

Abstract
Defective placentation and subsequent placental insufficiency lead to maternal and fetal adverse pregnancy outcome (APO), but their pathologic mechanisms are unclear, and treatment remains elusive. The mildly hypertensive BPH/5 mouse recapitulates many features of human APO with pregnancies characterized by fetal loss, growth restriction, abnormal placental development, and defects in maternal decidual arteries. Using this model, we show that recruitment of neutrophils triggered by complement activation at the maternal fetal interface leads to elevation in local TNF-α levels, reduction of the essential angiogenic factor VEGF and, ultimately, abnormal placentation and fetal death. Blockade of complement with inhibitors specifically targeted to sites of complement activation, depletion of neutrophils, or blockade of TNF-α improves spiral artery remodeling and rescues pregnancies. These data underscore the importance of innate immune system activation in the pathogenesis of placental insufficiency and identify novel methods for treatment of pregnancy loss mediated by abnormal placentation.
INTRODUCTION
Abnormal placentation is a leading cause of adverse pregnancy outcomes (APO) including fetal loss, intrauterine growth restriction and preeclampsia (1, 2). These disorders are characterized by shallow invasion of trophoblasts into the maternal decidua, inadequate spiral artery remodeling, underperfusion of the intervillous space and placental hypoxia (1). The effects of placental hypoperfusion on the fetus are growth restriction, and in some cases death. For the mother, anti-angiogenic factors released by the ischemic placenta lead to endothelial dysfunction and the clinical manifestations of preeclampsia, including hypertension and proteinuria, later in pregnancy.
Inflammation and innate immune system activation have been associated with abnormal placentation in both humans and rodents (3–8). In experimental models of pathologic pregnancies, altered placental development is attributed to abnormalities in immune responses to the semiallogenic fetal-placental unit and to exogenous immunologic triggers that initiate inflammation, some antibody dependent (9–11) and some antibody independent (5, 8, 12). Both uterine NK cells and regulatory T cells have been shown to be critical for normal placental development and maintenance of normal pregnancies, and their dysregulation, in genetically altered mice, is associated with abnormal placentation and fetal loss (13–16).
Complement activation is a common pathway of injury in many models of APO. The complement system is an integral component of innate immunity, a crucial element of host defense against invading organisms, and a trigger, as well as respondent, to “danger”, such as tissue inflammation, necrosis injury and ischemia (17–19). Both animal and human studies support the concept that complement activation is associated with APO (5, 10, 20–22). Complement components are produced by human first trimester trophoblasts, and their expression can be upregulated by inflammatory cytokines (23). Inability to regulate activation of complement has been implicated in fetal loss in animal models of disease (24). Complement activation products generated at sites of inflammation, like placenta, include anaphylatoxins that recruit and stimulate neutrophils (25), which infiltrate the placental tissue and release cytokines and proteases that enhance complement activity and lead to a feed-forward loop of innate immune system activation (26). Neutrophils have been shown to contribute to fetal loss in mouse models (27, 28) and to endothelial damage in preeclampsia (29).
TNF-α produced by the placenta and decidua modulates trophoblast proliferation and invasion, recruits inflammatory cells, including neutrophils, and stimulates those cells to produce more TNF-α (30–32). In rat models of inflammatory fetal loss and growth restriction, blockade of TNF-α activity prevents APOs (7, 33, 34). Elevated levels of TNF-α are present at the fetal-maternal interface in patients with growth restricted fetuses (35, 36), and in maternal blood and amniotic fluid in preeclampsia (37).
To assess the role of inflammation and define specific pathways of damage in a spontaneous mouse model of APO, we studied the BPH/5 mouse, a mildly hypertensive mouse with pregnancies characterized by fetal losses and growth restriction in association with abnormal placentation and defects in maternal decidual arteries (38, 39). Previous studies demonstrating that inflammation contributes to APO have used pregnant mice treated with pathogenic antibodies (antiphospholipid antibodies or anti-angiotensin receptor antibodies) (9–11), LPS (12) or non-syngeneic matings (CBA/JxDBA/2) (5) to induce APO. The spontaneous development of placental insufficiency in BPH/5 mice allows for study of early mediators of fetal loss that occur at implantation and in early gestation. Notably, vascular disease, specifically chronic hypertension, is a risk factor for APO in humans, and this phenomenon is recapitulated in the BPH/5 mouse, a syngeneic model of APO secondary to placental dysfunction (38, 39). We sought to determine whether pregnancy complications in the BPH/5 mouse are related to innate immune system activation and, specifically, if blockade of complement activation, neutrophil infiltration and TNF-α activity could prevent abnormal placentation, fetal loss and growth restriction.
MATERIALS AND METHODS
BPH/5 mice pregnancy model
Animal experiments were approved by the Hospital for Special Surgery Institutional Animal Care and Use Committee and were conducted in accordance with guidelines for the care and use of laboratory animals. As described in detail previously, BPH/5 is an inbred subline derived from the BPH/2, and C57BL/6J (C57) mice serve as controls (38–40). BPH/5 and C57 mice (8–12 weeks old) were obtained from in house colonies. Mice were housed in a barrier animal facility with a climate controlled environment and 10h light/14h dark cycle. Timed matings were performed by pairing strain matched virgin females and males. Mice were examined daily for the presence of a vaginal plug, and the day of plug detection designated embryonic day (E) 0.5. Except where indicated, mice were sacrificed at E12.5, uteri dissected, fetuses and placentas weighed, and fetal resorption rates calculated (number of resorptions/total number of formed fetuses and resorptions).
In experiments involving neutrophil depletion, mice were treated with rat anti-mouse granulocyte RB6-8C5 mAb (anti-GR1) (100 μg i.p.) (PharMingen,) or 1A8 (anti-Ly6G) (250 μg i.p.) (BioXCell) on E2.5. An IgG2b or IgG2a antibody, respectively, were the isotype controls. Depletion of neutrophils, confirmed by flow cytometry and peripheral blood smears, occurred by 48 hours after injection and persisted through day 10. For complement blockade, recombinant CR2-Crry (0.200 mg) or CR2-FH (0.250 mg), prepared as previously described (41, 42), was administered i.v. on E5.5. To inhibit TNF-α, etanercept (10 mg/kg) (Amgen) was administered s.c. at E4.5.
Immunohistochemistry and morphometric analysis of placentas
Mice were sacrificed with CO2 asphyxiation at E6.5, 8.5, or 12.5. The uterus was cut between implantation sites, briefly rinsed in cold PBS, fixed in 10% neutral buffered formalin, processed by standard cycle, and embedded in paraffin. Sagittal 5 μm serial sections were obtained through the uterus, embryo and placenta and mid-sagittal sections were used for analysis. Mid-sagittal sections were identified in early gestations (E6.5 and E8.5) by identification of the embryo, and at E12.5 by identification of the umbilical cord insertion site in the placenta. Sections were deparaffinized, rehydrated and endogenous peroxidase activity was quenched with H2O2 (Sigma-Aldrich). C3 deposition was identified with a polyclonal goat anti-mouse C3 antibody (ICN Pharmaceuticals) as described previously (43). Neutrophil infiltrate was identified with a monoclonal rat anti-mouse neutrophil antibody (Cedarlane clone 7/4) after sodium citrate antigen retrieval. Biotinylated Griffonia Simplicifolia isolectin B4 (Vector Laboratories,) and smooth muscle actin (SMA) (Abcam) were used to identify basement membrane of fetal endothelial cells in the labyrinth (39) and smooth muscle, respectively. Antibody detection was performed with Vectastain ABC peroxidase-conjugated streptavidin reagent (Vector laboratories) followed by DAB (Dako) detection. Appropriate isotype controls were used to determine antibody specificity. Photographs were taken with a light microscope equipped with a digital camera. Morphometric analysis was performed as described previously (39). All measurements were made by two investigators who were blind to the experimental groups. Mean values of the two independently obtained measurements were used.
