

Abstract
A phase 1/2 clinical trial evaluating dosing, safety, immunogenicity, and overall survival on metastatic colorectal cancer (mCRC) patients after immunotherapy with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine was performed. We report our extended observations on long-term overall survival and further immune analyses on a subset of treated patients including assessment of cytolytic T cell responses, T regulatory (Treg) to T effector (Teff) cell ratios, flow cytometry on peripheral blood mononuclear cells (PBMCs), and determination of HLA-A2 status. An overall survival of 20 % (median survival 11 months) was observed during long-term follow-up, and no long-term adverse effects were reported. Cytolytic T cell responses increased after immunizations, and cell-mediated immune (CMI) responses were induced whether or not patients were HLA-A2 positive or Ad5 immune. PBMC samples from a small subset of patients were available for follow-up immune analyses. It was observed that the levels of carcinoembryonic antigen (CEA)-specific CMI activity decreased from their peak values during follow-up in five patients analyzed. Preliminary results revealed that activated CD4+ and CD8+ T cells were detected in a post-immunization sample exhibiting high CMI activity. Treg to Teff cell ratios were assessed, and samples from three of five patients exhibited a decrease in Treg to Teff cell ratio during the treatment protocol. Based upon the favorable safety and immunogenicity data obtained, we plan to perform an extensive immunologic and survival analysis on mCRC patients to be enrolled in a randomized/controlled clinical trial that investigates Ad5 [E1-, E2b-]-CEA(6D) as a single agent with booster immunizations.
Keywords: Ad5 [E1-, E2b-]-CEA(6D); Colorectal cancer; Immunotherapy; Phase 1/2 trial
Introduction
As the knowledge on identification and function of tumor-associated antigens (TAA) increases, so has the development of immune-based therapies that target TAA to treat various cancers. Development of gene-based vaccines to treat cancer and/or prevent tumor recurrence has become an area of significant basic research and clinical evaluation. In contrast to therapeutic monoclonal antibodies that “passively immunize” to treat certain cancers, some anticancer vaccines are designed to “actively” stimulate cell-mediated immunity (CMI) and antibody responses following as a response to immunotherapy. Among the anticancer vaccines being investigated and developed, those utilizing recombinant-specific viral-based delivery platforms are being exploited because of their unique ability to also generate significant CMI responses, essential in achieving antitumor immune killing [1–3]. Early-generation recombinant non-replicating adenovirus serotype-5 (Ad5)-based vector platforms with deletions in the early 1 (E1) gene and early 3 (E3) gene regions (Ad5 [E1-]) emerged as prior leading candidates for such immunotherapy [3–6]. Moreover, Ad5-based vaccines are an important pharmaceutical consideration because they can be produced in large quantities and the viral genome is non-integrating remaining episomal, thus eliminating the possibility of permanent gene insertion in the host [6].
Unfortunately, one of the major challenges facing previous-generation Ad5 [E1-]-based vectors is the presence of preexisting immunity to Ad5 that mitigates their immunizing capability. The preponderance of humans exhibit neutralizing antibody against Ad5, the most widely used subtype for human vaccines, with two-thirds of humans studied having humoral and lympho-proliferative responses against Ad5 [7, 8]. This immunity inhibits immunization and especially re-immunization (boost) with Ad5-based vectors and precludes immunization of a vaccinee against a second disease antigen as well. Furthermore, previous Ad5 [E1-]-based vectors are not effective in cases where repeated homologous immunizations to maintain and/or increase anticancer-specific T cell (CD4+ and CD8+) and humoral immunity are required for continued killing of cancer cells. To avoid the Ad immunization barrier, we have constructed an improved and advanced-generation Ad5-based vector platform. The Ad5 [E1-, E2b-] vector platform is novel having additional deletions in the early gene 2b (E2b) region by removing the DNA polymerase (pol) and the pre-terminal protein (pTP) genes and is propagated in the E.C7 human cell line [9–12]. In various animal studies, we have reported that cancer- and infectious-disease-targeting vaccines based on the Ad5 [E1-, E2b-] vector platform can be used in multiple homologous immunization regimens designed to induce and increase CMI responses despite the presence of preexisting and/or vector-induced immunity [13–22].
We have previously reported on the development and clinical use in advanced-stage mCRC patients with the Ad5 [E1-, E2b-]-based cancer vaccine designed to target carcinoembryonic antigen (CEA)-expressing tumors [13, 14, 23]. In adult humans, CEA expression is normally low and mainly restricted to the mucosa of the colon [24, 25]; however, CEA is over expressed in all adenocarcinomas of the colon and rectum [26], and CEA has been classified as a priority cancer antigen biomarker [27]. Several immunotherapy platforms targeting CEA are being developed, and there is mounting clinical evidence indicating that targeting CEA can safely be overcome despite preexisting tolerance [28]. The vaccine we developed utilizes a modification in the nine amino acid sequence of the CAP1 region of CEA that is incorporated into our recombinant vector platform and referred to as Ad5 [E1-, E2b-]-CEA(6D) [23]. CAP1(6D) is a peptide analog of CAP1, and its sequence includes a heteroclitic (non-anchor position) mutation, resulting in an amino acid change from asparagine to aspartic acid, to enhance recognition by the T cell receptor without any change in binding to HLA-A2. Compared with the non-mutated CAP1 epitope, CAP1(6D) has been shown to enhance sensitization of cytotoxic T lymphocytes (CTL) by 100–1000 times [29, 30]. In preclinical studies by us and others, multiple homologous immunizations with Ad5 [E1-, E2b-]-CEA(6D) induced CEA-specific CMI responses with antitumor activity despite the presence of existing Ad5 immunity in mice [13, 14]. We have proceeded to perform a dose escalation/safety phase 1/2 trial and reported results on metastatic colorectal cancer (mCRC) patients treated with the Ad5 [E1-, E2b-]-CEA(6D) [23]. Escalating doses of vaccine up to 5X1011 viral particles (VP) of Ad5 [E1-, E2b-]-CEA(6D) were injected subcutaneously (SQ) three times and were shown to have minimal toxicity. Employing ELISpot assays on PBMC samples to detect interferon gamma (IFN-γ) secreting cells, CEA-specific CMI responses were induced. Importantly, these CMI responses were observed despite the presence of preexisting Ad5 immunity in a majority of patients and did not appear to affect the induction of CEA-specific CMI responses. At the time of publication, overall survival of mCRC patients was 48 % at 12 months. We now present further results on long-term overall survival in treated mCRC patients and report our extended findings on additional immune analyses of PBMC samples from treated patients.
