

Abstract
Muscle fibers in patients with sporadic inclusion-body myositis (s-IBM), the most common age-associated myopathy, are characterized by autophagic vacuoles and accumulation of ubiquitinated and congophilic multiprotein aggregates that contain amyloid-β and phosphorylated tau. Muscle fibers of autosomal-recessive hereditary inclusion-body myopathy due to the GNE mutation (GNE-h-IBM) display similar pathologic features, except with less pronounced congophilia. Accumulation of unfolded/misfolded proteins inside the ER lumen leads to ER stress, which elicits the unfolded protein response (UPR) as a protective mechanism. Here we demonstrate for the first time that UPR is activated in s-IBM muscle biopsies, since there was a) increased ATF4 protein and increased mRNA of its target CHOP, b) cleavage of the ATF6 and increased mRNA of its target GRP78, and c) an increase of the spliced form of XBP-1 and increased mRNA of EDEM, target of heterodimer of cleaved ATF6 and spliced XBP-1. In contrast, we did not find similar evidence of the UPR induction in GNE-h-IBM patient muscle, suggesting that different intracellular mechanisms might lead to the similar pathological phenotypes. Interestingly, cultured GNE-h-IBM muscle fibers had a robust UPR response to experimental ER stress stimuli, suggesting that the GNE mutation per se is not responsible for the lack of UPR in GNE-h-IBM biopsied muscle.
Keywords: Cultured human muscle fibers, Endoplasmic reticulum stress, GNE, Hereditary inclusion-body myopathy, Sporadic inclusion-body myositis, UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase, Unfolded protein response
INTRODUCTION
Endoplasmic reticulum (ER) plays a critical role in the processing, folding and exporting newly-synthesized normal, and unfolded/misfolded, proteins into the secretory pathway (1). Accumulation of the unfolded/misfolded proteins in the ER lumen leads to ER-stress, which subsequently elicits the unfolded protein response (UPR), a mechanism by which cells attempt to both protect themselves against the ER-stress and restore their folding capacity (1–3). The UPR involves a) attenuation of translation, which reduces protein overload and subsequent accumulation of unfolded proteins, b) refolding of unfolded/misfolded proteins, and c) removal of misfolded proteins from the ER through the retrograde transport coupled to their degradation by 26S proteasome (2, 4). In response to ER stress, the UPR mediators also play an important role in the upregulation of macroautophagy (5), a protein degradation pathway involving sequestration of proteins or cellular components destined for removal in autophagosomes, and their subsequent delivery into lysosomes.
There are 3 proximal sensors of the UPR localized to the ER membrane and activated upon ER-stress: inositol-requiring enzyme; protein kinase RNA-like ER protein kinase; and activating transcription factor 6 (ATF6) (1, 4). The ER sensors initiate different intracellular signaling cascades, which lead to activation of the set of transcription factors are X-box binding protein 1 (XBP-1), activating transcription factor 4 (ATF4), and p50ATF6. Those transcription factors execute the signal through coordinated upregulation of the genes encoding ER chaperones, whose role is to increase folding capacity of the ER and prevent protein aggregation (Fig. 1). ER-stress and markers of the UPR activation have been identified in various neurodegenerative diseases associated with accumulation of aggregated and misfolded proteins, including Alzheimer and Parkinson diseases (1, 3). We previously identified increased ER-stress chaperones BiP/GRP78, GRP94, ERp72, calnexin and calreticulin on the protein level, as well as their physical association with amyloid-β precursor protein (AβPP) in sporadic-inclusion body myositis (s-IBM) (6). Upregulation of Herp, the gene most highly inducible under ER-stress, is also increased both on the protein and mRNA levels (7). Those findings indicated that some aspects of ER-stress occur in s-IBM muscle.
Figure 1.
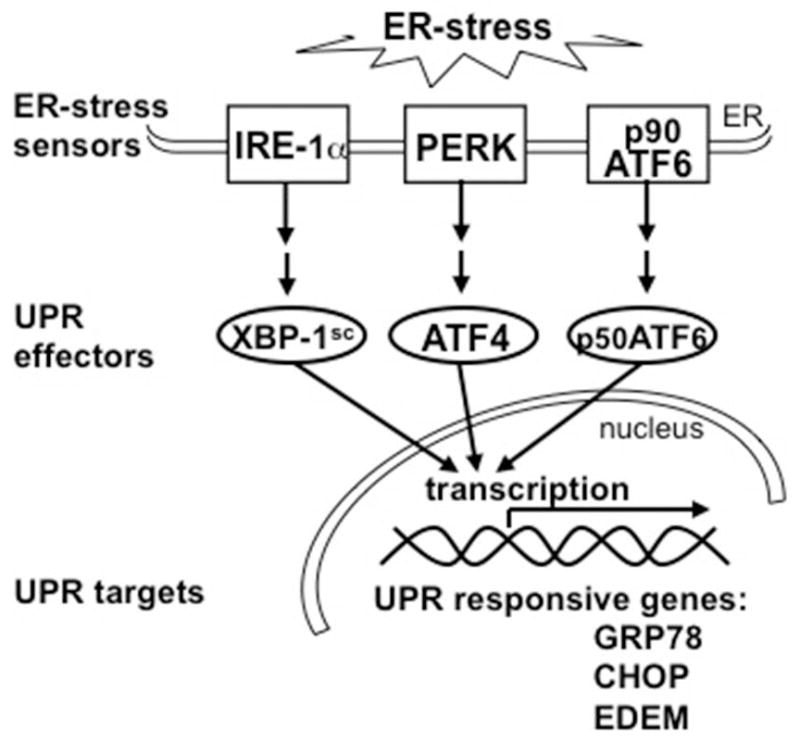
Diagram showing main components in the unfolded protein response (UPR) signaling cascade. Three endoplasmic reticulum (ER)-transmembrane sensors (inositol-requiring enzyme 1 [IRE-1α], protein kinase RNA-like ER protein kinase [PERK], and activating transcription factor 6 [ATF6]) detect ER-stress and pass the signal downstream. Induced transcription factors ATF4, spliced XBP-1 (XBP-1sc), and cleaved p50ATF6 execute the signal by upregulating various UPR-responsive genes including genes encoding ER chaperones.