The immunohistochemical detection of uNK cells with DBA lectin and macrophages with F4/80 was performed using Discovery XT processor (Ventana Medical Systems). The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems), sections were blocked for 30 minutes with Background Buster solution (Innovex) followed by blocking with avidin-biotin blocking reagents (Ventana Medical Systems) for 16 minutes. To detect uNK cells, biotinylated DBA lectin (Sigma-Aldrich) was applied and sections were incubated for 5 hours, followed by detection with DAB detection kit (Ventana Medical Systems) according to manufacturer instruction. Slides were counterstained with hematoxylin and coverslipped with Permount (Fisher Scientific). To detect macrophages, anti-F4/80 (Abcam) antibodies were applied and sections were incubated for 6 hours, followed by 60 minutes incubation with biotinylated goat anti-rat IgG (Vector labs). The detection was performed with DAB detection kit (Ventana Medical Systems) according to manufacturer instruction. Slides were counterstained with hematoxylin and coverslipped with Permount (Fisher Scientific). The number of DBA lectin and F4/80 positive cells was determined by counting a mid-sagittal section at E8.5 or E12.5 from untreated C57 and BPH/5 mice or BPH/5 mice treated with anti-GR1, CR2-Crry or etanercept.
Flow cytometry to characterize cellular infiltrates
Mice were sacrificed at E8.5, the uterine horn opened and individual placentas were removed and vigorously rinsed in three changes of ice cold PBS to remove maternal blood, then gently dissociated and passed through a 100 μM strainer. Trophoblasts and infiltrating lymphocytes were separated with a 30% percoll (Sigma-Aldrich) gradient and stained with anti-CD45 (eBiosciences), anti-CD11b (eBiosciences) and anti-GR1 (eBiosciences). Neutrophils were defined as CD45+CD11b+GR1hi. Neutrophil numbers were determined using counting beads (CountBright absolute counting beads; Invitrogen) according to manufacturer instruction. Analysis was performed with FlowJo (Treestar Software).
Measurement of angiogenic factor and cytokine levels
Blood was collected in EDTA containing tubes, spun at 5000 rpm, and plasma was collected and stored at -80°C. Placentas were dissected free from surrounding tissue, rinsed in ice cold PBS, snap frozen in liquid nitrogen, and thawed on ice. After 3 freeze thaw cycles, individual placentas were resuspended in PBS, homogenized, centrifuged at 8000 rpm for 10 minutes, and supernatants collected and stored at -80°C. A Bradford assay was performed to normalize for protein levels in individual placentas. Placenta and plasma levels of vascular endothelial growth factor (VEGF), and TNF-α were determined by commercial ELISA (R&D systems). Other cytokines and chemokines were measured using a Luminex mouse multiplex panel (EMD-Millipore)
First trimester human trophoblast cell response to neutrophils
HTR-8/SVneo cells (HTR8), an immortalized human first trimester trophoblast cell line that retains many characteristics of first trimester trophoblasts (44), were kindly provided by Dr. Charles Graham (Queens University, Kingston, ON, Canada). HTR8 cells (5×105) were cultured until confluent in RPMI supplemented with 10 mM Hepes, 0.2% sodium bicarbonate, 10% FBS, 0.1mM minimal essential medium non-essential amino acids, 1 mM sodium pyruvate, and 100 nm penicillin/streptomycin. Human neutrophils were isolated from freshly drawn venous blood by a two step discontinuous ficoll-hypaque gradient as previously described (45). Mouse neutrophils were isolated from the bone marrow of C57, BPH/5, and TNF−/− mice using the EasySep Mouse Neutrophil Enrichment Kit (Stem Cell Technologies) according to manufacturer instructions. HTR8 cells were cultured until confluent, and human or murine neutrophils were added at a concentration of 1×106 neutrophils/ml. For experiments to determine VEGF concentration, culture supernatants were collected after 24 hours and assayed by ELISA (R&D systems). For experiments to determine TNF-α concentration, supernatants were collected after 2 hours and assayed by ELISA for human (Invitrogen) and mouse (eBioscience) TNF-α. After collection of supernatants, cell viability was confirmed by trypan blue exclusion.
Statistical Analysis
Data were expressed as mean ± SEM and analyzed using Prism version 4 for macintosh (Graphpad software). Differences between two groups were analyzed with a two tailed Student’s t-test. Multiple comparisons were performed with an analysis of variance followed by the Tukey multiple comparison test. P < 0.05 was considered statistically significant.
RESULTS
Neutrophils infiltrate the placenta in the BPH/5 mouse model of APO
Fetal loss in mice lacking complement regulatory proteins and in mice treated with antiphospholipid antibodies show neutrophil infiltration in the placenta, and abnormal neutrophil activation is seen in the peripheral blood in patients with preeclampsia (46, 47). To test the hypothesis that infiltrating neutrophils early in pregnancy contribute to placental insufficiency in BPH/5 mice, we compared the number of neutrophils present in the developing BPH/5 placenta with that in C57 mice. Since neutrophils are transiently present in the decidua at the time of implantation on E4 to E5 (48), we assessed the extent of neutrophil infiltration in BPH/5 mice beginning at E6.5. Immunohistochemical studies did not demonstrate neutrophils in the ectoplacental cone, myometrium, or decidua of either BPH/5 or C57 at E6.5. In contrast, at E8.5 neutrophils were present in the ectoplacental cone of both strains, and they were markedly increased in the BPH/5 (Figure 1A, B). There was no difference in the number of uNK cells or macrophages in the decidua of C57 and BPH/5 at E8.5 (uNK: 750 ± 110 vs. 640 ± 230 per midsagittal section; n=6; macrophages: 52 ± 5 vs 51 ± 6 per midsagittal section; n=6, respectively).
Figure 1. Neutrophil infiltrate in placenta of BPH/5 mice.

Representative images of the ectoplacental cone (EPC) at E8.5 stained with an anti-GR1 antibody from C57 (A) and BPH/5 mice (B). (bars = 20μm) C. Infiltrating neutrophils from implantation sites or peripheral blood of C57 and BPH/5 mice at E8.5 were identified by flow cytometry as CD45+CD11b+GR1hi. D. Mean neutrophil number from implantation sites or peripheral blood of C57 and BPH/5 mice is shown (n=6/group) * p<0.05 D. Placental homogenates (n=4 for each group) were collected on E8.5 and assayed for the neutrophil chemoattactant CXCL1 by Luminex technology and normalized to protein concentration by Bradford assay **p<0.001.