Materials and methods
Patients and clinical protocol
The clinical trial was approved by all local institutional review boards where the study was conducted under an FDA-approved investigational new drug application (IND14325) and registered at ClinicalTrials.gov (NCT01147965). Informed consent was obtained from all individual participants included in the study. Details of the trial were previously reported [23]. Briefly, 32 patients with mCRC cancer, median age 57.5 (range 38–77) who had failed a median of three prior antitumor regimens (range 2–5), had a median Karnofsky performance status of 90 % (range 70–100 %), and had a range of 1–4 sites of metastatic disease, were enrolled. Eligibility requirements included metastatic cancer expressing CEA and adequate hematologic, renal, and hepatic function. Clinical activity was assessed according to Response Evaluation Criteria in Solid Tumors (RECIST 1.0 criteria) [31] using computed tomography (CT) or magnetic resonance imaging (MRI) scans obtained at baseline and after completion of treatments. Toxicity was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 [32].
A total of five cohorts were evaluated for dose-limiting toxicity (DLT). The Ad5 [E1-, E2b-]-CEA(6D) doses were delivered subcutaneously (SQ) to patients as follows: cohort 1: dose of 1 × 109 VP SQ in the same thigh every 3 weeks for three treatments; cohort 2: dose of 1 × 1010 VP SQ every 3 weeks for three treatments; cohort 3: dose of 1 × 1011 VP SQ every 3 weeks for three treatments; cohort 4—a phase 2 expansion—dose of 1 × 1011 VP SQ every 3 weeks for three treatments. After completing the phase 2 expansion cohort 4, an additional cohort (cohort 5) received a dose of 5 × 1011 VP SQ every 3 weeks for three treatments to determine safety of the highest dose. Serum and PBMC were collected from patients just prior to the immunizations at 0, 3, 6, and 3 weeks (week 9) following the last treatment. Blood was also collected from some patients up to 12 months following treatment. The serum was frozen at ≤−60°C and PBMC were frozen in liquid nitrogen until immune assays were performed.
Immunotherapeutic vaccine
Clinical grade Ad5 [E1-, E2b-]-CEA(6D) (product referred to as ETBX-011) was constructed as previously described [14, 23] and manufactured using the E.C7 human cell line (9–11) under GMP at Sigma-Aldrich Fine Chemicals (SAFC), Carlsbad, California. The vaccine was stored at −20 °C and thawed just prior to use.
ELISpot assays for IFN-γ and granzyme B-secreting cells
The ELISpot assay for IFN-γ-secreting lymphocytes was adapted from our previous studies and performed as described [14, 23]. An ELISpot assay for granzyme B-secreting lymphocytes to detect cytolytic T cell activity [33, 34] was also performed. Briefly, isolated PBMC (2 × 105 cells/well) from individual patient samples was incubated 36–40 h with a CEA peptide pool (15mers with 11 amino acid overlap covering full-length CEA with the CAP1-6D modification; 0.1 μg/well) to stimulate IFN-γ or granzyme B-secreting cells. Specificity of the responses was demonstrated by the lack of reactivity with the irrelevant antigens β-galactosidase and HIV-gag. Cells stimulated with concanavalin A (Con A) at a concentration of 0.25 μg/well served as positive controls. Colored spot-forming cells (SFC) were counted using an Immunospot ELISpot plate reader (Cellular Technology, Shaker Heights, OH), and responses were considered to be positive if 50 SFC were detected/106 cells after subtraction of the negative control number and SFC were twofold higher than those in the negative control wells.
Analyses of PBMC by immunofluorescence (flow) cytometry
To assess CD4+ and CD8+ T cell responses, PBMC samples from individual patients were assayed for IFN-γ and tumor necrosis factor alpha (TNF-α) expression using a BD Accuri™ C6 Flow Cytometer and intracellular cytokine staining methods previously described [35, 36]. Briefly, 106 PBMC cells/well were incubated 6 h with 2.0 μg/ml CEA peptide pool, 2.0 μg/ml SIV nef-negative control peptide pool, or media alone. A protein transport inhibitor (GolgiStop) was added for the final 4 h of the stimulation. After stimulation, cells were stained for CD4 and CD8, fixed, permeabilized, stained for IFN-γ and TNF-α, and then analyzed by flow cytometry.
Treg cells and Teff cells were analyzed by staining PBMC for CD4, CD25, and Foxp3 with the Human Regulatory T Cell Staining Kit #2 (eBiosciences). The ratio of regulatory T cells to effector T cells (Treg/Teff) was defined as the ratio of CD4+CD25+Foxp3+ cells to CD4+CD25−Foxp3− cells.
Flow cytometry was also employed to assess HLA-A2 expression on patient PBMC samples. Staining with fluorochrome-conjugated anti-HLA-A2 antibody was performed as recommended by the manufacturer (BD Biosciences).
