

Abstract
Rapid and efficient engulfment of apoptotic cells is an essential property of phagocytes for removal of the large number of apoptotic cells generated in multicellular organisms. To achieve this, phagocytes need to be able to continuously uptake apoptotic cells. It was recently reported that uncoupling protein 2 (Ucp2) promotes engulfment of apoptotic cells by increasing the phagocytic capacity, thereby allowing cells to continuously ingest apoptotic cells. However, the functions of Ucp2, beyond its possible role in dissipating the mitochondrial membrane potential, that contribute to elevation of the phagocytic capacity have not been determined. Here, we report that the anion transfer or nucleotide binding activity of Ucp2, as well as its dissipation of the mitochondrial membrane potential, is necessary for Ucp2-mediated engulfment of apoptotic cells. To study these properties, we generated Ucp2 mutations that affected three different functions of Ucp2, namely, dissipation of the mitochondrial membrane potential, transfer of anions, and binding of purine nucleotides. Mutations of Ucp2 that affected the proton leak did not enhance the engulfment of apoptotic cells. Although anion transfer and nucleotide binding mutations did not affect the mitochondrial membrane potential, they exerted a dominant-negative effect on Ucp2-mediated engulfment. Furthermore, none of our Ucp2 mutations increased the phagocytic capacity. We conclude that dissipation of the proton gradient by Ucp2 is not the only determinant of the phagocytic capacity and that anion transfer or nucleotide binding by Ucp2 is also essential for Ucp2-mediated engulfment of apoptotic cells.
Keywords: apoptotic cell, capacity, engulfment, membrane potential, phagocytosis
INTRODUCTION
Phagocytosis of apoptotic cells, also known as efferocytosis, is a fundamental process for the removal of aged, superfluous, and cancerous cells undergoing apoptosis. Clearance of apoptotic cells is important for embryonic development, tissue homeostasis, and preventing inflammation and autoimmune diseases (Henson and Hume, 2006; Maderna and Godson, 2003; Nagata et al., 2010; Ravichandran and Lorenz, 2007). Much progress has been made in understanding how phagocytes are recruited, how apoptotic cells are recognized, and how apoptotic cells are internalized into phagocytes (Erwig and Henson, 2008; Hochreiter-Hufford and Ravichandran, 2013; Lauber et al., 2004). Apoptotic cells actively recruit phagocytes by releasing attractants such as nucleotides, sphingosine-1-phosphate, and fractalkine (Elliott et al., 2009; Lauber et al., 2003). Apoptotic cells are then recognized by phagocytes via interactions between ligands on apoptotic cells and receptors on phagocytes. Phosphatidylserine is a hallmark of apoptosis and the best-known ligand of apoptotic cells for efferocytosis. Thus far, a few receptors that bind directly to this aminophospholipid have been identified (Miyanishi et al., 2007; Park et al., 2007; 2008). Upon recognition, apoptotic cells trigger intracellular signaling pathways, thereby stimulating cytoskeletal rearrangement to draw apoptotic cells into phagocytes (Albert et al., 2000; Lee et al., 2014; Park et al., 2007). Finally, ingested apoptotic cells within phagocytes are degraded in phagolysosomes by digestive enzymes derived from lysosomes. The sequence of events leading to phagocytosis of apoptotic cells is relatively well-established. However, the factors that influence the phagocytic capacity have not been identified.
