

Abstract
The proposed mechanism for tryptophan synthase shows βLys87 playing multiple catalytic roles: it bonds to the PLP cofactor, activates C4′ for nucleophilic attack via a protonated Schiff base nitrogen, and abstracts and returns protons to PLP-bound substrates (i.e. acid-base catalysis). ε-15N-lysine TS was prepared to access the protonation state of βLys87 using 15N solid-state nuclear magnetic resonance (SSNMR) spectroscopy for three quasi-stable intermediates along the reaction pathway. These experiments establish that the protonation state of the ε-amino group switches between protonated and neutral states as the β-site undergoes conversion from one intermediate to the next during catalysis, corresponding to mechanistic steps where this lysine residue has been anticipated to play alternating acid and base catalytic roles that help steer reaction specificity in tryptophan synthase catalysis.
Keywords: pyridoxal-5′-phosphate, tryptophan synthase, solid-state nuclear magnetic resonance, acid-base catalysis
1. Introduction
The bioactive form of vitamin B6, pyridoxal-5′-phosphate (PLP), acts as coenzyme in metabolic transformations of amino acids, including transamination, racemization, and α-, β-, γ-elimination/replacement.[1] Early in the study of PLP enzymes, it was shown that reduction of the holo-enzyme by NaBH4, followed by isolation and identification of modified peptide residues, invariably gave a peptide with PLP covalently linked to the ε-amino group of a lysine side chain, thus establishing that PLP is tethered to the active sites of virtually all PLP-dependent enzymes via a Schiff base linkage to this group.[2-13] Confirmation of the lysine linkage came later as X-ray crystal structures of PLP-dependent enzymes began to emerge. Based on the chemistry of the Schiff base functional group exhibited by small molecules in solution, it was deduced that a protonated Schiff base tautomer likely activates C4′ of PLP for nucleophilic attack by amino acid substrates.[3-6, 14-17] Thus, the lysine side chain not only covalently binds the PLP coenzyme, but is directly involved in subsequent stages of catalysis. Given the proximity of this residue to the coenzyme active site, it has long been suspected that it continues to play an acid-base catalytic role after its linkage to the coenzyme has been broken.[14-19] X-ray crystal structures show that it is well-positioned in many cases to supply or remove a proton during the enzymatic transformation.[20-24] Yet experimental evidence to support this continued catalytic role is indirect: the X-ray crystal structures generally do not report the protonation states of active site residues, and optical methods are a far less informative reporter on this group once the Schiff base linkage has been broken. Here, we detail the use of 15N solid-state nuclear magnetic resonance (SSNMR) spectroscopy to directly access the protonation state of the catalytic ε-amino group of the active site lysine that binds PLP in tryptophan synthase (TS). This allows us to map out its protonation state at several points in the catalytic cycle. These studies show that the protonation state of the ε-amino group switches between protonated and neutral states as the PLP-substrate complex undergoes conversion from one intermediate to the next during catalysis, and that these changes correlate to mechanistic steps in which this lysine residue has been anticipated to play alternating acid and base catalytic roles.
Tryptophan synthase presents an important paradigm for understanding enzyme structure-function relationships concerned with PLP-enzyme catalysis, including allosteric regulation of substrate channeling, C-C bond scission/bond synthesis, proton transfers to and from carbon centers, and multiple covalent transformations within a common catalytic site. The bacterial TS α2β2 bienzyme complex catalyzes the final two steps in the biosynthesis of L-tryptophan (Scheme 1), and the β-subunit of TS binds PLP in the holoenzyme (referred to as the internal aldimine form) through a Schiff base linkage to the ε-amino group of βLys87. Catalysis at the α-sites involves C-C bond scission to convert indole-3-glycerol phosphate (IGP) to indole and D-glyceraldehyde-3-phosphate (G3P). Catalysis at the β-sites involves the PLP-dependent replacement of the L-Ser hydroxyl by indole to give L-Trp and a water molecule (Scheme 1).[1, 19, 25-27] In Stage I of the multi-step β-reaction, L-Ser is converted to the quasi-stable α-aminoacrylate intermediate, E(A-A), via gem-diamine, E(GD1), external aldimine, E(Aex1), and L-Ser quinonoidal, E(Q1), intermediates. Stage II of the β-reaction is initiated by substrate channeling of indole from the α-site to the β-site through a 25 Å-long tunnel connecting the α- and β- active sites where it makes a nucleophilic attack on the Cβ of E(A-A) to form a C-C bond and give the first L-Trp quinonoid species, E(Q2).[20, 27-29] Formation of E(Q2) is followed by conversion to the second L-Trp quinonoid species, E(Q3), then the L-Trp external aldimine, and finally to L-Trp via the L-Trp gem-diamine, E(GD2).
Scheme 1.
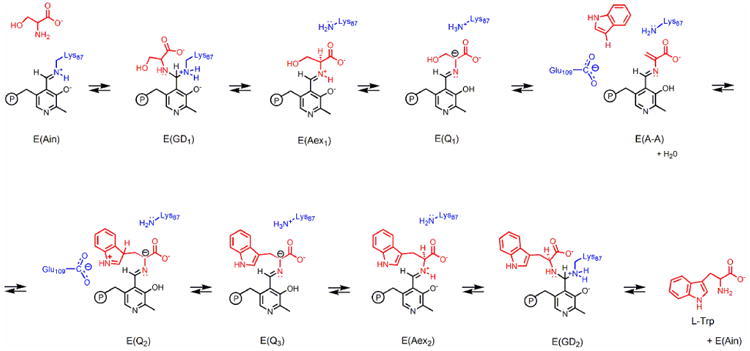
The PLP-dependent β-site reaction of tryptophan synthase.