Determination of Ad5 neutralizing antibody (NAb) titers in sera
End point Ad5 NAb titers were determined as previously described [14, 23]. Briefly, dilutions of heat-inactivated test sera in 100 μL of DMEM containing 10 % fetal calf serum were mixed with 4 × 107 VP of Ad5 [E1-]-null and incubated for 60 min at room temperature. The samples were added to microwells containing HEK293 cells cultured in DMEM containing 10 % heat-inactivated fetal calf serum at 2 × 103 cells/well for 24 h at 37° C in 5 % CO2. The mixture was incubated for an additional 72 h at 37° C in 5 % CO2. An MTS tetrazolium bioreduction assay (Promega Corp, Madison, WI) [37] was used to measure cell killing and end point Ad5 NAb titers.
Statistics
Statistical analyses were performed employing PRISM Software (GraphPad). The long-term survival curve was made employing Kaplan–Meier plots (PRISM Software, GraphPad).
Results
Summary of dose administration, adverse events, and initial survival
The majority of patients (28/32, 87.5 %) were able to receive all three immunizations. Four patients with rapid disease progression did not receive all three injections (one patient received only one dose, and three patients received two doses). There were no dose-limiting toxicities and no serious adverse effects (SAEs) that resulted in treatment discontinuation at any dose level of the vaccine, and there were no dropouts due to treatment. The most common adverse effect (AE) was a self-limited, injection site reaction. Other reactions occurred with less than a 10 % incidence and included fever, flu-like symptoms, anorexia, chills, nausea, and headache. These symptoms were self-limiting and did not require intervention other than symptomatic control measures such as acetaminophen. Routine hematology and chemistry studies showed no significant biologic changes during the immunization period, and these above results have been previously reported [23]. During our longer-term follow-up, no treatment-related adverse effects were reported.
Results on extended follow-up overall-survival data and post-treatment therapies
Survival data and post-treatment therapies that patients received are presented in Table 1. Kaplan–Meier overall-survival plots are presented in Fig. 1. Twenty-nine-month overall survival of the intent-to-treat population (32 patients) was 20 % (Fig. 1a) with a median survival time of 11 months from informed consent/first injection. For the subset of 28 patients that received all three immunizations, the 29-month survival was 23 % (Fig. 1b) with a median survival time of 13 months. For the 22 patients optimally dosed with the two highest doses of vaccine (1 and 5 × 1011) and receiving all three immunizations (see Table 1), the 28-month overall survival was 19 % (Fig. 1c). Median overall survival was 13 months in the optimally treated patients. Since there was no active control group in the study, comparisons for significance in survival time cannot be made. There were three stable disease events observed immediately after completion of treatment, and over the course of long-term follow-up, we observed that a total of six disease stabilizations were recorded.
Table 1.
Overall survival and post-treatment therapies
| Patient ID # | Cohort | Number of immunizations | Tumor type | Survival time (months)a | Post-treatment therapiesb |
|---|---|---|---|---|---|
| 002 | 1 | 3 | Colon | 2.75 (–) | None |
| 003 | 1 | 3 | Rectal | 8.75 (–) | R |
| 004 | 1 | 3 | Colon | 11.25 (–) | A, F, Xld |
| 005 | 2 | 3 | Colorectal | 17.8 (–) | OCT, R |
| 007 | 2 | 1 | Colon | 1.25 (–) | None |
| 008 | 2 | 3 | Colorectal | 29 (+) | None |
| 010 | 2 | 3 | Colon | 12 (–) | OCT |
| 011 | 3 | 3 | Rectal | 13.2 (–) | R |
| 012 | 3 | 3 | Colon | 28 (+) | OUC |
| 013 | 3 | 3 | Rectal | 3.5 (–) | N |
| 015 | 3 | 3 | Colorectal | 10 (–) | E |
| 016 | 3 | 3 | Colon | 5.75 (–) | OUC |
| 017 | 3 | 3 | Rectal | 2.75 (–) | None |
| 500 | 3 | 3 | Colon | 26.6 (–) | Not reported |
| 501 | 4 | 3 | Rectal | 25.5 (+) | OUC |
| 502 | 4 | 2 | Colon | 2.75 (–) | Not reported |
| 504 | 4 | 3 | Colon | 7.25 (–) | Not reported |
| 506 | 4 | 3 | Colon | 2.5 (–) | Not reported |
| 507 | 4 | 3 | Rectal | 24 (+) | Not reported |
| 019 | 4 | 3 | Colon | 17.5 (–) | A, Xli |
| 020 | 4 | 3 | Colon | 24.5 (+) | A |
| 021 | 4 | 3 | Colon | 25.25 (+) | A, Xld |
| 023 | 4 | 3 | Colon | 4.25 (–) | None |
| 024 | 4 | 3 | Colon | 23 (+) | None |
| 025 | 4 | 3 | Colon | 7.25 (–) | None |
| 026 | 4 | 2 | Colon | 4.2 (–) | None |
| 030 | 5 | 3 | Colorectal | 15.7 (–) | OCT |
| 031 | 5 | 3 | Rectal | 15.5 (+) | OUC |
| 032 | 5 | 3 | Rectal | 5.6 (–) | F |
| 033 | 5 | 3 | Rectal | 4.9 (–) | Not reported |
| 034 | 5 | 3 | Colon | 13.75 (+) | Xld |
| 035 | 5 | 2 | Colon | 2.0 (–) | Not reported |
a(+) Alive at last follow-up; (–) not alive
bPost-treatment therapy: A Avastin, E Erbitux, F FOLFOX, N Nexavar, OCT other clinical trial, OUC other unknown chemotherapy, R radiation, Xli Xeliri, Xld Xeloda
Fig. 1.