The phagocytic capacity is the ability of phagocytes to ingest multiple apoptotic cells in succession. This property of phagocytes is indispensable for the removal of large numbers of apoptotic cells rapidly and efficiently in multicellular organisms, thereby preventing secondary necrosis of apoptotic cells provoking an inflammatory response (A-Gonzalez and Hidalgo, 2014; Han and Ravichandran, 2011). Recently, uncoupling proteins (Ucps) were identified as possible contributors to the phagocytic capacity (Park et al., 2011). Ucps are mitochondrial inner membrane proteins belonging to the mitochondrial super-family of anion-carrier proteins. As their names imply, Ucps function to separate oxidative phosphorylation from ATP synthesis by dissipating the proton gradient in mitochondria. Although Ucps can dissipate the mitochondrial membrane potential in vitro, their roles in a more physiologically relevant setting remain unclear (Krauss et al., 2005; Nedergaard et al., 2005). Ucp1 is only detected in brown adipose tissue and helps to generate body heat by dissipating the proton gradient in mitochondria instead of producing ATP (Thomas and Palmiter, 1997). In addition, it was recently reported that Ucp2 enhances the engulfment of apoptotic cells by increasing the phagocytic capacity (Park et al., 2011). Expression of Ucp2 appears to increase during efferocytosis, causing the mitochondrial membrane potential to decrease, thereby affecting the phagocytic capacity. This implies that the expression level of Ucp2 protein in mitochondria determines whether a given phagocyte engulfs a few or many apoptotic cells. However, it remains to be determined whether the other functions of Ucp2 are linked to efferocytosis.
To analyze the structure-function relationship of Ucps more systematically, we generated mutations of Ucp2 affecting dissipation of the proton gradient in mitochondria, anion (e.g., chloride) transfer, and binding of purine nucleotides. Each mutation affected only one function of Ucp2 without influencing the others (Echtay et al., 2000; 2001; Urbankova et al., 2003). Mutations affecting anion transfer, nucleotide binding, and establishment of the proton gradient abolished Ucp2-mediated efferocytosis because phagocytes expressing these mutants failed to continuously ingest apoptotic cells. Moreover, the anion transfer and nucleotide binding mutations of Ucp2 exerted a dominant-negative effect on efferocytosis, suggesting a critical role for Ucp2-mediated efferocytosis. Therefore, disruption of the mitochondrial membrane potential by Ucp2 is not the only factor that determines the phagocytic capacity during efferocytosis.
MATERIALS AND METHODS
Cell culture and transfections
293T and NIH/3T3 cells were cultured with Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% FBS, and 1% penicillin-streptomycin-glutamine and LR73 cells were maintained in alpha-minimum essential medium (MEM) containing 10% FBS and 1% penicillin-streptomycin-glutamine. The Profection mammalian transfection system (Promega) was used to introduce plasmids into 293T cells, while LR73 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Plasmids and antibodies
Mouse Ucp2 gene (Open Biosystems) was amplified and cloned into a mammalian expression vector to generate pEBB-Ucp2-FLAG and pEBB-Ucp2. Amino acid residues at positions 25, 28, 30, 170, 185 and 212 of Ucp2 were mutated by site-directed mutagenesis to generate a proton leak mutation, a nucleotide binding mutation, and a anion transport mutation. All constructs were sequenced to confirm the fidelity of sequences and mutations. Anti-FLAG (M2 and M5), anti-AIF (D39D2) and anti-Porin (ab15895) were purchased from Sigma, Cell Signaling, and Abcam, respectively.
Immunoblotting and immunostaining
293T cells or LR73 cells were transiently transfected with indicated plasmids using calcium phosphate (Promega) or Lipofectamine 2000 (Invitrogen), respectively. After transfection, the cells were lysed and subjected to immunoblotting against the respective tag or proteins. To isolate mitochondria, cells were resuspended in 500 μl of TS buffer (10 mM Tris, pH 7.5, 250 mM sucrose, 2 μg·ml−1 DNase, and protease inhibitor cocktail), and then subjected to three cycles of 5-min freezing in liquid nitrogen followed by 10-min thawing at 37°C. Unbroken cells and nuclei were removed by centrifugation at 800 g for 10 min and mitochondria were collected from the supernatant by centrifugation at 10,000 g for 20 min. The isolated mitochondria were lysed in RIPA buffer and subjected to immunoblotting. To stain cells for microscopy, LR73 cells were plated and transfected with FLAG-tagged Ucp2. One day after transfection, the cells were incubated with Mitotracker Deep Red (100 nM, Molecular Probes) in the culture medium for 20 min. The cells were then fixed with 3% paraformaldehyde (Sigma) in PBS for 30 min, permeabilized with 0.1% Triton X-100 (Sigma) and blocked with 5% clarified milk. Antibody staining was then performed using antibodies to FLAG (Clone M5, Sigma) and AIF (D39D2, Cell Signaling Technologies). The stained cells were analyzed by Axio Imager D2 (Zeiss).