We confirmed the immunohistochemical findings of excess neutrophil infiltration with flow cytometry of leukocytes isolated from placentas. Both absolute number of CD45+CD11b+GR1hi and percentage of CD45+ that were CD11b+GR1hi were increased in BPH/5 (C57: 32.3±2.4% vs BPH/5: 50.4±5.8%, p<0.05) (Figure 1C, 1D). The difference between C57 and BPH/5 mice was due to infiltrating neutrophils and not from peripheral blood neutrophils, as the number and percentage of CD45+CD11b+GR1hi cells in the blood did not differ between strains (Figure 1C, 1D) (C57: 14.6±0.9% vs BPH/5: 16.2±3.4%, p=NS).
We assessed cytokines involved in neutrophil recruitment in placental lysates at E8.5 by Luminex multiplex assay. CXCL1 was higher in the BPH/5 lysates compared to C57 (Figure 1E). Levels of IL-17, IL1α, CCL3, CXCL2, or CXCL5 were not different between the two strains.
Neutrophils are required for abnormal placental and fetal development in BPH/5 mice
Neutrophils are effectors of fetal damage, and depletion of neutrophils has been shown to prevent pregnancy complications in antibody-mediated models of pregnancy loss (27). We sought to determine whether fetal loss and growth restriction in BPH/5 mice could be prevented by depletion of neutrophils. We treated mice at E2.5 with anti-GR1 to deplete neutrophils and assessed pregnancy outcome on E12.5. This time period was selected to ensure the absence of neutrophils prior to the time that we observed deposition of complement in BPH/5 placentas (see below). In BPH/5 mice treated with anti-GR1, there was a dramatic decrease in fetal loss and growth restriction; pregnancy outcomes in anti-GR1 treated mice were similar to those in C57 mice (Figure 2). Of note, treatment with anti-GR1 did not affect implantation number in BPH/5 or C57 (Figure S1) and did not alter pregnancy outcomes in C57 mice (Table S1). Because anti-GR1 depletes not only neutrophils, but subpopulations of other myeloid cells (49), we performed experiments to assess the effects of a different neutrophil specific monoclonal antibody anti-Ly6G (1A8) on APOs in BPH/5 mice. Treatment with anti-Ly6G rescued pregnancies in BPH/5 (Figure 2 A, B, C) confirming that neutrophils play a crucial role in APO in this mouse model.
Figure 2. Neutrophil depletion normalizes pregnancy phenotype of BPH/5 mice.
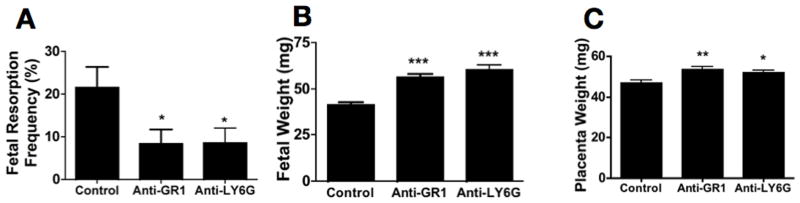
BPH/5 mice were treated with anti-GR1 or anti-Ly6G antibody to deplete neutrophils, or isotype control on E2.5. Mice were sacrificed on E12.5 and evaluated for resorption frequency and fetal and placental weight. Neutrophil depletion (A) protects mice from fetal resorption (control, n=13; anti-GR1, n=13; anti-Ly6G, n=9), (B) increases fetal weight (control, n=64; anti-GR1, n=54; anti-Ly6G, n=33) and (C) placental weight (control, n=28; anti GR-1, n=28; anti-Ly6G n=33). *p<0.05 **p<0.01 ***p< 0.0001
Decreased placental weights, altered invasion of the placental disc into the decidua, and defective spiral artery remodeling, as demonstrated by arteries with thick walls and retention of SMA positivity, are characteristics of BPH/5 placentas (39). It has been suggested that abnormal placentation defined by these anatomical features in BPH/5 mice leads to poor pregnancy outcomes. Consistent with the observed improved fetal outcomes, depletion of neutrophils in pregnant BPH/5 mice was associated with increased placental weight (Figure 2C) and normalized placental invasion and spiral artery remodeling (Figure 3). Both the proportional depth of the placental disc (Figure 3A, B, H) and relative area of the junctional zone (Figure 3C, D, I) were increased. The increased relative width of the decidual spiral arteries and absence of staining for SMA in the neutrophil depleted mice is consistent with normal spiral artery remodeling (Figure 3 E, F, G, J, K). Taken together, our findings demonstrate that neutrophils play a pivotal role in abnormal placental development and subsequent fetal loss in BPH/5 mice.
Figure 3. Neutrophil depletion improves placental morphology of BPH/5 mice.

BPH/5 mice were treated with anti-GR1 antibody to deplete neutrophils or isotype control on E2.5, sacrificed on E12.5, and placental histology examined. Representative images of intact feto-placental units from (A, C) isotype control and (B, D) anti-GR1 treated BPH/5 mice. (De, decidua; P, placental disc; JZ, junctional zone; L, labyrinth.). Neutrophil depletion normalizes (H) the proportional depth of the placental disc (P:P+De) and (I) the fractional area of the junctional zone relative to the placental disc (JZ:JZ+L). Representative images of decidual spiral arteries demonstrating thick walls in the (E) isotype treated BPH/5 mice and thin arterial walls in the (F) anti-GR1 treated mice (closed arrows=outer diameter, OD; open arrows=inner diameter, ID). (J) Data are summarized as ratios of the inner lumen to outer vessel diameter (ID/OD). Representative images of decidual spiral arteries demonstrating positive staining for SMA in (E, G) isotype treated BPH/5 mice (arrowhead) and loss of SMA staining in the (F) anti-GR-1 treated mice. (K) Data are summarized as a ratio of remodeled arteries relative to total number of decidual spiral arteries. Data are presented as mean SEM. For histologic studies a minimum of 9 feto-placental units were analyzed for each condition (3 implantation sites from three separate pregnancies). *p<0.05 ** p<0.01 ***p<0.001 Bars = 500μm (A, B), 200μm (C,D), 50μm (E, F), 25μm (G).
Consistent with previous findings, there is no difference in the number of uNK cells in the placentas of BPH/5 mice compared with C57 at E12.5 (39) (1000 ± 110 vs. 990 ± 120 per midsagittal section, respectively; n=4, p=NS). Similarly we found no difference in macrophage number in the placentas at E12.5 (14 ± 4 vs. 24 ± 21 per midsagittal section, respectively; n=4, p=NS). Furthermore, treatment with anti-GR1 did not alter the number of uNK cells (BPH/5: 1000±110 vs BPH/5 + anti-GR1 970±64 per midsagittal section; n=4, p=NS) or macrophages (BPH/5: 14 ± 41 vs BPH/5 + anti-GR1 6 ± 1 per midsagittal section; n=4, p=NS) in the placenta at E12.5.