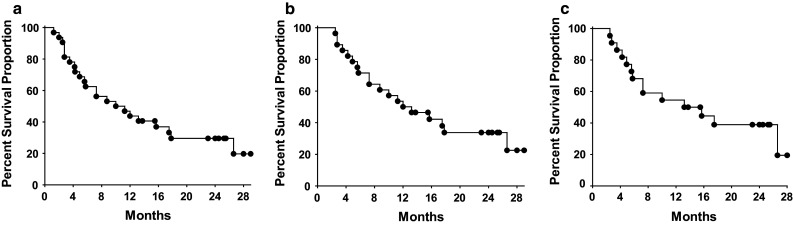
Kaplan–Meier survival plots on long-term overall survival of treated mCRC patients. a Represents all treated patients. b Represents patients that received all three treatments. c Represents patients immunized three times with the two highest doses of vaccine. There were 23 events during the study
Post-treatment therapies were also recorded during follow-up (see Table 1). For seven patients, follow-up therapy information was not available, for eight patients, there were no further therapies, four patients received other unknown chemotherapy, two patients received radiation treatment, one patient received radiation treatment and entered another clinical trial, two patients entered another clinical trial, one patient received treatment with Avastin, one patient received Avastin and Xeliri, one patient received Avastin and Xeloda, one patient received Avastin, Xeloda, and FOLFOX, one patient received Xeloda, one patient received FOLFOX, one patient received treatment with Nexavar, and one patient received Erbitux.
Immune analyses
We previously reported on ELISpot analyses of PBMC samples for CMI activity as assessed by numbers of IFN–γ-secreting cells after stimulation with CEA peptide pools [23]. We observed a dose–response effect with the highest magnitude CEA-specific CMI responses occurring in patients who received the highest dose of Ad5 [E1-, E2b-]-CEA(6D). Of the doses received, 0/3 (0 %) patients in cohort 1, 1/4 (25 %) patients in cohort 2, 10/19 (53 %) cohort 3/phase 2, and 4/6 (67 %) patients in cohort 5 exhibited positive CEA-directed CMI responses. As previously reported [23], the level of preexisting Ad5 neutralizing immunity did not appear to affect the induced CEA-directed CMI responses.
For this study, we performed additional flow cytometry analyses on patient PBMC. A sample from only one patient (patient 013, week 3) that exhibited high CMI activity (≥1000 IFN-γ-secreting SFC) during treatment was observed to be positive for polyfunctional T cells. Although the week 9 sample from patient 507 also exhibited high CMI activity, we were unable to detect polyfunctional T cells.
The flow cytometry ICS profile of the PBMC from patient 013 after exposure to CEA peptides showed the presence of:
CD8+/IFN-γ+ cells (3.5 %),
CD8+/IFN-γ+/TNF-α+ cells (1.0 %),
CD4+/IFN-γ+ cells (0.4 %), and
CD4+/IFN-γ+/TNF-α+ cells (0.2 %).
Specificity of the response was demonstrated by the lack of PBMC reactivity with SIV nef peptides.
In further follow-up, we obtained PBMC samples from a subset of five patients that could be tested post-treatment for extended CMI responses. It was observed that the levels of CEA-specific CMI activity decreased from their peak values during follow-up in the five patients analyzed (Fig. 2). We were able to perform further analyses on a large number of PBMC samples pre-treatment (week 0) and post-treatment (week 6–9) and assess CTL activity employing ELISpot assays for granzyme B secretion. Twenty-seven evaluable patient PBMC samples were tested. As shown in Fig. 3, significantly (P < 0.05, Wilcoxon test) elevated levels of granzyme B secretion activity were observed in post-immunization PBMC samples as compared to their respective baseline pre-immunization PBMC samples.
Fig. 2.
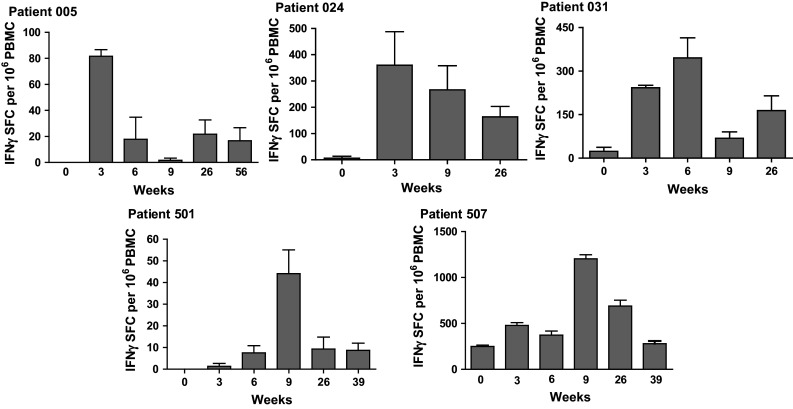
Profiles of CEA-directed CMI responses in five mCRC patients during and after the course of immunotherapy. Note the decrease in CMI responses from their peak values after immunizations ended. Values are mean ± SEM
Fig. 3.
