

Abstract
It has been previously shown that bilirubin prevents the up-regulation of inducible nitric oxide synthase (iNOS) in response to LPS. The present study examines whether this effect is exerted through modulation of Toll-Like Receptor-4 (TLR4) signaling. LPS-stimulated iNOS and NADPH oxidase (Nox) activity in RAW 264.7 murine macrophages was assessed by measuring cellular nitrate and superoxide () production, respectively. The generation of both nitrate and in response to LPS was suppressed by TLR4 inhibitors, indicating that activation of iNOS and Nox is TLR4-dependent. While treatment with superoxide dismutase (SOD) and bilirubin effectively abolished LPS-mediated production, hydrogen peroxide and nitrate release were inhibited by bilirubin and PEG-catalase, but not SOD, supporting that iNOS activation is primarily dependent upon intracellular H2O2. LPS treatment increased nuclear translocation of the redox-sensitive transcription factor Hypoxia Inducible Factor-1α (HIF-1α), an effect that was abolished by bilirubin. Cells transfected with murine iNOS reporter constructs in which the HIF-1α-specific hypoxia response element was disrupted exhibited a blunted response to LPS, supporting that HIF-1α mediates Nox-dependent iNOS expression. Bilirubin, but not SOD, blocked the cellular production of interferon-β, while interleukin-6 production remained unaffected. These data support that bilirubin inhibits the TLR4-mediated up-regulation of iNOS by preventing activation of HIF-1α through scavenging of Nox-derived reactive oxygen species. Bilirubin also suppresses interferon-β release via a ROS-independent mechanism. These findings characterize potential mechanisms for the anti-inflammatory effects of bilirubin.
Keywords: Aryl hydrocarbon receptor (AhR), Bilirubin, Hypoxia-inducible factor (HIF), NADPH oxidase, Nitric oxide synthase, Toll-like receptor 4 (TLR4), Superoxide
Abbreviations: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; Cyp1A1, cytochrome P450, family 1, subfamily A, polypeptide 1; HE, hydroethidine; HIF-1α, hypoxia inducible factor-1α; iNOS, inducible nitric oxide synthase; IL-6, interleukin-6; Inf-β, interferon-β; IRF3, interferon regulatory factor 3; ISRE, interferon-sensitive response element; MyD88, myeloid differentiation factor-88; Nox, NADPH oxidase; PMA, phorbol myristate acetate; ROS, reactive oxygen species; SOD, superoxide dismutase; TEMPOL, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; TLR4, toll-like receptor-4; TNFα, tumor necrosis factor-α
Graphical abstract
Highlights
-
●
Bilirubin blocks TLR4 signaling by scavenging NADPH oxidase-derived reactive oxygen species.
-
●
Bilirubin specifically inhibits the TRIF-dependent TLR4 signaling pathway.
-
●
LPS activation of inducible nitric oxide synthase is mediated by HIF-1α.
-
●
Macrophages exhibit reciprocal regulation of HIF-1α and aryl hydrocarbon receptor pathways.
-
●
Potential mechanisms underlying the anti-inflammatory effects of bilirubin are delineated.
1. Introduction
Expression of the inducible isoform of nitric oxide synthase (iNOS), which generates nitric oxide (NO) from the catalyzed conversion of l-arginine to l-citrulline, is triggered by the binding of LPS to Toll-Like Receptor 4 (TLR4) [1,2]. In rodent models of sepsis involving the intravenous administration of LPS, selective iNOS inhibitors prevent cardiovascular collapse and abrogate liver, lung, renal, and gastrointestinal injury [3], supporting that NO mediates many of the harmful consequences of endotoxemia [4]. We [5] and others [6] have demonstrated that the administration of bilirubin, an endogenous product of heme catabolism, to LPS-treated rats ameliorates tissue injury, reduces serum nitrate concentrations, and attenuates the expression of iNOS message in the liver. Physiologically relevant concentrations of bilirubin (≤50 µM≈3 mg/dL) also have been shown to suppress LPS-stimulated iNOS up-regulation and nitrate production by murine macrophages in vitro [5,6]. However, the mechanism(s) by which bilirubin exerts these effects remains poorly delineated.
LPS binding to TLR4 triggers two distinct intracellular signaling pathways [7]. The adaptor protein myeloid differentiation factor (MyD88)-dependent pathway culminates in the early activation of MAPK and nuclear factor-κB (NF-κB), leading to increased expression of inflammatory cytokines, such as tumor necrosis factor-α (TNFα) and interleukin-6 (IL-6) [8]. The MyD88-independent Toll/IL-1 receptor (TIR) domain-containing adaptor (TRIF)-dependent pathway is characterized by the late-phase activation of NF-κB, up-regulation of iNOS and interferon-β (Inf-β), and downstream modulation of numerous interferon-inducible genes [7,9]. In previous studies, bilirubin has not been found to influence the early activation of NF-κB or MAPK in response to LPS [5,6], suggesting that it does not exert an effect on the MyD88-dependent TLR4 pathway. On the other hand, the ability of bilirubin to modulate TRIF-dependent TLR4 signaling has not previously been investigated.
It is postulated that NADPH oxidases (Nox), which generate superoxide anion () from molecular oxygen, play an important role in LPS-stimulated iNOS up-regulation. Support for this conclusion is derived from the observation that the increased expression of iNOS in response to LPS is abolished by Nox inhibitors, such as apocynin [6,10,11]. It has further been shown that treatment of macrophages with LPS stimulates the cellular production of reactive oxygen species (ROS) [12,13], a process that also is efficiently abrogated by Nox inhibitors [6,14,15]. Specific evidence that superoxide mediates TLR4 signaling is provided by the demonstrated co-localization of TLR4 with the Nox4 isoform, and by the fact that knockdown of Nox4 prevents both LPS-induced ROS generation and NF-κB activation [16]. It is notable that bilirubin is able to efficiently scavenge Nox-derived ROS [6,17], likely due to its potent antioxidant properties [18] and its ability to undergo redox cycling within the cell [19]. To assess whether bilirubin suppresses LPS-stimulated up-regulation of iNOS by inhibiting Nox activity, we investigated the effect of bilirubin on TLR4-mediated iNOS expression in RAW 264.7 murine macrophages. Our findings indicate that bilirubin disrupts TRIF-dependent TLR4 signaling and modulates iNOS expression by scavenging NADPH oxidase-derived superoxide, an effect that is mediated by the hypoxia-inducible transcription factor-1α (HIF-1α).
2. Materials and methods
2.1. Materials
Bilirubin (bilirubin IXα) was obtained from Porphyrin Products (Logan, UT) and further purified according to the method of McDonagh and Assisi [20]. Hydroethidine (dihydroethidium) and Accutase were purchased from Life Technologies. Apocynin was obtained from Cayman Chemical. Superoxide dismutase (SOD; bovine erythrocyte, BioUltra, ≥97% SDS-PAGE), catalase (bovine liver), catalase-polyethylene glycol (PEG-catalase), (±)-α-tocopherol (synthetic, ≥96% HPLC), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and the TLR-4 inhibitors cinnamaldehyde, mastoparan, and resveratrol were purchased from Sigma-Aldrich.