s-IBM, the most common degenerative muscle disease of persons over age 50, is relentlessly progressive, and it leads to pronounced muscle weakness and atrophy, causing severe, crippling disability (8, 9). There is no enduring treatment currently available (9–12). Histological hallmarks of s-IBM muscle biopsies include vacuolar degeneration and atrophy of muscle fibers, accompanied by intra-muscle-fiber accumulations of congophilic, ubiquitinated, multi-protein aggregates, and T-cell lymphocytic inflammation (8, 12). There is also an impairment of both the proteasomal and lysosomal protein degradations (13, 14).
Hereditary inclusion body myopathies (h-IBMs) encompass a group of either autosomal recessive or dominant myopathies, having various clinical presentations, but strikingly similar muscle-fiber pathology to that of s-IBM (15, 16). GNE-h-IBM, a quadriceps sparing autosomal-recessive myopathy, is associated with the mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE) gene, which encodes a bifunctional enzyme involved in the synthesis of sialic acid (15, 17–19). Various GNE mutations have been described worldwide, including 3 founder mutations: p.M712T in Persian-Jewish patients, and p.V572L and p.D176V in Japanese patients (17, 20, 21). Although the disease-associated gene has been identified, the exact role of the mutated protein in the pathogenesis of GNE-h-IBM is not understood (17–20). GNE-h-IBM generally begins clinically in early adulthood and is manifested by weakness of distal limbs muscles and relative sparing of the quadriceps (15, 17–19). Pathologically, the GNE-h-IBM muscle biopsy has vacuolated muscle fibers very similar to those present in s-IBM (Fig. 2). However, in contrast to s-IBM, which has very pronounced intra-myofiber congophilia (which indicates proteins in misfolded β-pleated amyloid configuration), GNE-h-IBM muscle fibers have only very small, rounded congophilic inclusions, or no congophilia and p62-immunopositive aggregates are somewhat less abundant (Fig. 2) (22). Moreover, muscle biopsies of the majority of the GNE-h-IBM patients do not exhibit mononuclear cell inflammation (21, 23).
Figure 2.
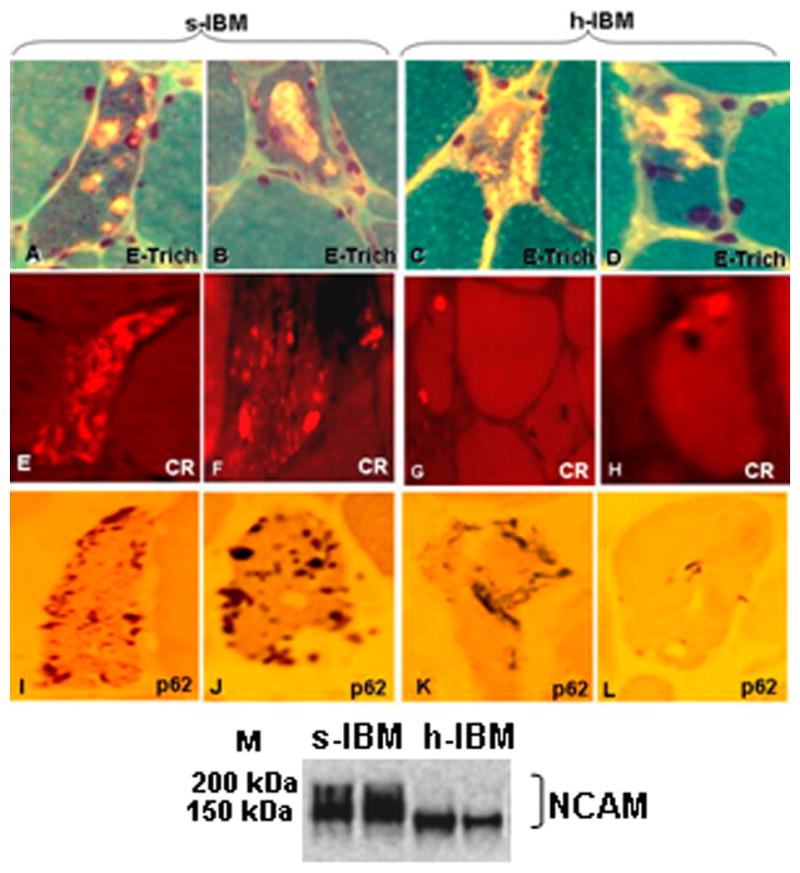
Diagnostic findings in muscle biopsies of patients with sporadic inclusion-body myositis (s-IBM) and hereditary IBM due to the GNE mutation (GNE-h-IBM). (A–D) Engel-trichrome staining (E-Trich) demonstrates typical vacuolated muscle fibers. (E–H) Congo red (CR) staining visualized through Texas-red filters and epifluorescence shows typical abundant congophilic inclusions in s-IBM (E, F), and occasional small, round inclusions in GNE-h-IBM (G, H). (I–L) p62 immunoreactivity indicates numerous various-sized brown inclusions in s-IBM (I, J), and less abundant inclusions in GNE-h-IBM (K, L). All are x1,400. (M) Representative immunoblot shows hypoglycosylated neural cell adhesion molecule (NCAM) in GNE-h-IBM patients.