Neutrophil infiltration is associated with reduced VEGF levels in vivo and in vitro
Placental insufficiency in BPH/5 mice is characterized by angiogenic imbalance in the dams. Circulating levels of VEGF, an angiogenic factor required for normal placental development, are decreased in the BPH/5 mice compared to C57 (38). To determine whether angiogenic imbalance occurs in the absence of neutrophils, we measured peripheral and placental levels of VEGF in BPH/5 mice treated with anti-GR1 at E2.5, as described above. Depletion of neutrophils resulted in higher levels of peripheral VEGF, as well as VEGF in the placenta (Figure 4A, B). That restoration of homeostatic levels of placental VEGF (C57: 34 ± 4pg/mg protein; BPH/5: 18 ± 4 pg/mg protein; BPH/5 + anti-GR1: 33 ± 4 pg/mg protein) is associated with normal placentation and pregnancy outcomes is consistent with the finding that adeno-VEGF improves placental function in the BPH/5 (50).
Figure 4. Neutrophil depletion is associated with normalization of VEGF levels.
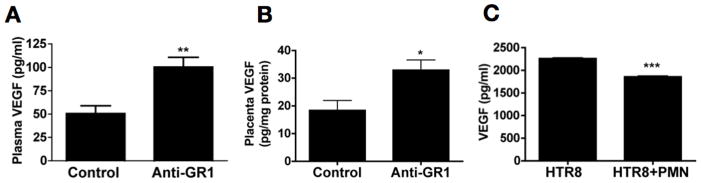
BPH/5 mice were treated with anti GR-1 antibody or isotype control. (A) Plasma (n=6 for each condition) or (B) placental homogenates (n=9 for each condition) were collected on E12.5, and VEGF levels were assayed by ELISA. (C) The first trimester trophoblast cells line (HTR8) were cultured alone or with 1×106 neutrophils/ml for 24 hrs. Cell free supernatants were collected and assayed for VEGF levels by ELISA. Data are pooled from three individual experiments done in duplicate. Data are presented as mean SEM. *p<0.05 ** p<0.01 ***p<0.0001
To determine whether neutrophils can directly affect levels of VEGF produced by trophoblast, we performed in vitro studies with HTR8 trophoblast cells cultured in the presence and absence of human neutrophils, and measured VEGF in supernatants. Incubation with neutrophils decreased release of VEGF (Figure 4C). These data support our in vivo findings showing that neutrophil depletion increased placental VEGF, and they suggest direct effects on the availability of VEGF.
Complement deposition precedes pathogenic neutrophil infiltration in vivo
Excessive complement activation is associated with APO in animal models and humans (9, 24, 51, 52), and products of the complement cascade recruit and stimulate neutrophils which, in turn, amplify activation of complement. To test the hypothesis that complement activation precedes neutrophil recruitment, we examined the kinetics of complement deposition in the BPH/5 mouse. C3 deposition was initially observed in the ectoplacental cone at E6.5 in the BPH/5; at this gestational age there is no C3 seen in the C57 (Figure 5A, B). By E8.5, there was extensive complement deposition in the ectoplacental cone of BPH/5 with minimal staining of C57 (Figure 5C, D). Notably, neutrophil infiltrates were first evident at E8.5 (Figure 1) and not before. Thus, complement activation precedes infiltration of neutrophils. To exclude the possibility that systemic complement activation occurs in BPH/5 pregnancies, similar to that noted in humans with preeclampsia, we measured hemolytic complement activity and found no differences in levels between BPH/5 and C57 (Figure S2). These data argue that complement is activated locally in BPH/5 mice.
Figure 5. Complement deposition in developing BPH/5 mice.
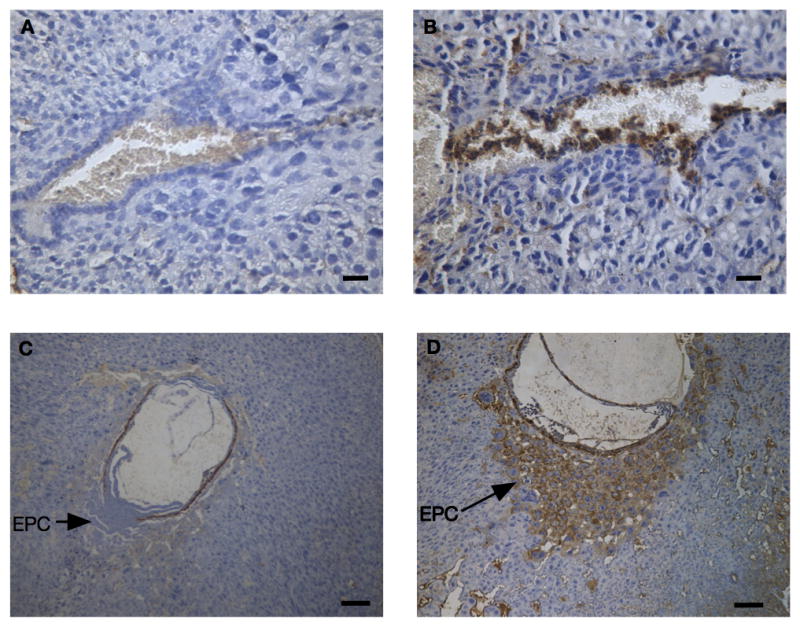
Uteri from (A, C) C57 and (B, D) BPH/5 mice were stained with an anti-C3 mAb. Representative images of the ectoplacental cone at E6.5 (A, B) and E8.5 (C, D) demonstrate increased complement deposition in the ectoplacental cone of the BPH/5 mice but not the C57. Bar = 20μm (A,B) 100μm (C,D).
Inhibition of complement activation prevents fetal loss and growth restriction in BPH/5 mice
To determine whether blockade of complement activation can prevent abnormal placentation and APOs, we treated BPH/5 mice with targeted complement inhibitors: CR2-Crry or CR2-FH. These agents are fusion proteins of either Crry, a pan-C3 convertase inhibitor, or factor H (FH), a regulator of the alternative complement pathway, with complement receptor 2 (CR2), which binds C3 degradation products and thus localizes the protein to sites of complement deposition. CR2-Crry inhibits the classical, lectin and alternative complement pathways at the C3 activation step, whereas CR2-FH is specific for the alternative pathway (41, 42). At the doses used, CR2-Crry and CR2-FH have minimal effects on systemic complement activity. They target cell bound products of complement activation, accumulate in tissues at sites of complement activation, and remain there for prolonged periods (53). Blockade of complement activation with either targeted inhibitor prevented fetal loss (Figure 6A) and growth restriction (Figure 6B). These results were comparable to those seen with neutrophil depletion.
Figure 6. Complement inhibition rescues BPH/5 pregnancies.