2.2. Cell culture
RAW 264.7 murine macrophages and MOVAS, an SV-40 immortalized murine aortic smooth muscle cell line, were obtained from ATCC (Rockville, MD). Cells were grown in Dulbecco’s Modified Eagle Medium containing 4 mM l-glutamine, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, and 10% FBS at 37 °C in 5% CO2. Bilirubin stock solutions were prepared freshly in 50 mM potassium phosphate (pH 12), as described previously [5]. The addition of an aliquot (≤1% v:v) of bilirubin solubilized in this manner had no appreciable effect on the pH of the medium, and neither bilirubin nor the potassium phosphate vehicle altered cell viability.
2.3. Assays for cellular nitrate and cytokine production
RAW cells (~2×106) were seeded and grown in 12-well plates containing 1 mL media. Nitrate accumulation in the cell supernatant was determined 24 h following the addition of the indicated treatments using a Nitrate/Nitrite Colorimetric Assay kit (Cayman Chemical, Ann Arbor, MI), as previously described [5]. Briefly, nitrite is produced from the catalyzed conversion of nitrate and then quantified colorimetrically using Griess reagent, based on a standard curve. IL-6, Inf-β, and TNFα levels in the cell culture medium were quantified using commercially available mouse ELISA kits (Thermo Scientific, Rockford, IL). Data are normalized to cellular protein, as determined by Bio-Rad protein assay, which trivially impacted results.
2.4. Measurement of cellular superoxide production
Cells were grown on 6 well-plates, lifted with Accutase, washed, and suspended in 5 mL of phenol red-free RPMI at a concentration of 2×106 cells/mL. Cell suspensions were loaded with 10 µM hydroethidine (HE) at 37 °C for 5 min and then maintained on ice until 5 min prior to the initiation of the experiments (when the indicated treatments are added), which were performed at 37 °C. Fetal bovine serum (10%) was added to the cell suspension in order to maintain bilirubin solubility. Cells were activated by the addition of 16 µM phorbol myristate acetate (PMA) or 1 µg/mL LPS, along with various inhibitor compounds, as described. Superoxide production was quantified by the time-dependent increase in mean fluorescence intensity (ex: 488 nm, em: 610 nm long pass filter) of the cell population, as measured by flow cytometry.
2.5. Determination of cellular hydrogen peroxide production
Cellular hydrogen peroxide was quantified using an Amplex Red assay kit (Molecular Probes), according to the manufacturer’s instructions. Briefly, RAW 264.7 cells were seeded in 96-well plates (~30,000 cells per well) and grown in phenol red-free RPMI. Cells were activated by the addition of PMA or LPS, along with 50 µM Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) and 0.1 U/mL HRP at a total volume of 100 µL. Hydrogen peroxide production was quantified from the time-dependent changes in absorbance at 560 nm, using a standard curve.
2.6. Quantitative PCR analysis
Total cellular RNA was extracted and RT-PCR was performed using an Mx3000P system (Stratagene, Cedar Creek, TX). Primers for murine iNOS (sense: 5′-acatgcagaatgagtaccgg-3′; antisense: 5′-tcaacatctcctggtggaac-3′), cytochrome P450, family 1, subfamily A, polypeptide 1 (Cyp1A1; sense: 5′-cgggatccttacagcccaagcagc-3′, 5′-ggggtacccagagcactcttcaggag-3′) and GAPDH (sense: 5′-tcaacagcaactcccactcttcca-3′; antisense: 5′-accctgttgctgtagccgtattca-3′) were employed, with the latter used to control for amplification.
2.7. Determination of HIF-1α activation
HIF-1α DNA binding activity was measured using an ELISA-based HIF-1α Transcription Factor Assay Kit (Cayman Chemical), according to the manufacturer’s instructions. Briefly, 1×107 cells were grown in a 100 mm culture disk and, following the indicated treatments and incubation times, nuclear proteins were isolated using a Cayman Nuclear Extraction Kit. The nuclear extracts were incubated with HIF-1α response elements immobilized on a 96-well plate, and the HIF transcription factor complex detected using an HIF-1α-specific primary antibody and HRP-conjugated secondary antibody.
2.8. Plasmid preparation and luciferase reporter assay
A 1192-bp fragment from the 5′-flanking region of the wild-type (WT) murine iNOS gene (kindly provided by R. Lyons, University of New Mexico Health Science Center, Albuquerque, NM) was inserted into a pGL3-basic vector containing a luciferase reporter (Promega). Plasmid HRE-del, possessing a deletion of the 6-bp hypoxia response element (HRE) sequence (−226 to −221), and HRE-TT, containing a 2-bp exchange (TACGTG to TATTTG) within the HRE of the iNOS promoter, also were created. RAW 264.7 cells were transiently transfected with the indicated reporter constructs using Effectene Transfection Reagent (Qiagen, Inc.). Promoter activity was quantified utilizing a dual Luciferase Reporter Assay System (Promega) and employing a co-transfected Renilla reporter vector to control for transfection efficiency.
2.9. Statistical methods
Data were analyzed using a statistical program (SSI SigmaStat, San Jose, CA). Mean values were compared by ANOVA with t-test to assess for significance. For data that were not normally distributed, a Kruskal–Wallis analysis of variance on ranks was performed.
3. Results
3.1. Role of NADPH oxidases and reactive oxygen species in mediating TLR4 signaling
To evaluate the contribution of NADPH oxidase-derived superoxide to TLR4 signaling, we employed a fluorescent assay using hydroethidine, a redox-sensitive probe with specificity for [21]. RAW 264.7 cells were pre-loaded with HE and superoxide production was quantified by measuring the time-dependent changes in cellular fluorescence intensity using FACS. We first validated the assay using the classical Nox activator phorbol myristate acetate (PMA). As expected, incubation of cells in the presence of PMA induced a sharp increase in HE fluorescence that was markedly attenuated by simultaneous treatment with a maximally effective dose (500 U) of SOD (Fig. 1A), which catalyzes the conversion of superoxide to H2O2 and O2. As shown in Fig. 1B, treatment of RAW 264.7 cells with LPS also increases cellular superoxide generation, albeit at substantially lower levels than PMA. Notably, apocynin, failed to suppress superoxide production by RAW 264.7 cells in response to stimulation with either PMA or LPS (Fig. 1B), indicating that this compound did not inhibit Nox activity. While surprising, these findings are consistent with previous reports that apocynin does not impede the function of NADPH oxidase in cells lacking significant myeloperoxidase activity, which is required for apocynin activation [22,23].
Fig. 1.

Modulation of PMA- and LPS-induced cellular superoxide production. RAW 264.7 cells were preloaded with 10 µM HE, and then activated with PMA (16 µM) or LPS (1 µg/mL). Superoxide production, as reflected in the time-dependent changes in cellular fluorescence intensity (a.u., arbitrary units), was measured using FACS. In panel A, cells were treated with PMA (triangles) or vehicle (circles) in the presence (gray symbols) or absence (dark symbols) of SOD (500 U). Panel B shows the effect of apocynin (1 mM; gray symbols) on cells treated with PMA (triangles), LPS (squares), or vehicle (circles). Panel C compares the influence of 500 U SOD (gray squares) or 50 µM bilirubin (gray inverted triangles) with vehicle (solid squares) on superoxide production by LPS-treated cells, and also shows the effect of 50 µM bilirubin (solid triangles) on vehicle-treated cells (solid circles) in the absence of LPS stimulation. In panel D, the effect of 100 µM cinnamaldehyde (gray symbols) on LPS-treated (squares) and untreated (circles) cells is displayed. Symbols reflect the mean (±S.E.M.) of 5 experiments. *p<0.05 vs. all other curves, **p<0.05 vs. control.