The proposed mechanism for TS shows βLys87 playing multiple catalytic roles: it bonds to the PLP cofactor, activates C4′ for nucleophilic attack via a protonated Schiff base nitrogen, and abstracts and returns protons to PLP-bound substrates (i.e. acid-base catalysis). Experimental support for these integral contributions comes primarily from X-ray crystallography, which identifies βLys87 as the acid-base residue in closest proximity to the coenzyme C4′ and the substrate Cα (see Figure 1), and mutagenesis, which shows that βLys87 is critically important for enzyme activity.[19-23, 26, 30-32] However, the proximity of other amino acid residues with acid-base catalytic potential (e.g., His, Glu, Asp) can make the assignment of the acid-base catalytic role(s) ambiguous. Indeed, it now seems likely that acid-base catalysis in some PLP enzymes requires both the active site Lys and an additional acid-base group.[22, 33] For example, in TS proton transfer at Cα is likely mediated by βLys87, but either βLys87 or βGlu109 could act as an acid catalyst facilitating scission of the hydroxyl C-O bond to give the α-aminoacrylate species. Furthermore, βGlu109 is well-positioned to stabilize the developing positive charge on the indole ring during nucleophilic attack of the indole C3 at Cβ of the α-aminoacrylate species (Figure 1).[34, 35]
Figure 1.
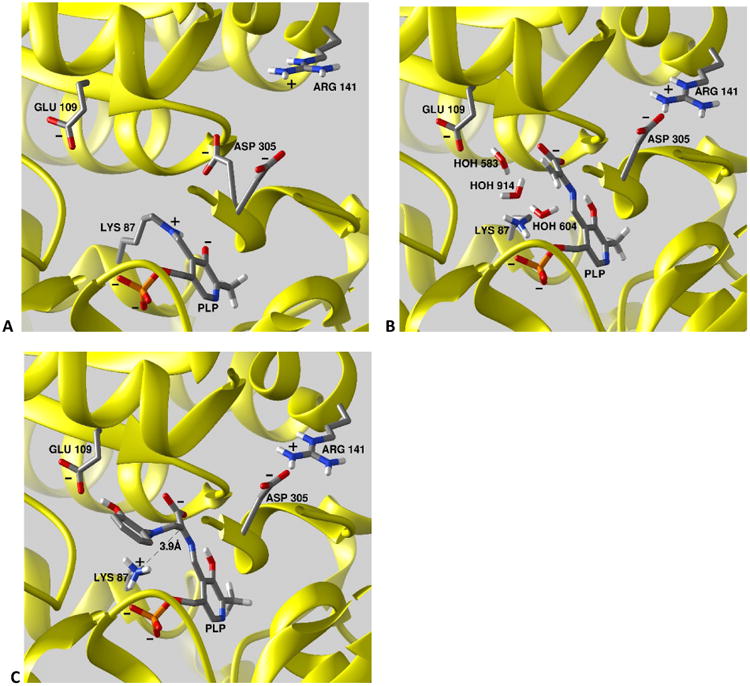
Comparison of the structures of the β-active site intermediates investigated in this work. (A) the internal aldimine, E(Ain) (PDBID 4HT3); (B) the α-aminoacrylate Schiff base, E(A-A) PDBID 4HN4); and (C) the 2-aminophenol quinonoid, E(Q)2AP (PDBID 4HPJ). Hydrogen atoms have been added to the structure using standard bond lengths and angles, and for βLys87 correspond to the charge states determined in this study. All stick structures are rendered in CPK colors. The β-subunits are depicted as a yellow cartoon ribbon in each panel. For clarity, portions of the β-subunit are cut away to provide views into the β-site. Structures prepared using Chimera.
Tryptophan synthase catalysis requires conformational changes that switch the α- and β-subunits from inactive, open conformations to active, closed conformations. These conformational gymnastics shuttle potential acid-base catalytic residues (specifically βGlu109) into close proximity of the substrate during catalysis.[26, 28, 36, 37] The open β-subunit conformation of the L-Ser external aldimine intermediate shows that the side chain of βAsp305 hydrogen bonds to the β-hydroxyl of the L-Ser substrate, stabilizing this species.[21, 22, 33, 38, 39] However, in the closed conformation, βAsp305 shifts far away to form a salt bridge with βArg141 and cannot play an acid-base role in catalysis.[21-23, 26, 32, 33, 39, 40] X-ray structures also confirm that βHis115 remains too far from the substrate-coenzyme complex to be an acid-base catalyst.[21-23, 33] Indeed, the X-ray structures of TS in closed conformations reveal that the ε-amino group of βLys87 is always nearest the substrate Cα, while βGlu109 is positioned to facilitate charge stabilization at N1 of the indole ring, indicating these two residues play the primary acid-base and charge stabilization roles in the synthesis of L-Trp.[19-22, 27] Further evidence for an essential role of βLys87 in catalysis comes from mutation of this residue to Thr, which renders the β-site inactive.[31] While this variant is still able to form L-Ser and L-Trp external aldimine species by reaction of these amino acids with apo-enzyme and PLP, no interconversion of these intermediates is detected. These findings provide strong support for the involvement of βLys87 as a key acid-base catalyst. Nevertheless, many single amino acid replacements in the β-subunit render the β-site inactive, and therefore the effects of the βK87T mutation are not conclusive.[41, 42]
Recent X-ray crystal structures solved by our group help to disentangle the acid-base roles played by βLys87.[33, 39] The internal aldimine structure (PDB ID: 4HT3, Figure 1A) shows the β-subunit in the open conformation; the ε-amino N of βLys87 is covalently bonded to PLP and is modeled as the protonated Schiff base form. The locations of the side chain groups of βGlu109, βAsp305 and βArg141 are also shown in relation to the PLP ring. Notice that two conformations of the βAsp305 side chain are observed in this internal aldimine intermediate: one folds in toward the active site, the other folds away from the site toward, but distant from, βArg141. In the open β-subunit conformation, the site is solvent accessible and the opening into the site from solution is sufficiently large to accommodate entry/exit of substrate L-Ser and product L-Trp. The site is filled with six crystallographic waters (not shown). The α-aminoacrylate structure (PDB ID: 