Intriguing features of both the s-IBM and GNE-h-IBM vacuolated muscle fibers are intracellular accumulations of a group of the same proteins that are accumulated in the brains of patients with Alzheimer disease; these include AβPP and amyloid-β (Aβ), as well as phosphorylated tau in the form of paired helical filaments, even though phosphorylation of tau appears less advanced in GNE-h-IBM as compared to s-IBM (15, 24). There is growing evidence that Aβ is an important upstream pathogenic factor leading to muscle-fiber degeneration, atrophy and weakness in s-IBM (9, 25, 26); this is supported by various experimental studies employing cultured human muscle fibers (CHMFs) and transgenic mouse models (27). It has been postulated that similar mechanism might be involved in GNE-h-IBM pathogenesis (23) because Aβ accumulation preceded the development of vacuoles and other detectable abnormalities in CHMFs of GNE-h-IBM patients of Persian Jewish origin (28), and in the GNE-knock-out mouse overexpressing a mutated human GNE D176V (29).
Even though ER-stress chaperones have been studied in s-IBM muscle (6, 7), the activation of the UPR pathways leading to their increase has never been reported. The current study has focused on expression of the UPR-activated transcription factors in muscle biopsies of s-IBM and GNE-h-IBM patients, and under experimentally induced ER-stress in normal and GNE-h-IBM CHMFs. The UPR-activated transcription factors and their downstream genes studied here include ATF4 and its target gene CHOP, ATF6 and its target GRP78, and XBP-1 mRNA and its downstream target EDEM.
MATERIALS AND METHODS
Muscle Biopsies
The studies were performed on fresh-frozen diagnostic muscle biopsies obtained (with informed consent) from 14 s-IBM patients (ages 56–79 years, median 66 years), 8 h-IBM patients (ages 34–65 years, median 52 years), and 28 age-matched control patients (ages 22–86 years, median 55 years), who, after all tests performed, were considered free of muscle disease. Both s- and h-IBM muscle biopsies had their own age-matched controls. Patient diagnoses were based on clinical and laboratory investigations, including our routinely performed 16-reaction diagnostic histochemistry of their muscle biopsies, (9, 10). All IBM patient biopsies had muscle fibers with vacuoles on Engel trichrome staining (Fig. 2A–D), p62-immunopositive inclusions (Fig. 2I–L) and paired helical filaments by either or phosphorylated tau immunoreactivity or electron microscopy.
s-IBM patients had also pronounced Congo-red-positivity using fluorescence enhancement (30) (Fig. 2E, F), whereas h-IBM patients had only occasional, small-sized Congo-red positive rounded foci (Fig. 2G, H). s-IBM patient biopsies displayed various-sized foci of mononuclear cell inflammation, which was not present in any of the GNE-h-IBM patients studied (not shown). By immunoblots, in contrast to s-IBM, all our h-IBM patients had hyposialylated neural cell adhesion molecule (NCAM) with a molecular weight below 150kDa (Fig. 2M), as previously described by others (31, 32).
All of the h-IBM patients had GNE mutations detected on DNA isolated either from peripheral blood lymphocytes (QIAamp DNA Blood kit, Qiagen, Valencia, CA), or from muscle biopsies (GeneJET Genomic DNA Purification kit, Fermentas/ThermoFisher Scientific, Pittsburgh, PA). Isolated DNA was subjected to PCR amplification using specific GNE primers (described in [33]), except for exon 12, for which we designed specific primers utilizing the Primer3 website:
GNE_ex12_F/TTTCTGCCTTCCTCTTTGG;
GNE_ex12_R/TGATTGTTAAGAAACGGTCATCC).
All of the h-IBM patients, 6 of Persian-Jewish origin and 2 Caucasian brothers of Spanish origin (and possibly of Persian-Jewish background), had mutations in the GNE kinase domain (Table), which were previously described by others (31, 32).
Table.
Hereditary Inclusion-Body Myopathy Patients and Their GNE Mutations
| Patient | Age/Gender | GNE mutation (exon: protein change) | Affected GNE domain |
|---|---|---|---|
| 1 | 53/M | Ex12: pM712T | kinase |
| 2 | 34/F | Ex12: pM712T | kinase |
| 3 | 65/M | Ex12: pM712T | kinase |
| 4 | 63/M | Ex12: pM712T | kinase |
| 5 | 50/M | Ex12: pM712T | kinase |
| 6 | 58/M | Ex12: pM712T | kinase |
| 7 | 33/M | Ex9: pN519S | kinase |
| 8 | 45/M | Ex9: pN519S | kinase |
Patients: #1–6 were of Persian-Jewish origin; patients #7, 8 were 2 Caucasian brothers of Spanish origin. F, female; M male.
Immunoblotting
Muscle biopsies of s-IBM, h-IBM and control patients were immunoblotted as recently detailed (6, 14, 34). The proper amount of protein was loaded onto NuPAGE gels (Invitrogen, Carlsbad, CA), electrophoresed, transferred to nitrocellulose membranes, and immuno-probed with the following specific primary antibodies to: NCAM (123C3, 1:100), ATF4/CREB2 (C-2; 1:200), XBP-1 (M-186; 1:500) (all from Santa Cruz Biotechnology, Santa Cruz, CA), XBP-1 (1:500, BioLegend, San Diego, CA), GRP78 (1:500, BD Transduction Laboratory, San Jose CA, or 1:500, Abcam, Cambridge, MA) and ATF6 (1:350) (35). Blots were developed using WesternBreeze Immunodetection Kit (Invitrogen). Protein-loading was evaluated by actin bands (C-20; Santa Cruz Biotechnology). Quantification of immunoreactive bands of all control and disease muscle biopsies was performed by densitometric analysis using NIH Image 1.310 software. Intensity of the band of interest was calculated in relation to intensity of the actin band.