BPH/5 mice were treated with CR2-Crry or CR2-FH to inhibit complement or control (PBS) on E5.5. Mice were sacrificed on E12.5 and evaluated for resorption frequency, fetal and placental weight and placental histology. The effect of complement inhibition on (A) fetal resorption (minimum of 6 mice/group), (B) fetal weight (minimum 28 fetuses/group) and (C) placental weight (minimum of 28 fetuses/group) is shown. (D–G) Placental morphology was assessed after treatment with CR2-Crry or control: (D) the fractional area of the junctional zone relative to the placental disc (JZ:JZ+L), (E) and the depth of the placental disc (P:P+De) are shown. Spiral artery remodeling as measured by (F) ratios of the inner lumen to outer vessel diameter (ID/OD) of decidual spiral arteries and (G) the ratio of SMA negative (remodeled arteries) relative to total number of decidual spiral arteries after treatment with CR2-Crry or control is shown. (H) Pregnant mice were treated with CR2-Crry at E5.5 and infiltrating cells were separated from trophoblast at E8.5. The number of infiltrating GR1+ cells in C57 or BPH/5 with or without CR2-Crry treatment are shown. (I) The effect of complement inhibition on VEGF levels from placental homogenates was determined by ELISA. Data are presented as mean SEM. For histologic studies a minimum of 9 feto-placental units were analyzed for each condition (3 implantation sites from three separate pregnancies). *p<0.05 ** p<0.01 ***0.001
Because abnormal placentation is a cause of fetal loss and growth restriction, we examined the effect of complement inhibition on placental phenotype. Local complement inhibition was associated with increased weight of the placenta in the CR2-Crry treated animals but not in those treated with CR2-FH (Figure 6C). We performed histologic analyses of placentas from CR2-Crry treated animals because this agent was most effective in preserving placental weight. Complement inhibition with CR2-Crry normalized the junctional zone without affecting the ratio of placenta to decidua (Figure 6D, E) and normalized placental spiral artery morphology to that of low resistance vessels: arterial walls were thinner and fewer were positive for SMA (Figure 6F, G). Targeted inhibitors of complement improved placental architecture in BPH/5 mice to the same extent as depletion of neutrophils.
Complement inhibition decreases neutrophil recruitment and angiogenic imbalance in vivo
Given our immunohistochemical evidence that complement activation precedes infiltration of neutrophils into the placenta, we hypothesized that blockade of complement activation would prevent recruitment of neutrophils. We treated BPH/5 mice with the targeted complement inhibitor CR2-Crry at E5.5 and quantified placental neutrophil infiltration by flow cytometry at E8.5, the time point when it was prominent in BPH/5 (Figure 1C). When complement activation was blocked, neutrophils were not recruited into the placenta; the number of neutrophils in BPH/5 placentas was comparable to that of C57 (Figure 6H). Importantly, treatment with CR2-Crry did not alter the number of uNK cells (control: 1000 ± 110 vs CR2-Crry: 1000 ± 60 per midsagittal section; n=4; p=NS) or macrophages (control: 14 ± 4 vs CR2-Crry: 10 ± 1 per midsagittal section; n=4; p=NS) in the placenta at E 12.5.
Inhibition of complement activation also increased VEGF concentration in the placenta. We observed a nearly twofold increase in placental VEGF in BPH/5 mice treated with either CR2-Crry or CR2-FH (Figure 6I). There was a more modest effect on peripheral blood levels of VEGF (control 63 ± 3.4 pg/ml vs CR2-Crry: 76 ± 3.1 pg/ml; vs CR2-FH: 72 ± 4.2 pg/ml; p=NS.) Taken together, these data support a critical role of local complement activation as a proximal mediator in the pathogenesis of APOs in BPH/5.
TNF-α is a mediator of adverse outcomes in pregnant BPH/5 mice
TNF-α has been shown to mediate APO in LPS and anti-phospholipid antibody-treated rodent models (7, 33). Because complement activation products trigger release of TNF-α by neutrophils, and TNF-α stimulates neutrophils in an autocrine and paracrine manner to amplify damage, we performed studies to determine whether TNF-α contributes to inflammation and fetal loss in BPH/5 mice. We measured TNF-α in placental lysates obtained at E8.5, the time when neutrophil infiltration and complement deposition were increased in the BPH/5 compared to C57, and found markedly higher TNF-α levels in the BPH/5 mice (Figure 7A). Furthermore, depletion of neutrophils with anti-GR1 decreased TNF-α, implicating neutrophils in the pathway leading to elevations in TNF-α (Figure 7A). To investigate the source of TNF-α, we cultured HTR8 human trophoblasts with neutrophils from C57, BPH/5, TNF−/− or humans, collected supernatants after 2 hours, and assayed for mouse TNF-α (Figure 7B) and human TNF-α (Figure 7C). Mouse neutrophils cultured in the presence of HTR8 produced TNF-α (Figure 7B), and there was no difference in TNF-α production between C57 and BPH/5 mice (Figure 7B and Figure S3A). Human neutrophils were also stimulated to release TNF-α in response to HTR8 (Figure 7C). Neither HTR8 alone (Figure 7C) or stimulated with C5a (Figure S3B) produce TNF-α. Although the triggers for TNF-α production are not clear, these data indicate that neutrophils are a likely source of TNF-α in inflammatory sites such as BPH/5 placentas. We observed no difference in TNF-α production by neutrophils from BPH/5 and C57 suggesting that excess TNF-α in BPH/5 placenta is due to recruitment of greater numbers of neutrophils.
Figure 7. Elevated TNF-α in placentas from BPH/5 mice and supernatants from neutrophils cultured with HTR8 cells.
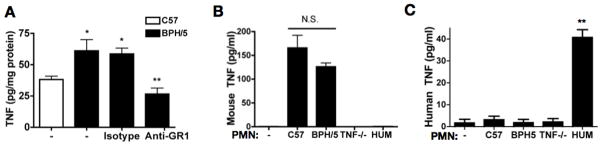
(A) TNF-α levels were assayed from placental homogenates at E8.5 from untreated C57 and BPH/5 mice, and from BPH/5 mice treated at E2.5 with anti-GR1 antibody to deplete neutrophils or isotype control (n=9 for each condition) *p< 0.05 vs C57. **p<0.001 vs isotype treated. (B) Levels of mouse TNF-α were assayed from supernatants of neutrophils from C57, BPH/5, TNF−/− mice or humans (HUM) cocultured with HTR8 cells. (C) Levels of human TNF-α were assayed from supernatants of neutrophils from C57, BPH/5, TNF−/− mice or humans cocultured with HTR8 cells.
Treatment with etanercept, an available biological therapeutic that blocks TNF-α activity, reduced fetal loss in BPH/5 mice to levels of C57 (Figure 8A), normalized placental weight (Figure 8C) and restored all studied metrics of placentation: junctional zone ratio (Figure 8D), placental invasion (Figure 8E), thinner spiral artery walls (Figure 8F) and loss of SMA staining (Figure 8G). There was no significant change in the weight of the surviving fetuses (Figure 8B). Similar to treatment with the complement inhibitors, etanercept increased placental VEGF levels (Figure 8H). Of note, etanercept did not alter numbers of uNK cells (control: 1000 ± 110 vs. etanercept: 790 ± 340 per midsagittal section; n=4; p=NS) or macrophages (control: 14 ± 4 vs etanercept: 4 ± 2 per midsagittal section; n=4; p=NS) in the placenta at E12.5.
Figure 8. TNF-α blockade prevents APO and normalizes placental development in BPH/5 mice.