Phagocytosis assay
The 1 × 105 LR73 cells were plated on a 24-well culture plate and transfected with GFP alone or GFP and the indicated plasmids using Lipofectamine 2000. One day after transfection, the cells were incubated with 1 μl of carboxylate-modified red fluorescent beads (2 μm in diameter) (Invitrogen) or 2 × 106 TAMRA stained apoptotic thymocytes for 2 h, trypsinized, and analyzed by flow cytometry (Canto II, BD). GFP and red-fluorescent double-positive cells were classified as phagocytes engulfing carboxylate beads or apoptotic thymocytes. To make TAMRA-stained apoptotic thymocytes, thymocytes were prepared from thymi of 5 to 8 week-old C57/B6 wild type mice, and stained with 50 μM TAMRA. To induce apoptosis of thymocytes, cells cultured in RPMI 1640 medium (containing 10% FBS and 1% penicillin-streptomycin-glutamine) were stimulated with 50 μM dexamethasone (Calbiochem) in a 5% CO2 incubator for 4 h, washed with phagocyte culture medium twice, and resuspended with phagocyte culture medium at a concentration of 2 × 106 cells/300 μl.
Measuring mitochondrial membrane potential
To detect mitochondrial membrane potential, LR73 cells were stained with Mitotracker Deep Red FM (Invitrogen) according to the manufacturer’s instructions. For TMRE staining, cells were stained with 50 nM TMRE in culture medium in a 5% CO2 incubator for 30 min and fluorescence intensity was measured by flow cytometry (Canto II, BD). Mean fluorescence intensity was compared to that of a control experiment and mitochondrial membrane potential was represented as a value relative to that of a control experiment.
RESULTS
Ucp2 promotes the engulfment of apoptotic cells but not of indigestible surrogate targets
Ucp2 is a mitochondrial inner membrane protein that dissipates the mitochondrial membrane potential (Arsenijevic et al., 2000; Fleury et al., 1997; Flier and Lowell, 1997). Our first task was to determine whether exogenously expressed Ucp2 was localized appropriately in mitochondria. Therefore, Ucp2-FLAG was expressed in LR73 cells, which were used as phagocytes in this study, and was detected only in the mitochondrial fraction, not in the cytosolic fraction (Fig. 1A). The co-localization of Ucp2-FLAG and apoptosis-inducing factor (AIF), a protein that localizes to the mitochondrial inner membrane, indicated that Ucp2 was localized properly, despite being exogenously expressed in LR73 cells (Fig. 1B). Next, we tested whether Ucp2 reduces the mitochondrial membrane potential. For this purpose, Ucp2-FLAG was expressed in LR73 cells, and these cells were stained with MitoTracker, a dye whose accumulation in mitochondria is proportional to the mitochondrial membrane potential. Ucp2-positive cells incorporated far less MitoTracker than Ucp2-negative cells (Fig. 1C), indicating that the mitochondrial membrane potential was reduced in the former cells. This observation was not limited to LR73 cells, because NIH/3T3 cells expressing Ucp2 were also less able to incorporate MitoTracker (Fig. 1D), indicating that overexpression of Ucp2 in mammalian cells reduces the mitochondrial membrane potential. An alternative method to detect disruption of the proton gradient is to stain cells with tetramethylrhodamine ethyl ester perchlorate (TMRE), another dye widely used to measure the mitochondrial membrane potential. The mean fluorescence intensity of TMRE was approximately 35% lower in LR73 cells expressing Ucp2-FLAG than in control cells expressing an empty vector (Fig. 1E), providing strong evidence that Ucp2 expression in phagocytes decreases the mitochondrial membrane potential.
Fig. 1.