RNA Isolation and RT-PCR
Total RNA was isolated using an RNA isolation kit (BD Pharmingen, San Diego, CA) as previously described (7). One μg of RNA was subjected to genomic DNA removal, and to cDNA synthesis using a QuantiTect Reverse Transcription Kit (Qiagen). Real-time PCR was performed in duplicate at a total volume of 25 μl using QuantiFastSYBR Green PCR Master mix (Qiagen), and QuantiTect Primers (Qiagen) for GRP78, CHOP, EDEM, and GAPDH; for spliced-XBP1 previously published primers were used (36). PCR runs were performed on an Eppendorf Mastercycler Realplex2. Cycling conditions were 95°C for 5 minutes, followed by 40 cycles of 95°C for 10 seconds and 60°C (59°C for XBP1) for 30 seconds. Relative gene expression was calculated using the 2−ΔΔCt method, in which the amount of mRNA was normalized to an endogenous reference gene GAPDH. The results are expressed as fold-induction relative to controls. Correct PCR products were confirmed by agarose-gel electrophoresis and melting-curve analysis.
Cultured human muscle fibers
Primary cultures of normal human muscle were established as routinely performed in our laboratory (37–39), from archived satellite cells of portions of diagnostic muscle biopsies from: a) 3 h-IBM patients carrying M712T and 1 patient carrying N519S GNE mutation, and b) 5 different patients who, after all tests had been performed, were considered free of muscle disease. Each experiment was established from satellite cells derived from a different muscle biopsy but not all experiments were performed on each sample. Ten to 15 days after myoblast fusion, the well-differentiated myotubes were exposed for 24 hours to ER-stress inducers tunicamycin, an N-glycosylation inhibitor (4 μg/ml), or thapsigargin, an inhibitor of ER-calcium-ATPase (300 nM) (9, 40) (both from Sigma-Aldrich, St. Louis, MO), as previously described (7, 41). After treatment, experimental cultures and their untreated-sister-controls were processed for RNA isolation and immunoblotting.
Statistical Analysis
The statistical significance of differences between 2 groups was determined by Student’s t-test. The statistical significance of differences between more than two groups was determined by one-way analysis of variance (ANOVA) followed by Tukey-Kramer post-hoc multiple comparison test using GraphPad InStat software. The level of significance was set at p < 0.05. Data are presented as means ± SE.
RESULTS
Induction of the UPR in s-IBM Muscle Biopsies
In s-IBM patient biopsies, ATF4 protein was increased 7-fold (p < 0.01) as compared to their age-matched controls (Fig. 3A,A′). Increased translation of ATF4 is known to be selectively induced during ER-stress (4). p90ATF6, which was present in both s-IBM and control muscle homogenates, was increased 15-fold in s-IBM as compared to controls (p < 0.05) (Fig. 3B,B′). p50ATF6, which occurs after the cleavage of the full-length p90ATF6 into a shorter p50ATF6 isoform, was detected exclusively in s-IBM samples (Fig. 3B,B′), providing strong evidence of the ER-stress (36). The third pathway of UPR involves splicing of XBP-1 mRNA, which results in the synthesis of the active transcription factor (1). A 28-kDa band representing a non-spliced XBP-1 protein was detected in control and s-IBM muscle fibers at a similar level (Fig. 3C); however, the active, higher molecular weight spliced XBP-1 was prominently increased (5.8-fold, p < 0.05) in s-IBM muscle biopsies, whereas in controls, only a weak band was present (Fig. 3C,C′). Those results corresponded with a significant increase of spliced XBP-1 mRNA (7.8-fold, p < 0.01) in s-IBM muscle (Fig. 3G). Because activation of ATF6, ATF4 and XBP-1 should lead to upregulation of the UPR-responsive genes, we studied mRNA of their important target genes, such as glucose regulated protein 78 (GRP78), C/EBP homologous protein (CHOP), and ER degradation-enhancing α-mannosidase-like protein (EDEM) (4, 42). In s-IBM muscle fibers, as compared to controls, all studied mRNAs were increased as follows: GRP78 mRNA 6.3-fold (p < 0.001); CHOP mRNA 3.3-fold (p < 0.01); and EDEM mRNA 6.6-fold (p < 0.001) (Fig. 3G). These results corroborate our results demonstrating activation of ATF4, ATF6 and XBP1 in s-IBM. In addition, in s-IBM muscle fibers, GRP78 protein was increased 8-fold (p < 0.01) (Fig. 3H,H′), corresponding to the increase of its mRNA. Those results provide a strong evidence of the activation of the UPR signaling cascade in s-IBM.
Figure 3.

Comparison of the unfolded protein response (UPR) induction in sporadic inclusion-body myositis (s-IBM) and hereditary IBM due to the GNE mutation (GNE-h-IBM) muscle. (A–F) Representative immunoblots and densitometric analyses of the blots of muscle homogenates of normal-control [C], s-IBM and h-IBM muscle biopsies. The actin band indicates protein loading in each sample. Data are indicated as mean ± SE. ATF4 is significantly increased in s-IBM compared to normal controls (controls [C], n = 6, s-IBM, n = 5) (A,A′), but not changed h-IBM (C, n = 4, h-IBM n = 4) (D,D′). There is a substantial increase of full-length ATF6 precursor and of its active shorter isoform in s-IBM (C, n = 4, s-IBM, n = 4) (B,B′); (E,E′) shows an increase of full-length ATF6 and no shorter isoform present in h-IBM (C, n = 4, h-IBM, n = 4). The spliced isoform of XBP-1 is significantly increased in s-IBM (C, n = 6, s-IBM, n = 10) (C,C′), but not changed in h-IBM (C, n = 6, h-IBM, n = 6)( F,F′). (G) Real-time PCR shows increase of mRNA of spliced XBP-1, as well as of GRP78, CHOP and EDEM in s-IBM, and no increase in h-IBM (C, n = 6–8, s-IBM, n = 4–6, h-IBM n = 7–8). (H,H′) Representative immunoblot shows a significant increase of GRP78 in s-IBM. (I,I′) There is no increase in h-IBM (n = 6 for all). Details in text. *p < 0.05, **p < 0.01, ***p < 0.001.