4HN4, Figure 1B) shows the β-subunit in the closed conformation with a salt bridge between βAsp305 and βArg141. The ε-amino nitrogen of βLys87 and the carboxylate of βGlu109 are depicted as neutral and ionized, respectively. Three crystallographic waters found in the vicinity of Cβ are also shown. Notice that both the carboxylate of βGlu109 and the ε-amino of βLys87 are sufficiently close to one or another of these waters to qualify as a candidate for catalysis of water attack on Cβ of the α-aminoacrylate moiety to give the L-Ser quinonoid species (the reverse of the water elimination step). The 2-aminophenol quinonoid structure (PDB ID: 4HPJ, Figure 1C) also shows the β-subunit in the closed conformation with the side chains of βAsp305 and βArg141 hydrogen-bonded in a salt bridge that stabilizes the closed subunit conformation. 2-aminophenol (2AP) acts as an analogue for indole; reaction of 2AP with the α-aminoacrylate intermediate gives a long-lasting quinonoid intermediate that does not appear to turn over to product (Scheme 2, showing progression from the α-aminoacrylate through a putative E(Q2)2AP intermediate to the stable E(Q3)2AP form).[41] The ε-N of βLys87 is shown protonated and located in close proximity (3.9 Å) to Cα of the quinonoid intermediate and only 3.0 Å from the 2AP amino group. The waters seen in (B) are displaced by the 2AP group. The amino group of 2AP is bonded to the Ser substrate through an N-C bond formed by nucleophilic attack of the 2AP amino group on Cβ. The carboxylate of βGlu109 is hydrogen bonded to the phenolic oxygen of 2AP.
Scheme 2.
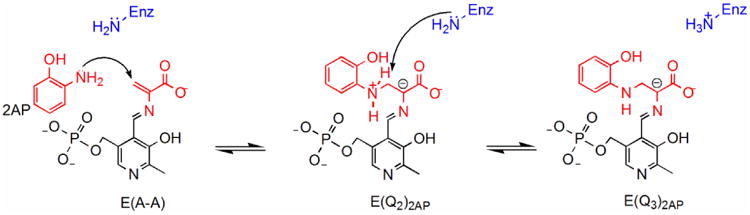
The 2AP reaction.
While X-ray and mutagenesis studies suggest the acid-base roles of βLys87, atomic-resolution probes such as NMR spectroscopy have not yet been applied to follow the protonation/charge state of the ε-amino nitrogen at multiple points along the catalytic path. The chemical shift in NMR is an extremely sensitive reporter of chemical and charge state, and the ε-Lys-N in the Schiff base is a particularly attractive target, as changes in chemical shift of up to 100 ppm are observed upon protonation or deprotonation.[43] This marker has been used to determine the protonation state of the Lys Schiff base linkage in model compounds,[43-46] the retinal Schiff base in bacteriorhodopsin,[47] and for Schiff base linkages from substrates to PLP in tryptophan synthase[23, 39] and alanine racemase.[48] Upon liberation of the lysine residue, the chemical shift of the ε-amino nitrogen shows a much less dramatic, but nonetheless informative, dependence on protonation state, varying from 33 to 24 ppm when switched from the protonated to neutral forms, respectively.[49, 50] Recently, we reported the 15N chemical shift measurement for the linking imine nitrogen in the internal aldimine state of tryptophan synthase, the first such measurement for the internal aldimine state of a PLP-dependent enzyme.[39] Here we extend this initial work and employ 15N SSNMR combined with enzyme containing 15N enriched ε-amino Lys residues to directly probe the protonation states of βLys87 for two additional stable intermediates in the β-subunit catalytic cycle.
2. Materials and Methods
ε-15N- lysine tryptophan synthase was prepared as previously described through addition of 250 mg ε-15N- Lysine•2HCl to 1 L minimal media during the induction phase of overexpression of S. typhimurium TS in E. coli, resulting in an enzyme sample in which only the ε-N of each lysine residue contains the NMR-active label.[39] Microcrystals were prepared as described previously[39] and were collected and washed with a 50 mM Cs-bicine solution, pH 7.8, containing 8% PEG-8000 and 1.5 mM spermine, and packed into a Bruker 4 mm magic-angle spinning (MAS) rotor with an approximate volume of 80 μL. When present, serine was introduced by direct addition of 5 μL of 1.2 M L-serine to the packed MAS rotors, while 2AP was introduced by addition of 8 μL of a concentrated stock 2AP in acetonitrile. Solid-state NMR experiments were performed at 9.4 T (400.37 MHz 1H, 40.57 MHz 15N) on a Bruker AVIII spectrometer equipped with an 1H-13C-15N triple resonance 4 mm MAS probe spinning at a MAS rate of 8 kHz and with the bearing gas cooled to -15 °C, giving an effective sample temperature of -5 °C. Cross-polarization was accomplished at a 1H spin-lock field of 45 kHz, an 15N spin-lock field of 37 kHz (ramped ±2 kHz), and a contact time of 2 ms; 85 kHz Spinal64 1H decoupling[51] was used throughout. Each spectrum consists of the sum of 81,920 transients acquired with a relaxation delay of 4 s, for a total acquisition time of 3 days 19 h. 15N chemical shifts were referenced indirectly to NH3(l) via an external solid-state sample of 15NH4Cl (δ[NH3(l)] = 39.3 ppm) calibrated under MAS conditions.