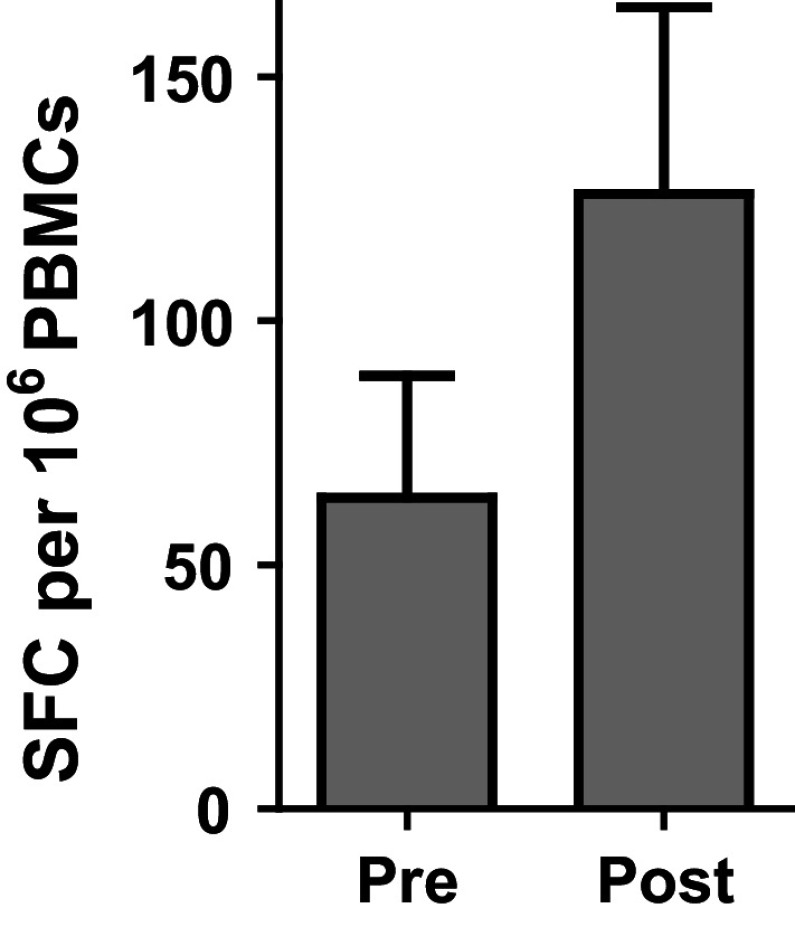
CTL responses (ELISpot granzyme B secretion) were assessed pre-treatment (week 0) and post-treatment (week 6–9). Note the increase in CTL activity after immunizations of mCRC patients (P < 0.05 Wilcoxon test). Values are mean ± SEM
Analysis of peripheral blood T regulatory (Treg) to T effector (Teff) cell ratios pre- and post-treatment (week 9) in 15 patients was examined employing flow cytometry. No change in Treg to Teff cell ratio occurred over the course of the 9-week treatment evaluation period. The mean Treg to Teff cell ratio at baseline was 0.062 ± 0.02 (SD), and the mean Treg to Teff cell ratio at week 9 was 0.07 ± 0.03. Of the five-patient PBMC samples available for longer-term assessment, one patient exhibited an increase in Treg to Teff cell ratio, one patient exhibited an increase in Treg to Teff cell ratio that decreased to near baseline level at last follow-up, and samples from three patients exhibited a decrease in Treg to Teff cell ratio during the treatment protocol with one of these patients showing a return to near baseline ratio after 52 weeks (Fig. 4).
Fig. 4.
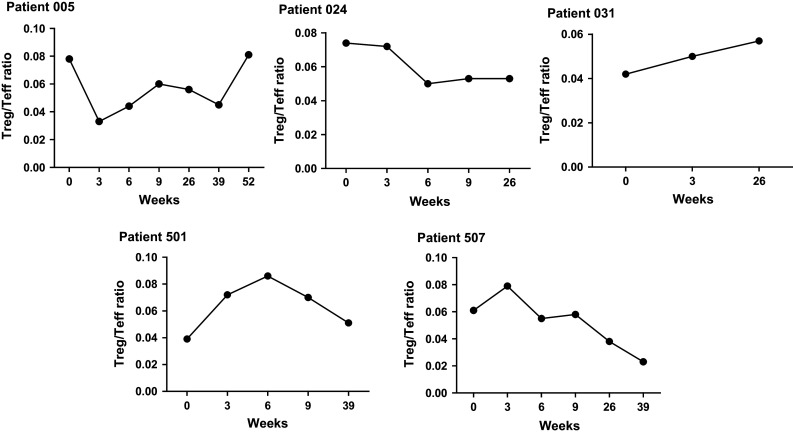
Treg/Teff ratios in five mCRC patients during and after immunotherapy. Ratios increased in one patient (031) and increased with a return to baseline ratio in another patient (501). Ratios decreased with a return to baseline ratio at 52 weeks in one patient (005). Ratios were decreased in two of the other five patients (024 and 507)
Patient PBMC samples were tested for HLA-A2 positivity by flow cytometry, and 61 % of the samples tested were HLA-A2 positive. When the highest CMI response level (as measured by IFN–γ-secreting cells) achieved per patient during treatment was assessed in association with the presence of HLA-A2, there was no significant difference observed between HLA-A2+ and HLA-A2− patients (HLA-A2+ = 264.6 ± 119.0 IFN–γ-secreting cells versus HLA-A2− = 165.6 ± 108.1 IFN–γ-secreting cells; P = 0.5809).
Ad5 NAb titers were assessed in follow-up serum samples from six patients. Patients 005, 024, 501, and 507 had preexisting Ad5 end point titers of 200, 200, 100, and 50, respectively. Patients 031 and 500 did not exhibit Ad5 NAb activity at baseline. As shown in Fig. 5, Ad5 NAb titers increased at week 9 and then decreased thereafter. In patient 507, Ad5 NAb titers decreased to below baseline titer but were within one titer of the baseline value. In the remaining five patients (005, 024, 031, 500, and 501), the Ad5 NAb titers decreased but remained above baseline levels at last follow-up.
Fig. 5.