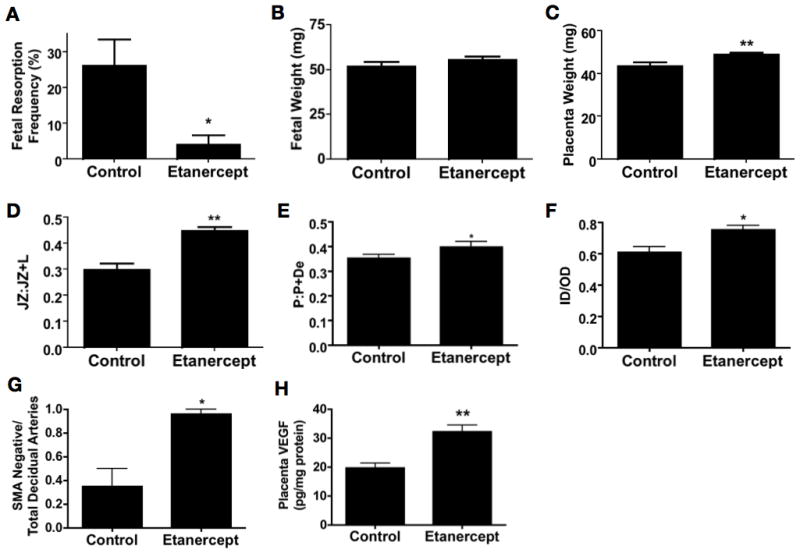
The effect of TNF-α blockade on (A) fetal resorption (n= 6 mice/group), (B) fetal weight (minimum 28 fetuses/group), and (C) placental weight (minimum 28 fetuses/group) is shown. Placental morphology was assessed after TNF-α blockade or control: (D) the fractional area of the junctional zone relative to the placental disc (JZ:JZ+L) and (E) depth of the placental disc (P:P+De) are shown. Spiral artery remodeling as measured by (F) ratios of the inner lumen to outer vessel diameter (ID/OD) of decidual spiral arteries and (G) the ratio of SMA negative (remodeled arteries) relative to total number of decidual spiral arteries after treatment with etanercept or control is shown. (H) Placental VEGF levels were assayed in the BPH/5 after TNF-α blockade with etanercept. Data are presented as mean SEM. For histologic studies a minimum of 9 feto-placental units were analyzed for each condition (3 implantation sites from three separate pregnancies). *p<0.05 ** p<0.01
DISCUSSION
We have shown that complement activation at the maternal fetal interface leads to recruitment of neutrophils, elevation in local TNF-α levels, reduction of the essential angiogenic factor VEGF and ultimately, abnormal placentation. We provide the first evidence that complement activation and the ensuing infiltration of neutrophils into the placenta leads to abnormal spiral artery remodeling and angiogenic dysregulation. These findings, the first in a syngeneic spontaneous model of abnormal placental development, support work from our lab and others that complement is an essential proximal mediator in antibody-dependent and antibody-independent mouse models of APO (9, 10, 27). Here, we demonstrate the critical role of local complement activation as an initiator of APO in BPH/5 mice by showing that features of abnormal placental development and its consequences on the fetus are reversed by inhibiting the complement cascade, specifically at the maternal-fetal interface.
Defective placentation is associated with preeclampsia, growth restriction and other obstetric syndromes (2). The BPH/5 mouse has placental findings, including inadequate spiral artery remodeling, similar to those seen in humans. As in patients with preeclampsia, not all spiral arteries are inadequately remodeled in BPH/5 (2). Inadequate spiral artery remodeling is believed to contribute to fetal growth restriction and placental ischemia (1, 2). Inhibition of complement, depletion of neutrophils and blockade of TNF-α improves spiral artery remodeling in BPH/5 pregnancies.
Pregnancy in BPH/5 mice is characterized by a maternal syndrome including hypertension and proteinuria late in gestation. Our studies focused on early changes at the maternal-fetal interface that precede, and perhaps cause, manifestations of preeclampsia. Although we observed complement deposition, neutrophil infiltration, TNF-α elevation and decreased VEGF in the placenta, there was no evidence of systemic alterations of these mediators in the second trimester, a point before clinically apparent maternal responses to placental insufficiency in BPH/5 mice or humans. That inflammation is restricted to the placenta, before clinically apparent disease, allows for the insidious progression of disorders of placental dysfunction, which, as in humans, variably lead to maternal or fetal abnormalities.
To attenuate complement activation at the maternal-fetal interface, we used CR2-Crry, which blocks all pathways of complement activation (classical, alternative and lectin) and CR2-FH, which selectively inhibits the alternative pathway (54). The CR2 domain of both compounds binds covalently bound complement activation fragments iC3b, C3dg, and C3d on tissue (55) and thereby localizes the complement regulators, Crry or FH, to the sites of complement activation. Both compounds prevented fetal loss and growth restriction but did so to different extents. The efficacy of CR2-FH underscores the importance of the alternative pathway in this pathway. Such targeted inhibition is especially attractive because it allows for specific inhibition at the site and time of injury without generalized immune suppression of the host (41).
We show that local complement activation triggers neutrophil recruitment, but complement activation in the absence of neutrophils is insufficient to cause APO. Depletion of neutrophils early in pregnancy was able to reduce all studied metrics of APO in BPH/5 mice. Although neutrophil depletion is not a clinically viable option, the importance of infiltrating leukocytes in driving fetal loss and growth restriction provides a framework for understanding the pathogenesis of these conditions. In our experiments, mice were treated on day 2.5 with anti-GR1 antibody and neutrophils were depleted by day 5.5 and returned to circulation by day 12.5. Taken together these data indicate that prevention of the initial inflammatory insult by neutrophils may have long-term benefits on pregnancy outcome and that blockade of inflammation may not be necessary throughout pregnancy.
Evidence for elevated levels of the neutrophil chemoattractant CXCL1 in BPH/5 placenta is consistent with our model that early inflammation leads to APO. CXCL1 recruits neutrophils and is released from neutrophils in response to TNF-α (56). In addition, CXCL1 is secreted from trophoblast in response to damage (57). Thus, production of CXCL1 by injured trophoblasts or activated neutrophils may initiate a positive feedback loop in which placental injury drives neutrophil infiltration and TNF-α release, which then increases CXCL1 and amplifies inflammation.
Activated neutrophils release mediators of tissue damage, including TNF-α, which has been shown to cause abnormal placentation and growth restriction, and to recruit and activate other effectors of inflammation (58–60). In our studies, depletion of neutrophils normalized placental TNF-α and VEGF in BPH/5 mice; blockade of TNF-α also normalized placental VEGF. Both animal models and studies in people have identified release of anti-angiogenic factors and pro-inflammatory cytokines to be key mediators of placental insufficiency and the associated fetal and maternal complications, typically preeclampsia (61–63). Our work defines a pathway that precedes this response and offers a target for treatment before manifestation of impaired placentation and maternal end-organ dysfunction. The salutary effects of TNF-α blockade may be sufficient to justify this therapy to prevent placental inflammation associated with fetal hypoperfusion and APO in high-risk pregnancies.
The present work focused on mechanisms of placental insufficiency. We examined fetal loss and growth restriction, which are significant APOs. We did not directly examine the contribution of complement, neutrophils and TNF-α to the maternal features of preeclampsia, because these clinical manifestations occur later in this model. Nonetheless, maternal disease is highly associated with abnormal placental development and it is likely that inhibition of complement or TNF-α also prevents maternal disease. Taken together our findings provide the rationale for trials with agents that modulate innate pathways early in pregnancy to prevent APOs in those women at high risk.
Supplementary Material
Acknowledgments
We are grateful to Jenny Sones, PhD, Christa Heyward, Ph.D., Heinrich Lob, Ph.D., and Xiaoping Qing, M.D., Ph.D. for valuable discussions.
