rNP = 5.3–7.5 nm, reactions were carried out in a 250 mL round-bottom flask. The reaction mixture was heated to 320 °C at a rate of 200 °C/h and kept at that temperature for 30 min. The resulting solution was cooled to room temperature and washed three times with acetone. After each acetone addition, the product was centrifuged for 10 min at 7000 rpm and redissolved in hexane. After the final wash, the nanoparticles were dissolved in chloroform and centrifuged at low speed (3000 rpm) to remove large aggregates. The diameters of nanoparticles used were determined to be 4.6 ± 0.5, 5.6 ± 0.5, 6.4 ± 0.5, 10.5 ± 1.3, 10.8 ± 0.7, and 14.9 ± 0.9 nm by TEM (see Supporting Information).
Self-Assembly of Iron Oxide Nanoparticles and Polymers
Co-assemblies of iron oxide nanoparticles and polymers were prepared by the slow addition of water to the mixture of nanoparticles and polymers dispersed in either DMF or THF. In typical experiments to form magneto-core/shell assemblies at 16 np wt %, a THF solution of nanoparticles (50 μL, 1.0 mg/mL) was mixed with a DMF solution of PAA38-b-PS154 (70.0 μL, 3.77 mg/mL) or PAA38-b-PS247 (30.0 μL, 5.15 mg/mL). Then, the total volume of the mixture was adjusted to a constant volume (1.55 mL) by adding additional DMF. While stirring, water (600 μL) was slowly added (10 μL per 30 s) to the mixture of nanoparticles and block copolymers. The mixture was kept under stirring for 15 h before adding additional water (1.5 mL) over 15 min. Then, the samples were dialyzed against water for 24 h and further purified by a series of centrifugations. The samples were centrifuged at 14 000 rpm (30 min) to collect precipitates of isolated assemblies. Magneto-micelles were assembled similarly, except that the polymer was initially dissolved in THF. In the absence of nanoparticles, PAA38-PS154 self-assembled into micelles for both DMF and THF initial solvents. On the other hand, PAA38-PS247 assembled into worm-like micelles in DMF and vesicles in THF, respectively, without nanoparticles.
Supplementary Material
Acknowledgments
S.J.P. acknowledges the support from the NSF Career Award (DMR 0847646) and the Camille Dreyfus Teacher Scholar Award. P.Z. acknowledges the support from the National Institutes of Health grants to P.Z. (GM085043). The STEM, EELS, and EDS measurements were carried out using instrumentation at the Penn Regional Nanotechnology Facility. The authors would like to acknowledge Prof. Rob Riggleman for helpful discussions.
References
- 1.Mai Y, Eisenberg A. Selective Localization of Preformed Nanoparticles in Morphologically Controllable Block Copolymer Aggregates in Solution. Acc Chem Res. 2012;45:1657–1666. doi: 10.1021/ar2003144. [DOI] [PubMed] [Google Scholar]
- 2.Sánchez-Iglesias A, Grzelczak M, Altantzis T, Goris B, Pérez-Juste J, Bals S, Van Tendeloo G, Donaldson SH, Chmelka BF, Israelachvili JN, et al. Hydrophobic Interactions Modulate Self-Assembly of Nanoparticles. ACS Nano. 2012;6:11059–11065. doi: 10.1021/nn3047605. [DOI] [PubMed] [Google Scholar]
- 3.Pöselt E, Kloust H, Tromsdorf U, Janschel M, Hahn C, Maßlo C, Weller H. Relaxivity Optimization of a PEGylated Iron-Oxide-Based Negative Magnetic Resonance Contrast Agent for T2-Weighted Spin–Echo Imaging. ACS Nano. 2012;6:1619–1624. doi: 10.1021/nn204591r. [DOI] [PubMed] [Google Scholar]
- 4.Sanson C, Diou O, Thévenot J, Ibarboure E, Soum A, Brûlet A, Miraux S, Thiaudière E, Tan S, Brisson A, et al. Doxorubicin Loaded Magnetic Polymersomes: Theranostic Nanocarriers for MR Imaging and Magneto-Chemotherapy. ACS Nano. 2011;5:1122–1140. doi: 10.1021/nn102762f. [DOI] [PubMed] [Google Scholar]