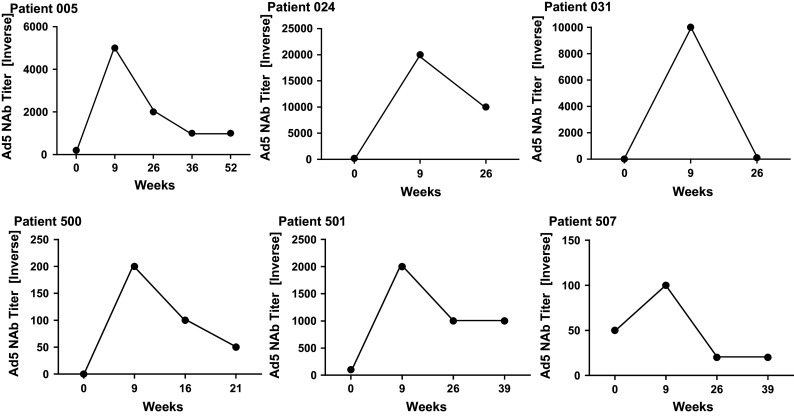
Ad5 NAb titers over time in six mCRC patients immunized with Ad5 [E1-, E2b-]-CEA(6D). Titers increased in all patients by week 9 (3 weeks after the last immunization) and then decreased thereafter
Discussion
We previously reported our initial study findings on the dosing, safety, immunogenicity, and clinical findings of a phase 1/2 clinical trial, evaluating the use of an Ad5 [E1-, E2b-]-CEA(6D) in mCRC patients [23]. Patient demographics were similar to those reported in other clinical trials with mCRC patients [38–40]. Study findings indicated that the Ad5 [E1-, E2b-]-CEA(6D) immunotherapeutic product as a single agent was well tolerated and could easily be administered. It was reported that CEA-specific CMI responses were induced and increased over the course of three immunizations. In addition, a dose–response effect was observed. Preexisting Ad5 immunity was present in 61 % of patients and the presence of Ad5 immunity did not appear to affect the CEA immunogenicity. Although no objective antitumor responses occurred by RECIST, there were three stable disease events observed immediately after completion of treatment. Over the course of long-term follow-up, we noted that a total of six disease stabilizations were recorded. Previously, we reported a 48 % survival at 12-month follow-up [23], and in our extended follow-up for overall survival, we noted that on an intent-to-treat analysis the 29-month survival was 20 % with a favorable median overall survival of 11 months. However, one should note that these overall-survival data are confounded by the fact that the patients received other anticancer treatment(s) after they went off protocol. Also, it is of interest to note that the FDA-approved cancer vaccine sipuleucel-T did not show a significant antitumor impact but did demonstrate overall-survival benefit [41–43]. Whether or not we observe similar results can only be demonstrated by testing the Ad5 [E1-, E2b-]-CEA(6D) immunotherapeutic as a single agent to determine its true clinical benefit in a larger randomized/controlled phase 2b study wherein patients are followed long term and which we plan to perform.
In our further analyses of patient PBMC samples, ELISpot assays were employed to determine CEA-directed CTL activity pre- and post-immunization. ELISpot granzyme B assays were used since this assay provides a measure of functional CTL activity [33, 34]. We observed a significant increase in CTL activity after immunizations with Ad5 [E1-, E2b-]-CEA(6D), indicating that CEA-specific CTL activity was induced. Also, we observed granzyme B-secreting activity in baseline PBMC samples, and we speculate that this reflects endogenous CTL activity induced by the presence of CEA-expressing tumors. Thus, in addition to the induction of CEA-directed CMI responses as previously reported [23], we also observed increases in CEA-directed CTL activity in treated patients.
Our flow cytometry data indicated that a high level of CMI activity, as measured by ELISpot IFN–γ-secreting cells, must be achieved before we could detect the presence of activated T cells by ICS. In two patients, PBMC samples were sufficiently evaluable and we were able to detect activated CD4+ and CD8+ T cells in a post-immunization sample from one patient. Although the sample size is very small, these data may provide evidence that the Ad5 [E1-, E2b-]-CEA(6D) can induce robust CMI responses and we plan to study this on a larger patient sample size in a phase 2b trial. Moreover, if one were to treat mCRC patients earlier in the course of their disease, more robust CD4+ and CD8+ populations might be achieved.
Because of the importance of Treg in influencing antitumor CMI responses [44], Treg to Teff ratios were determined in patient PBMC samples. Of the five-patient samples tested, three patients exhibited a decrease in the Treg to Teff cell ratio during the immunization period. Since we also observed that levels of CMI activity decreased during follow-up in the five patients analyzed, it may well be that we need to administer additional booster immunizations in order to maintain elevated levels of CEA-directed CMI responses that generate activated T cells with beneficial changes in Treg to Teff cell ratios. Based upon the observation that CMI responses waned by 26 weeks (23 weeks after the last immunization), we plan to administer “booster” immunizations at 4-month intervals in our next phase 2b study to determine whether we can maintain induced CMI responses. Immune tests including ELISpot assays for IFN-γ and granzyme B-secreting cells and flow cytometry tests for activated CD4+, CD8+, Treg, and Teff cells will be studied as surrogate parameters of immune function in planned future trials. We expect that a larger randomized mCRC patient trial will give us much needed additional information and provide a greater understanding of the immune mechanisms(s) generated by immunizations with Ad5 [E1-, E2b-]-CEA(6D).
Since the modified immunogenic HLA-A2-restricted CAP1(6D) epitope of CEA was incorporated into our vaccine, we investigated the association between induced CMI responses and the presence or the absence of HLA-A2 in PBMC samples tested. Although the average CMI responses were relatively higher in HLA-A2+ PBMC samples, the difference was not significant when compared to values obtained in HLA-A2− samples. Thus, in this small study, CMI responses did not appear to be HLA-A2+ restricted. Again a larger trial may elucidate this difference.
In our analyses of Ad5 NAb titers on follow-up serum samples from six patients, we observed that the Ad5 NAb titers decreased from their peak values after immunizations ended. This is similar to our findings with CMI responses in which the responses declined after treatment was stopped. We previously reported on the analyses of Ad5 NAb titers in non-human primate (NHP) studies investigating Ad5 [E1-, E2b-]-HIV/SIV vaccines [18, 21]. In these prior studies employing NHP with preexisting Ad5 immunity, we observed that multiple homologous immunizations with Ad5 [E1-, E2b-]-HIV/SIV vaccines resulted in an increase in Ad5 NAb titers during vaccinations; however, after immunizations ended, Ad5 NAb titers decreased from their peak values. Thus, the present observations are in agreement with our earlier findings using this platform-based vaccine on NHP.