Lack of the UPR Induction in h-IBM
In contrast to s-IBM, we did not find any evidence of induction of the UPR in h-IBM muscle. In h-IBM muscle biopsies, ATF4 protein was not changed as compared to controls (Fig. 3D,D′). Although in h-IBM muscle as compared to controls p90ATF6 was increased 2-fold (p < 0.05), its active form p50ATF6 was not detectable in either h-IBM or control muscles (Fig. 3E,E′). (Interestingly, although p90ATF6 is a N-glycosylated protein (43), in h-IBM it migrated at the same molecular weight level as in controls, suggesting that its glycosylation was normal.) The amount of protein of both XBP-1 isoforms in h-IBM samples was comparable to controls (Fig. 3F,F′), and the mRNAs of the spliced XBP-1, GRP78, CHOP or EDEM were not changed in h-IBM as compared to controls (Fig. 3G). Consequently, GRP78 protein was not changed in h-IBM (Fig. 3I,I′). Therefore, it appears that in contrast to s-IBM, in h-IBM muscle biopsies UPR activation does not occur.
h-IBM CHMFs Respond to Experimental ER-Stress Induction
To evaluate whether the lack of UPR induction observed in the h-IBM muscle biopsies was caused by their inability to respond to the ER-stress, possibly related to their GNE mutation, we compared the response of CHMFs grown from h-IBM vs. control satellite cells to 2 experimental ER-stress inducers. In both h-IBM and control cultures, exposure to tunicamycin or thapsigargin resulted in comparable magnitudes of UPR induction, as evidenced by real-time PCR showing: a) significant increase of spliced XBP-1 mRNA, in normal CHMFs treated with tunicamycin 4-fold, and with thapsigargin 6-fold (p < 0.05 and p < 0.01 respectively), and in h-IBM CHMFs respectively 3-fold, and 7-fold ( p < 0.05); b) significantly increased GRP78 mRNA, in normal CHMFs treated with tunicamycin 24-fold (p < 0.05), and with thapsigargin 35-fold (p < 0.01), and in h-IBM CHMFs respectively 27-fold and 68-fold (p < 0.05 and p < 0.01 respectively); c) increased CHOP mRNA, in normal CHMFs treated with tunicamycin 16-fold, and with thapsigargin 21-fold (both p < 0.05), and in h-IBM CHMFs respectively 9-fold and 20-fold (p < 0.05); and d) increased EDEM mRNA, in normal CHMFs treated with tunicamycin 5-fold, and with thapsigargin 10-fold (p < 0.05), and in h-IBM CHMFs respectively 5-fold and 16-fold (p < 0.05) (Fig. 4A, B). In addition, by immunoblots, the spliced XBP-1 was significantly increased in normal and h-IBM CHMFs exposed to ER-stress inducers (Fig. 4C, D). Exposure to tunicamycin caused 2.5-fold XBP-1 and to thapsigargin 7.2-fold increase in control CHMFs (p < 0.01)(Fig. 4C) and in h-IBM CHMFs respectively 3.7-fold (p < 0.05) and 7.3-fold (p < 0.001) increase (Fig. 4D). Similarly, GRP78 protein was substantially increased in normal CHMFs exposed to tunicamycin 2.8-fold (p < 0.05) and to thapsigargin 3.8-fold (p < 0.01) (Fig. 4E), and in h-IBM CHMFs exposed to tunicamycin 2.9-fold (p < 0.05) and thapsigargin 2.6-fold increase (Fig. 4F) (possibly because of the small number of samples, no significant p value was obtained with the ANOVA test, despite an obvious tendency toward the increased value of GRP78, but a significant p value [p < 0.05] was obtained with Student t-test). These results demonstrate that GNE-h-IBM CHMFs are able to respond to experimentally induced ER-stress.
Figure 4.

Response of normal and hereditary inclusion-body myopathy (h-IBM) cultured human muscle fibers (CHMFs) to experimentally induced endoplasmic reticulum (ER)-stress. (A, B) Real-time PCR shows significant increase of mRNA of spliced XBP-1, GRP78, CHOP and EDEM in normal (A) and h-IBM CHMFs (B) due to experimental induction of ER-stress by tunicamycin [Tm] and thapsigargin [Tg], as compared to non-treated control sister cultures [C] (n = 4 for all). (C–F) Representative immunoblots and densitometric analysis of the blots of control [C] (non-treated), tunicamycin [Tm]- and thapsigargin [Tg]-treated normal and h-IBM CHMFs. The actin band indicates protein loading in each sample. Spliced XBP-1 is significantly increased by ER-stress in normal (C) (n= 4) and in h-IBM (n=4) (D) CHMFs. GRP78 is significantly increased by ER-stress in normal (n=5) (E), and it is significantly increased in Tm-treated in h-IBM CHMFs (n=3) (F). (In Tg-treated h-IBM CHMFs, there is a substantial increase of GRP78, but probably because of the small number of samples, a significant p value was not achieved with the ANOVA test). Data are indicated as mean ± SE. Details in text. *p < 0.05, **p < 0.01.