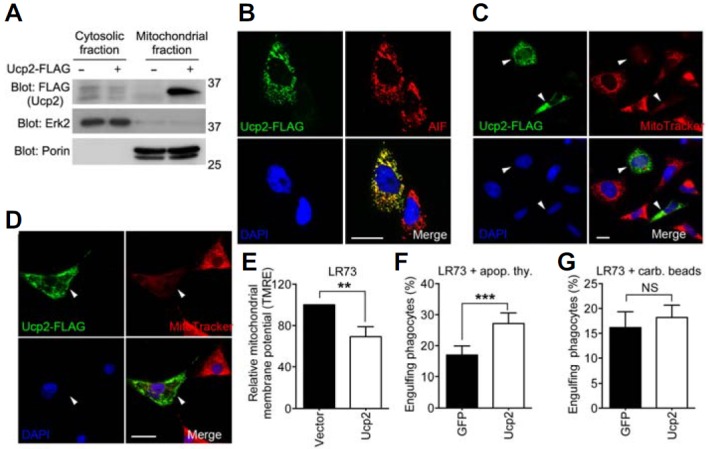
Ucp2 facilitates engulfment of apoptotic cells. (A) 293T cells were transfected with Ucp2-FLAG, lysed, and separated into the cytoplasmic and the mitochondrial fractions. Erk2 and Porin were used to confirm the cytosolic and the mitochondrial fractions, respectively. (B) LR73 cells were transfected with Ucp2-FLAG and stained with anti-FLAG and anti-AIF antibody. Scale bar, 20 μm. (C, D) LR73 (C) or NIH/3T3 (D) cells transfected with Ucp2-FLAG were stained with anti-FLAG antibody and Mitotracker. Arrowheads indicate the same cells expressing Ucp2-FLAG in all four panels. Scale bar, 20 μm. (E) Ucp2-FLAG was expressed in LR73 cells, stained with TMRE, and analyzed by flow cytometry. Mean fluorescence intensity of TMRE was normalized to the control. (F, G) LR73 cells were transfected with the indicated plasmids and incubated with TAMRA-stained apoptotic thymocytes (F) or 2 μm carboxylate-modified red fluorescent beads (G) for 2 h. The cells were analyzed by flow cytometry. NS, not significant; **P < 0.01, ***P < 0.001.
Ucps can enhance phagocytosis of apoptotic cells by increasing the phagocytic capacity. To examine the ability of Ucp2 to mediate engulfment of apoptotic cells or carboxylate-modified beads (as surrogates of apoptotic cells), we transfected LR73 cells with Ucp2 and measured the ability of these cells to phagocytose the targets. More Ucp2-expressing LR73 cells ingested apoptotic cells in comparison to control cells not expressing Ucp2 (Fig. 1F), although there was no significant difference in the engulfment of beads (Fig. 1G), as reported previously. Together, these data indicate that Ucp2 specifically promotes the engulfment of apoptotic cells, but not of other indigestible surrogate targets such as carboxylate-modified beads.
Ucp2 mutants are properly localized in mitochondria
Within the mitochondrial inner membrane, Ucp2 functions to dissipate the membrane potential, transport anions such as chloride or molecules such as fatty acids, and bind to purine nucleotides (Bouillaud, 1999; Krauss et al., 2005). It appears that Ucp2 promotes the engulfment of apoptotic cells by decreasing the mitochondrial membrane potential; however, the other functions of Ucp2, aside from its ability to decrease the proton gradient, that affect efferocytosis have not been determined. To address this issue, we generated mutations of Ucp2 designed to disrupt only one function without affecting the others (Fig. 2A). The proton leak mutants, Ucp2D28N and Ucp2D212N, cannot reduce the mitochondrial membrane potential, because of an inability to allow the passage of protons from the intermembrane space to the mitochondrial matrix, but do not affect nucleotide binding or anion transport by Ucp2 (Echtay et al., 2000; Urbankova et al., 2003). Two other mutants of Ucp2, Ucp2E170Q and Ucp2R185Q, are unable to transport anions or bind nucleotides, respectively, but do not affect the passage of protons through Ucp2, thereby allowing the mitochondrial membrane potential to be reduced (Echtay et al., 2000; 2001). When Ucp2 and the various constructs were expressed in LR73 cells, their expression levels were comparable (Fig. 2B), and all were localized properly within the mitochondria, as confirmed by the co-localization of AIF and the various forms of Ucp2 (Fig. 2C). Together, these results rule out the possibility that any abnormalities in efferocytosis are caused by different levels of protein expression or by the improper subcellular localization of the various Ucp2 constructs.