In conclusion, the clinical data obtained using the Ad5 [E1-, E2b-]-CEA(6D) demonstrate that this product can be easily and safely administered to mCRC patients to induce CEA-directed CMI responses. Although no objective antitumor responses were observed, there appeared to be favorable survival with an observed median overall survival of 11 months. A small number of patient PBMC samples were available for follow-up immune analyses, and although the sample size was small, the results obtained indicate that the generated CEA-directed CMI responses induced might require additional booster immunizations to maintain the induced immune response. This will be an important factor to consider especially in the context of immunotherapy vaccines. We plan to conduct a randomized/controlled phase 2b clinical trial to evaluate immunogenicity and include booster immunizations to determine the effect of Ad5 [E1-, E2b-]-CEA(6D) as a single agent on overall survival in mCRC patients. Also, it may be important to investigate the use of this immunotherapeutic agent in an adjuvant setting, enrolling recently resected early-stage colorectal cancer patients to assess the time to progression. The results of such clinical trials will allow us to evaluate the true clinical benefit of using Ad5 [E1-, E2b-]-CEA(6D) as an immunotherapeutic.
Acknowledgments
The authors wish to thank Susan Thorburn for her assistance in the patient data collection and Ms. Carol Jones for her excellent assistance with grants management. This study was funded by National Cancer Institute Small Business Innovative Research Grant 1R43CA134063, National Cancer Institute Small Business Innovative Research Grant 2R44CA134063, National Cancer Institute Small Business Innovative Research Contract HHSN261200900059C, and National Cancer Institute Small Business Innovative Research Contract HHSN261201100097C.
Conflict of Interest
Joseph P. Balint is a shareholder and employee of Etubics and has stock options in the Company. Elizabeth S. Gabitzsch is a shareholder and employee of Etubics and has stock options in the Company. Adrian Rice is an employee of Etubics and has stock options in the Company. Yvette Latchman is an employee of Etubics and has stock options in the Company. Younong Xu was an employee of Etubics during performance of the study and has stock options in the Company. Gerry L. Messerschmidt is an employee of Etubics and has stock options in the Company. Frank R. Jones is a shareholder and employee of Etubics and has stock options in the Company. Arvind Chaudhry and Michael Morse have no conflicts of interest related to this work.
Abbreviations
- Ad5
Adenovirus serotype-5
- Ad5 [E1-]
Adenovirus serotype-5 (Ad5)-based vector platforms with deletions in the early 1 (E1) gene and early 3 (E3) gene regions
- Ad5 [E1-E2b-]
Ad5 [E1-] with additional deletions in the early 2 (E2) gene region
- Ad5 [E1-E2b-]-CEA(6D)
Ad5 [E1-, E2b-] containing the CAP1(6D) gene
- AE
Adverse effects
- CAP1
Nine amino acid sequence of CEA
- CAP1(6D)
Peptide analog sequence of CAP1
- CEA
Carcinoembryonic antigen
- CMI
Cell-mediated immune
- Con A
Concanavalin A
- CT
Computed tomography
- CTCAE
Common terminology criteria for adverse events
- CTL
Cytotoxic T lymphocytes
- E1
Early 1 gene region
- E2b
Early 2b gene region
- E3
Early 3 gene region
- IFN-γ
Interferon gamma
- mCRC
Metastatic colorectal cancer
- MRI
Magnetic resonance imaging
- NAb
Neutralizing antibody
- NHP
Non-human primates
- PBMCs
Peripheral blood mononuclear cells
- pol
Polymerase gene
- pTP
Pre-terminal protein gene
- RECIST
Response evaluation criteria in solid tumors
- SAEs
Serious adverse effects
- SAFC
Sigma-Aldrich Fine Chemicals
- SFC
Spot-forming cells
- SQ
Subcutaneously
- TAA
Tumor-associated antigens
- Teff
T effector
- Treg
T regulatory
- TNF-α
Tumor necrosis factor alpha
- VP
Viral particles
References
- 1.Vergati M, Intrivici C, Huen N-Y, Schlom J, Tsang KY. Strategies for cancer vaccine development. J Biomed Biotechnol. 2010 doi: 10.1155/2010/596432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palena C, Schlom J. Vaccines against human carcinomas: strategies to improve antitumor immune responses. J Biomed Biotechnol. 2010 doi: 10.1155/2010/380697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlom J. Therapeutic cancer vaccines: current status and moving forward. J Natl Cancer Inst. 2012;104:599–613. doi: 10.1093/jnci/djs033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Mol Ther. 2004;10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bangari DS, Mittal SK. Development of nonhuman adenoviruses as vaccine vectors. Vaccine. 2006;24:849–862. doi: 10.1016/j.vaccine.2005.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campos SK, Barry MA. Current advances and future challenges in adenoviral vector biology and targeting. Curr Gene Ther. 2007;7:189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chirmule N, Propert K, Magosin S, Qian Y, Qian R, Wilson J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999;6:1574–1583. doi: 10.1038/sj.gt.3300994. [DOI] [PubMed] [Google Scholar]
- 8.Barouch DH, Kikc SV, Weverling GJ, et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine. 2011;29:5203–5209. doi: 10.1016/j.vaccine.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amalfitano A, Chamberlain JS. Isolation and characterization of packaging cell lines that coexpress the adenovirus E1, DNA polymerase, and preterminal proteins: implications for gene therapy. Gene Ther. 1997;4:258–263. doi: 10.1038/sj.gt.3300378. [DOI] [PubMed] [Google Scholar]
- 10.Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. J Virol. 1998;72:926–933. doi: 10.1128/jvi.72.2.926-933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartigan-O’Connor D, Amalfitano A, Chamberlain JS. Improved production of gutted adenovirus in cells expressing adenovirus preterminal protein and DNA polymerase. J Virol. 1999;73:7835–7841. doi: 10.1128/jvi.73.9.7835-7841.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seregin SS, Amalfitano A. Overcoming pre-existing adenovirus immunity by genetic engineering of adenovirus based vectors. Expert Opin Biol Ther. 2009;9:1521–1531. doi: 10.1517/14712590903307388. [DOI] [PubMed] [Google Scholar]