Concordant with our observations in PMA-treated cells, incubation of RAW 264.7 cells with SOD abrogated LPS-stimulated superoxide production (Fig. 1C). Similarly, 50 µM bilirubin was found to efficiently suppress superoxide production in both LPS-activated and in non-stimulated cells (Fig. 1C), in the absence of any alteration in cell viability (data not shown). We speculate that bilirubin appears to be more effective than SOD in this assay system because it also scavenges hydrogen peroxide, which is able to react with HE to a limited extent [21]. Additionally, we show that cinnamaldehyde, which inhibits TLR4 signaling by preventing receptor oligomerization [24], completely suppresses LPS-stimulated production (Fig. 1D), supporting that TLR4 mediates the increase in Nox activity induced by LPS.
Since superoxide undergoes both catalyzed and spontaneous dismutation to form hydrogen peroxide, we further assessed cellular H2O2 by Amplex Red colorimetric assay. In a similar manner to superoxide, both PMA and LPS were observed to induce a time-dependent increase in hydrogen peroxide production by RAW 264.7 cells, with PMA producing a more robust response (Fig. 2A). However, in contrast with its marked inhibitory effect on superoxide production (Fig. 1A and C), SOD did not significantly alter cellular ROS levels (Fig. 2B), most likely because it generates H2O2 from . In support of this proposition, the SOD mimetics [25] Tiron (disodium 4,5-dihydroxy-1,3-benzenedisulfonate) and TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl) also did not alter cellular hydrogen peroxide generation in response to PMA (Fig. 2B). Notably, treatment with the cell permeant polyethylene-glycolated form of catalase (PEG-catalase) [26], an enzyme that degrades hydrogen peroxide to form water and oxygen, significantly reduced PMA-stimulated H2O2 formation, while the non-PEG form of the enzyme had minimal effect, suggesting that PMA-stimulated H2O2 is primarily intracellular. As anticipated, bilirubin, which rapidly and spontaneously traverses cellular membranes [27], also reduced cellular H2O2 production in response to PMA (Fig. 2B) and to LPS (C), providing further support that it acts as a potent scavenger of Nox-derived ROS. In contrast, despite a lack of effect on PMA- or LPS-stimulated superoxide production, apocynin caused a marked reduction in cellular hydrogen peroxide (Fig. 2B and C) supporting, as previously proposed by Heumüller et al. [22], that it acts as a H2O2 scavenger.
Fig. 2.

Modulation of cellular hydrogen peroxide production. RAW 264.7 cells were activated with PMA (16 µM) or LPS (1 µg/mL), and hydrogen peroxide levels determined by Amplex Red assay. Panel A shows the time course of hydrogen peroxide production following treatment with PMA (triangles), LPS (squares), or vehicle (Veh; circles). Symbols reflect the mean (±S.E.M.) of 8 experiments. In panel B, the influence of SOD (500 U), the SOD mimetics Tiron (10 mM) and TEMPOL (Temp; 5 mM), catalase (Cat; 100 U), PEG-catalase (Peg-cat; 100 U), bilirubin (Bili; 50 µM), and apocynin (Apo; 1 mM) on PMA-induced hydrogen peroxide production at 1 h is depicted. Panel C summarizes the effect of SOD, apocynin, and bilirubin on LPS-stimulated RAW 264.7 cells at 1 h. Bars reflect the mean (±S.E.M.) of 5 experiments. *p<0.05 Vs. vehicle; **p<0.05 vs. PMA+vehicle; ***p<0.05 vs. LPS+vehicle.
3.2. Mechanism of LPS-stimulated nitrate production by murine macrophages
When RAW 264.7 macrophages were incubated in the presence of LPS, a marked increase in cellular nitrate production (indicative of iNOS activity) was observed (Fig. 3A). Consistent with previous reports [5,6], this effect was dose-dependently attenuated by simultaneous treatment with bilirubin or apocynin (Fig. 3A). To determine whether the activation of iNOS by LPS is mediated through TLR4, cells were incubated in the presence of the TLR4 inhibitors cinnamaldehyde [24], mastoparan [28], or resveratrol [29]. Each of these agents effectively inhibited cellular nitrate production in response to LPS (Fig. 1B), validating an essential role for TLR4 in mediating LPS-induced iNOS activity [30,31]. Since treatment with LPS previously has been shown to activate NADPH oxidase (Nox) and generate reactive oxygen species in RAW 264.7 macrophages [13,32], we sought to determine whether LPS-stimulated iNOS activation is ROS-dependent. We found that treatment with PEG-catalase, but not SOD, effectively blocked cellular nitrate production (Fig. 3C), supporting that hydrogen peroxide (rather than superoxide) is the primary ROS mediator of LPS-induced iNOS activity. Consistent with these findings, the antioxidant α-tocopherol also was found to exhibit an, albeit modest, dose-dependent inhibitory effect on nitrate release (Fig. 3C). The observation that nitrate production is similar between cells stimulated with 16 µM PMA or 1 µg/mL LPS (Fig. 3C) suggests that iNOS expression is regulated by relatively low-levels (~1 µM) of ROS (Fig. 2C).
Fig. 3.

LPS-stimulated nitrate production by murine macrophages. RAW 264.7 cells were incubated in the presence or absence of 1 µg/mL LPS, and nitrate levels in the medium were assessed at 24 h by colorimetric assay. In panel A, cells were simultaneously incubated for 24 h with bilirubin (Bili; 20, 50 µM), apocynin (Apo; 0.2, 1.0 mM), or vehicle (Veh). Panel B displays the effect of co-incubating cells with the TLR4 inhibitors cinnamaldehyde (Cin; 50, 100 µM), mastoparan (Mas; 20, 50 µM), and resveratrol (Res; 50, 100 µM), or the vehicle. Panel C shows the influence of PEG-catalase (Peg-cat; 100 U), SOD (500 U), α-tocopherol (α-toc; 40, 80 µM), on LPS-stimulated nitrate production and compares the effect with that of PMA (16 µM). Bars reflect the mean (±S.E.M.) of 6 experiments. *p<0.001 vs. all other LPS treatments; **p<0.05 vs. LPS+vehicle.
3.3. Effect of bilirubin on the activation of HIF-1α by LPS
We have validated that LPS stimulates iNOS activity in RAW 264.7 cells through a mechanism that involves TLR4-dependent ROS production. As there exists a growing body of evidence that Nox-derived ROS modulate the activity of hypoxia inducible factor-1α [33] and, as activation of HIF-1α induces iNOS expression through binding to a hypoxia response element (HRE) on the iNOS promoter [34,35], we postulated that bilirubin may inhibit LPS-stimulated iNOS up-regulation by scavenging Nox-generated ROS and thereby prevent activation of HIF-1α. To test this hypothesis, we created a luciferase reporter construct containing a 1.2-kb fragment of the wild-type murine iNOS promoter, as well as two constructs in which the 6-bp HRE was either deleted (HRE-del) or disrupted by a 2-bp (CG to TT) exchange (HRE-TT) (Fig. 4A).
Fig. 4.