3. Results and Discussion
Observation of the key βLys87 chemical shift in the enzyme active site involves expression of ε-15N- lysine tryptophan synthase. Use of the labeled enzyme in conjunction with strategically labeled 13C,15N- L-serine substrate aids in the determination of the protonation state of βLys87. Furthermore, the protein NMR sample was prepared in the microcrystalline state under catalytically active conditions,[23] making it possible to probe kinetically competent, stable intermediates that lie on or near the reaction coordinate path of the physiological αβ-reaction and their interactions with the βLys87 side chain in three of the intermediates shown in Scheme 1. Stabilization of the active species is made possible through intermediate trapping or by greatly slowing conversion to other species. Trapping arises due to the combined use of low temperature and stabilizing ligands which bind relatively tightly to the α- and β-subunits and stabilize the active, closed αβ-dimeric unit. These ligands consist of N-(4′-trifluoro-methoxybenzenesulfonyl)-2-amino-ethyl phosphate (F9), a tight binding analogue of IGP which binds to the α-site,[21, 22] and Cs+, an analogue of Na+/K,+ which binds tightly to the monovalent cation site in the β-subunit.[52-54] The internal aldimine form is the resting state of the enzyme and exhibits an open conformation for the β-subunit. The α-aminoacrylate intermediate is a relatively stable species along the reaction pathway chemically poised to react with indole, but protected from many other nucleophiles, especially water/hydroxide ion, by its sequestration within the confines of the closed αβ-dimeric unit. Even so, its reaction with water to give pyruvate ion and ammonium ion is a significant side reaction.[55, 56] 2AP is an analogue of indole which binds tightly to the β-site and reacts with E(A-A) to form E(Q)2AP, the analogue of E(Q3), which does not turn over to any product.
Figure 2 shows 15N solid-state NMR spectra for TS with F9 bound to the α-site under the following experimental conditions: (A) microcrystals of TS prepared at natural abundance 15N isotopomer concentration; (B) microcrystals of selectively enriched ε-15N-lysine TS; (C) ε-15N-Lys TS microcrystals reacted with 30 mM L-Ser; and (D) ε-15N-Lys TS microcrystals reacted with 30 mM L-[U-13C,15N]Ser and ∼50 mM 2-aminophenol. In Figure 2A, only signals from the large number of protein backbone nitrogens are observed near 120 ppm, while the spectrum of the ε-15N- Lysine TS in Figure 2B reveals a large feature centered at 33 ppm, corresponding to the 26 charged ε-lysine side chains in each αβ subunit. Also evident in Figure 2B is a smaller peak at 202.3 ppm, the expected resonance of a protonated Schiff base. We have previously assigned this resonance to the ε-N of βLys87 using double resonance experiments.[39] 15N SSNMR experiments using 15N-labeled PLP have also established that the pyridine nitrogen is deprotonated in the internal aldimine form;[39] preliminary data suggest that it remains deprotonated throughout. The observation that βLys87 is indeed protonated in the internal aldimine species confirms a long-standing structural hypothesis for PLP enzymes and is consistent with the mechanistic hypothesis that a protonated imine enhances reactivity toward nucleophiles at C4′, thereby activating catalysis.
Figure 2.
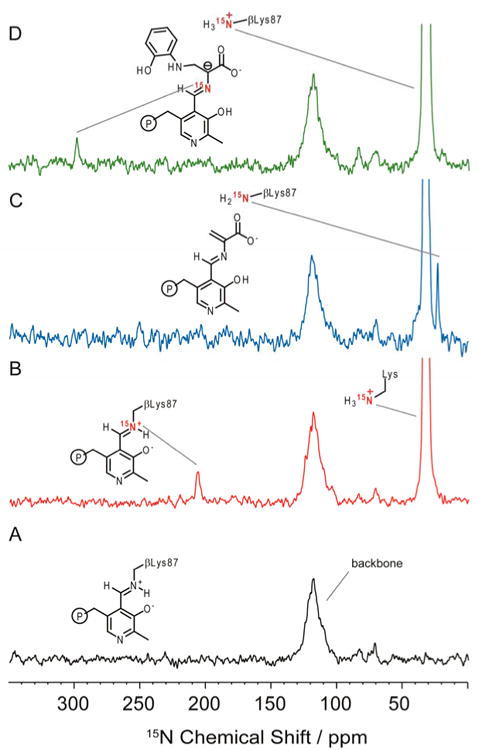
15N SSNMR cross-polarization magic-angle-spinning (CPMAS) spectra of S. typhimurium tryptophan synthase with N-(4′-trifluoro-methoxybenzenesulfonyl)-2-amino-ethyl phosphate (F9) bound to the α-site under the following experimental conditions: (A) Natural abundance 15N isotopomer concentration TS microcrystals, (F9)(Cs+)E(Ain); (B) ε-15N-Lys TS microcrystals; (C) ε-15N-Lys TS microcrystals reacted with 30 mM L-Ser, giving E(A-A)F9; (D) ε-15N-Lys TS microcrystals reacted with 30 mM L-[U-13C,15N]Ser and ∼50 mM 2-aminophenol, giving (F9)(Cs+)E(Q3)2AP. Experimental conditions described in Section 2. In (A), only signals from the large number of protein backbone nitrogen atoms are observed, centered at 120 ppm. The spectrum in (B) reveals a large feature at 33 ppm, corresponding to charged lysine side chains throughout the protein, and a resonance at 202.3 ppm that is assigned to the protonated Schiff base linkage from βLys87 to the PLP cofactor. In (C), this linkage has been broken and the ɛ-amino group of βLys87 is found to be neutral, while in (D), only charged lysine residues are observed.