DISCUSSION
Because of very similar pathological findings in s-IBM and GNE-h-IBM muscle fibers, including intra-myofiber accumulations of the same proteins (e.g. Aβ and phosphorylated tau), we have previously proposed that similar downstream pathogenic mechanism might participate in both disorders (15). Most recently, intra-myofiber p62 has also been shown accumulated in both disorders (27, 44). Accordingly, we decided to compare the expression of the UPR-involved transcription factors in both s-IBM and GNE-h-IBM. Here we show for the first time that the 3 main UPR transcription factors that are activated in response to ER-stress are induced in s-IBM muscle biopsies, as evidenced by a) increased ATF4 protein level and increased mRNA of its target gene CHOP, b) cleavage of the ATF6 accompanied by increased mRNA of its target GRP78, and c) the increase of the spliced form of XBP-1 mRNA and protein, followed by an increase of mRNA of EDEM (EDEM is induced by heterodimer of cleaved p50ATF6 and spliced XBP-1 [45]). In contrast, we did not find similar evidence of the UPR induction in GNE-h-IBM muscle biopsies.
Lack of the UPR induction in our GNE-h-IBM muscle biopsies, suggesting lack of ER-stress, was unexpected. GNE mutation per se does not seem responsible, since in GNE-h-IBM cultured muscle fibers UPR was robustly induced due the experimentally provoked ER.
Disturbances in protein glycosylation are known to induce ER-stress and to activate the UPR, and tunicamycin, an N-glycosylation inhibitor, is widely used experimentally to induce ER-stress (36). The genetic defect in GNE impairing synthesis of sialic acid, an important component of glycoconjugates, should theoretically influence the glycosylation status of proteins, which, in turn, should cause ER-stress and activate UPR. However, even though activity of the mutated GNE has been reported by others to be reduced, neither unified pattern of protein hyposialylation, nor correlation of diminished GNE activity with a clinical phenotype has been found, suggesting that impairment of protein sialylation might not be a direct/main cause of the muscle fiber degeneration (17–19).
Our results differ from those of Li et al who reported upregulation of the ER chaperones in patients with GNE-h-IBM (46). The discrepancies may be caused by a very limited number of control patients used for some analyses by Li et al, and the fact that their patients had a different genetic background and different GNE mutations from the patients in our studies. Our group of patients is very homogenous, i.e. they all carry a homozygous mutation in the kinase domain of GNE, whereas Li et al had a more diverse group of patients (46). It is known that different GNE mutations cause different clinical phenotypes (21, 47). Accordingly, it is conceivable that different mutations in the GNE gene may cause different intracellular consequences, considering that additional functions beyond its enzymatic involvement in sialic acid synthesis have been proposed (48).
Whether lack of UPR activation in muscle biopsies of GNE-h-IBM bearing mutations in the kinase domain of GNE aggravates the disease is currently not known. In gastrocnemius muscle of GneM712T/M712T knock-in mice, mRNA of the markers of UPR activation GRP78, CHOP and spliced XBP-1 was increased (49). Because the mice did not display any pathological muscle phenotype (in contrast to humans bearing the same GNE mutation), the phenomenon has been postulated to be related to a protective role of the ER-stress-induced UPR in muscle; it is possibly relevant that in severely affected kidneys of those mice, UPR activation has not been detected (49).
The upregulation of the UPR markers in the GneM712T/M712T knock-in mice (49), and in our experiment involving pharmacological induction of the ER-stress in our GNE-h-IBM CHMFs, confirm that myofibers with a mutated kinase domain of GNE are able to respond to the ER-stress insult; thus, those mutations do not impair UPR signaling. Accordingly, in the muscle biopsies of patients with those mutations there might be an absence of the ER-stress providing the signal for the UPR induction, or sensing of it might be diminished or insufficient. We previously postulated that ER-stress is an important part of the s-IBM pathogenic cascade, that it occurs in response to the accumulation of numerous abnormally unfolded or misfolded proteins, and that the UPR is an attempt to facilitate proper folding of malfolded proteins and/or their disposal (6). Although in h-IBM, muscle fibers many of the same proteins accumulate as in s-IBM muscle fibers (15), some differences have also been described, including the paucity of β-pleated-sheet amyloid (23), as indicated by very rare congophilic inclusions (Fig. 2E–H). Thus, the presumed lack of an ER-stress signal in h-IBM as compared to s-IBM might be related to: a) different conformation of the accumulated proteins, e.g. h-IBM paired helical filaments lack some immunoreactive epitopes of phosphorylated tau (24); b) different composition of the inclusions, i.e. some of the proteins accumulated in s-IBM in the form of aggregates are diffusely stained in h-IBM (50); c) the amounts of accumulated misfolded proteins are less than in s-IBM; and d) there may possibly be more efficient proteasome-ubiquitin and authophagic-lysosomal pathways of protein degradation in h-IBM vs. s-IBM (8).
Because the role of ER-stress in inducing inflammation is well established (3), it is possible that the lack of UPR activation could be responsible for the lack of inflammatory response in h-IBM. NF-κB, a pro-inflammatory transcription factor known to be induced by ER-stress (3), including in CHMFs with experimentally induced ER-stress (51), and whose activation and acetylation are increased in s-IBM muscle fibers (51), may play a central role in this process. Interestingly, no hyperacetylation of NF-κB was detected in h-IBM muscle fibers as compared to controls (data not shown). On the other hand, it is also possible that the lack of inflammation in h-IBM muscle biopsies might repress UPR activation; for example, in a mouse model in vivo and in vitro, MHC I overexpression induced aspects of the UPR (52).
In summary, our results demonstrate a coordinated activation of three branches of the UPR in s-IBM muscle fibers. The lack of UPR, implying no ER-stress, in GNE-h-IBM suggests that different intracellular mechanisms might be associated with very similar pathological phenotypes.