Fig. 2.
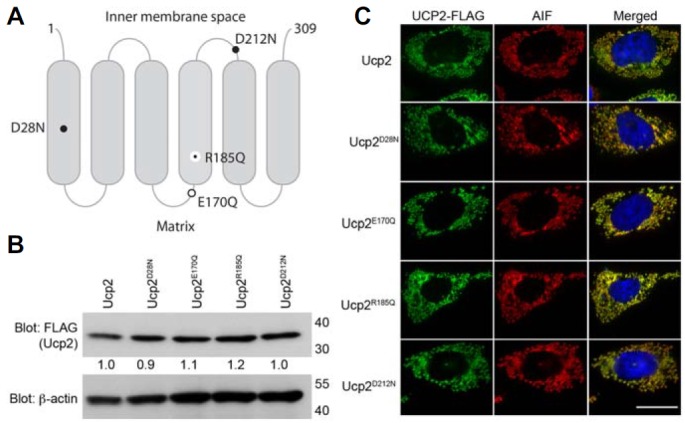
Subcellular localization of the Ucp2 mutants. (A) Schematic diagram of Ucp2 illustrating the positions of Ucp2 mutations. Large black dot, proton leak mutations; open circle, chloride transport mutation; small black dot, nucleotide-binding mutation. (B) LR73 cells were transfected with the indicated plasmids, lysed, and protein expression analyzed by immunoblotting. The expression of Ucp2 and mutated Ucp2 proteins were detected by immunoblotting. β-actin was used as a loading control. The numbers under the FLAG blot indicate relative expressions of the proteins to Ucp2. (C) LR73 cells, expressing the indicated proteins, were stained with anti-FLAG and AIF antibody. Scale bar, 20 μm.
Ucp2E170Q and Ucp2R185Q, but not Ucp2D28N or Ucp2 D212N, disrupt the mitochondrial membrane potential
Having determined that none of the mutations affected the expression or subcellular localization of Ucp2, we examined whether the mutations affected the mitochondrial membrane potential. For this purpose, LR73 cells expressing either Ucp2 or specific mutants were stained with MitoTracker. Cells expressing the proton leak mutants, Ucp2D28N and Ucp2D212N, showed strong incorporation of MitoTracker, indicating that these mutations disrupt or inhibit proton transport across the inner membrane, thereby allowing the membrane potential to be maintained (Fig. 3A). Cells expressing Ucp2E170Q (anion transport defect) or Ucp2R185Q (nucleotide binding defect) were very weakly stained with MitoTracker, comparable to those expressing Ucp2, indicating that these mutants still reduced the mitochondrial membrane potential and that protons could flow properly through these Ucp2 proteins (Fig. 3A). Changes in the mitochondrial membrane potential were also detected by the incorporation of TMRE, as described above. Ucp2, Ucp2E170Q, and Ucp2R185Q showed similar levels of TMRE incorporation, indicating that the flow of protons across the inner membrane was not affected (Fig. 3B). However, the proton leak mutants did not allow the membrane potential to be reduced (Fig. 3C). Therefore, our data clearly indicate that Ucp2E170Q (anion transport defect) and Ucp2R185Q (nucleotide binding defect) still lower the proton gradient across the mitochondrial inner membrane.
Fig. 3.