Contribution of HIF-1α to LPS-stimulated iNOS expression. Panel A shows schematic representations of the wild-type (WT) murine iNOS luciferase (Lucif) reporter, along with promoter constructs containing a 6-bp deletion (HRE-del) and 2-bp exchange (HRE-TT) of the hypoxia response element (HRE). Panel B depicts the effect of 50 µM bilirubin (Bili) on HIF-1α nuclear binding activity in RAW 264.7 cells incubated in the presence or absence of LPS (1 µg/mL). Bars reflect relative HIF-1α binding (±S.E.M.) with respect to vehicle-treated cells (Veh) at 24 h. Panel C displays the relative luciferase activity of the wild-type murine iNOS reporter construct in RAW 264.7 cells treated with 1 µg/mL LPS in the presence of 50 µM bilirubin or vehicle for 24 h. The results of parallel experiments in which cells were transfected with the HRE-TT or HRE-del constructs are shown in panels D and E, respectively (n=5 experiments). *p<0.05 vs. all other groups.
We first examined the effect of bilirubin on LPS-stimulated HIF-1α nuclear binding activity in RAW 264.7 cells. As shown in Fig. 4B, treatment with bilirubin abolishes the LPS-induced increase in HIF-1α nuclear binding, supporting that bilirubin prevents activation of HIF-1α. Consistent with the known inhibitory effect of bilirubin on iNOS expression [5,6], we found that treatment of transiently transfected RAW 264.7 cells with bilirubin caused a significant reduction in the LPS-stimulated increase in luciferase activity of the wild-type iNOS promoter construct (Fig. 4B). The responses of both the HRE-TT (Fig. 4C) and HRE-del (D) reporter constructs to LPS also were blunted as compared with that of the wild-type construct, confirming a contribution of HIF-1α to TLR4-mediated iNOS up-regulation. Deletion of the HRE also abolished the inhibitory effect of bilirubin on the iNOS promoter response to LPS (Fig. 4D), consistent with the proposition that bilirubin suppresses iNOS up-regulation by preventing HIF-1α activation.
3.4. Role of the aryl hydrocarbon receptor in modulating iNOS expression
HIF-1α binds to the hypoxia response element as a heterodimeric complex with the transcription cofactor aryl hydrocarbon nuclear translocator (ARNT), a constitutively expressed protein that resides within the cell nucleus and mediates several signal transduction pathways. In addition to HIF-1α, ARNT also binds to the aryl hydrocarbon receptor (AhR), which facilitates its association with xenobiotic-responsive elements to mediate biological effects of halogenated aromatic hydrocarbons. A functional interference between the hypoxia (HIF-1α-dependent) and AhR pathways, through competition for ARNT binding, has previously been proposed [36,37]. Since it has been reported that bilirubin serves as an activator ligand for AhR [38,39], we speculated that bilirubin might modulate iNOS expression by activating AhR which, in turn, would compete with HIF-1α for binding to ARNT.
To assess whether activation of AhR could cause reciprocal inhibition of HIF-1α-mediated iNOS expression, we examined the effect of TCDD, a prototypic AhR activator, on LPS-stimulated nitrate production in RAW 264.7 cells. As shown in Fig. 5A, treatment with TCDD suppressed nitrate production in a dose-dependent manner, supporting that activation of AhR can lead to reciprocal inhibition of LPS-mediated (HIF-1α-dependent) iNOS up-regulation. Because of an inability to detect expression of either of the classic AhR inducible genes, Cyp1A1 or Cyp1B1, in RAW 264.7 cells, we utilized murine smooth muscle cells, which previously have been shown to express AhR, Cyp1A1, and iNOS [40,41]. Treatment of MOVAS with LPS was found to up-regulate the expression of iNOS mRNA, while co-incubation with TCDD significantly attenuated this response (Fig. 5B), consistent with the proposed reciprocal inhibition of HIF-1α by AhR. In a parallel manner, the TCDD-stimulated expression of Cyp1A1 was inhibited by simultaneous treatment of MOVAS with LPS (Fig. 5C). On the other hand, bilirubin, even at pathophysiological concentrations (100 µM), exhibited a rather modest ability to induce Cyp1A1 expression in comparison with a half-maximal dose of TCDD (Fig. 5D).
Fig. 5.

Reciprocal effect of LPS and TCDD on the expression of iNOS and Cyp1A1. In panel A, the influence of co-incubating RAW 264.7 cells for 24 h with 1 µg/mL LPS and increasing concentrations of TCDD on nitrate levels in the media is displayed. Bars reflect the mean (±S.E.M.) of 3 experiments. Panel B shows the reciprocal effect of TCDD (50 nM) and LPS (1 µg/mL) on the relative expression of iNOS in MOVAS cells at 24 h, as determined by RT-PCR, while panel C displays the results of parallel studies of Cyp1A1 expression. Panel D compares the relative potency of bilirubin (25–100 µM) to that of TCDD (50 nM) in inducing the expression of Cyp1A1 mRNA in MOVAS. Bars indicate mRNA expression (±S.E.M.) relative to vehicle-treated cells. *p<0.05 vs. all other groups; **p<0.05 vs. LPS+vehicle; ***p<0.05 vs. TCDD.
3.5. Influence of bilirubin on TLR-4 signaling pathways
The binding of LPS to TLR4 activates two principal downstream signaling pathways [7]. The MyD88-dependent pathway leads to activation of NF-κB and the production of pro-inflammatory cytokines such as IL-6 and TNFα, while the TRIF-dependent pathway induces interferon-β through activation of interferon regulatory factor 3 (IRF3). To determine the effect of bilirubin on these two TLR4 pathways, we treated RAW 264.7 cells with LPS and measured the production of IL-6, TNFα, and Inf-β (Fig. 6). Co-incubation of cells in the presence of bilirubin resulted in a marked reduction in Inf-β production (Fig. 6B), in the absence of an effect on IL-6 (A) or TNFα (data not shown), suggesting that bilirubin specifically inhibits TRIF-dependent TLR4 signaling. In contrast, apocynin inhibited both IL-6 and Inf-β production (Fig. 6A and B), supporting a distinct mechanism of action. The induction of IL-6 and Inf-β also was effectively abolished by treatment with cinnamaldehyde (Fig. 6C and D), mastoparan, or resveratrol (data not shown), validating that LPS-induced cytokine production is mediated through TLR4. While treatment of cells with SOD, PEG-catalase, or α-tocopherol did not significantly alter LPS-stimulated cytokine release (Fig. 6E and F), the combination of SOD plus PEG-catalase was found to exert a modest inhibitory effect on Inf-β production. These data suggest a limited role for ROS in modulating the TLR4-dependent induction of cytokines.
Fig. 6.

Effect of bilirubin, TLR4 inhibitors, and ROS scavengers on LPS-stimulated IL-6 and Inf-β production. RAW 264.7 cells were incubated in the presence or absence of 1 µg/mL LPS and the indicated inhibitors for 24 h, after which time the concentrations of IL-6 (left panels) and Inf-β (right panels) in the media were determined by ELISA. Panels A and B show the effect of 50 µM bilirubin (Bili), 1 mM apocynin (Apo), or vehicle (Veh). Panels C and D demonstrate the dose-dependent effects of cinnamaldehyde (Cin). In panels E and F, the effect of PEG-catalase (Peg cat; 100 U), SOD (500 U), the combination of SOD and PEG-catalase, and α-tocopherol (toc; 80 µM) are shown. Bars reflect the mean (±S.E.M.) of 6 experiments. *p<0.05 vs. LPS+vehicle.