- 5.Kim B-S, Qiu J-M, Wang J-P, Taton TA. Magnetomicelles: Composite Nanostructures from Magnetic Nanoparticles and Cross-Linked Amphiphilic Block Copolymers. Nano Lett. 2005;5:1987–1991. doi: 10.1021/nl0513939. [DOI] [PubMed] [Google Scholar]
- 6.Moffitt M, McMahon L, Pessel V, Eisenberg A. Size Control of Nanoparticles in Semiconductor-Polymer Composites. 2. Control via Sizes of Spherical Ionic Microdomains in Styrene-Based Diblock Ionomers. Chem Mater. 1995;7:1185–1192. [Google Scholar]
- 7.Kang Y, Taton TA. Core/Shell Gold Nanoparticles by Self-Assembly and Crosslinking of Micellar, Block-Copolymer Shells. Angew Chem Int Ed. 2005;44:409–412. doi: 10.1002/anie.200461119. [DOI] [PubMed] [Google Scholar]
- 8.Kim B-S, Taton TA. Multicomponent Nanoparticles via Self-Assembly with Cross-Linked Block Copolymer Surfactants. Langmuir. 2006;23:2198–2202. doi: 10.1021/la062692w. [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Tang Z. Design and Application of Inorganic Nanoparticle Superstructures: Current Status and Future Challenges. Small. 2011;7:2133–2146. doi: 10.1002/smll.201100474. [DOI] [PubMed] [Google Scholar]
- 10.Gong J, Li G, Tang Z. Self-Assembly of Noble Metal Nanocrystals: Fabrication, Optical Property, and Application. Nano Today. 2012;7:564–585. [Google Scholar]
- 11.Xia YS, Nguyen TD, Yang M, Lee B, Santos A, Podsiadlo P, Tang ZY, Glotzer SC, Kotov NA. Self-Assembly of Self-Limiting Monodisperse Supraparticles from Polydisperse Nanoparticles. Nat Nanotechnol. 2011;6:580–587. doi: 10.1038/nnano.2011.121. [DOI] [PubMed] [Google Scholar]
- 12.Berret J-F, Schonbeck N, Gazeau F, El Kharrat D, Sandre O, Vacher A, Airiau M. Controlled Clustering of Superparamagnetic Nanoparticles Using Block Copolymers: Design of New Contrast Agents for Magnetic Resonance Imaging. J Am Chem Soc. 2006;128:1755–1761. doi: 10.1021/ja0562999. [DOI] [PubMed] [Google Scholar]
- 13.Hickey RJ, Haynes AS, Kikkawa JM, Park S-J. Controlling the Self-Assembly Structure of Magnetic Nanoparticles and Amphiphilic Block-Copolymers: From Micelles to Vesicles. J Am Chem Soc. 2011;133:1517–1525. doi: 10.1021/ja1090113. [DOI] [PubMed] [Google Scholar]
- 14.Jun Y-w, Huh Y-M, Choi J-S, Lee J-H, Song H-T, Kim S, Yoon S, Kim K-S, Shin J-S, Suh J-S, et al. Nanoscale Size Effect of Magnetic Nanocrystals and Their Utilization for Cancer Diagnosis via Magnetic Resonance Imaging. J Am Chem Soc. 2005;127:5732–5733. doi: 10.1021/ja0422155. [DOI] [PubMed] [Google Scholar]
- 15.Brooks RA. T2-Shortening by Strongly Magnetized Spheres: A Chemical Exchange Model. Magn Reson Med. 2002;47:388–391. doi: 10.1002/mrm.10064. [DOI] [PubMed] [Google Scholar]
- 16.Gillis P, Moiny F, Brooks RA. On T2-Shortening by Strongly Magnetized Spheres: A Partial Refocusing Model. Magn Reson Med. 2002;47:257–263. doi: 10.1002/mrm.10059. [DOI] [PubMed] [Google Scholar]
- 17.Lee N, Choi Y, Lee Y, Park M, Moon WK, Choi SH, Hyeon T. Water-Dispersible Ferrimagnetic Iron Oxide Nanocubes with Extremely High r2 Relaxivity for Highly Sensitive in Vivo MRI of Tumors. Nano Lett. 2012;12:3127–3131. doi: 10.1021/nl3010308. [DOI] [PubMed] [Google Scholar]