Acknowledgments
Supported by NIH (AG16768 Merit Award), MDA (all to Valerie Askanas), and the Helen Lewis Research Fund.
Anna Nogalska was on leave from Department of Biochemistry, Medical University of Gdansk, Gdansk, Poland. We are grateful to Dr. Michael Jakowec from the USC Department of Neurology for allowing us to use his real-time PCR equipment.
Footnotes
The authors declare no conflict of interest.
References
- 1.Roussel BD, Kruppa AJ, Miranda E, et al. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105–18. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- 2.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–4. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 3.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 5.Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci. 2013;70:2425–41. doi: 10.1007/s00018-012-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vattemi G, Engel WK, McFerrin J, et al. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nogalska A, Engel WK, McFerrin J, et al. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem. 2006;96:1491–9. doi: 10.1111/j.1471-4159.2006.03668.x. [DOI] [PubMed] [Google Scholar]
- 8.Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol. 2012;71:680–93. doi: 10.1097/NEN.0b013e31826183c8. [DOI] [PubMed] [Google Scholar]
- 9.Askanas V, Engel WK. Sporadic inclusion-body myositis: Conformational multifactorial ageing-related degenerative muscle disease associated with proteasomal and lysosomal inhibition, endoplasmic reticulum stress, and accumulation of amyloid-beta42 oligomers and phosphorylated tau. Presse Med. 2011;40:e219–35. doi: 10.1016/j.lpm.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 10.Engel WK, Askanas V. Inclusion-body myositis: clinical, diagnostic, and pathologic aspects. Neurology. 2006;66:S20–29. doi: 10.1212/01.wnl.0000192260.33106.bb. [DOI] [PubMed] [Google Scholar]
- 11.Dimachkie MM, Barohn RJ. Inclusion body myositis. Curr Neurol Neurosci Rep. 2013;13:321. doi: 10.1007/s11910-012-0321-4. [DOI] [PubMed] [Google Scholar]
- 12.Dalakas MC. Review: An update on inflammatory and autoimmune myopathies. Neuropathol Appl Neurobiol. 2011;37:226–42. doi: 10.1111/j.1365-2990.2010.01153.x. [DOI] [PubMed] [Google Scholar]
- 13.Fratta P, Engel WK, McFerrin J, et al. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in AbPP-overexpressing cultured human muscle fibers. Am J Pathol. 2005;167:517–26. doi: 10.1016/s0002-9440(10)62994-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nogalska A, D’Agostino C, Terracciano C, et al. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol. 2010;177:1377–87. doi: 10.2353/ajpath.2010.100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Askanas V, Engel WK. Inclusion-body myositis and myopathies: different etiologies, possibly similar pathogenic mechanisms. Curr Opin Neurol. 2002;15:525–31. doi: 10.1097/00019052-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Askanas V, Engel WK. New advances in inclusion-body myositis. Curr Opin Rheumatol. 1993;5:732–41. doi: 10.1097/00002281-199305060-00007. [DOI] [PubMed] [Google Scholar]
- 17.Argov Z, Mitrani-Rosenbaum S. The hereditary inclusion body myopathy enigma and its future therapy. Neurotherapeutics. 2008;5:633–7. doi: 10.1016/j.nurt.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broccolini A, Gidaro T, Morosetti R, et al. Hereditary inclusion-body myopathy with sparing of the quadriceps: the many tiles of an incomplete puzzle. Acta Myol. 2011;30:91–5. [PMC free article] [PubMed] [Google Scholar]
- 19.Malicdan MC, Noguchi S, Nishino I. Recent advances in distal myopathy with rimmed vacuoles (DMRV) or hIBM: treatment perspectives. Curr Opin Neurol. 2008;21:596–600. doi: 10.1097/WCO.0b013e32830dd595. [DOI] [PubMed] [Google Scholar]
- 20.Huizing M, Krasnewich DM. Hereditary inclusion body myopathy: a decade of progress. Biochim Biophys Acta. 2009;1792:881–7. doi: 10.1016/j.bbadis.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broccolini A, Mirabella M. Hereditary inclusion-body myopathies. Biochim Biophys Acta - Mol Basis Dis. 2014 doi: 10.1016/j.bbadis.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Askanas V, Engel WK. Hereditary inclusion body myopathies. In: Rosenberg RN, Di Mauro S, Paulson HL, Ptácek L, Nestler EJ, editors. The molecular and genetic basis of neurologic and psychiatric disease. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. pp. 524–31. [Google Scholar]
- 23.Askanas V, Engel WK. Hereditary inclusion-body myopathies. In: Rosenberg R, Di Mauro S, Paulson HL, Ptácek L, editors. The Molecular and Genetic Basis of Neurologic and Psychiatric Disease. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 24.Mirabella M, Alvarez RB, Bilak M, et al. Difference in expression of phosphorylated tau epitopes between sporadic inclusion-body myositis and hereditary inclusion-body myopathies. J Neuropathol Exp Neurol. 1996;55:774–86. doi: 10.1097/00005072-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Dalakas MC. Interplay between inflammation and degeneration: using inclusion body myositis to study “neuroinflammation”. Ann Neurol. 2008;64:1–3. doi: 10.1002/ana.21452. [DOI] [PubMed] [Google Scholar]