4. Conclusions
It was first speculated over 75 years ago that bilirubin exerts anti-inflammatory effects when it was observed that patients with rheumatoid arthritis experienced a remission of symptoms after developing jaundice from superimposed liver disease [42]. More recently, bilirubin has been shown to prevent tissue injury by inhibiting the up-regulation of iNOS by LPS [5,6]. In the present studies, we attempt to elucidate the molecular mechanism(s) underlying this effect. Employing RAW 264.7 murine macrophages, we validated that physiological concentrations of bilirubin significantly inhibit iNOS expression and cellular nitrate production in response to LPS treatment. Our finding that TLR4 receptor antagonists effectively abrogate nitrate production (Fig. 3B), are consistent with prior reports that cells [30] and animals [31] deficient in TLR4 fail to up-regulate iNOS in response to LPS. We further show that TLR4 inhibitors suppress LPS-mediated superoxide production (Fig. 1D), also in line with previous investigations demonstrating that TLR4 interacts directly with NADPH oxidase [16] and that TLR4 activation causes an increase in intracellular ROS [43,44]. We found that both PEG-catalase and α-tocopherol, but not SOD, suppress LPS-stimulated nitrate production (Fig. 3C), suggesting that hydrogen peroxide (rather than superoxide) serves as the primary ROS signaling intermediate involved in the regulation TLR4-mediated iNOS expression. Finally, we demonstrate that bilirubin, a potent chain-breaking antioxidant that undergoes intracellular redox cycling [18,19], is an efficient scavenger of LPS-induced and H2O2 (Figs. 1C and 2C). Taken together, these data support that bilirubin suppresses TLR4-mediated up-regulation of iNOS by scavenging Nox-derived reactive oxygen species.
As LPS activates HIF-1α through an ROS-dependent mechanism [45] and, since the iNOS promoter contains a hypoxia responsive element [34,35], we speculated that bilirubin inhibits the LPS-stimulated induction of iNOS by suppressing ROS production, and thereby preventing HIF-1α activation. This hypothesis is supported by our finding that bilirubin blocks LPS-stimulated nuclear binding of HIF-1α and that the induction of iNOS by LPS is significantly blunted in cells transfected with promoter constructs where the hypoxia response element has been modified or deleted (Fig. 4D and E). The observation that bilirubin exerts no additional effect on iNOS expression beyond that which occurs when the HRE is deleted from the promoter, further supports that HIF-1α is the predominant mechanism through which bilirubin acts to suppress LPS-mediated iNOS induction.
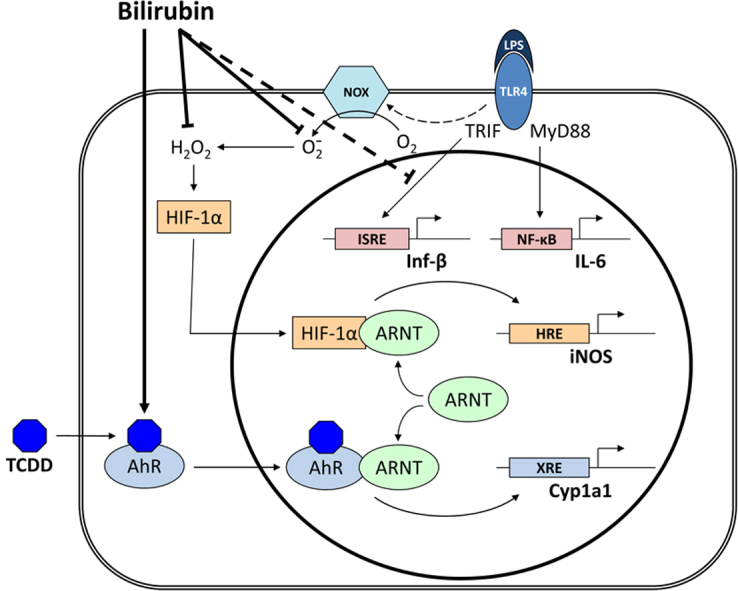
Because bilirubin has been postulated to serve as an endogenous ligand for the aryl hydrocarbon receptor [38,39], we speculated that it may also regulate iNOS expression by activating AhR, which could compete with HIF-1α for ARNT binding. While we were able to demonstrate that the simultaneous activation of HIF-1α with LPS and AhR with TCDD resulted in the reciprocal regulation of message for iNOS and Cyp1A1, bilirubin was noted to exert a quite modest effect on Cyp1A1 expression at all but supraphysiologic concentrations (Fig. 5D). These findings are reasonably concordant with those of Sinal and Bend [38], who demonstrated a dose-dependent increase in Cyp1A1 expression in Hepa1c1c7 cells exposed to 100 µM bilirubin, reaching approximately one-third the level achieved with TCDD. Phelan et al. reported a more robust effect of 50 µM bilirubin on Cyp1A1 induction in liver and intestinal cell lines, approaching that of TCDD [39]; however, the absence of albumin in the culture medium in these studies could have resulted in supraphysiologic free bilirubin concentrations. Given its relatively modest influence on AhR activation, we suspect that bilirubin is unlikely to regulate iNOS expression through ARNT-dependent reciprocal inhibition of HIF-1α, at least under physiological conditions. A proposed schema via which bilirubin may modulate LPS-dependent iNOS up-regulation is depicted in Fig. 7.
Fig. 7.

Proposed mechanisms of bilirubin modulation of TLR4 signaling. The interaction of LPS with TLR4 activates the MyD88-dependent pathway (leading to the up-regulation of IL-6), and the TRIF-dependent pathway (which induces the expression of Inf-β). LPS further stimulates the TLR4-dependent activity of Nox, resulting in the generation of superoxide that dismutates to form hydrogen peroxide. Intracellular hydrogen peroxide stimulates the nuclear translocation and interaction of HIF-1α with ARNT, which binds to the HRE in the iNOS promoter and induces gene expression. TCDD activates the translocation and binding of AhR to ARNT, causing up-regulation of Cyp1A1 through binding to the xenobiotic response element (XRE). Reciprocal activation of Cyp1A1 and iNOS appears to occur as a result of competition for ARNT binding. Bilirubin inhibits the LPS-stimulated induction of iNOS by scavenging Nox-derived ROS, and also suppresses the TLR4-dependent up-regulation of Inf-β. Due to the relatively weak activation of AhR by physiological concentrations of bilirubin, it is unclear whether this mechanism plays a significant role in iNOS regulation under normal circumstances.
Although apocynin is commonly utilized as a specific inhibitor of NADPH oxidase, it is well-established that this effect is dependent upon activation by cellular myeloperoxidases [46]. In cells lacking myeloperoxidase activity, apocynin not only fails to block the generation of , but has variably been shown to scavenge [22] or even augment [23] the production of H2O2. Consistent with prior studies in HEK293 cells [22] and vascular fibroblasts [23], we found that apocynin had no effect on either PMA or LPS-stimulated superoxide production as measured by hydroethidine fluorescence. These findings suggest that apocynin does not inhibit Nox activity in RAW 264.7 cells, at least over the 45 min incubation period employed. Despite its lack of effect on superoxide production, apocynin effectively blocked cellular hydrogen peroxide generation (as assessed by Amplex Red), supporting the hypothesis originally put forth by Heumüller et al. that apocynin acts as a scavenger for peroxide-dependent ROS [22]. These data highlight the caution that is required when employing apocynin as a Nox inhibitor.