Upon addition of L-Ser to the ε-15N-lysine internal aldimine sample, the Schiff base linkage to the βLys87 side chain is broken as stage I of the β-reaction unfolds. Formation of the external aldimine intermediate, conversion to the L-Ser quinonoid species, followed by loss of the β-hydroxyl and a proton from L-Ser produces the next long-lived intermediate in the cycle, the α-aminoacrylate species, E(A-A) (Scheme 1; Figure 1B). The 15N spectrum of E(A-A) shows a significant change: the peak at 202.3 ppm is lost, and a new peak at 24 ppm, indicative of a neutral amine, arises (Figure 2C). These changes indicate that the Schiff base linkage to βLys87 in the enzyme active site has been broken at the same time that a neutral amine has been formed. Based on this correlation, we assign this new resonance to the neutral ε-NH2 of βLys87. The neutral βLys87 in the α-aminoacrylate intermediate is poised to act as the base in the next stage of catalysis, which consists of proton abstraction from the attacking nucleophile to give the next stable intermediate, the carbanionic quinonoid species.
Reaction of the nucleophile 2AP with the α-aminoacrylate gives the 2AP quinonoid analogue of E(Q3), (Figure 1C; Scheme 2). The 24 ppm peak of the neutral βLys87 side chain nitrogen now expires, and the only signals detected in the spectrum arise from natural abundance protein backbone amides, centered at 120 ppm, and charged lysine side chains, centered at 33 ppm. To ensure the quinonoid intermediate has indeed formed in situ, the experiment was repeated using 15N-L-Ser. This gave rise to an additional peak at 298.6 ppm (Figure 2D), which is assigned to the Schiff base linkage of the substrate to PLP. The resonance of the Schiff base peak is indicative of a (mostly) deprotonated imine nitrogen and is in good agreement with the previously reported chemical shift of the Schiff base linkage for the indoline quinonoid analogue.[23] The Schiff base peak gives perspective on the expected intensity for the ε-N resonance of βLys87, which appears to have shifted to join the other charged lysine residues centered at 33 ppm. We take these findings as a strong indication that βLys87 is protonated in the quinonoid intermediate state. A protonated ε-amino nitrogen at this stage allows βLys87 to play an effective role as an acid catalyst in the next step of the mechanism, formation of the L-Trp external aldimine (Scheme 1).
These results indicate that βLys87 plays the role of both acid and base during catalysis, activating C4′ and facilitating proton transfers at Cα throughout the catalytic cycle. In TS, reaction specificity relies on the ability of the enzyme to fine-tune the microenvironment of the active site to accommodate the charged or neutral βLys87 side chain specifically for each intermediate. It appears that one consequence of these stabilizing interactions is to select the predominant protonation state of the βLys87 ε-amino group for each of the intermediates investigated. Thus the ε-N of βLys87 switches from a protonated Schiff base in the internal aldimine species to a neutral ε-amino group in the α-aminoacrylate intermediate and to a protonated ε-amino group in the 2AP quinonoid species. It is interesting to note that the UV/vis spectrum of the reaction mixture derived from reaction of L-Ser with α2β2 TS shows a remarkable pH dependence. Yet, the sensitivity to pH arises from a shifting of the β-subunit between open and closed conformations that in turn selectively stabilize the L-Ser external aldimine species (open conformation) and the α-aminoacrylate species (closed conformation). The chemical structures of the active site species do not undergo changes in protonation; the pH dependence arises from deprotonation of protein residues that affect the stability of the open and closed subunit conformations, and, while the distribution of species changes, the protonation states of the intermediates present (internal aldimine, α-aminoacrylate, and quinonoid species) are invariant. This confirms that the active sites are shielded from the effects of bulk solvent over a wide range (pH 6 to 10).[57, 58] The SSNMR results support this finding, as we see a dominant species rather than an equilibrating mixture of protonated and ionized intermediates.
4. Conclusions
The measured 15N chemical shifts of βLys87 in these intermediates provide snapshots of the changing acid-base roles performed by βLys87 during catalysis at the enzyme active site. Initially, the ɛ-amino group forms a protonated Schiff-base linkage to the PLP cofactor, activating C4′ for nucleophilic addition. Once released, the ɛ-amino group of the free side chain is found to act as both a base and acid catalyst, with neutral and charged amino groups, respectively. Understanding how the microenvironment of the β-subunit active site fine-tunes the acid-base properties of βLys87, the PLP cofactor, and substrate as the site switches from one intermediate to the next is the subject of our ongoing investigations.
Highlights.
Protonation state of the active site lysine in tryptophan synthase determined
15N solid-state NMR of ε-15N-Lys enzyme
βLys87 activates C4′ of PLP for substrate nucleophilic attack
βLys87 abstracts and returns protons to PLP-bound substrates
The active site lysine ε-amino group plays alternating acid/base catalytic roles
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM097569.