Abbreviations used in this article
- APO
adverse pregnancy outcome
- C57
C57Bl/6
- E
embryonic day
- SMA
smooth muscle actin
- CR2
complement receptor 2
- FH
factor H
Footnotes
This work was supported by the March of Dimes and NICHD (NIHK12HD000849) and Clinical and Translation Science Center at WCMC (UL1RR024996) to SEG, the NIH (R01HL086576), and VA (1I01RX001141, 5I01BX001218) to ST, Qatar National Research Fund, NPRP09-1099-279 to RLD. Immunohistochemistry was performed at the Molecular Cytology Core Facility of MSKCC which is supported by NCI (P30 CA008748)
References
- 1.Roberts DJ, Post MD. The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol. 2008;61:1254–1260. doi: 10.1136/jcp.2008.055236. [DOI] [PubMed] [Google Scholar]
- 2.Brosens I, Pijnenborg R, Vercruysse L, Romero R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. YMOB. 2011;204:193–201. doi: 10.1016/j.ajog.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li M, Huang SJ. Innate immunity, coagulation and placenta-related adverse pregnancy outcomes. Thromb Res. 2009;124:656–662. doi: 10.1016/j.thromres.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hahn S, Gupta AK, Troeger C, Rusterholz C, Holzgreve W. Disturbances in placental immunology: ready for therapeutic interventions? Springer Semin Immunopathol. 2006;27:477–493. doi: 10.1007/s00281-006-0016-5. [DOI] [PubMed] [Google Scholar]
- 5.Qing X, Redecha PB, Burmeister MA, Tomlinson S, D’Agati VD, Davisson RL, Salmon JE. Targeted inhibition of complement activation prevents features of preeclampsia in mice. Kidney Int. 2011;79:331–339. doi: 10.1038/ki.2010.393. [DOI] [PubMed] [Google Scholar]
- 6.Redecha P, Tilley R, Tencati M, Salmon JE, Kirchhofer D, Mackman N, Girardi G. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. 2007;110:2423–2431. doi: 10.1182/blood-2007-01-070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Renaud SJ, Cotechini T, Quirt JS, Macdonald-Goodfellow SK, Othman M, Graham CH. Spontaneous Pregnancy Loss Mediated by Abnormal Maternal Inflammation in Rats Is Linked to Deficient Uteroplacental Perfusion. J Immunol. 2011;186:1799–1808. doi: 10.4049/jimmunol.1002679. [DOI] [PubMed] [Google Scholar]
- 8.Scharfe-Nugent A, Corr SC, Carpenter SB, Keogh L, Doyle B, Martin C, Fitzgerald KA, Daly S, O’Leary JJ, O’Neill LAJ. TLR9 provokes inflammation in response to fetal DNA: mechanism for fetal loss in preterm birth and preeclampsia. J Immunol. 2012;188:5706–5712. doi: 10.4049/jimmunol.1103454. [DOI] [PubMed] [Google Scholar]
- 9.Girardi G, Yarilin D, Thurman JM, Holers VM, Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. 2006;203:2165–2175. doi: 10.1084/jem.20061022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W, Irani RA, Zhang Y, Ramin SM, Blackwell SC, Tao L, Kellems RE, Xia Y. Autoantibody-mediated complement c3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension. 2012;60:712–721. doi: 10.1161/HYPERTENSIONAHA.112.191817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, Espinola R, Xiaowei LE, Mao D, Vialpando CG, Salmon JE. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. 2002;195:211–220. doi: 10.1084/jem.200116116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carpentier PA, Dingman AL, Palmer TD. main. Am J Pathol. 2011;178:2802–2810. doi: 10.1016/j.ajpath.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol. 2005;175:4084–4090. doi: 10.4049/jimmunol.175.6.4084. [DOI] [PubMed] [Google Scholar]
- 14.Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. 2012;150:29–38. doi: 10.1016/j.cell.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee AJ, Kandiah N, Karimi K, Clark DA, Ashkar AA. Interleukin-15 is required for maximal lipopolysaccharide-induced abortion. J Leukoc Biol. 2013;93:905–912. doi: 10.1189/jlb.0912442. [DOI] [PubMed] [Google Scholar]
- 16.Murphy SP, Hanna NN, Fast LD, Shaw SK, Berg G, Padbury JF, Romero R, Sharma S. Evidence for participation of uterine natural killer cells in the mechanisms responsible for spontaneous preterm labor and delivery. Am J Obstet Gynecol. 2009;200:308.e1–9. doi: 10.1016/j.ajog.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pangburn MK, V, Ferreira P, Cortes C. Discrimination between host and pathogens by the complement system. Vaccine. 2008;26(Suppl 8):I15–21. doi: 10.1016/j.vaccine.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynch AM, Murphy JR, Byers T, Gibbs RS, Neville MC, Giclas PC, Salmon JE, Holers VM. Alternative complement pathway activation fragment Bb in early pregnancy as a predictor of preeclampsia. Am J Obstet Gynecol. 2008;198:385.e1–9. doi: 10.1016/j.ajog.2007.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch AM, Salmon JE. Dysregulated complement activation as a common pathway of injury in preeclampsia and other pregnancy complications. Placenta. 2010;31:561–567. doi: 10.1016/j.placenta.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buurma A, Cohen D, Veraar K, Schonkeren D, Claas FH, Bruijn JA, Bloemenkamp KW, Baelde HJ. Preeclampsia Is Characterized by Placental Complement Dysregulation. Hypertension. 2012 doi: 10.1161/HYPERTENSIONAHA.112.194324. [DOI] [PubMed] [Google Scholar]
- 23.Bulla R, Bossi F, Agostinis C, Radillo O, Colombo F, De Seta F, Tedesco F. Complement production by trophoblast cells at the feto-maternal interface. J Reprod Immunol. 2009;82:119–125. doi: 10.1016/j.jri.2009.06.124. [DOI] [PubMed] [Google Scholar]
- 24.Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- 25.Wetsel RA. Structure, function and cellular expression of complement anaphylatoxin receptors. Curr Opin Immunol. 1995;7:48–53. doi: 10.1016/0952-7915(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 26.Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, Halbwachs-Mecarelli L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 27.Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, Lambris JD, Holers VM, Salmon JE. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112:1644–1654. doi: 10.1172/JCI18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. 2008;118:3453–3461. doi: 10.1172/JCI36089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, Resink TJ. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584:3193–3197. doi: 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Hunt JS, Chen HL, Miller L. Tumor necrosis factors: pivotal components of pregnancy? Biol Reprod. 1996;54:554–562. doi: 10.1095/biolreprod54.3.554. [DOI] [PubMed] [Google Scholar]
- 31.Haider S, KnOfler M. Human Tumour Necrosis Factor: Physiological and Pathological Roles in Placenta and Endometrium. Placenta. 2009;30:111–123. doi: 10.1016/j.placenta.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otun HA, Lash GE, Innes BA, Bulmer JN, Naruse K, Hannon T, Searle RF, Robson SC. Effect of tumour necrosis factor-α in combination with interferon-γ on first trimester extravillous trophoblast invasion. J Reprod Immunol. 2011;88:1–11. doi: 10.1016/j.jri.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Berman J, Girardi G, Salmon JE. TNF-alpha is a critical effector and a target for therapy in antiphospholipid antibody-induced pregnancy loss. J Immunol. 2005;174:485–490. doi: 10.4049/jimmunol.174.1.485. [DOI] [PubMed] [Google Scholar]