One notable observation is that treatment of RAW 264.7 cells with bilirubin results in a marked reduction in LPS-stimulated interferon-β production, in the absence of an effect on cellular IL-6 release. These data suggest that bilirubin exerts a specific inhibitory effect on the TRIF-dependent TLR4 signaling pathway, which also is believed to modulate iNOS up-regulation [47]. However, unlike iNOS, it appears unlikely that the influence of bilirubin on Inf-β is mediated through HIF-1α, since the murine Inf-β promoter does not possess a hypoxia response element [48]. Seleme et al. have shown that TLR3-mediated induction of Inf-β is abolished in macrophages derived from mice possessing a spontaneous mutation in the Ncf1 gene encoding the p47phox subunit of Nox, while production of IL-6 remained unaltered [49]. These investigators further reported a reduction in TRIF message and protein levels in TLR3-stimulated macrophages derived from these superoxide-deficient animals, supporting that Nox is required for activation of the TRIF-dependent pathway, at least with respect to TLR3. However, our finding that incubation of RAW 264.7 cells in the presence of SOD had no effect on LPS-mediated Inf-β production suggests that this mechanism may not be relevant to TLR4-mediated processes. Nevertheless, our data clearly show that bilirubin disrupts TLR4-dependent Nox activation and interferon-β production, which likely represents the molecular mechanism(s) underlying its inhibitory effect on iNOS expression in response to LPS. As Inf-β previously has been shown to act in a paracrine manner to augment LPS-stimulated iNOS expression by murine macrophages [50,51], the ability of bilirubin suppress interferon-β production (in conjunction with its influence on HIF-1α activation) could contribute to its observed inhibitory effect on iNOS up-regulation.
Bilirubin is produced as part of the normal physiological degradation of heme via the sequential activity of heme oxygenase (HO) and biliverdin reductase. Although markedly elevated serum bilirubin concentrations (>20 mg/dL≈340 µM) can cause neurologic injury (kernicterus) in newborns, toxicity in adults is negligible, and bilirubin has been administered intravenously to patients without sequellae [52], reaching serum levels as high as 22 mg/dL (normal≤1.2 mg/dL≈20 µM). More modest (<3-fold) elevations in serum bilirubin, at the levels employed in the present studies, are commonly encountered in individuals with Gilbert’s syndrome, a benign condition resulting from polymorphisms in the gene encoding UGT1A1, the principal bilirubin conjugating enzyme. Indeed, we have shown that oral [53] or intraperitoneal [17] administration of bilirubin to rodents produces significant physiological effects at serum levels that are 3- to 4-fold baseline (≤0.4 mg/dL) [5]. Nitric oxide generated by the activity iNOS has been implicated in the pathogenesis of a number of clinical disorders, including inflammatory bowel disease, cardiovascular disease, rheumatoid arthritis, and cancer [54]. In animal models of sepsis and intestinal malignancy, treatment with bilirubin has been shown to ameliorate tissue injury and suppress iNOS expression in the liver, intestine, kidney, heart, and aortic tissues [5,6,53]. Although human macrophages, even when stimulated, produce little to no nitric oxide [55] as a result of epigenetic modifications that effectively silence the iNOS gene [56], the fact that epidemiological studies support an inverse association between serum bilirubin levels and cardiovascular disease [57,58], inflammatory bowel disease [59], colorectal cancer [60], and rheumatoid arthritis [61], raise the possibility of a potential therapeutic benefit of bilirubin through its ability to inhibit iNOS up-regulation.
Acknowledgments
The authors wish to thank Megan Vogel for her critical reading of the manuscript. This study was supported by National Institutes of Health research Grant DK063954 (SDZ). No financial disclosures are necessary for any of the authors.
References
- 1.Guha M., Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13(2):85–94. doi: 10.1016/s0898-6568(00)00149-2. 11257452 [DOI] [PubMed] [Google Scholar]
- 2.Miyake K. Endotoxin recognition molecules, Toll-like receptor 4-MD-2. Semin. Immunol. 2004;16(1):11–16. doi: 10.1016/j.smim.2003.10.007. 14751758 [DOI] [PubMed] [Google Scholar]
- 3.Feihl F., Waeber B., Liaudet L. Is nitric oxide overproduction the target of choice for the management of septic shock? Pharmacol. Ther. 2001;91(3):179–213. doi: 10.1016/s0163-7258(01)00155-3. 11744067 [DOI] [PubMed] [Google Scholar]
- 4.Laroux F.S., Pavlick K.P., Hines I.N., Kawachi S., Harada H., Bharwani S., Hoffman J.M., Grisham M.B. Role of nitric oxide in inflammation. Acta Physiol. Scand. 2001;173(1):113–118. doi: 10.1046/j.1365-201X.2001.00891.x. 11678733 [DOI] [PubMed] [Google Scholar]
- 5.Wang W.W., Smith D.L.H., Zucker S.D. Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology. 2004;40(2):424–433. doi: 10.1002/hep.20334. 15368447 [DOI] [PubMed] [Google Scholar]
- 6.Lanone S., Bloc S., Foresti R., Almolki A., Taillé C., Callebert J., Conti M., Goven D., Aubier M., Dureuil B., El-Benna J., Motterlini R., Boczkowski J. Bilirubin decreases NOS2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J. 2005;19(13):1890–1892. doi: 10.1096/fj.04-2368fje. 16129699 [DOI] [PubMed] [Google Scholar]
- 7.Zughaier S.M., Zimmer S.M., Datta A., Carlson R.W., Stephens D.S. Differential induction of the Toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect. Immun. 2005;73(5):2940–2950. doi: 10.1128/IAI.73.5.2940-2950.2005. 15845500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dauphinee S.M., Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 2006;86(1):9–22. doi: 10.1038/labinvest.3700366. 16357866 [DOI] [PubMed] [Google Scholar]
- 9.Takeda K., Akira S. TLR signaling pathways. Semin. Immunol. 2004;16(1):3–9. doi: 10.1016/j.smim.2003.10.003. 14751757 [DOI] [PubMed] [Google Scholar]
- 10.Sumi D., Ignarro L.J. Regulation of inducible nitric oxide synthase expression in advanced glycation end product-stimulated raw 264.7 cells: the role of heme oxygenase-1 and endogenous nitric oxide. Diabetes. 2004;53(7):1841–1850. doi: 10.2337/diabetes.53.7.1841. 15220209 [DOI] [PubMed] [Google Scholar]
- 11.Muijsers R.B.R., van den Worm E., Folkerts G., Beukelman C.J., Koster A.S., Postma D.S., Nijkamp F.P. Apocynin inhibits peroxynitrite formation by murine macrophages. Br. J. Pharmacol. 2000;130(4):932–936. doi: 10.1038/sj.bjp.0703401. 10864902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uchikura K., Wada T., Hoshino S., Nagakawa Y., Aiko T., Bulkley G.B., Klein A.S., Sun Z. Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287(3):G620–G626. doi: 10.1152/ajpgi.00314.2003. 15087279 [DOI] [PubMed] [Google Scholar]