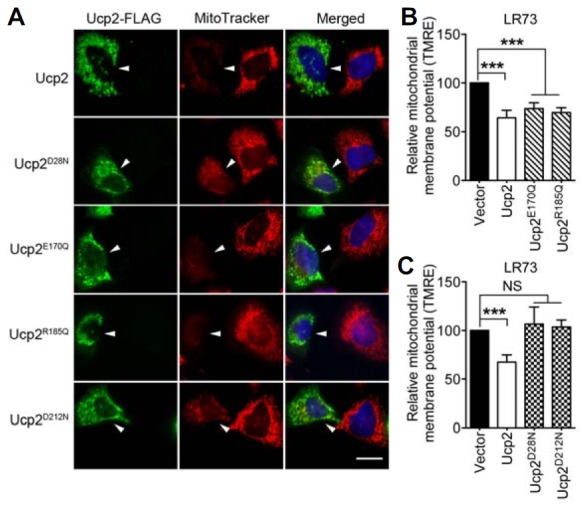
The effects of the Ucp2 mutants on the mitochondrial membrane potential. (A) LR73 cells, expressing the indicated proteins, were stained with anti-FLAG antibody and MitoTracker. The arrowheads point to the cell expressing Ucp2-FLAG. Scale bar, 20 μm. (B, C) LR73 cells were transfected with the indicated plasmids and then stained with TMRE. Mean fluorescence intensity of TMRE was measured by flow cytometry and compared to that of control cells transfected with empty vectors. NS, not significant; ***P < 0.001.
Ucp2E170Q and Ucp2 R185Q are dominant-negative regulators of efferocytosis
Next, it was important to determine whether phagocytes use Ucp2 to regulate the mitochondrial membrane potential in order to influence the phagocytic capacity. To examine this possibility, Ucp2 and the various Ucp2 mutants were expressed in LR73 cells, and the ability of these cells to phagocytose apoptotic thymocytes and carboxylate-modified beads was tested. As expected, expression of Ucp2, but not of the proton leak mutants, allowed efficient phagocytosis of apoptotic cells but not of the beads (Figs. 4A and 4B). Surprisingly, cells expressing Ucp2E170Q and Ucp2R185Q were extremely inefficient at phagocytosing apoptotic cells (Fig. 4C). Furthermore, it appeared as though both these Ucp2 mutants reduced phagocytosis of apoptotic cells to below the basal level, although phagocytosis of beads was no different from that in cells expressing Ucp2 (Figs. 4C and 4D). These results were unexpected and implied that Ucp2-mediated efferocytosis is affected not only by the mitochondrial membrane potential but also by its ability to transport anions and bind to purine nucleotides. Finally, the ability of cells expressing Ucp2 and each of the Ucp2 mutants to continuously phagocytose apoptotic cells was tested. For this purpose, LR73 cells expressing Ucp2 or its mutants were allowed to phagocytose apoptotic thymocytes over 5 h (Fig. 4E). After 1 h of incubation and every hour thereafter, we measured the mean fluorescence intensity, which serves as an indicator of the relative number of apoptotic cells ingested by a single phagocyte. Over the course of 5 h, Ucp2-expressing cells were able to continuously ingest apoptotic cells, while cells expressing the Ucp2 mutants essentially stopped phagocytosing apoptotic cells after 2 h, suggesting that a threshold level had been reached (Fig. 4E). Therefore, it is apparent that reduction of the mitochondrial membrane potential by Ucp2 is not the sole determinant of Ucp2-mediated efferocytosis or the phagocytic capacity. In conclusion, anion transport and nucleotide binding, together with disruption of the mitochondrial membrane potential, are essential Ucp2-mediated functions for increased phagocytic capacity.
Fig. 4.