- 26.Askanas V, Engel WK. Does overexpression of betaAPP in aging muscle have a pathogenic role and a relevance to Alzheimer’s disease? Clues from inclusion body myositis, cultured human muscle, and transgenic mice. Am J Pathol. 1998;153:1673–7. doi: 10.1016/s0002-9440(10)65680-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Askanas V, Engel WK, Nogalska A. Pathogenesis of sporadic inclusion-body myositis; role of ageing and muscle-fibre degeneration, and accumulation of the same proteins as in Alzheimer and Parkinson brains. In: Askanas V, Engel WK, editors. Muscle Ageing, Inclusion-Body Myositis and Myopathies. Oxford, UK: Wiley-Blackwell; 2012. pp. 111–45. [Google Scholar]
- 28.McFerrin J, Engel WK, Askanas V. Impaired innervation of cultured human muscle overexpressing betaAPP experimentally and genetically: relevance to inclusion-body myopathies. Neuroreport. 1998;9:3201–5. doi: 10.1097/00001756-199810050-00013. [DOI] [PubMed] [Google Scholar]
- 29.Malicdan MC, Noguchi S, Nonaka I, et al. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. 2007;16:2669–82. doi: 10.1093/hmg/ddm220. [DOI] [PubMed] [Google Scholar]
- 30.Askanas V, Engel WK, Alvarez RB. Enhanced detection of congo-red-positive amyloid deposits in muscle fibers of inclusion body myositis and brain of Alzheimer’s disease using fluorescence technique. Neurology. 1993;43:1265–7. doi: 10.1212/wnl.43.6.1265-a. [DOI] [PubMed] [Google Scholar]
- 31.Ricci E, Broccolini A, Gidaro T, et al. NCAM is hyposialylated in hereditary inclusion body myopathy due to GNE mutations. Neurology. 2006;66:755–8. doi: 10.1212/01.wnl.0000200956.76449.3f. [DOI] [PubMed] [Google Scholar]
- 32.Broccolini A, Gidaro T, Tasca G, et al. Analysis of NCAM helps identify unusual phenotypes of hereditary inclusion-body myopathy. Neurology. 2010;75:265–72. doi: 10.1212/WNL.0b013e3181e8e8f1. [DOI] [PubMed] [Google Scholar]
- 33.Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–7. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 34.D’Agostino C, Nogalska A, Cacciottolo M, et al. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol. 2011;122:627–36. doi: 10.1007/s00401-011-0874-3. [DOI] [PubMed] [Google Scholar]
- 35.Haze K, Yoshida H, Yanagi H, et al. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Back SH, Schrèoder M, Lee K, et al. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods. 2005;35:395–416. doi: 10.1016/j.ymeth.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Askanas V, Engel WK. A new program for investigating adult human skeletal muscle grown aneurally in tissue culture. Neurology. 1975;25:58–67. doi: 10.1212/wnl.25.1.58. [DOI] [PubMed] [Google Scholar]
- 38.Desnuelle C, Askanas V, Engel WK. Insulin enhances development of functional voltage-dependent Ca2+ channels in aneurally cultured human muscle. J of Neurochem. 1987;49:133–8. doi: 10.1111/j.1471-4159.1987.tb10003.x. [DOI] [PubMed] [Google Scholar]
- 39.Pegolo G, Askanas V, Engel WK. Expression of muscle-specific isozymes of phosphorylase and creatine kinase in human muscle fibers cultured aneurally in serum-free, hormonally/chemically enriched medium. Int J Devl Neuroscience. 1990;8:299–308. doi: 10.1016/0736-5748(90)90036-2. [DOI] [PubMed] [Google Scholar]
- 40.Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–81. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 41.Nogalska A, Wojcik S, Engel WK, et al. Endoplasmic reticulum stress induces myostatin precursor protein and NF-kappaB in cultured human muscle fibers: relevance to inclusion body myositis. Exp Neurol. 2007;204:610–8. doi: 10.1016/j.expneurol.2006.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong M, Luo S, Baumeister P, et al. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J Biol Chem. 2004;279:11354–63. doi: 10.1074/jbc.M309804200. [DOI] [PubMed] [Google Scholar]
- 44.Nogalska A, Terracciano C, D’Agostino C, et al. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009;118:407–13. doi: 10.1007/s00401-009-0564-6. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell. 2007;13:365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 46.Li H, Chen Q, Liu F, et al. Unfolded protein response and activated degradative pathways regulation in GNE myopathy. PLoS ONE. 2013;8:e58116. doi: 10.1371/journal.pone.0058116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori-Yoshimura M, Monma K, Suzuki N, et al. Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. J Neurol Sci. 2012;318:100–105. doi: 10.1016/j.jns.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Sun Z, Li AV, et al. Roles for UDP-GlcNAc 2-epimerase/ManNAc 6-kinase outside of sialic acid biosynthesis: modulation of sialyltransferase and BiP expression, GM3 and GD3 biosynthesis, proliferation, and apoptosis, and ERK1/2 phosphorylation. J Biol Chem. 2006;281:27016–28. doi: 10.1074/jbc.M604903200. [DOI] [PubMed] [Google Scholar]
- 49.Sela I, Yakovlev L, Becker Cohen M, et al. Variable phenotypes of knockin mice carrying the M712T Gne mutation. Neuromolecular Med. 2013;15:180–91. doi: 10.1007/s12017-012-8209-7. [DOI] [PubMed] [Google Scholar]
- 50.Askanas V, Engel WK. Newest approaches to diagnosis and pathogenesis of sporadic inclusion-body myositis and hereditary inclusion-body myopathies, including molecular-pathologic similarities to Alzheimer disease. In: Askanas V, Serratrice G, Engel WK, editors. Inclusion-Body Myositis and Myopathies. Cambridge, UK: University Press; 1998. pp. 3–78. [Google Scholar]
- 51.Nogalska A, D’Agostino C, Engel WK, et al. Decreased SIRT1 deacetylase activity in sporadic inclusion-body myositis muscle fibers. Neurobiol Aging. 2010;31:1637–48. doi: 10.1016/j.neurobiolaging.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 52.Nagaraju K1, Casciola-Rosen L, Lundberg I, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–35. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]