Abbreviations
- PLP
pyridoxal-5′-phosphate
- TS
tryptophan synthase
- SSNMR
solid-state nuclear magnetic resonance
- MAS
magic-angle-spinning
- F9
N-(4′-trifluoro-methoxybenzenesulfonyl)-2-amino-ethyl phosphate
- 2AP
2-aminophenol
- IGP
indole-3-glycerol phosphate
- G3P
D-glyceraldehyde-3-phosphate
- E(Ain)
enzyme internal aldimine state
- E(A-A)
enzyme-substrate α-aminoacrylate intermediate
- E(Q)
enzyme-substrate quinonoid intermediate
References
- 1.Yanofsky C, Crawford IP. Tryptophan Synthetase. In: Boyer PD, editor. The Enzymes. VII. Academic Press; New York, New York: 1972. [Google Scholar]
- 2.Umbreit WW, Wood WA, Gunsalus IC. The activity of pyridoxal phosphate in tryptophane formation by cell-free enzyme preparations. J Biol Chem. 1946;165:731. [PubMed] [Google Scholar]
- 3.Braunshtein AE, Shemyakin MM. A Theory of Amino Acid Metabolic Processes Catalyzed by Pyridoxal-Dependent Enzymes. Biochemistry - USSR. 1953;18:393–411. [PubMed] [Google Scholar]
- 4.Metzler DE, Ikawa M, Snell EE. A General Mechanism for Vitamin-B6-Catalyzed Reactions. J Am Chem Soc. 1954;76:648–652. [Google Scholar]
- 5.Yanofsky C. Tryptophan Synthetase from Neurospora. Method Enzymol. 1955;2:233–238. [Google Scholar]
- 6.Crawford IP, Yanofsky C. On the Separation of the Tryptophan Synthetase of Escherichia-Coli into 2 Protein Components. P Natl Acad Sci USA. 1958;44:1161–1170. doi: 10.1073/pnas.44.12.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schirch L, Jenkins WT. Serine Transhydroxymethylase. Properties of the Enzyme-Substrate Complexes of D-Alanine and Glycine. J Biol Chem. 1964;239:3801–3807. [PubMed] [Google Scholar]
- 8.Wilson DA, Crawford IP. Purification and properties of the B component of Escherichia coli tryptophan synthetase. J Biol Chem. 1965;240:4801–4808. [PubMed] [Google Scholar]
- 9.Kagamiya H, Morino Y, Snell EE. Chemical Structure of Tryptophanase from Escherichia-Coli .1. Isolation and Structure of a Pyridoxyl Decapeptide from Borohydride-Reduced Holotryptophanase. J Biol Chem. 1970;245:2819–&. [PubMed] [Google Scholar]
- 10.Miles EW, Hatanaka M, Crawford IP. A New Thiol-Dependent Transamination Reaction Catalyzed by B Protein of Escherichia Coli Tryptophan Synthetase. Biochemistry. 1968;7:2742–&. doi: 10.1021/bi00848a008. [DOI] [PubMed] [Google Scholar]
- 11.Miles EW, Houck DR, Floss HG. Stereochemistry of Sodium-Borohydride Reduction of Tryptophan Synthase of Escherichia-Coli and Its Amino-Acid Schiffs Bases. J Biol Chem. 1982;257:4203–4210. [PubMed] [Google Scholar]
- 12.Snell EE, Di Mari SJ. Schiff Base Intermediates in Enzyme Catalysis. In: Boyer PD, editor. The Enzymes. II. Academic Press; New York, New York: 1970. [Google Scholar]
- 13.Davis L, Metzler DE. Pyridoxal-Linked Elimination and Replacement Reactions. In: Boyer PD, editor. The Enzymes. VII. Academic Press; New York, New York: 1972. [Google Scholar]
- 14.Metzler DE. Biochemistry: The Chemical Reactions of Living Cells. 2. Academic Press; New York, New York: 1977. [Google Scholar]
- 15.Bruice TC, Benkovic SJ. Bioorganic Mechanisms. W.A. Benjamin; New York, New York: 1966. [Google Scholar]
- 16.Jencks WP. Catalysis in Chemistry and Enzymology. McGraw-Hill; New York, New York: 1969. [Google Scholar]
- 17.Walsh C. Enzymatic Reaction Mechanisms. W.H. Freeman and Company; New York, New York: 1979. [Google Scholar]
- 18.Drewe WF, Jr, Dunn MF. Characterization of the reaction of L-serine and indole with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry. 1986;25:2494–2501. doi: 10.1021/bi00357a032. [DOI] [PubMed] [Google Scholar]
- 19.Miles EW. Structural basis for catalysis by tryptophan synthase. Adv Enzymol Relat Areas Mol Biol. 1991;64:93–172. doi: 10.1002/9780470123102.ch3. [DOI] [PubMed] [Google Scholar]
- 20.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 21.Ngo H, Harris R, Kimmich N, Casino P, Niks D, Blumenstein L, Barends TR, Kulik V, Weyand M, Schlichting I, Dunn MF. Synthesis and characterization of allosteric probes of substrate channeling in the tryptophan synthase bienzyme complex. Biochemistry. 2007;46:7713–7727. doi: 10.1021/bi700385f. [DOI] [PubMed] [Google Scholar]
- 22.Ngo H, Kimmich N, Harris R, Niks D, Blumenstein L, Kulik V, Barends TR, Schlichting I, Dunn MF. Allosteric regulation of substrate channeling in tryptophan synthase: Modulation of the L-Serine reaction in stage I of the beta-reaction by alpha-site ligands. Biochemistry. 2007;46:7740–7753. doi: 10.1021/bi7003872. [DOI] [PubMed] [Google Scholar]