- 13.Dikalov S.I., Dikalova A.E., Mason R.P. Noninvasive diagnostic tool for inflammation-induced oxidative stress using electron spin resonance spectroscopy and an extracellular cyclic hydroxylamine. Arch. Biochem. Biophys. 2002;402(2):218–226. doi: 10.1016/S0003-9861(02)00064-4. 12051666 [DOI] [PubMed] [Google Scholar]
- 14.Hsu H.Y., Wen M.H. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J. Biol. Chem. 2002;277(25):22131–22139. doi: 10.1074/jbc.M111883200. 11940570 [DOI] [PubMed] [Google Scholar]
- 15.Bai S.K., Lee S.J., Na H.J., Ha K.S., Han J.A., Lee H., Kwon Y.G., Chung C.K., Kim Y.M. beta-Carotene inhibits inflammatory gene expression in lipopolysaccharide-stimulated macrophages by suppressing redox-based NF-kappaB activation. Exp. Mol. Med. 2005;37(4):323–334. doi: 10.1038/emm.2005.42. 16155409 [DOI] [PubMed] [Google Scholar]
- 16.Park H.S., Jung H.Y., Park E.Y., Kim J., Lee W.J., Bae Y.S. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 2004;173(6):3589–3593. doi: 10.4049/jimmunol.173.6.3589. 15356101 [DOI] [PubMed] [Google Scholar]
- 17.Keshavan P., Deem T.L., Schwemberger S.J., Babcock G.F., Cook-Mills J.M., Zucker S.D. Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J. Immunol. 2005;174(6):3709–3718. doi: 10.4049/jimmunol.174.6.3709. 15749910 [DOI] [PubMed] [Google Scholar]
- 18.Stocker R., Yamamoto Y., McDonagh A.F., Glazer A.N., Ames B.N. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. doi: 10.1126/science.3029864. 3029864 [DOI] [PubMed] [Google Scholar]
- 19.Barañano D.E., Rao M., Ferris C.D., Snyder S.H. Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U. S. A. 2002;99(25):16093–16098. doi: 10.1073/pnas.252626999. 12456881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDonagh A.F., Assisi F. The ready isomerization of bilirubin IX- in aqueous solution. Biochem. J. 1972;129(3):797–800. doi: 10.1042/bj1290797. 4659001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nauseef W.M. Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. Biochim. Biophys. Acta. 2014;1840(2):757–767. doi: 10.1016/j.bbagen.2013.04.040. 23660153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heumüller S., Wind S., Barbosa-Sicard E., Schmidt H.H.H.W., Busse R., Schröder K., Brandes R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51(2):211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. 18086956 [DOI] [PubMed] [Google Scholar]
- 23.Vejražka M., Mícek R., Štípek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim. Biophys. Acta. 2005;1722(2):143–147. doi: 10.1016/j.bbagen.2004.12.008. 15716123 [DOI] [PubMed] [Google Scholar]
- 24.Youn H.S., Lee J.K., Choi Y.J., Saitoh S.I., Miyake K., Hwang D.H., Lee J.Y. Cinnamaldehyde suppresses Toll-like receptor 4 activation mediated through the inhibition of receptor oligomerization. Biochem. Pharmacol. 2008;75(2):494–502. doi: 10.1016/j.bcp.2007.08.033. 17920563 [DOI] [PubMed] [Google Scholar]
- 25.Krishna M.C., Russo A., Mitchell J.B., Goldstein S., Dafni H., Samuni A. Do nitroxide antioxidants act as scavengers of or as SOD mimics? J. Biol. Chem. 1996;271(42):26026–26031. doi: 10.1074/jbc.271.42.26026. 8824242 [DOI] [PubMed] [Google Scholar]
- 26.Beckman J.S., Minor R.L., White C.W., Repine J.E., Rosen G.M., Freeman B.A. Superoxide dismutase and catalase conjugated to polyethylene glycol increases endothelial enzyme activity and oxidant resistance. J. Biol. Chem. 1988;263(14):6884–6892. 3129432 [PubMed] [Google Scholar]
- 27.Zucker S.D., Goessling W., Hoppin A.G. Unconjugated bilirubin exhibits spontaneous diffusion through model lipid bilayers and native hepatocyte membranes. J. Biol. Chem. 1999;274(16):10852–10862. doi: 10.1074/jbc.274.16.10852. 10196162 [DOI] [PubMed] [Google Scholar]
- 28.Lentschat A., Karahashi H., Michelsen K.S., Thomas L.S., Zhang W., Vogel S.N., Arditi M. Mastoparan, a G protein agonist peptide, differentially modulates TLR4- and TLR2-mediated signaling in human endothelial cells and murine macrophages. J. Immunol. 2005;174(7):4252–4261. doi: 10.4049/jimmunol.174.7.4252. 15778388 [DOI] [PubMed] [Google Scholar]
- 29.Youn H.S., Lee J.Y., Fitzgerald K.A., Young H.A., Akira S., Hwang D.H. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J. Immunol. 2005;175(5):3339–3346. doi: 10.4049/jimmunol.175.5.3339. 16116226 [DOI] [PubMed] [Google Scholar]
- 30.Wang M.J., Jeng K.C., Shih P.C. Differential expression of inducible nitric oxide synthase gene by alveolar and peritoneal macrophages in lipopolysaccharide-hyporesponsive C3H/HeJ mice. Immunology. 1999;98(4):497–503. doi: 10.1046/j.1365-2567.1999.00908.x. 10594680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cauwels A., Bultinck J., De Zwaef R., Vandendriessche B., Magez S., Brouckaert P. Nitric oxide production by endotoxin preparations in TLR4-deficient mice. Nitric Oxide. 2014;36:36–43. doi: 10.1016/j.niox.2013.11.001. 24269486 [DOI] [PubMed] [Google Scholar]
- 32.Kim J.S., Yeo S., Shin D.G., Bae Y.S., Lee J.J., Chin B.R., Lee C.H., Baek S.H. Glycogen synthase kinase 3beta and beta-catenin pathway is involved in toll-like receptor 4-mediated NADPH oxidase 1 expression in macrophages. FEBS J. 2010;277(13):2830–2837. doi: 10.1111/j.1742-4658.2010.07700.x. 20528914 [DOI] [PubMed] [Google Scholar]
- 33.Kietzmann T., Görlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005;16(4–5):474–486. doi: 10.1016/j.semcdb.2005.03.010. 15905109 [DOI] [PubMed] [Google Scholar]
- 34.Melillo G., Musso T., Sica A., Taylor L.S., Cox G.W., Varesio L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 1995;182(6):1683–1693. doi: 10.1084/jem.182.6.1683. 7500013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi Z., Rapisarda A., Taylor L., Brooks A., Creighton-Gutteridge M., Melillo G., Varesio L. Synergystic induction of HIF-1alpha transcriptional activity by hypoxia and lipopolysaccharide in macrophages. Cell Cycle. 2008;7(2):232–241. doi: 10.4161/cc.7.2.5193. 18212534 [DOI] [PubMed] [Google Scholar]
- 36.Gradin K., McGuire J., Wenger R.H., Kvietikova I., fhitelaw M.L., Toftgård R., tora L., Gassmann M., Poellinger L. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the ARNT transcription factor. Mol. Cell. Biochem. 1996;16(10):5221–5231. doi: 10.1128/mcb.16.10.5221. 8816435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nie M., Blankenship A.L., Giesy J.P. Interactions between aryl hydrocarbon receptor (AhR) and hypoxia signaling pathways. Environ. Toxicol. Pharmacol. 2001;10(1–2):17–27. doi: 10.1016/s1382-6689(01)00065-5. 11382553 [DOI] [PubMed] [Google Scholar]