- 18.Vuong QL, Gillis P, Gossuin Y. Monte Carlo Simulation and Theory of Proton NMR Transverse Relaxation Induced by Aggregation of Magnetic Particles Used as MRI Contrast Agents. J Magn Reson. 2011;212:139–148. doi: 10.1016/j.jmr.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 19.Yoon T-J, Lee H, Shao H, Hilderbrand SA, Weissleder R. Multicore Assemblies Potentiate Magnetic Properties of Biomagnetic Nanoparticles. Adv Mater. 2011;23:4793–4797. doi: 10.1002/adma.201102948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paquet C, de Haan HW, Leek DM, Lin H-Y, Xiang B, Tian G, Kell A, Simard B. Clusters of Superparamagnetic Iron Oxide Nanoparticles Encapsulated in a Hydrogel: A Particle Architecture Generating a Synergistic Enhancement of the T2 Relaxation. ACS Nano. 2011;5:3104–3112. doi: 10.1021/nn2002272. [DOI] [PubMed] [Google Scholar]
- 21.Kamps AC, Sanchez-Gaytan BL, Hickey RJ, Clarke N, Fryd M, Park S-J. Nanoparticle-Directed Self-Assembly of Amphiphilic Block Copolymers. Langmuir. 2010;26:14345–14350. doi: 10.1021/la1025558. [DOI] [PubMed] [Google Scholar]
- 22.Hooper JB, Schweizer KS. Contact Aggregation, Bridging, and Steric Stabilization in Dense Polymer–Particle Mixtures. Macromolecules. 2005;38:8858–8869. [Google Scholar]
- 23.Chen Z, Moore J, Radtke G, Sirringhaus H, O’Brien S. Binary Nanoparticle Superlattices in the Semiconductor–Semiconductor System: CdTe and CdSe. J Am Chem Soc. 2007;129:15702–15709. doi: 10.1021/ja076698z. [DOI] [PubMed] [Google Scholar]
- 24.Landman U, Luedtke WD. Small is Different: Energetic, Structural, Thermal, and Mechanical Properties of Passivated Nanocluster Assemblies. Faraday Discuss. 2004;125:1–22. doi: 10.1039/b312640b. [DOI] [PubMed] [Google Scholar]
- 25.Mueggenburg KE, Lin XM, Goldsmith RH, Jaeger HM. Elastic Membranes of Close-Packed Nanoparticle Arrays. Nat Mater. 2007;6:656–660. doi: 10.1038/nmat1965. [DOI] [PubMed] [Google Scholar]
- 26.Luo Q, Hickey RJ, Park S-J. Controlling the Location of Nanoparticles in Colloidal Assemblies of Amphiphilic Polymers by Tuning Nanoparticle Surface Chemistry. ACS Macro Lett. 2013;2:107–111. doi: 10.1021/mz3006044. [DOI] [PubMed] [Google Scholar]
- 27.Akcora P, Liu H, Kumar SK, Moll J, Li Y, Benicewicz BC, Schadler LS, Acehan D, Panagiotopoulos AZ, Pryamitsyn V, et al. Anisotropic Self-Assembly of Spherical Polymer-Grafted Nanoparticles. Nat Mater. 2009;8:354–359. doi: 10.1038/nmat2404. [DOI] [PubMed] [Google Scholar]
- 28.Nikolic MS, Olsson C, Salcher A, Kornowski A, Rank A, Schubert R, Frömsdorf A, Weller H, Förster S. Micelle and Vesicle Formation of Amphiphilic Nanoparticles. Angew Chem Int Ed. 2009;48:2752–2754. doi: 10.1002/anie.200805158. [DOI] [PubMed] [Google Scholar]
- 29.Kang Y, Taton TA. Controlling Shell Thickness in Core–Shell Gold Nanoparticles via Surface-Templated Adsorption of Block Copolymer Surfactants. Macromolecules. 2005;38:6115–6121. [Google Scholar]
- 30.Hickey RJ, Sanchez-Gaytan BL, Cui W, Composto RJ, Fryd M, Wayland BB, Park S-J. Morphological Transitions of Block-Copolymer Bilayers via Nanoparticle Clustering. Small. 2010;6:48–51. doi: 10.1002/smll.200901266. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez-Gaytan BL, Cui W, Kim Y, Mendez-Polanco MA, Duncan TV, Fryd M, Wayland BB, Park S-J. Interfacial Assembly of Nanoparticles in Discrete Block-Copolymer Aggregates. Angew Chem Int Ed. 2007;46:9235–9238. doi: 10.1002/anie.200703032. [DOI] [PubMed] [Google Scholar]
- 32.Bishop KJM, Wilmer CE, Soh S, Grzybowski BA. A Nanoscale Forces and Their Uses in Self-Assembly. Small. 2009;5:1600–1630. doi: 10.1002/smll.200900358. [DOI] [PubMed] [Google Scholar]
- 33.Lin S, Li M, Dujardin E, Girard C, Mann S. One-Dimensional Plasmon Coupling by Facile Self-Assembly of Gold Nanoparticles into Branched Chain Networks. Adv Mater. 2005;17:2553–2559. [Google Scholar]
- 34.Thomas JR. Preparation and Magnetic Properties of Colloidal Cobalt Particles. J Appl Phys. 1966;37:2914–2915. [Google Scholar]