- 23.Lai JF, Niks D, Wang YC, Domratcheva T, Barends TRM, Schwarz F, Olsen RA, Elliott DW, Fatmi MQ, Chang CEA, Schlichting I, Dunn MF, Mueller LJ. X-ray and NMR Crystallography in an Enzyme Active Site: The Indoline Quinonoid Intermediate in Tryptophan Synthase. J Am Chem Soc. 2011;133:4–7. doi: 10.1021/ja106555c. [DOI] [PubMed] [Google Scholar]
- 24.Christen P, Metzler DE. Transaminases. In: Christen PM, D E, editors. Transaminsases. Vol. 2. John Wiley & Sons; New York, New York: 1985. [Google Scholar]
- 25.Miles EW. Tryptophan Synthase: Structure, Function, and Subunit Interaction. Adv Enzymol Relat Areas Mol Biol. 1979;49:127–186. doi: 10.1002/9780470122945.ch4. [DOI] [PubMed] [Google Scholar]
- 26.Dunn MF, Niks D, Ngo H, Barends TR, Schlichting I. Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem Sci. 2008;33:254–264. doi: 10.1016/j.tibs.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Dunn MF. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch Biochem Biophys. 2012;519:154–166. doi: 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunn MF, Aguilar V, Brzovic P, Drewe WF, Jr, Houben KF, Leja CA, Roy M. The tryptophan synthase bienzyme complex transfers indole between the alpha- and beta-sites via a 25-30 A long tunnel. Biochemistry. 1990;29:8598–8607. doi: 10.1021/bi00489a015. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 30.Weyand M, Schlichting I. Crystal structure of wild-type tryptophan synthase complexed with the natural substrate indole-3-glycerol phosphate. Biochemistry. 1999;38:16469–16480. doi: 10.1021/bi9920533. [DOI] [PubMed] [Google Scholar]
- 31.Rhee S, Parris KD, Hyde CC, Ahmed SA, Miles EW, Davies DR. Crystal structures of a mutant (beta K87T) tryptophan synthase alpha(2)beta(2) complex with ligands bound to the active sites of the alpha- and beta-subunits reveal ligand-induced conformational changes. Biochemistry. 1997;36:7664–7680. doi: 10.1021/bi9700429. [DOI] [PubMed] [Google Scholar]
- 32.Barends TR, Domratcheva T, Kulik V, Blumenstein L, Niks D, Dunn MF, Schlichting I. Structure and mechanistic implications of a tryptophan synthase quinonoid intermediate. Chembiochem. 2008;9:1024–1028. doi: 10.1002/cbic.200700703. [DOI] [PubMed] [Google Scholar]
- 33.Niks D, Hilario E, Dierkers A, Ngo H, Borchardt D, Neubauer TJ, Fan L, Mueller LJ, Dunn MF. Allostery and Substrate Channeling in the Tryptophan Synthase Bienzyme Complex: Evidence for Two Subunit Conformations and Four Quaternary States. Biochemistry. 2013;52:6396–6411. doi: 10.1021/bi400795e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brzovic PS, Kayastha AM, Miles EW, Dunn MF. Substitution of Glutamic Acid-109 by Aspartic-Acid Alters the Substrate-Specificity and Catalytic Activity of the Beta-Subunit in the Tryptophan Synthase Bienzyme Complex from Salmonella-Typhimurium. Biochemistry. 1992;31:1180–1190. doi: 10.1021/bi00119a030. [DOI] [PubMed] [Google Scholar]
- 35.Ruvinov SB, Ahmed SA, McPhie P, Miles EW. Monovalent cations partially repair a conformational defect in a mutant tryptophan synthase alpha 2 beta 2 complex (beta-E109A) J Biol Chem. 1995;270:17333–17338. doi: 10.1074/jbc.270.29.17333. [DOI] [PubMed] [Google Scholar]
- 36.Pan P, Woehl E, Dunn MF. Protein architecture, dynamics and allostery in tryptophan synthase channeling. Trends Biochem Sci. 1997;22:22–27. doi: 10.1016/s0968-0004(96)10066-9. [DOI] [PubMed] [Google Scholar]
- 37.Fatmi MQ, Ai R, Chang CEA. Synergistic Regulation and Ligand-Induced Conformational Changes of Tryptophan Synthase. Biochemistry. 2009;48:9921–9931. doi: 10.1021/bi901358j. [DOI] [PubMed] [Google Scholar]
- 38.Miles EW, Fan YX, Jhee KH, Ro HS, McPhie P, Rhee S, Davies DR. Structure and Function of Tryptophan Synthase. In: Iriarte A, Kagan HM, Martinez-Carrion M, editors. Biochemistry and Molecular Biology of Vitamin B6 and PQQ-Dependent Proteins. Birkhäuser Verlag; 2000. pp. 145–150. [Google Scholar]
- 39.Caulkins BG, Bastin B, Yang C, Neubauer TJ, Young RP, Hilario E, Huang YM, Chang CE, Fan L, Dunn MF, Marsella MJ, Mueller LJ. Protonation states of the tryptophan synthase internal aldimine active site from solid-state NMR spectroscopy: direct observation of the protonated Schiff base linkage to pyridoxal-5′-phosphate. J Am Chem Soc. 2014;136:12824–12827. doi: 10.1021/ja506267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dierkers AT, Niks D, Schlichting I, Dunn MF. Tryptophan Synthase: Structure and Function of the Monovalent Cation Site. Biochemistry. 2009;48:10997–11010. doi: 10.1021/bi9008374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blumenstein L, Domratcheva T, Niks D, Ngo H, Seidel R, Dunn MF. I. Schlichting, beta Q114N and beta T110V mutations reveal a critically important role of the substrate alpha-carboxylate site in the reaction specificity of tryptophan synthase. Biochemistry. 2007;46:14100–14116. doi: 10.1021/bi7008568. [DOI] [PubMed] [Google Scholar]