- 34.Cotechini T, Komisarenko M, Sperou A, Macdonald-Goodfellow S, Adams MA, Graham CH. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. Journal of Experimental Medicine. 2014;211:165–179. doi: 10.1084/jem.20130295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holcberg G, Huleihel M, Sapir O, Katz M, Tsadkin M, Furman B, Mazor M, Myatt L. Increased production of tumor necrosis factor-alpha TNF-alpha by IUGR human placentae. Eur J Obstet Gynecol Reprod Biol. 2001;94:69–72. doi: 10.1016/s0301-2115(00)00321-3. [DOI] [PubMed] [Google Scholar]
- 36.Pijnenborg R, McLaughlin PJ, Vercruysse L, Hanssens M, Johnson PM, Keith JC, Van Assche FA. Immunolocalization of tumour necrosis factor-alpha (TNF-alpha) in the placental bed of normotensive and hypertensive human pregnancies. Placenta. 1998;19:231–239. doi: 10.1016/s0143-4004(98)90054-6. [DOI] [PubMed] [Google Scholar]
- 37.Kupferminc MJ, Peaceman AM, Wigton TR, Rehnberg KA, Socol ML. Tumor necrosis factor-alpha is elevated in plasma and amniotic fluid of patients with severe preeclampsia. YMOB. 1994;170:1752–7. discussion 1757–9. [PubMed] [Google Scholar]
- 38.Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, Merrill DC, Sethi S, Weiss RM, Bates JN. Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension. 2002;39:337–342. doi: 10.1161/hy02t2.102904. [DOI] [PubMed] [Google Scholar]
- 39.Dokras A, Hoffmann DS, Eastvold JS, Kienzle MF, Gruman LM, Kirby PA, Weiss RM, Davisson RL. Severe Feto-Placental Abnormalities Precede the Onset of Hypertension and Proteinuria in a Mouse Model of Preeclampsia. Biol Reprod. 2006;75:899–907. doi: 10.1095/biolreprod.106.053603. [DOI] [PubMed] [Google Scholar]
- 40.Schlager G. Biometrical genetic analysis of blood pressure level in the genetically hypertensive mouse. Clin Exp Hypertens. 1994;16:809–824. doi: 10.3109/10641969409078027. [DOI] [PubMed] [Google Scholar]
- 41.Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, Tsokos GC, Tomlinson S. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest. 2005;115:2444–2453. doi: 10.1172/JCI25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol. 2008;181:8068–8076. doi: 10.4049/jimmunol.181.11.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–1912. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 44.Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp Cell Res. 1993;206:204–211. doi: 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- 45.Pricop L, Gokhale J, Redecha P, Ng SC, Salmon JE. Reactive oxygen intermediates enhance Fc gamma receptor signaling and amplify phagocytic capacity. J Immunol. 1999;162:7041–7048. [PubMed] [Google Scholar]
- 46.Wang Y, Gu Y, Philibert L, Lucas MJ. Neutrophil Activation Induced by Placental Factors in Normal and Pre-eclamptic Pregnancies In Vitro. Placenta. 2001;22:560–565. doi: 10.1053/plac.2001.0691. [DOI] [PubMed] [Google Scholar]
- 47.Butterworth BH, I, Greer A, Liston WA, Haddad NG, Johnston TA. Immunocytochemical localization of neutrophil elastase in term placenta decidua and myometrium in pregnancy-induced hypertension. Br J Obstet Gynaecol. 1991;98:929–933. doi: 10.1111/j.1471-0528.1991.tb13516.x. [DOI] [PubMed] [Google Scholar]
- 48.McMaster MT, Dey SK, Andrews GK. Association of monocytes and neutrophils with early events of blastocyst implantation in mice. J Reprod Fertil. 1993;99:561–569. doi: 10.1530/jrf.0.0990561. [DOI] [PubMed] [Google Scholar]
- 49.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 50.Woods AK, Hoffmann DS, Weydert CJ, Butler SD, Zhou Y, Sharma RV, Davisson RL. Adenoviral delivery of VEGF121 early in pregnancy prevents spontaneous development of preeclampsia in BPH/5 mice. Hypertension. 2011;57:94–102. doi: 10.1161/HYPERTENSIONAHA.110.160242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salmon JE, Heuser C, Triebwasser M, Liszewski MK, Kavanagh D, Roumenina L, Branch DW, Goodship T, Frémeaux-Bacchi V, Atkinson JP. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med. 2011;8:e1001013. doi: 10.1371/journal.pmed.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch AM, Gibbs RS, Murphy JR, Giclas PC, Salmon JE, Holers VM. Early elevations of the complement activation fragment C3a and adverse pregnancy outcomes. Obstet Gynecol. 2011;117:75–83. doi: 10.1097/AOG.0b013e3181fc3afa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banda NK, Levitt B, Glogowska MJ, Thurman JM, Takahashi K, Stahl GL, Tomlinson S, Arend WP, Holers VM. Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice. J Immunol. 2009;183:5928–5937. doi: 10.4049/jimmunol.0901826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sekine H, Kinser TTH, Qiao F, Martinez E, Paulling E, Ruiz P, Gilkeson GS, Tomlinson S. The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice. Arthritis Rheum. 2011;63:1076–1085. doi: 10.1002/art.30222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song H, Qiao F, Atkinson C, Holers VM, Tomlinson S. A complement C3 inhibitor specifically targeted to sites of complement activation effectively ameliorates collagen-induced arthritis in DBA/1J mice. J Immunol. 2007;179:7860–7867. doi: 10.4049/jimmunol.179.11.7860. [DOI] [PubMed] [Google Scholar]
- 56.Gasperini S, Calzetti F, Russo MP, De Gironcoli M, Cassatella MA. Regulation of GRO alpha production in human granulocytes. J Inflamm. 1995;45:143–151. [PubMed] [Google Scholar]
- 57.Mulla MJ, Brosens JJ, Chamley LW, Giles I, Pericleous C, Rahman A, Joyce SK, Panda B, Paidas MJ, Abrahams VM. ORIGINAL ARTICLE: Antiphospholipid Antibodies Induce a Pro-Inflammatory Response in First Trimester Trophoblast Via the TLR4/MyD88 Pathway. American Journal of Reproductive Immunology. 2009;62:96–111. doi: 10.1111/j.1600-0897.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu B, Nakhla S, Makris A, Hennessy A. TNF-α inhibits trophoblast integration into endothelial cellular networks. Placenta. 2011;32:241–246. doi: 10.1016/j.placenta.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Renaud SJ, Sullivan R, Graham CH. Tumour necrosis factor alpha stimulates the production of monocyte chemoattractants by extravillous trophoblast cells via differential activation of MAPK pathways. Placenta. 2009;30:313–319. doi: 10.1016/j.placenta.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 60.Cotechini T, Komisarenko M, Sperou A, Macdonald-Goodfellow S, Adams MA, Graham CH. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. Journal of Experimental Medicine. 2014;211:165–179. doi: 10.1084/jem.20130295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 63.LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep. 2007;9:480–485. doi: 10.1007/s11906-007-0088-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.