- 13.Osada T, Yang XY, Hartman ZC, et al. Optimization of vaccine responses with an E1, E2b and E3-deleted Ad5 vector circumvents pre-existing anti-vector immunity. Cancer Gene Ther. 2009;16:673–682. doi: 10.1038/cgt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabitzsch ES, Xu Y, Balint JP, Jr, Hartman ZC, Lyerly HK, Jones FR. Anti-tumor immunity despite immunity to adenovirus using a novel adenoviral vector Ad5 [E1-, E2b-]-CEA. Cancer Immunol Immunother. 2010;59:1131–1135. doi: 10.1007/s00262-010-0847-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabitzsch ES, Xu Y, Yoshida LH, Balint J, Gayle RB, Amalfitano A, Jones FR. A preliminary and comparative evaluation of a novel Ad5 [E1-, E2b-] recombinant based vaccine used to induce cell mediated immune responses. Immunol Lett. 2009;122:44–51. doi: 10.1016/j.imlet.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabitzsch ES, Yu X, Yoshida LH, Balint J, Amalfitano A, Jones FR. Novel adenovirus type 5 vaccine platform induces cellular immunity against HIV-Gag, Pol, Nef despite the presence of Ad5 immunity. Vaccine. 2009;27:6394–6398. doi: 10.1016/j.vaccine.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabitzsch ES, Xu Y, Balcaitis S, Balint JP, Jr, Jones FR. An Ad5 [E1-, E2b-]-HER2/neu vector induces immune responses and inhibits HER2/neu expressing tumor progression in Ad5 immune mice. Cancer Gene Ther. 2011;18:326–335. doi: 10.1038/cgt.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabitzsch ES, Xu Y, Balint JP, Jr, Balcaitis S, Sanders-Beer B, Jones FR. Induction and comparison of SIV immunity in Ad5 Naïve and Ad5 immune non-human primates using an Ad5 [E1-, E2b-] based vaccine. Vaccine. 2011;29:8101–8107. doi: 10.1016/j.vaccine.2011.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones FR, Gabitzsch ES, Xu Y, et al. Prevention of influenza virus shedding and protection from lethal H1N1 challenge using a consensus 2009 H1N1 HA and NA adenovirus vector vaccine. Vaccine. 2011;29:7020–7026. doi: 10.1016/j.vaccine.2011.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabitzsch ES, Jones FR. New recombinant Ad5 vector overcomes Ad5 immunity allowing for multiple safe, homologous immunizations. J Clin Cell Immunol. 2011;S4:001. [Google Scholar]
- 21.Gabitzsch ES, Balint JP, Jr, Xu Y, et al. Control of SIV infection and subsequent induction of pandemic H1N1 immunity in rhesus macaques using an Ad5 [E1-, E2b-] vector platform. Vaccine. 2012;30:7265–7270. doi: 10.1016/j.vaccine.2012.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wieking BG, Vermeer DW, Spanos WC, et al. A non-oncogenic HPV 16 E6/E7 vaccine enhances treatment of HPV expressing tumors. Cancer Gene Ther. 2012;19:667–674. doi: 10.1038/cgt.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morse MM, Chaudhry A, Gabitzsch ES, et al. Novel adenoviral vector induces T cell responses despite anti-adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol Immunother. 2013;62:1293–1301. doi: 10.1007/s00262-013-1400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal. 1991;5:344–366. doi: 10.1002/jcla.1860050510. [DOI] [PubMed] [Google Scholar]
- 25.Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 26.Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002;20:2197–2207. doi: 10.1200/JCO.2002.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gameiro SR, Jammeh ML, Hodge JW. Cancer vaccines targeting carcinoembryonic antigen: state-of-the-art and future promise. Expert Rev Vaccines. 2013;12:617–629. doi: 10.1586/erv.13.40. [DOI] [PubMed] [Google Scholar]
- 29.Zaremba S, Barzaga E, Zhu M, Soares N, Tsang KY, Schlom J. Identification of an enhancer agonist cytotoxic T lymphocyte peptide from human carcinoembryonic antigen. Cancer Res. 1997;57:4570–4577. [PubMed] [Google Scholar]
- 30.Tangri S, Ishioka GY, Huang X, et al. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild type peptide. J Exp Med. 2001;194:833–846. doi: 10.1084/jem.194.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumors: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 32.CTEP Cancer Therapy Evaluation Program (2010) CTCAE and CTC website http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Accessed 10 Feb 2012
- 33.Rininsland FH, Helms T, Asaad RJ, Boehm BO, Tary-Lehmann M. Granzyme B ELISPOT assay for ex vivo measurements of T cell immunity. J Immunol Methods. 2000;240:143–155. doi: 10.1016/S0022-1759(00)00191-5. [DOI] [PubMed] [Google Scholar]
- 34.Shafer-Weaver K, Sayers T, Strobl S, et al. The Granzyme B ELISPOT assay: an alternative to the 51Cr-release assay for monitoring cell-mediated cytotoxicity. J Transl Med. 2003;1:14–23. doi: 10.1186/1479-5876-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hobeika AC, Clay TM, Mosca PJ, Lyerly HK, Morse MA. Quantitating therapeutically relevant T-cell responses to cancer vaccines. Crit Rev Immunol. 2001;21:287–297. doi: 10.1615/CritRevImmunol.v21.i1-3.190. [DOI] [PubMed] [Google Scholar]
- 36.Hobeika AC, Morse MA, Osada T, et al. Enumerating antigen-specific T-cell responses in peripheral blood: a comparison of peptide MHC tetramer, ELISpot, and intracellular cytokine analysis. J Immunother. 2005;28:63–72. doi: 10.1097/00002371-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Comm. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 38.Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 39.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 40.Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 41.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 42.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 43.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 44.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–811. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]