- 35.Kim BY, Shim I-B, Monti OLA, Pyun J. Magnetic Self-Assembly of Gold Nanoparticle Chains Using Dipolar Core-Shell Colloids. Chem Commun. 2011;47:890–892. doi: 10.1039/c0cc02375b. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Gao Y, Cao D, Zhang L, Guo Z. Nanoparticle Dispersion and Aggregation in Polymer Nanocomposites: Insights from Molecular Dynamics Simulation. Langmuir. 2011;27:7926–7933. doi: 10.1021/la201073m. [DOI] [PubMed] [Google Scholar]
- 37.Jancar J, Douglas JF, Starr FW, Kumar SK, Cassagnau P, Lesser AJ, Sternstein SS, Buehler MJ. Current Issues in Research on Structure–Property Relationships in Polymer Nanocomposites. Polymer. 2010;51:3321–3343. [Google Scholar]
- 38.Arruebo M, Fernandez-Pacheco R, Ibarra MR, Santamaria J. Magnetic Nanoparticles for Drug Delivery. Nano Today. 2007;2:22–32. [Google Scholar]
- 39.Josephson L, Perez JM, Weissleder R. Magnetic Nanosensors for the Detection of Oligonucleotide Sequences. Angew Chem Int Ed. 2001;40:3204–3206. doi: 10.1002/1521-3773(20010903)40:17<3204::AID-ANIE3204>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 40.Jun YW, Seo JW, Cheon A. Nanoscaling Laws of Magnetic Nanoparticles and Their Applicabilities in Biomedical Sciences. Acc Chem Res. 2008;41:179–189. doi: 10.1021/ar700121f. [DOI] [PubMed] [Google Scholar]
- 41.Na HB, Song IC, Hyeon T. Inorganic Nanoparticles for MRI Contrast Agents. Adv Mater. 2009;21:2133–2148. [Google Scholar]
- 42.Pankhurst QA, Connolly J, Jones SK, Dobson J. Applications of Magnetic Nanoparticles in Biomedicine. J Phys D: Appl Phys. 2003;36:R167–R181. [Google Scholar]
- 43.Perez JM, Josephson L, O’Loughlin T, Hogemann D, Weissleder R. Magnetic Relaxation Switches Capable of Sensing Molecular Interactions. Nat Biotechnol. 2002;20:816–820. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]
- 44.Sun C, Lee JSH, Zhang MQ. Magnetic Nanoparticles in MR Imaging and Drug Delivery. Adv Drug Delivery Rev. 2008;60:1252–1265. doi: 10.1016/j.addr.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park J, An KJ, Hwang YS, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Ultra-Large-Scale Syntheses of Monodisperse Nanocrystals. Nat Mater. 2004;3:891–895. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- 46.Ballard DGH, Wignall GD, Schelten J. Measurement of Molecular Dimensions of Polystyrene Chains in the Bulk Polymer by Low Angle Neutron Diffraction. Eur Polym J. 1973;9:965–969. [Google Scholar]
- 47.Russell TP, Hjelm RP, Seeger P. A Temperature Dependence of the Interaction Parameter of Polystyrene and Poly(methyl methacrylate) Macromolecules. 1990;23:890–893. [Google Scholar]
- 48.Bates FS. Polymer-Polymer Phase-Behavior. Science. 1991;251:898–905. doi: 10.1126/science.251.4996.898. [DOI] [PubMed] [Google Scholar]
- 49.Mackay ME, Tuteja A, Duxbury PM, Hawker CJ, Van Horn B, Guan Z, Chen G, Krishnan RS. General Strategies for Nanoparticle Dispersion. Science. 2006;311:1740–1743. doi: 10.1126/science.1122225. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez-Gaytan BL, Li S, Kamps AC, Hickey RJ, Clarke N, Fryd M, Wayland BB, Park S-J. Controlling the Radial Position of Nanoparticles in Amphiphilic Block-Copolymer Assemblies. J Phys Chem C. 2011;115:7836–7842. [Google Scholar]
- 51.Mai Y, Eisenberg A. Self-Assembly of Block Copolymers. Chem Soc Rev. 2012;41:5969–5985. doi: 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- 52.Kremer JR, Mastronarde DN, McIntosh JR. Computer Visualization of Three-Dimensional Image Data Using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 53.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.