- 42.Ahmed SA, Ruvinov SB, Kayastha AM, Miles EW. Mechanism of mutual activation of the tryptophan synthase alpha and beta subunits. Analysis of the reaction specificity and substrate-induced inactivation of active site and tunnel mutants of the beta subunit. J Biol Chem. 1991;266:21548–21557. [PubMed] [Google Scholar]
- 43.Limbach HH, Chan-Huot M, Sharif S, Tolstoy PM, Shenderovich IG, Denisov GS, Toney MD. Critical hydrogen bonds and protonation states of pyridoxal 5′-phosphate revealed by NMR. BBA - Proteins Proteom. 2011;1814:1426–1437. doi: 10.1016/j.bbapap.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Sharif S, Denisov GS, Toney MD, Limbach HH. NMR studies of coupled low- and high-barrier hydrogen bonds in pyridoxal-5′-phosphate model systems in polar solution. J Am Chem Soc. 2007;129:6313–6327. doi: 10.1021/ja070296+. [DOI] [PubMed] [Google Scholar]
- 45.Sharif S, Schagen D, Toney MD, Limbach HH. Coupling of functional hydrogen bonds in pyridoxal-5′-phosphate-enzyme model systems observed by solid-state NMR spectroscopy. J Am Chem Soc. 2007;129:4440–4455. doi: 10.1021/ja066240h. [DOI] [PubMed] [Google Scholar]
- 46.Chan-Huot M, Sharif S, Tolstoy PM, Toney MD, Limbach HH. NMR studies of the stability, protonation States, and tautomerism of (13)C- AND (15)N-labeled aldimines of the coenzyme pyridoxal 5′-phosphate in water. Biochemistry. 2010;49:10818–10830. doi: 10.1021/bi101061m. [DOI] [PubMed] [Google Scholar]
- 47.Harbison GS, Herzfeld J, Griffin RG. Solid-state nitrogen-15 nuclear magnetic resonance study of the Schiff base in bacteriorhodopsin. Biochemistry. 1983;22:1–4. doi: 10.1021/bi00270a600. [DOI] [PubMed] [Google Scholar]
- 48.Copie V, Faraci WS, Walsh CT, Griffin RG. Inhibition of alanine racemase by alanine phosphonate: detection of an imine linkage to pyridoxal 5′-phosphate in the enzyme-inhibitor complex by solid-state 15N nuclear magnetic resonance. Biochemistry. 1988;27:4966–4970. doi: 10.1021/bi00414a002. [DOI] [PubMed] [Google Scholar]
- 49.Poon DKY, Schubert M, Au J, Okon M, Withers SG, McIntosh LP. Unambiguous determination of the ionization state of a glycoside hydrolase active site lysine by H-1-N-15 heteronuclear correlation spectroscopy. J Am Chem Soc. 2006;128:15388–15389. doi: 10.1021/ja065766z. [DOI] [PubMed] [Google Scholar]
- 50.Andre I, Linse S, Mulder FA. Residue-specific pKa determination of lysine and arginine side chains by indirect 15N and 13C NMR spectroscopy: application to apo calmodulin. J Am Chem Soc. 2007;129:15805–15813. doi: 10.1021/ja0721824. [DOI] [PubMed] [Google Scholar]
- 51.Khitrin A, Fung BM. Design of heteronuclear decoupling sequences for solids. J Chem Phys. 2000;112:2392–2398. [Google Scholar]
- 52.Woehl E, Dunn MF. Mechanisms of monovalent cation action in enzyme catalysis: the first stage of the tryptophan synthase beta-reaction. Biochemistry. 1999;38:7118–7130. doi: 10.1021/bi982918x. [DOI] [PubMed] [Google Scholar]
- 53.Woehl E, Dunn MF. Mechanisms of monovalent cation action in enzyme catalysis: the tryptophan synthase alpha-, beta-, and alpha beta-reactions. Biochemistry. 1999;38:7131–7141. doi: 10.1021/bi982919p. [DOI] [PubMed] [Google Scholar]
- 54.Peracchi A, Mozzarelli A, Rossi GL. Monovalent cations affect dynamic and functional properties of the tryptophan synthase alpha 2 beta 2 complex. Biochemistry. 1995;34:9459–9465. doi: 10.1021/bi00029a022. [DOI] [PubMed] [Google Scholar]
- 55.Drewe WF, Jr, Dunn MF. Detection and identification of intermediates in the reaction of L-serine with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry. 1985;24:3977–3987. doi: 10.1021/bi00336a027. [DOI] [PubMed] [Google Scholar]
- 56.Miles EW, Bauerle R, Ahmed SA. Tryptophan synthase from Escherichia coli and Salmonella typhimurium. Methods Enzymol. 1987;142:398–414. doi: 10.1016/s0076-6879(87)42051-x. [DOI] [PubMed] [Google Scholar]
- 57.Mozzarelli A, Peracchi A, Rovegno B, Dale G, Rossi GL, Dunn MF. Effect of pH and monovalent cations on the formation of quinonoid intermediates of the tryptophan synthase alpha(2)beta(2) complex in solution and in the crystal. J Biol Chem. 2000;275:6956–6962. doi: 10.1074/jbc.275.10.6956. [DOI] [PubMed] [Google Scholar]
- 58.Peracchi A, Bettati S, Mozzarelli A, Rossi GL, Miles EW, Dunn MF. Allosteric regulation of tryptophan synthase: Effects of pH, temperature, and alpha-subunit ligands on the equilibrium distribution of pyridoxal 5′-phosphate-L-serine intermediates. Biochemistry. 1996;35:1872–1880. doi: 10.1021/bi951889c. [DOI] [PubMed] [Google Scholar]