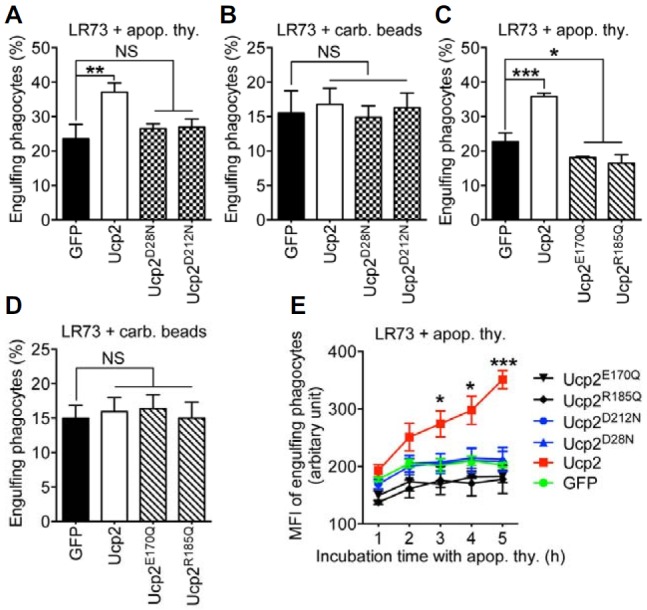
The anion transport mutant exerts a dominant negative effect on efferocytosis. (A, B) LR73 cells expressing the proton leak mutations of Ucp2, Ucp2D28N and Ucp2D212N, were incubated with apoptotic thymocytes (A) or 2 μm carboxylate-modified beads (B) for 2 h and analyzed by flow cytometry. (C, D) LR73 cells were transfected with the indicated plasmids and incubated with apoptotic thymocytes (C) or 2 μm carboxylate-modified beads (D) for 2 h. Phagocytes ingesting apoptotic thymocytes (C) or the beads (D) were evaluated by flow cytometry. (E) The Ucp2 and mutated Ucp2 proteins were expressed in LR73 cells and then the cells were incubated with TAMRA-stained apoptotic thymocytes for 1 h to 5 h. The mean fluorescence intensity (MFI) of TAMRA of the cells ingesting apoptotic cells was measured by flow cytometry. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
Apoptotic cells should be removed promptly and efficiently because large numbers of apoptotic cells are generated in multicellular organisms, and any delay or defect in the clearance of apoptotic cells gives rise to inflammatory responses. To clear many apoptotic cells as rapidly and effectively as possible, phagocytes must increase their phagocytic capacity. Nonetheless, the mechanisms by which a phagocyte constantly uptakes multiple apoptotic cells and the cellular factors that contribute to the phagocytic capacity have not been elucidated. It was recently reported that Ucp2 promotes efferocytosis by increasing the phagocytic capacity of a cell. In addition, disruption of the mitochondrial membrane potential by Ucp2 was suggested to be the mechanism by which phagocytes enhance the engulfment of apoptotic cells and increase the phagocytic capacity (Park et al., 2011). However, it is unclear whether disruption of the mitochondrial membrane potential by Upc2 is the only factor that contributes to the increased rate of efferocytosis.
We addressed this issue by using Ucp2 mutations that specifically nullify one function of Ucp2 without affecting the others. Surprisingly, Ucp2E170Q (anion transport defect) and Ucp2R185Q (nucleotide binding defect) exerted a dominant-negative effect on the engulfment of apoptotic cells, meaning that they inhibit even a basal level of apoptotic cell uptake. This was not observed for the proton leak mutations, which only abrogated Ucp2-promoted engulfment of apoptotic cells. These different effects might be caused by different alterations in the structure of Ucp2. Ucps work as a homodimer (Klingenberg and Appel, 1989; Lin et al., 1980). Thus, it is plausible that Ucp2E170Q and Ucp2R185Q abolish the activity upon dimerization with endogenous Ucp2, whereas the proton leak mutants do not. It is also possible that the proton leak mutants have a stronger affinity for each other, which facilitates mutant-mutant dimer formation. Therefore, expression of the proton leak mutants does not impair the activity of endogenous Ucp2. By contrast, Ucp2E170Q and Ucp2R185Q have a comparable affinity for endogenous Ucp2 and thus disrupt the activity of endogenous Ucp2 by dimerizing with it. It will be interesting to determine whether the Ucp2 mutations cause each subunit to have different binding affinities for each other upon dimerization.
Ucp2 transports anions, dissipates the mitochondrial membrane potential, and binds purine nucleotides. Unexpectedly, the anion transport and nucleotide- binding activities of Ucp2 appear to be indispensable for Ucp2-mediated engulfment of apoptotic cells. However, these functions of Ucp2 do not have any effect on the engulfment of carboxylate-modified beads. These observations strongly hint that Ucp2 can directly sense metabolic materials such as fatty acids and nucleotides derived from apoptotic cells and that these molecules influence how many apoptotic cells can be successively engulfed by phagocytes.