- 38.Sinal C.J., Bend J.R. Aryl hydrocarbon receptor-dependent induction of Cyp1a1 by bilirubin in mouse hepatoma Hepa 1c1c7 cells. Mol. Pharmacol. 1997;52(4):590–599. doi: 10.1124/mol.52.4.590. 9380021 [DOI] [PubMed] [Google Scholar]
- 39.Phelan D., Winter G.M., Rogers W.J., Lam J.C., Denison M.S. Activation of the AH receptor signal transduction pathway by bilirubin and biliverdin. Arch. Biochem. Biophys. 1998;357(1):155–163. doi: 10.1006/abbi.1998.0814. [DOI] [PubMed] [Google Scholar]
- 40.Anazawa T., Dimayuga P.C., Li H., Tani S., Bradfield J., Chyu K.Y., Kaul S., Shah P.K., Cercek B. Effect of exposure to cigarette smoke on carotid artery intimal thickening: the role of inducible NO synthase. Arterioscler. Thromb. Vasc. Biol. 2004;24(9):1652–1658. doi: 10.1161/01.ATV.0000139925.84444.ad. 15271786 [DOI] [PubMed] [Google Scholar]
- 41.Kerzee J.K., Ramos K.S. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: role of the Ahr bHLH/PAS transcription factor. Circ. Res. 2001;89(7):573–582. doi: 10.1161/hh1901.097083. 11577022 [DOI] [PubMed] [Google Scholar]
- 42.Sidel N., Abrams M.I. Jaundice in arthritis: its analgesic action. N. Engl. J. Med. 1934;210(4):181–182. [Google Scholar]
- 43.Asehnoune K., Strassheim D., Mitra S., Kim J.Y., Abraham E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J. Immunol. 2004;172(4):2522–2529. doi: 10.4049/jimmunol.172.4.2522. 14764725 [DOI] [PubMed] [Google Scholar]
- 44.Sanlioglu S., Williams C.M., Samavati L., Butler N.S., Wang G., McCray P.B., Ritchie T.C., Hunninghake G.W., Zandi E., Engelhardt J.F. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J. Biol. Chem. 2001;276(32):30188–30198. doi: 10.1074/jbc.M102061200. 11402028 [DOI] [PubMed] [Google Scholar]
- 45.Nishi K., Oda T., Takabuchi S., Oda S., Fukuda K., Adachi T., Semenza G.L., Shingu K., Hirota K. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species–dependent manner. Antioxid. Redox Signal. 2008;10(5):983–996. doi: 10.1089/ars.2007.1825. [DOI] [PubMed] [Google Scholar]
- 46.Simons J.M., Hart B.A., Ip Vai Ching T.R., Van Dijk H., Labadie R.P. Metabolic activation of natural phenols into selective oxidative burst agonists by activated human neutrophils. Free Radic. Biol. Med. 1990;8(3):251–258. doi: 10.1016/0891-5849(90)90070-y. 2160411 [DOI] [PubMed] [Google Scholar]
- 47.Lee J.Y., Lowell C.A., Lemay D.G., Youn H.S., Rhee S.H., Sohn K.H., Jang B., Ye J., Chung J.H., Hwang D.H. The regulation of the expression of inducible nitric oxide synthase by Src-family tyrosine kinases mediated through MyD88-independent signaling pathways of Toll-like receptor 4. Biochem. Pharmacol. 2005;70(8):1231–1240. doi: 10.1016/j.bcp.2005.07.020. 16140274 [DOI] [PubMed] [Google Scholar]
- 48.Bonnefoy E., Bandu M.T., Doly J. Specific binding of high-mobility-group I (HMGI) protein and histone H1 to the upstream AT-rich region of the murine beta interferon promoter: HMGI protein acts as a potential antirepressor of the promoter. Mol. Cell. Biol. 1999;19(4):2803–2816. doi: 10.1128/mcb.19.4.2803. 10082546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seleme M.C., Lei W., Burg A.R., Goh K.Y., Metz A., Steele C., Tse H.M. Dysregulated TLR3-dependent signaling and innate immune activation in superoxide-deficient macrophages from nonobese diabetic mice. Free Radic. Biol. Med. 2012;52(9):2047–2056. doi: 10.1016/j.freeradbiomed.2012.01.027. 22361747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X., Alley E.W., Russell S.W., Morrison D.C. Necessity and sufficiency of beta interferon for nitric oxide production in mouse peritoneal macrophages. Infect. Immun. 1994;62(1):33–40. doi: 10.1128/iai.62.1.33-40.1994. 8262648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujihara M., Ito N., Pace J.L., Watanabe Y., Russell S.W., Suzuki T. Role of endogenous interferon-beta in lipopolysaccharide-triggered activation of the inducible nitric-oxide synthase gene in a mouse macrophage cell line, J774. J. Biol. Chem. 1994;269(17):12773–12778. [PubMed] [Google Scholar]
- 52.Thompson H.E., Wyatt B.L. Experimentally induced jaundice (hyperbilirubinemia): report of animal experimentation and of the physiologic effect of jaundice in patients with atrophic arthritis. Arch. Intern. Med. 1938;61(3):481–500. [Google Scholar]
- 53.Smith D.L.H., Keshavan P., Avissar U., Ahmed K., Zucker S.D. Sodium taurocholate inhibits intestinal adenoma formation in APCMin/+ mice, potentially through activation of the farnesoid X receptor. Carcinogenesis. 2010;31(6):1100–1109. doi: 10.1093/carcin/bgq050. 20194350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanwar J.R., Kanwar R.K., Burrow H., Baratchi S. Recent advances on the roles of NO in cancer and chronic inflammatory disorders. Curr. Med. Chem. 2009;16(19):2373–2394. doi: 10.2174/092986709788682155. 19601787 [DOI] [PubMed] [Google Scholar]
- 55.Albina J.E. On the expression of nitric oxide synthase by human macrophages. Why no NO? J. Leukoc. Biol. 1995;58(6):643–649. doi: 10.1002/jlb.58.6.643. 7499961 [DOI] [PubMed] [Google Scholar]
- 56.Gross T.J., Kremens K., Powers L.S., Brink B., Knutson T., Domann F.E., Philibert R.A., Milhem M.M., Monick M.M. Epigenetic silencing of the human NOS2 gene: rethinking the role of nitric oxide in human macrophage inflammatory responses. J. Immunol. 2014;192(5):2326–2338. doi: 10.4049/jimmunol.1301758. 24477906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hunt S.C., Kronenberg F., Eckfeldt J.H., Hopkins P.N., Myers R.H., Heiss G. Association of plasma bilirubin with coronary heart disease and segregation of bilirubin as a major gene trait: the NHLBI family heart study. Atherosclerosis. 2001;154(3):747–754. doi: 10.1016/s0021-9150(00)00420-2. 11257278 [DOI] [PubMed] [Google Scholar]
- 58.Schwertner H.A., Jackson W.G., Tolan G. Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem. 1994;40(1):18–23. 8287538 [PubMed] [Google Scholar]
- 59.de Vries H.S., te Morsche R.H.M., Jenniskens K., Peters W.H.M., de Jong D.J. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn’s disease. J. Crohns Colitis. 2012;6(5):597–602. doi: 10.1016/j.crohns.2011.11.010. 22398043 [DOI] [PubMed] [Google Scholar]
- 60.Zucker S.D., Horn P.S., Sherman K.E. Serum bilirubin levels in the U.S. population: gender effect and inverse correlation with colorectal cancer. Hepatology. 2004;40(4):827–835. doi: 10.1002/hep.20407. 15382174 [DOI] [PubMed] [Google Scholar]
- 61.Fischman D., Valluri A., Gorrepati V.S., Murphy M.E., Peters I., Cheriyath P. Bilirubin as a protective factor for rheumatoid arthritis: an NHANES study of 2003–2006 data. J. Clin. Med. Res. 2010;2(6):256–260. doi: 10.4021/jocmr444w. 22043258 [DOI] [PMC free article] [PubMed] [Google Scholar]