In conclusion, although significant progress has been made to understand the mechanisms by which phagocytes recognize and ingest apoptotic cells, knowledge of what contributes to the phagocytic capacity is lacking. The data presented here shed light on this conundrum. In addition, defects in the clearance of apoptotic cells and Ucp2 are linked to many autoimmune and metabolic diseases. Thus, insights from this study could be used to help develop therapeutic strategies to combat these diseases.
Acknowledgments
This work was supported by the National Research Foundation of Korea grant funded by the Korea government (MSIP) (2012R1A5A1048236 and 2013R1A1A1004518), by the Bio Imaging Research Center at GIST and by a grant from the National R&D Program for Cancer Control and the Korean Heath Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (1420120 and HI12C08920000).
REFERENCES
- A-Gonzalez N, Hidalgo A. Nuclear receptors and clearance of apoptotic cells: stimulating the macrophage's appetite. Front. Immunol. 2014;5:211. doi: 10.3389/fimmu.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat. Cell Biol. 2000;2:899–905. doi: 10.1038/35046549. [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- Bouillaud F. UCP1, UCP2 and UCP3: are they true uncouplers of respiration? Int. J. Obes. Relat. Metab. Disord. 1999;23(Suppl 6):S19–23. doi: 10.1038/sj.ijo.0800938. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Winkler E, Bienengraeber M, Klingenberg M. Site-directed mutagenesis identifies residues in uncoupling protein (UCP1) involved in three different functions. Biochemistry. 2000;39:3311–3317. doi: 10.1021/bi992448m. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Bienengraeber M, Klingenberg M. Role of intrahelical arginine residues in functional properties of uncoupling protein (UCP1) Biochemistry. 2001;40:5243–5248. doi: 10.1021/bi002130q. [DOI] [PubMed] [Google Scholar]
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008;15:243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF, Surwit RS, Ricquier D, et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- Flier JS, Lowell BB. Obesity research springs a proton leak. Nat. Genet. 1997;15:223–224. doi: 10.1038/ng0397-223. [DOI] [PubMed] [Google Scholar]
- Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell. 2011;147:1442–1445. doi: 10.1016/j.cell.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27:244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 2013;5:a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M, Appel M. The uncoupling protein dimer can form a disulfide cross-link between the mobile C-terminal SH groups. Eur. J. Biochem. 1989;180:123–131. doi: 10.1111/j.1432-1033.1989.tb14622.x. [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 2005;6:248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mo. Cell. 2004;14:277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- Lee J, Park B, Kim G, Kim K, Pak J, Kim K, Ye MB, Park SG, Park D. Arhgef16, a novel Elmo1 binding partner, promotes clearance of apoptotic cells via RhoG-dependent Rac1 activation. Biochim. Biophys. Acta. 2014;1843:2438–2447. doi: 10.1016/j.bbamcr.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Lin CS, Hackenberg H, Klingenberg EM. The uncoupling protein from brown adipose tissue mitochondria is a dimer. A hydrodynamic study. FEBS Lett. 1980;113:304–306. doi: 10.1016/0014-5793(80)80614-4. [DOI] [PubMed] [Google Scholar]
- Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim. Biophys. Acta. 2003;1639:141–151. doi: 10.1016/j.bbadis.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Ricquier D, Kozak LP. Uncoupling proteins: current status and therapeutic prospects. EMBO Rep. 2005;6:917–921. doi: 10.1038/sj.embor.7400532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, Ravichandran KS. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477:220–224. doi: 10.1038/nature10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat. Rev. Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Palmiter RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature. 1997;387:94–97. doi: 10.1038/387094a0. [DOI] [PubMed] [Google Scholar]
- Urbankova E, Hanak P, Skobisova E, Ruzicka M, Jezek P. Substitutional mutations in the uncoupling protein-specific sequences of mitochondrial uncoupling protein UCP1 lead to the reduction of fatty acid-induced H+ uniport. Int. J. Biochem. Cell Biol. 2003;35:212–220. doi: 10.1016/s1357-2725(02)00131-0. [DOI] [PubMed] [Google Scholar]